國 立 交 通 大 學
材 料 科 學 與 工 程 系 所
碩士論文
施體受體型窄能隙高分子的製備暨太陽能
電池材料開發與熱電材料上之應用
The Preparation of Donor-Acceptor Type
Narrow Band Gap Polymers and Their
Applications on Thermoelectrics and
Photovoltaics
研 究 生:何明益
指導教授:林宏洲 教授
中文摘要
共軛導電型高分子經過數十餘年發展,已經在電晶體、有機電激發光顯 示(OLED)、太陽能電池(Solar cell)…等方面,受到廣泛的研究與應用。 作為有機熱電材料基本條件為1.具有導電性 2. 穩定在空氣環境中 3. 合理的使用時效。為了達到上述條件,我們採用了施體-受體共軛高分子的 策略來設計高分子結構。我們使用雙噻吩環戊烷和吡咯作為施體結構、吡嗪 做受體結構分別進行聚合。雙噻吩環戊烷結構和吡嗪有強烈的得失電子的作 用力外,又有併雜環的特性利於高分子的堆積,以期達到好的載子傳遞。而 吡咯與吡嗪間因為可以形成氫鍵,更有利於共平面化。我們也藉由有機合成 使吡嗪帶有不同的官能基,來探討其在物性上與化性上對於熱電效率整體的 影響。最後我們調控摻雜的時間和適當摻雜物濃度,藉由稱重計算出摻雜程 度來找出最佳的性能指標值(ZT)。其 P3 為本實驗中有最高的性能指標,約 8.11×10-5。 我們除了把材料應用在熱電部分,亦把實驗高分子進行太陽能電池的量 測。我們採用混摻異質接面型太陽能電池的製作方式來製成元件,而初步量 測可以將能量轉換效率可達到2.57×10-2%Abstract
Organic polymer may offer a route to thermoelectric function when characteristics such as light weight, flexibility, low thermoconductivity, and potentially low coast. Key organic polymer material requirements for thermoelectric application are: (1) in the level of conducting or semiconducting, (2)stable in the air condition. , and(3) a reasonable life time in the temperature range of hot and cold sides. We reported the preparation and characterization of several novel thiophene derivative polymers and identified their thermoelectric properties. The electron donor-acceptor conjugation concept was used to manipulate the energy band structure to get high electric conductivity and high Seebeck coefficient at the same time. We chose 4,4dialkylcyclopentadithiophene which is the Donor part and thieno[3,4-b]pyrazine which is the accptor part to polymerization. In order to enhance electrons mobility of organic semiconducting materials, we use supermolecular design to arrange the polymers
in order. So we change the Donor part to pyrrole which can produce the H bond to thieno[3,4-b]pyrazine.
In the experiment, P3 has the highest value of ZT. The value is about 8.11 x 10-5. We use the process of bulk heterojunction solar cells to prepare photovoltaic devices. Preliminary measurements have revealed a power conversion efficiency (PCE) up to2.57×10-2%.
目錄
中文摘要 ... I 英文摘要 ... II 目錄 ... III 表目錄 ...VI 圖目錄 ... VII 附圖目錄 ... X壹、緒論 ..
... 1 1-1、前言...
………..………. 1 1-2、共軛高分子簡介... 2 1-2-1、共軛高分子的半導體性質 ... 2 1-2-2、導電高分子之導電機制... ...5 1-2-3、 能帶理論、偏極子、雙偏極子和孤立子………... 6 1-2-4、導電高分子的摻雜分類: ……… ……….10 1-2-5、受體-給體系統(Donor-Acceptor Syste)………12 1-2-6 導電高分子當前發展與應用………...……….….14 1-3、熱電性質簡介………..……….14 1-3-1、熱電材料主要性質之一:Seebeck 效應...14 1-3-2、熱電材料主要性質之二:Peltier 效應...161-3-3、熱電材料主要性質之三:Thomson 效應...18 1-3-4、熱電材料性能指標(Figure of merit):ZT……….20 1-3-5、共軛高分子應用在熱電材料...23 1-4 、共軛高分子應用在太陽能電池...23 1-4-1、前言...23 1-4-2、有機太陽能材料的優勢...24 1-4-3、有機高分子發展史...25 1-5、研究目的...30
貳、共軛高分子的合成與量測...34
2-1、合成實驗...34 2-1-1、實驗藥品...34 2-1-2、實驗儀器...35 2-2、合成流程...36 2-2-1、施體單體(1)...36 2-2-2、前驅施體單體(2)...37 2-2-3、受體單體...37 2-2-4、高分子聚合流程圖...38 2-3、合成步驟...39 2-3-1、施體結構(1)...392-3-2、施體前驅物結構(2)...43 2-3-3、受體結構...45 2-3-4、高分子聚合...50 2-3-4-1、針對 P1、 P2、 P3 聚合步驟...51 2-3-4-2、針對 IP4、IP5、IP6 聚合步驟...51 2-4、有機熱電元件量測步驟...52 2-4-1、電阻量測...52 2-4-2、Seebeck 係數量測...54
參、結果與討論...57
3-1、高分子的基礎性質(GPC、TGA、DSC)...57 3-2、高分子吸收光譜及電化學性質(UV-vis-NIR、CV)...61 3-3、太陽能電池之性質...67 3-4、有機熱電性質...71 3-4-1、試片製作的方式...71 3-4-2、各高分子的熱電性質...75肆、結論...84
伍、未來展望...86
陸、參考文獻...87
柒、附錄...91
表目錄
表1-2-1-1 常見的有機共軛高分子結構………...…….……… ....4 表1-2-1-2 經典的導電高分子之結構和導電度(摻雜後)………..5 表1-3-4-1 不同溫度對應不同熱電材料……….. 22 表1-4-2-1 近期各式太陽能電池之效率...25 表1-4-3-1 各波長區段佔總光子通量的百分比...29 表2-1-1-1 實驗使用藥品...35 表2-1-1-2 實驗使用溶液...35 表3-1-1 高分子分子量、裂解溫度...57 表3-1-2 tert-butyl 碳鏈的酯基官占高分子重覆單元比例...60 表3-2-1 P1、P2、P3 高分子之 UV-vis-NIR 之吸收波長及光學能隙...61 表3-2-2 P4、P5、P6 高分子之 UV-vis-NIR 之吸收波長及光學能隙...63 表3-2-3 各個高分子的電化學數據...66表
3-3-1
高分子和PCBM 經過 1:1 混摻後所測得的太陽能電池數據...68 表 3-3-2 P2 高分子調整不同轉速下,元件效率變化...69 表 3-4-2-1 P1 高分子不同摻雜程度的熱電量測數據...75 表 3-4-2-2 P2 高分子不同摻雜程度的熱電量測數據...77 表 3-4-2-3 P3 高分子不同摻雜程度的熱電量測數據...79 表 3-4-2-4 P4 高分子不同摻雜程度的熱電量測數據...82圖目錄
圖1-2-1 聚乙炔的結構………...……. 2 圖1-2-2 1,2-丁二烯的立體鍵結示意圖……….……….. ...3 圖1-2-3-1 能帶與物質電性示意圖………..………. .7 圖1-2-3-2 聚乙炔隨鏈長增加能階分布圖………...…….. ...8 圖1-2-3-3 偏極子、雙偏極子和孤立子能階示意圖………... 9 圖1-2-3-4 反式聚乙炔孤立子與能階圖...10 圖1-2-5-1 π-共軛高子之能階示意圖……….12 圖1-2-5-2 施體(D)與受體(A)的分子軌域交互作用……….…………...14 圖1-3-1-1 Seebeck 效應之簡示圖………...…… 15 圖1-3-1-2 利用 Seebeck 效應將廢熱回收之應用簡示圖………...………...17 圖1-3-2-2 利用Peltier效應的熱電致冷之應用簡示圖…...………….… ....18 圖1-3-3-1 Thomson 效應早期實驗圖…...19 圖1-3-3-2 Thomoson 效應示意圖………...…...……… 19 圖1-3-4-1 p-type材料的ZT值隨溫度變化之示意圖…...………...….. 21 圖1-3-4-2 n-type 材料的 ZT 值隨溫度變化之示意圖……… .21 圖1-4-3-1 有機太陽能電池元件演進圖………..… ...27 圖1-4-3-2 混摻異質接面型太陽能電池之示意圖………. .28 圖1-4-3-3 Alan J. Heeger 所用的高分子之吸收光譜圖…….…...28圖1-4-3-4 在 AM 1.5 下之太陽光譜圖………..…...… 29 圖1-4-3-5 串疊型高分子元件結構示意圖……….. 30 圖1-5-1 單體結構作用力示意圖……….. 31 圖1-5-2 目標高分子 P1、P2、P3 結構………...………….. 31 圖 1-5-3 目標高分子 P4、P5、P6 結構………...32 圖 2-2-1-1 施體單體(1)之合成...…...36 圖 2-2-2-1 施體單體(2)之合成……...………..………… 37 圖 2-2-3-1 受體單體之合成………...…………. 37 圖 2-2-4-1 高分子聚合圖...………..……… 38 圖2-4-1-1 電阻儀器量測圖示一……….………. 53 圖2-4-1-2 電阻儀器量測圖示二……….………….. ...53 圖 2-4-2-1 Seebeck 係數量測儀器之圖示一...………...…….55 圖2-4-2-2 Seebeck 係數量測儀器之圖示二……….………….. ..55 圖2-4-2-3 Seebeck 係數量測示意圖………...……….. 56 圖3-1-1 高分子 P1、P2、P3 熱重分析圖…………...………. 58 圖3-1-2 文獻中相似結構的聚合步驟兩步………...……..… 59 圖3-1-3 IP4、IP5、IP6 熱重分析圖...… 59 圖 3-2-1 P1、P2、P3 的溶液狀態吸收光譜圖...…………...61 圖 3-2-2 P1、P2、P3 薄膜態吸收光譜圖...… 62
圖 3-2-3 IP4、IP5、IP6 薄膜態吸收光譜圖………...…...63 圖 3-2-4 P4、P5、P6 薄膜態吸收光譜圖...64 圖 3-2-5 tert-butyl 碳鏈的酯基官能基為拉電官能基...64 圖 3-2-6 tert-butyl 碳鏈的酯基官脫去...65 圖3-2-7 各個高分子的 HOMO 和 LUMO 之能階示意圖...67 圖3-3-1 P2 高分子的 I-V 曲線圖...68 圖 3-3-2 高分子 P2 暗電流 I-V 曲線圖...70 圖 3-4-1-1 早期的試片做法...71 圖3-4-1-2 試片製作步驟...73
圖3-4-2-1 Electrical conductivity, Seebeck coefficient, and power factor for FeCl3-doped P1 films as a function of the doping level...76
圖3-4-2-2 Electrical conductivity, Seebeck coefficient, and power factor for FeCl3-doped P2 films as a function of the doping level...78
圖3-4-2-3 Electrical conductivity, Seebeck coefficient, and power factor for FeCl3-doped P3 films as a function of the doping level...80
圖3-4-2-4 Electrical conductivity, Seebeck coefficient, and power factor for FeCl3-doped P4 films as a function of the doping level...82
附圖目錄
附圖1 化合物 6 之氫譜………..………. ..91 附圖2 化合物 6 之碳譜…....………. 92 附圖3 化合物 9 之氫譜………...93 附圖4 化合物 9 之碳譜……….………...94 附圖5 化合物 16 之氫譜………....95 附圖6 化合物 16 之 MS-EI 圖………...……….. 96 附圖7 化合物 17 之氫譜……….………..97 附圖8 化合物 17 之 MS-FAB 圖………...……….. ...98 附圖9 化合物 18 之氫譜………..……….………99 附圖10 化合物 18 之 MS-FAB 圖……….…………. 100壹、緒論
1-1、前言
呈現出物質基本特性的最小單位是分子,而當許多相同的分子進行鍵 結反應,則稱為聚合反應(polymerization),所得產物我們稱之為聚合物。 當聚合物分子量大小在數百到數千時,我們稱之寡聚物(oligomer)。而當聚 合物分子量已經達到數萬以上時,此時聚合物已經連接了千百個、數萬個 聚合單體,故稱之為高分子(polymer)。高分子又因為其組成元素不同可分 為無機、有機高分子,而高分子早期因為絕緣效果不錯,所以常被拿來當 作絕緣材料使用。當物質的分子量越大,其機械強度越大,也呈現高度可 塑性、熱穩定性及伸縮性。 早在西元 1812 年,化學家用酸水解木屑、樹皮、澱粉等植物的實驗中 得到了葡萄糖,證明澱粉、纖維素都由葡萄糖組成。1826 年,法拉第通過 元素分析發現橡膠的單體分子是 C5H8,後來人們測出 C5H8的結構是異戊 二烯。但是直到 1922 年才由史坦汀格首先提出高分子是由小原子團或小分 子片段藉由共價鍵結合而成的高分子量化合物的概念。經過數年學術上的 辯論,在 1926 年瑞士科學家施維德貝格在實驗上証實高分子的分子量確實 是數萬到數百萬間,成為高分子理論成立的重要證據。1932 年,史坦汀格 總結了自己的高分子理論,出版了劃時代的巨著《高分子有機化合物》成 為高分子科學誕生的標誌。到了 20 世紀初期人工合成塑膠、橡膠、化學纖維三大合成工業基礎已 經奠定。加上第二次世界大戰後,由於廣泛的發展纖維、彈性物、膠合劑 及人工樹脂,使得製造產品廣泛增加。因為這些聚合物大多是以含有碳原 子的有機物,故一般所指的高分子通常為有機高分子。
1-2、共軛高分子簡介
1-2-1、共軛高分子的半導體性質 常見的有機高分子例如塑膠、橡膠均為絕緣體,其原因在於碳氫化合 物所組成的共價單鍵長鏈分子並沒有可自由移動的電荷。而所有可導電的 高分子皆是共軛高分子,這種高分子具有本質導電性(intrinsic conductivity), 有別於一般摻入金屬粉或導電級碳黑的高分子複合體。導電高分子最早在 1958 年,Natta 等人使用催化劑將乙炔(Acetylene)製成聚乙炔(Polyacetylene), 雖然具有高度結晶體性,但是形成的黑色粉末具有空氣不穩性且溶解度很 低性質,因此難以做為成熟的材料 1。 圖 1-2-1 聚乙炔的結構 聚乙炔是由長鏈的碳分子以 sp2鍵鏈結而成,如圖 1-2-1 所示。由於 sp2 鍵結使得每一個碳原子有一個價電子未配對且在垂直於 sp2面上形成未配對鍵,可以猜想出相鄰原子之未配對鍵的電子雲互相接觸,導致未配對電 子沿著長鏈移動,如圖 1-2-2 所 示。 圖 1-2-2 1,2-丁二烯的立體鍵結示意圖 但實際可能未必如此,因未配對電子很容易和隔壁配對而成單鍵-雙鍵 交替出現的結構,這種轉變稱為配對化(Dimerization),在物理上稱為 Peirels 不穩定性 2。為了使共軛高分子達到好的導電效果,就必頇進行摻雜,這 和半導體經過摻雜後可以經由荷電載子提高導電度類似。於是在 1974 年, Shirakawa 等合成出全順式聚乙炔(All-cis-polyacetylene),進一步加工製成 銀色的全反式聚乙炔(All-trans-polyacetylene),這時高分子導電性已提高至 全順式聚乙炔的 10-8 ~10-7(S·m-1)及全反式聚乙炔的 10-3~10-2(S·m-1)1但是並 沒有真正成為導體。到了 1975 年,當正在研究共價無機高分子金屬性的
Heeger 及 MacDiarmid 教授遇到 Shirakawa 後3,便在聚乙炔中添加鹵元素 碘,將導電係數提高到 3000(S·m-1 )。接下去的科學家以類似的技術導入, 其中德國 BASF 科學家宣稱可達 107 (S·m-1),相較於銅金屬的 108(S·m-1), 在聚乙炔中添加鹵素可達到金屬特性的高分子導體,即對共軛高分子領域 展開了另一條新的研究道路。這三位教授又因導電塑膠的傑出成就而獲得
2000 年的諾貝爾化學獎。 名稱 高分子結構 Polyacetylene(PA) Polypyrrole(PPY) Polyfuran(PF) Polythiophene(PT) Poly-p-phenylene(PPP) Poly-p-phenyl vinylene(PPV) Polyaniline(PAN) 表 1-2-1-1 常見的有機共軛高分子結構 摻雜後的聚乙炔在空氣中或濕氣的環境之下十分不穩定,沒有實際應 用價值4,但由於導電高分子在成膜性上有突出的表現、易於加工,因而 引起許多學者們的興趣,紛紛投注心力研究,使得新結構之導電高分子陸 續的被發現或重新提出來研究,如聚吡咯(Polypyrrole,PPy)、聚苯胺 (Polyaniline,PAni)、聚噻吩(Polythiophene,PT)、聚對位苯(Polyparaphenylene,
PPP)、聚苯基乙烯(Polyphenylenevinylene,PPV) 和聚噻吩乙烯 (Polythienylenevinylene)等,經典高分子結構及摻雜後的導電度如表 1-2-1-25所示。 表 1-2-1-2 經典的導電高分子之結構和導電度(摻雜後) 1-2-2、導電高分子之導電機制 導電高分子的主鏈是由碳─碳單、雙鍵交替組成的共軛鍵結,主鏈有π
電子可以移動,使之具有導電特性。並且可以藉由化學方法或電化學方摻 雜(doping)後產生電荷的載體,使其導電度可以大幅度提升並其導電度可以 介於半導體和導體之間。有機導電高分子的導電機制到目前為止尚未完全 了解,但可知在中性態(未參雜前),大多是絕緣體或低導電性的半導體。 而其原因可以利用古典半導體的能帶(band structure)理論和物理學的偏極 子(polaron)、雙偏極子(bipolaron)和孤立子(soliton)來概略說明之。 1-2-3、能帶理論、偏極子、雙偏極子和孤立子 我們常用能帶理論來解釋材料的導電性。其中能帶是指分子中由原子 軌域重疊組成能量密集分布幾乎連續之分子軌域的能量區域,最外層的價 電子所佔有的能帶成為共價帶(valance band, VB),在基態時沒有電子占有 之能帶叫做傳導帶(conduction band, CB),而傳導帶的最低能位與共價帶最
高高能位之間隔,我們稱作能帶間隙或是能隙(band gap,Bg;energy gap,Eg)。
絕緣體的能隙很大,在室溫的熱能無法將電子從共價帶激發到傳導帶,故 導電度十分低。對於金屬而言因共價帶與傳導帶重疊,故能隙為零,電子 可以自由傳遞而具有高的導電性。對於半導體則因能隙不如絕緣體大,故 在室溫下能有電子可以躍遷至傳導帶形成些微的導電,其導電度介於絕緣 體和金屬之間。一般來說,當能隙為零是導體,大於 3.0eV 時為絕緣體, 最後能隙介在 0 ─ 3eV 便叫做半導體,如圖 1-2-3-1;而導電高分子的能隙 多落在1.0 ─ 3.5 eV,所以有時又被稱為有機導體。
圖 1-2-3-1 能帶與物質電性示意圖 導電高分子之電子結構中,分子軌域之 π-π*能隙隨著分子共軛長度的 增加而降低6,故對聚乙炔(PA)而言,若結構上沒有缺陷之無限長共軛系統, 其電子結構應與石墨相似,導電帶與共價帶重疊而具高導電度。實際上 PA 會因主鏈上兩相鄰重複單位之二極化(dimerization)出現結構上的缺陷而造 成有限的共軛長度,導致能隙最低只達到 1.4 eV,如圖 1-2-3-2 所示。此種 電子結構類似前述之半導體,因而從 1980 年代早期就借用物理學的偏極子 (polaron)、雙偏極子(bipolaron)和孤立子(soliton)之觀念,來解釋摻雜態的 導電高分子之電子結構。 價帶 ﹞0-3eV >3eV (a)導體 (b)半導體 (C)絕緣體 傳導帶
圖 1-2-3-2 聚乙炔隨鏈長增加能階分布圖 7 當一個電子從共軛高分子的價帶上方被移走時,則產生一個空孔 (Vacancy),稱為電洞(Hole)或自由基陽離子, 此空孔是部分不定域化 (Delocalized),可共振擴展至幾個單體單位,並造成高分子結構改變。由於 自由基陽離子為不安定性鍵結軌域,其能階比價帶的能階高,在固態物理 學上稱此自由基陽離子或電子− 電洞對(Electron-hole Pair)為偏極子 (Polaron)。如果另一個電子從已含偏極子的高分子鏈被移去時,將有兩種 情形發生:1.從高分子鏈上的不同鍵結移去電子,產生另 一偏極子;2.從 先前形成的偏極子能階移去電子,產生雙陽離子(Bication),在固態物理學
上稱此雙偏極子(Bipolaron),此雙偏極子同樣 會造成高分子結構變形。雙 偏極子的兩個正電荷並非各自獨立,而是形成一對。偏極子與雙偏極子皆 為可移動的載子,而且可藉著共軛系統在電場下的單雙鍵重排而延著高分 子鏈移動。如果較多數的雙偏極子形成(在高摻雜度時),則它們的能階可 以重疊,而在能帶間隙中產生較窄的雙偏極子能帶8、9。如圖 1-2-3-3 圖 1-2-3-3 偏極子、雙偏極子和孤立子能階示意圖 孤立子及其能帶圖示於圖 1-2-3-4,圖中上方是反式聚乙炔高分子鏈上 的正中央有缺陷,此缺陷右側與左側的結構一樣且分子的鍵結組態的總能 量和原本無缺陷反式聚乙炔相比並未增加;圖中央表中性態的孤立子,其 能階位於能隙的正中央,故可以產生新的電子轉移而有新的吸收譜帶;而 經過 n-或 p-態摻雜後,孤立子可帶電荷,圖下方表帶正電荷的孤立子。孤 CB VB Eg LUMO HOMO Undoped ploymer Natural solition solitem Slightly Doped polymer polaron bipolaron Heavy Doped polymer Bipolaron Bands Bipolaron energy Levels
立子並不會發生在高分子本身是 non-degenerate ground state 者,諸如聚吡
咯、聚苯胺、聚噻吩,這是因為此類高分子的芳香型(aromatic form)及醌型
(quinoid form)共振形式的能量並不相同所致,所以孤立子的傳導現象為具
degenerate ground state 高分子的電荷傳遞之主要機構;而偏極子與雙偏極
子則為具 non-degenerate ground state 高分子的電荷傳遞之主要機構,此亦
可解釋共軛高分子被摻雜後導電度增加與光學吸收的變化。
圖 1-2-3-4 反式聚乙炔孤立子與能階圖
1-2-4、導電高分子的摻雜分類:
鹵素可提高導電性,亦可選擇添加鹼金屬,前者稱為 p-doping,使聚乙炔 碳鏈中的碳原子失去電子,故屬於氧化摻混(Oxidative doping);後者則相 反稱為 n-doping,還原摻混(Reductive doping),其化學反應式(以聚乙炔為 例)如下: [CH]n + 3x/2 I2 [CH]nx+ +xI3-(氧化摻混) [CH]n + xNa [CH]nx- + xNa+ (還原摻混) 被摻混的高分子形成鹽類,在高分子鏈上移動的帶電載子並不是帶電 的鹵素離子或鹼金屬離子,而是產生於共軛高分子之碳鏈上被激化的載子 因外加電場傳輸產生電流導通的現象。一般來說,帶電載子的運動是有方 向性的,因此朝電場方向的導電性稱為異向性(Anisotropic)。 依照不同的摻雜方式可分為液相摻雜、氣相摻雜、電化學摻雜10、自 身摻雜(self-doping)11和二次摻雜 12等五種。液相摻雜是將高分子放在含有 摻雜物的溶液當中,與高分子材料進行直接接觸式的摻雜。氣相摻雜則是 先把摻雜物氣化,再把高分子材料放置在有摻雜物的氣氛下進行。電化學 摻雜主要特色為把高分子材料當作電極至於含有摻雜物的電解液當中,在 施加特定的電壓後進行摻雜。自身摻雜則是可摻雜高分子的摻雜物已經由 化學鍵結方式鍵結在高分子上形成特定的官能基團,進行摻雜。最後一種 二次摻雜則是將另一種溶劑加入已經過摻雜的高分子,使得高分子鏈與溶 劑、共溶劑或對離子基間的作用力不同,改變高分子鏈的結構使導電度上
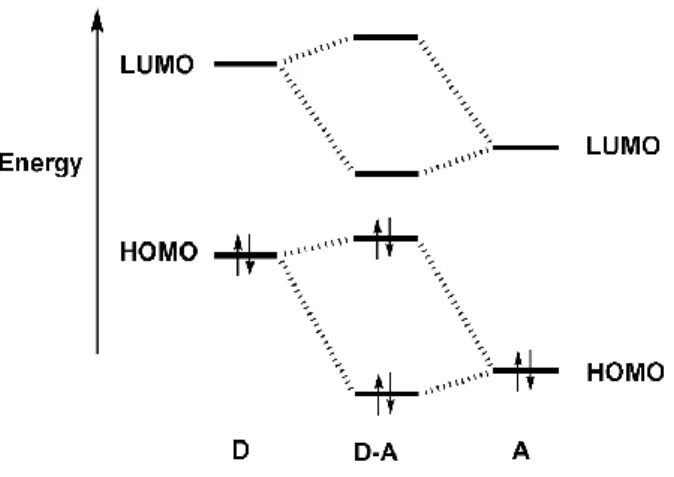
升。 1-2-5、受體-給體系統(Donor-Acceptor System) 若降低高分子的能隙將會增進傳導帶的熱量總數(Thermal population) 並且增加本質的載子數目,形成真正的"有機型金屬"(Orangic metals)。另 一方面,窄能隙有較低的氧化電位時,將會導致摻雜狀態穩定,而且對於 以共軛高分子為基礎的超高電容器,其安定的還原摻雜在適度的電位是具 有關鍵的重要性。因此藉由化學合成技巧修飾高分子來控制 HOMO-LUMO 之間的間隙,對於窄能隙共軛高分子來說是極其重要的。 共軛高分子的電性和光學性質是來自位於 HOMO 和 LUMO 周圍的能 階所影響的。根據能帶理論來說,最高填滿帶來自於每個單體的 HOMO, 也就是所謂的價帶;而最低未填滿帶則是來自於每個單體的的 LUMO,可 當成傳導帶。在共軛高分子中,能帶的形成是來自於聚合時每個單體的參 與,如圖 1-2-5-113。 圖 1-2-5-1 π-共軛高子之能階示意圖
在吸收光譜裡若要得到共軛高分子的 Eg,則是將吸收峰進行外插法而
得,因此窄能隙的共軛高分子其吸收應在近紅外光區(Near infrared region),
等同於吸收長波長。 許多分子工程策略用來設計窄能隙的共軛高分子已發展多年,本論文 中的所合成之共軛高分子是選用施體-受體系統(Donor-Acceptor)的概念來 降低能隙。而此系統中合成策略是利用強推電子部分和強拉電子部分進行 結合並以一種連續的單元形成共軛高分子,使得單元和單元之間的鍵結扭 轉程度降到最低。由於受體和施體之間的作用力,使得單元之間的鍵結更 具有雙鍵的特性,讓窄能隙共軛高分子的主鏈上形成類似醌環的穩定結 構。 由於近來電腦的處理速度倍增,使得模擬運算快速發展演進。現今我 們已可用分子軌域計算來呈現施體和受體能階經過混成的分子軌域分布情
形。D-A 系統大致上具有較低的 HOMO 和 LUMO 間隔,如果再加上施體
的 HOMO 和受體的 LUMO 能階彼此很接近,就會形成窄能隙,如圖
圖 1-2-5-2 施體(D)與受體(A)的分子軌域交互作用
1-2-6、導電高分子當前發展與應用
在過去 30 多年來,在半導體的研究與應用領域中,以研究單晶矽
(Monocrystal silicon)與非晶矽(Amorphous silicon)化合物為主要的課題,但
目前的導電高分子也具有半導體之性質,因此利用各種不同 π-共軛的芳香 族之分子結構的有機半導體材料來取代目前的矽材料以及有半導體性質的 金屬材料,並製造低成本又較簡易製備的有機積體電路之物理元件以及各 種應用的元件,如有機發光二極體(OLED)、有機薄膜電晶體(OTFT)、光伏 打電池(Photovoltaic cell)、生物感應器(Biosensor)以及有機熱電材料(OTE), 目前已經有許多研究單位正積極的研發。 1-3 、熱電性質簡介 1-3-1、熱電材料主要性質之一:Seebeck 效應 德國物理學家 T. J. Seebeck 於西元 1821 年在實驗中發現,若將一指南
針放置在一個由兩個不同導體所組成的封閉迴路旁時,同時在其中的一端 接點處加熱,會看到指針出現偏轉的現象,而其偏轉是由於電流生磁所致。 圖 1-3-1-1 Seebeck 效應之簡示圖 如圖 1-3-1-1 所示,當一個導體或半導體的兩端有溫度差異時,會產生 電位差異進而有電流的流通之現象就稱為 Seebeck 效應,此現象可以定義 出 Seebeck 係數14: dT dV S 上式中,dV 為電位差,dT 為溫度差,若 S 大於零則為 p-type 材料,S 小 於零則為 n-type 材料。一開始 Seebeck 是利用兩種不同的導體,但雙金屬 材料由於其效應微弱,只能被利用做溫度、輻射能量測量用的雙金屬電偶 等開路電壓量測,如在工廠和實驗室中普遍使用的熱電偶( thermo couples ) 溫度計來量測溫度梯度以及熱電產生器( thermoelectric generator ),都是熱 電原理應用的例子。到了 1950 年代末,某些半導體材料的高熱電效應被發 現後,其實用價值才獲得重視,主要是利用正( P )型半導體與負( N )型半 導體串聯組成的熱電發電元件,經由 Seebeck 效應並利用所供應之熱源造 成溫度差產生電流,如圖 1-3-1-2 所示。1977 年美國旅行家無人太空船升
空,其中部分電能便是利用放射性熱電產生器( radioisotope thermoelectric generators )產生,這為 Seebeck 效應之應用寫下光明的前景。 圖 1-3-1-2 利用 Seebeck 效應將廢熱回收之應用簡示圖 利用熱電材料發電具有設備簡單、無傳動部件、低噪音、無排放污染、 取用方便、安全可靠、壽命長、不需維修等優點,可置於室內並適合個人 或家庭發電、工廠或發電廠排放低階熱能發電使用。起初是以一些偏遠地 區發電、戰場上之緊急電源、無人看管裝置發電、以天然氣或丙烷為燃料 燃燒產生熱源等小型發電產品為主。 近年來由於在技術上熱電材料性能的不斷提升,及環保議題上溫室效 應的二氧化碳減量等因素,因此利用熱電轉換技術,進一步將大量廢熱回 收轉為電能的方式,普遍得到日、美、歐等先進國家的重視。低溫餘熱、 特別是 140℃以下的廢熱再利用,增加了熱電發電的競爭力,一些新興應 用研究諸如垃圾焚燒餘熱、煉鋼廠的餘熱、利用汽車以及發動機尾氣的餘 熱進行熱電發電,為汽車提供輔助電源的研究也正在進行,並且有部份成
果已實際應用,相信在不久的將來會廣泛使用。
1-3-2、熱電材料主要性質之二:Peltier 效應
西元 1834 年法國物理學家 Jean-Charles-Athanase Peltier 觀察到如果電
流通過兩種不同導電材料所構成的迴路時,兩端接點上,其中一端會吸熱 而另一端則會有放熱現象產生,如圖 1-3-2-1 所示。這種因電流而有吸熱放 熱的現象,即是 Peltier 效應。 圖 1-3-2-1 Peltier 效應之簡示圖 這種 Peltier 效應可以定義出 Peltier 係數,Πab14: I Qab 而 I 為電流,Q 為吸收或釋放的熱量。下標 ab 表示兩種材料的相對 Peltier 係數。 Goldsmid 和 Douglas 於 1954 年將具有較高熱電效應的半導體材料取代 先前所應用的導電材料( 如圖 1-3-2-2 ),使得所研發的熱電致冷器成功地 冷卻至 0O C,因此引起全球性的研發熱潮。
圖 1-3-2-2 利用 Peltier 效應的熱電致冷之應用簡示圖 若使用這種熱電致冷器,則可以不必使用氟氯碳化物(CFC’s)當作冷煤,有 助於降低破壞臭氧層的機會,但目前利用熱電材料製成的裝置其效率仍比 傳統冰箱或發電機小。所以若能大幅提升這些熱電材料的效率,將對廣泛 用於露營的手提式致冷器、太空應用和半導體晶片冷卻等產生相當重要的 影響。 1-3-3、熱電材料主要性質之三:Thomson 效應
英國物理學家William Thomson( Lord Kelvin )於 1851 年做了一個有趣
的實驗,他在一個馬蹄鐵形的銅金屬導通電流,並在銅棒的兩邊架設兩個
平衡的惠思同電橋後,在銅棒的下方加熱(如圖 1-3-3-1),此時兩個惠思同
電橋不再平衡而有電流導通,證明了在銅棒兩端出現了吸放熱的情況,此
稱作 Thomson 效應。其效應在描述帶電導體因溫度梯度的關係,而有加熱
圖 1-3-3-1 Thomson 效應早期實驗圖 15 任何帶電導體(除了超導體)當兩端點有溫度差時,將會因材料的不同而 有吸放熱的現象。如果一個電流密度,J,經過一個勻相的導體,則每單位 體積會產生的熱(q)為: dx dT J J q 2 其中 ρ 是材料的電阻率,dT/dx 是金屬線的溫度梯度,μ 是 Thomson 係數。 第一項ρJ2只是焦耳熱( Joule heating ),其不可逆;第二項是湯木生熱 (Thomson heating),當 J 改變方向時,符號需要改變。如圖 1-3-3-2 圖 1-3-3-2 Thomoson 效應示意圖 在金屬當中,像鋅與銅,其較熱端是位於高電位處,較冷端是位於低
電位處,當電流從較熱端流經較冷端時,就是從高電位移動到低電位,所
以是放熱,這就叫做正 Thomson 效應(Positive Thomson effect);而像鈷,鎳,
鐵,其較冷端在較高電位,較熱端在較低電位,當電流從較熱端流到較冷
端時,就是從低電位移動到高電位,所以是吸熱,這就叫做負 Thomson 效
應(Negative Thomson effect)。
在 1954 年時,他推導出 Seebeck 係數( Sab )以及 Peltier 係數( Πab )之間 的關係,稱為 Thomson 或者 Kelvin 關係:
ab ab S
其中 T 為決對溫度,Πab是 Peltier 係數,Sab是 Seebeck 係數。而和 Thomson 係數有關的公式即是第二 Thomson 關係: dT TdS 1-3-4、熱電材料性能指標(Figure of merit):ZT 材料的熱電效率可以從定義一個值來評估,ZT: T S ZT 2
其中 S 為 Seebeck 係數,T 為決對溫度,σ 為電導率(electrical conductivity),
κ 為熱傳導係數(thermal conductivity)。為了得到較高的 ZT 值,其材料必頇
具備較高的 Seebeck 係數,較高的電導率,以及較低的熱導率,但要增加
普通材料的 ZT 值相當困難,因為當電導性增加時,熱導性也會跟著增加,
而 Seebeck 係數會降低,使得 ZT 值沒辦法有效的提高。目前電熱材料的
見圖 1-3-4-1、圖 1-3-4-2: 圖 1-3-4-1 p-type 材料的 ZT 值隨溫度變化之示意圖 圖 1-3-4-2 n-type 材料的 ZT 值隨溫度變化之示意圖 1、碲化鉍(Bismuth telluride)及其合金 16:這是時下被廣為使用於熱電 致冷器的材料,低溫其最佳運作溫度(< 4500 C)。 2、碲化鉛(Lead telluride)及其合金17:這是時下被廣為使用於熱電產生
器的材料,其最佳運作溫度大約為 10000 C。
3、矽鍺合金(Silicon germanium):此材料亦常應用於熱電產生器,其最
佳運作溫度大約為 13000
C。整理如下表 1-3-4-1:
Class of materials Temperature
range materials Low-temperature Tmax < 600K Bismuth telluride Middle-temperature Tmax < 1000K Lead telluride Hight-temperature Tmax > 1000K Silicon germanium 表 1-3-4-1 不同溫度對應不同熱電材料 事實上,碲化鉍(Bi2Te3)一直是具有最高之 ZT 值,如 Bi2Te3在室溫下 之 ZT~0.52,而銻 doped 之 Bi2Te3-Bi0.5Sb1.5Te3的 ZT 值則為 1.0。碲化鉍固 溶液如同碲化鉍和硒化鉍都是層狀化合物,他們皆含由共價鍵結的五原子 網狀結構(Te-Bi-Te-Bi-Te 或 Se-Bi-Se-Bi-Se)層,而層與層之間的鍵結僅是
微弱的凡得瓦耳力(Van der Waals)。碲化鉍和硒化鉍乃固態熱電致冷器所
用之主要物質,若能其 ZT 值提高數倍以上,則固態熱電致冷器就可以與
1-3-5、共軛高分子應用在熱電材料 有效率的熱電材料需要高的性能指標(Figure of merit),ZT 值: T S ZT 2 其中 S2σ 又可稱為 Power factor。而高分子本身就具有低熱傳導性,對於傳 統 的 熱 電 材 料 而 言 是 一 個 重 要 的 物 質 特 性 。 在 文 獻 中 指 出 , 聚 乙 炔 (Polyacetylenes)、聚苯胺(Polyanilines)、聚吡咯(Polypyrroles)以及聚噻吩 (Polythiophenes)等導電高分子的熱電性質之數據已被發表過 18~33,到目前 為止,熱電性質最好的共軛高分子仍然是聚乙炔,其 ZT 值最高可到 6,主 要是因為有很好的導電率和很高的 Seebeck 係數。但即使在惰性環境中進 行摻雜時不穩定,然而其他高分子雖然在參雜狀態時很穩定,但 Seebeck 卻很低,限制了性能指標的數值,甚至更少於 10-2。因此找到一個在摻雜 狀態下能穩定存在並具有高 ZT 值的高分子是目前最重要的方向。
Xing Gao 等人在 2005 年 Computational Materials Science 有提到利用窄
能隙的共軛高分子當作半導體材料 34,35經過摻雜來找到最佳化的載子濃度
(Carrier concentration)以及移動能力(Mobility),並且利用對高分子的修飾來
得 到 最 好 的 有 效 質 量 (Effective mass) 以 及 電 子 能 階 密 度 (Density of
electronic states),將有助於得到較佳的 ZT 值。
1-4 、共軛高分子應用在太陽能電池
1-4-1、前言
人類過渡開發,石油過量使用,導致了 1973 年發生了石油危機,讓世 界各國察覺到能源開發的重要性。由於太陽光是取之不盡,用之不竭的天 然資源,除了沒有能源耗盡的疑慮之外,也可以避免能源被壟斷的問題, 因此各國也積極地發展太陽能的應用科技,期望由增加太陽能源的利用來 減低對化石能源的依賴性,使得太陽能成為再生能源的最佳選擇。照射到 地球表面的太陽能量(輸出功率為 2.86 X 1023千瓦)約為地球所需能源的一 萬倍以上,如此龐大的能量是來自於核心的核融合反應。太陽能電池同時 具備可隨身攜帶、太陽光直接照射由光能轉換成電能、不需添加任何補充 物、不受地理限制,及產生能源過程中不會產生任何副產物等優點而受到 高度重視。拜今日技術能力的提升、新型材料的研發以及半導體產業的進 步,已有效提升太陽能電池之功率轉換效率,故有效利用太陽能電池發電 不再只是空談。 1-4-2、有機太陽能材料的優勢 有機太陽能電池利用現有發展完備之旋轉塗佈、噴墨列印、滾筒壓印 或網版印刷技術製作於塑膠基版達成可繞曲、輕薄、快速製作、大面積製 作及常溫製程等優點,相對於無機太陽能電池製作成本大幅降低且生產迅 速,增加了有機太陽能電池成為未來替代性能源的潛力。 國外許多產業界知名公司如:Kodak、Sharp、Sony、Siemens、CDT 及 Toshiba 也開始對有機太陽能電池進行研究,其中 Siemens 已於 2005 年
以印刷方式製作出效率高達 5% 的可繞曲式有機太陽能電池原型。當前 種不通材料的光電轉換效率如表1-4-2-1 太陽能電池總類 半導體材料 模組轉換效率 矽 結晶矽 單晶矽 (晶圓型) 13~20% 多晶矽 (晶圓型、薄膜型) 10~15% 非晶矽 α-Si、α-SiO、α-SiGe 5~10% 化合物半導體 二元素 GaAs(晶圓型) GaAs18~30% CdS、CdTe(薄膜型) 7~10% 三元素 CuInSe2(薄膜型) 8~10% 染料型電池 TiO2 (Dye Senzitized Solar Cell)
10% 有機半導體 (有機薄膜太陽能電池) ~6.1% Single Cell 6.5% Tandem Cell 表 1-4-2-1 近期各式太陽能電池之效率 1-4-3、有機高分子發展史
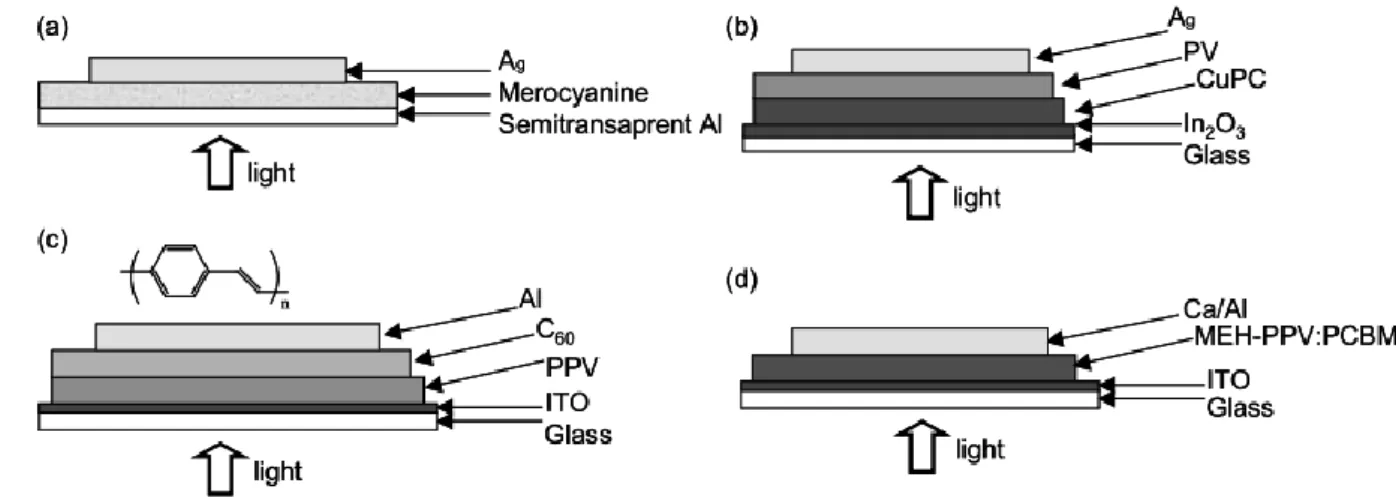
1959 年 H. Kallmann 和 M. Pope 發現單晶 anthrathcene 具有照光
特性的研究,早期關於有機太陽能電池之研究,多以小分子為基材。 1978 年 T. Feng 等人提出以光敏性染料-merocyanine 為材料,製作出一單 層結構之小分子有機太陽能電池,元件結構如圖 1-4-3-1 所示,由於其單層 結構,且陰、陽兩極皆使用金屬材料,因單一種類之小分子所能涵蓋之吸 收光波範圍長有限,及陰陽極金屬之穿透度不佳,使其完成之元件光電轉 換效率只達到 0.62%。接著 1986 年,C. W. Tang 首次提出以 CuPC 為電子
予體(donor, D)及 perylene 衍生物為電子受體(acceptor, A),組成電子予體/
受體(D/A)異質接面的元件結構 37,如圖 1-4-3-1 (b)所示。又經過了十年 R.H. Friend 團隊才提出使用 PPV 為電子予體材料,Fullerene (C60)為電子受體 材料,形成 D/A 結構之高分子有機太陽能電池38,光電轉換效率提升為 0.5%, 如圖 1-4-3-1 (c)所示。在同期間 Heeger 團隊提出以 MEH-PPV 予體及 PCBM 受體相互摻混,形成單層異質接面結構39,如圖 1-4-3-1 (d)所示,電子予 體-受體間之接觸面積較大,使激發態分子形成後能有效被分離,防止電 子、電洞再結合,其光電轉換效率達到 1.5%。到這裡奠定了以 PCBM 混 摻高分子的有機太陽能電池元件的設計。
圖 1-4-3-1 36 (a)單層結構 (b)有機予體/受體之雙層異質接面結構 (c)有機高分子予體/受體太陽能電池 (d)電子予體摻混受體之單層異質接面結構 由於高分子太陽能電池具有易加工、質量小、成本低等優點,使得各 國越來越重視高分子太陽能電池的研究。但可由表 1-4-2-1 得知高分子太陽 能電池之所以一直沒有大規模的實際運用,是因為光電轉換效率較低,至 今效率最高約 6.5%,但其為串疊型的電池,而非串疊型電池最高效率接近 6.1%40。 就非串疊型太陽能電池的次高效率所使用之材料來看41,42,43,目前最常 用的是Poly(3-hexylthiophene) (P3HT)當做施體,與[6,6]-phenyl-C61 butyric acid methyl ester (PCBM)當做受體混合成電池中的主動層(Active layer),再
把混合物經過旋轉塗佈在基板上,就會成為最常見到的混摻異質接面型太
圖1-4-3-2 混摻異質接面型太陽能電池之示意圖 雖然影響太陽能電池效率的因素很多,但主要原因是大多數活性材料 如上述的 P3HT 之薄膜為例,最大吸收波段界於 350~650nm 之間(如圖 1-4-3-3 所示)。這個區間我們可以由表 1-4-3-1 和圖 1-4-3-4 得知其佔不到 太陽光總光子通量的一半,因此近年來開始有人試著用兩種不同波段的高 分子串疊在同一個電池裡,讓這兩個高分子各盡其責,吸收不同波段的太 陽光能量,而形成串疊型太陽能電池。
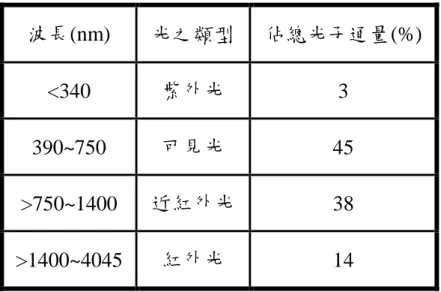
表 1-4-3-1 各波長區段佔總光子通量的百分比
圖 1-4-3-4 在 AM 1.5 下之太陽光譜圖
Alan J. Heeger 等人發表在 2007 年 Science44的串疊型高分子太陽能電 池是最具代表性的研究,其元件結構如圖 1-4-3-5 所示,所用的高分子材料 有兩種,一種是 P3HT;而另一種是 Poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b'] 波長(nm) 光之類型 佔總光子通量(%) <340 紫外光 3 390~750 可見光 45 >750~1400 近紅外光 38 >1400~4045 紅外光 14
dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)],簡稱 PCPDTBT,是比 P3HT 的能隙還要窄,吸收波長越紅位移的高分子,可以由圖 1-4-3-3 知道其薄膜 最大吸收峰比 P3HT 之薄膜最大吸收峰還要紅位移,負責吸收局部近紅外 光區的能量。此串疊型電池的效率突破以往高分子太陽能電池的效率,高 達了 6.5%的轉換效率,因此激勵了各國的研究人員相繼合成出新穎的窄能 隙高分子,為的是要讓此種太陽能電池達到最好的效率。 圖1-4-3-5 串疊型高分子元件結構示意圖 1-5、研究目的 使用施體和受體結構的混成軌域形成低能隙的共軛高分子,又因為施體 結構給電子能力強與受體結構的搶電子強的作用結果,導致了單體結構間 的鍵結更具有雙鍵的特性(如圖 1-5-1)45故不易扭轉而利於載體傳導。
圖 1-5-1 單體結構作用力示意圖 除了施體受體結構在得電子能力強弱的考量之外,我們也導入了併雜環 (Fused ring)的構想46,其效益在於若有堅硬的結構結構主幹,會更有益於 高分子堆疊、提高載子的移動率和熱穩定性。 實 驗 第 一 部 分 所 中 採 用 的 受 體 單 體 是 2,3-Substituted-thieno[3,4-b]pyrazines ; 施 體 單 體 是 4,4-Dioctyl-4H-cyclopenta[2,1-b;3,4-b']dithiophene。高分子結構下圖 1-5-2 所示: S S R R R=C8H17 S N N * * n S S R R R=C8H17 S N N * * n S S R R R=C8H17 S N N * * n S S P1 P2 P3 圖 1-5-2 目標高分子 P1、P2、P3 結構 實驗中利用有機合成的技巧將受體的側鏈接上不同官能基團,進而找 出對於導電性以及溶解性都有幫助之最佳化有機材料。
由於上述之施體與受體結構之間,除了推拉電子的作用力之外,並沒 有其他更大的作用力,來維持分子共平面性,故在實驗上更增加了超分子 的設計概念。其方法為更換施體結構為 pyrrole 結構。在文獻的記載中47,48, pyrrole 在氮原子上的氫原子可以和 thieno[3,4-b]pyrazine 上的氮原子形成氫 鍵,使整個高分子骨架更趨於平面化,利於載子的傳輸。文獻49也指出在 類似的結構中,當聚合度越高時,能間隙也會下降至窄能隙高分子的範圍。 實驗上的第二個部份的高分子結構如圖 1-5-3 為: S N N * H H N H * n S N N * N H * n S N N * N H * n S S P4 P5 P6 圖 1-5-3 目標高分子 P4、P5、P6 結構 若側鏈軟段部分越長,導致溶解性會提高而有利於製程,但導電性勢 必會因高分子的有效共軛長度(ECL)被迫減少而降低,導電性是熱電的性 能指標重要參數,故間接造成 ZT 值的降低 ;反之若側鏈軟段過短,則難 溶於泛用的低沸點有機溶劑,甚至不溶,將不利於元件的製備與量測50。 接續先前的研究可知,受體若再接上軟段則十分不穩定、不易製備,完成 高分子之後也因軟段影響而不利於高分子導電,所以只採用有機合成的技 巧將單體的側鏈接上不同的官能基團,希望找出對於導電性以及溶解性都 有幫助之最佳化有機熱電材料。
太陽能電池的部分 51,52,53,在近紅外光區吸收的高分子太陽能電池性 之量測和可見光吸收的高分子相比是少之又少,而可見光吸收的高分子之 能量轉換效率又一直無法突破 6.5%以上,到達可生產的效率,但 Alan J. Heeger 在 2007 年發表於 Sceince54的串疊型高分子太陽能電池就彌補了只 有吸收可見光區的高分子之缺陷。因此給予學生想要量測這吸收在近紅外 光區的窄能隙高分子之太陽能性質的動機,若先能將此區的能量轉換效率 提高,相信有助於串疊型高分子太陽能電池的整理效率提升。
貳、共軛高分子的合成與量測
2-1、合成實驗
2-1-1、實驗藥品 藥品 純度/濃度 廠牌 3-Bromothiophene 97% Aldrich 2,5-Dibromothiophene 95% Aldrich2-Ethylhexyl bromoide 95% Acros
3-Thiophenealdehyde HPLC TCI
Tetrakis(triphenylphosphine)palladium(0) 99% Aldrich
Oxalyl chloride 98% Aldrich
Pyridinium chlorochromate 98% Aldrich
1-Bromohexane 98% Aldrich
n-Butyllithium 2.5M Chemetall
Trimethyltin chloride in hexane 1M Aldrich
Sodium carbonate anhydrous 99.8% RDH
Copper powder 99.5% RDH
Potassium iodide 99.5% Showa
Magnesium powder 99% Showa
Magnesium sulfate anhydrous 99% Showa
Ammonium chloride 99.5~100.5% RDH
Potassium hydroxide pellets 87.9% J.T.Baker
Tin powder 99% RDH
Copper(I) iodide 99% RDH
Benzil ACS Sigma
N-Bromosuccinimide 95% Fluka
Sodium sulfite anhydrous 98.1% TEDIA
Glyoxal solution 38~42% RDH
Iodine 99.8~100.5% RDH
Lithium bromide 99% RDH
Hydrazine monohydrate 98% 98% Aldrich
Fuming sulfuric acid 87% SO3 87% RDH
Hydrochloric acid 37% RDH
Fuming nitric acid ACS
Sulfuric acid 95~97% ACS Fluka
Bis(triphenylphosphine) palladium(Ⅱ) chloride
97% Fluka
Calcium hydride 98% Aldrich
Pyrrole-1-carboxylic acid tert-butyl ester 97% Aldrich
表 2-1-1-1 實驗使用藥品
溶劑名稱 廠牌
N,N-Dimethylformamide
100% J.T.Baker
Methyl alcohol
anhydrous 100% Mallinckrodt Chemicals
Acetonitrile 99.99% ECHO Toluene 99.8% TEDIA Dimethyl sulfoxide 99.8% Scharlau Chloroform 99~99.4% RDH Ethylene glycol 99.5% RDH 表 2-1-1-2 實驗使用溶液 2-1-2、實驗儀器
1.核磁共振光譜儀(Nuclear Magnetic Resonance Spectrometer):Varian 300型
2.元素分析儀(Elemental Analyzer):Perkin-Elmer 240C型
3.循環伏安電化學儀(Cyclic Voltammeter,CV):AutoLab
4. 紫 外 光 - 可 見 光 - 近 紅 外 光 光 譜 儀 (Ultraviolet-Visible) Infrared Spectrophotometer)
5.熱重量分析儀(Thermogravimetric Analyszer,TGA):TA Q500型
Pyris 7型
7.凝膠滲透層析儀(Gel Permeation Chromatography,GPC):Waters 2414型
2-2、合成流程
2-2-1、施體結構(1)4,4-Bis-(octyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene
圖 2-2-1-1 施體單體(1)之合成
A.n-BuLi, Dry Et2O, -78℃, 3hr;B. 3-ThCHO, Dry Et2O, -78℃, 0.5hrr.t., 0.5hr ;C. n-BuLi, -78℃, 2hrr.t., 1hr;D. I2, Dry Et2O ; E. Na2SO3(aq); F. PCC, CH2Cl2, r.t., 12hr;
G.Cu(powder), DMF, reflux 1500C, 15hr ;
H.NH2NH2, KOH, Ethylene glycol;
I. R2Br, KOH, KI, DMSO; K. n-BuLi, Trimethyltin chloride, Dry THF
S Br S S OH I I S S O I I S S O S S S S R R R=C8H17 S S R R R=C8H17 Sn Sn A~E F G H I K 1 2 3 4 5 6
2-2-2、前驅施體單體(2)2,5-Bis-trimethylstannanyl-pyrrole-1-carboxylic acid tert-butyl ester
圖 2-2-2-1 施體單體(2)之合成
2-2-3、受體單體:2,3-Substituted-thieno[3,4-b]pyrazines
圖 2-2-3-1 受體單體之合成
A.Conc.H2SO4, Fum.H2SO4, Fum.HNO3, 20-30 0
C;
B. a)Sn, HCl; b)Na2CO3;C.a)Mg b)CuBr, LiBr; c) Oxalyl chloride; D. EtOH;H2O, Na2CO3;E. NBS, DMF N Boc N Boc Br Br N Boc Me3Sn SnMe3 Boc= R O Ot-Bu NBS,THF -75oC→0oC 1. 2eq.nBuLi THF.75min 2.Me3SnCl/ THF -75oC 1 h →r.t. 12 h 7 8 9 S Br Br NO2 NO2 S Br Br NH3+ NH3+ S Br Br NH2 NH2 S N N S N N S S S N N O O S S O O S Br S N N Br Br S N N S S S N N Br Br Br Br D C A B E S Br Br B 10 11 12 13 14 15 16 17 18
2-2-4、高分子聚合流程圖 圖 2-2-4-1 高分子聚合圖 S S R R R=C8H17 N Boc Me3Sn SnMe3 Me3Sn SnMe3 S N N Br Br S N N Br Br S N N S S Br Br S N N S S Br Br S N N Br Br S N N Br Br Pd(pph3)4 800C,5 day Pd(pph3)4 800C,5 day Pd(pph3)4 800C,5 day Pd(pph3)2Cl2 750C,5 day Pd(pph3)2Cl2 750C,5 day Pd(pph3)2Cl2 750C,5 day N Boc * S N N * * N Boc S N N * N Boc * S N N * S S S S R R R=C8H17 * S N N * S S R R R=C8H17 * S N N * S S R R R=C8H17 * S N N * S S P1 P2 P3 IP4 IP5 IP6
2-3、合成步驟 2-3-1、施體結構(1)4,4-Bis-(octyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene A. Bis-(2-iodo-thiophen-3-yl)-methanol 1 取 3-Bromothiophene(4.68ml, 50mmol)溶在無水乙醚(50ml) 中,氮氣下注入三頸圓底燒瓶,並冷卻至-780 C,隨後慢慢加入 n-Butyllithium(n-BuLi) 20ml,磁石攪拌反應 3 小時後,再將 Thiophene-3-carbaldehyde(4.38ml, 50mmol)均勻溶在無水乙醚 (30ml)中,慢慢注入反應瓶內,反應半小時後,再回到室溫反 應半小時。 將反應瓶的溫度再降低至-780 C,慢慢注入 n-BuLi(40ml), 磁石攪拌反應 2 小時後,放置室溫反應 1 小時。再降低溫度至 -780C,氮氣下加入含碘溶液(溶在無水乙醚中),加完放置室溫 反應半小時,用飽和 Na2SO3水溶液做終止反應。 S Br BuLi S Li S O Li S S O Li S O Li Li S S O Li Li Li S OI I S S IO I I S OH I S S HO I I BuBr (i) n-BuLi, Dry Et2O
-780C, 3hr
(ii) 3-ThCHO, Dry Et2O -780C, 0.5hr->r.t., 0.5hr (iii) n-BuLi, -780C 2hr->r.t., 1hr (iv) I2, Dry Et2O -780C, 0.5hr->r.t., 0.5hr (v) Na2SO3(aq) 1
用乙酸乙酯進行萃取,用 MgSO4除水,濃縮後用乙酸乙酯 : 正己烷等於 1:6 當沖提液進行層析,取得棕褐色固體 14.11 克, 產率 63%。1 H NMR δ 2.37 (d, 1H), 5.75 (d, 1H), 6.92 (d, 2H), 7.43 (d, 2H)。 B. Bis-(2-iodo-thiophen-3-yl)-methanone 2 S S HO I I S S O I I 1 PCC, CH2Cl2 r.t., 12hr 2 取化合物 1(14.7g, 32.7mmol)溶在二氯甲烷,並將 Pyridinium chlorochromate(PCC)溶在 CH2Cl2中,慢慢注入至反應瓶,氮氣 下反應 12 小時後,用二氯甲烷萃取,MgSO4除水,濃縮後,用 乙酸乙酯 : 正己烷等於 1:5 當沖提液進行管柱層析,取得黃色 固體 13.13 克,產率 90%,可再用熱甲醇進行再結晶而純化。1 H NMR δ 7.05 (d, 2H), 7.47 (d, 2H)。 C. Cyclopenta[2,1-b;3,4-b']dithiophen-4-one 3 S S O S S O I I 2 3 Cu(powder), DMF reflux 1500C, 15hr 取化合物 2(6.69g, 15mmol)溶在 Dimethylformamide(DMF), 將銅粉慢慢加入反應瓶中,氮氣下回流加熱至 1500 C,反應 15
小時後,過濾掉 Cu,用水和二氯甲烷進行萃取,用 MgSO4除水, 濃縮後用正己烷當沖提液進行管柱層析,而得紫紅色針狀固體 2.4 克,產率 83%,可再用熱正己烷進行再結晶純化。1H NMR δ 6.97 (d, 2H), 7.02 (d, 2H)。 D. 4H-Cyclopenta[2,1-b;3,4-b']dithiophene 4 S S S S O 3 NH2NH2, KOH Ethylene glycol 4 取化合物 3(3g, 15.6mmol)置於三頸圓底燒瓶並注入 150ml
的 Ethylene glycol , 並 加 入 Hydrazine hydrate(NH2NH2·H2O, 14.56ml, 18.655mmol),在 800C 下反應一個小時,當溶液呈紫紅 色時,再將溫度升到 2000 C 進行加熱回流一個小時,直到溶液 呈橘紅色。 將 熱 的 橘 紅 色 溶 液 冷 卻 到 室 溫 , 將 氫 氧 化 鉀 水 溶 液 (4.46g/20ml)慢慢滴入溶液會成暗橘紅色。注射完後,加熱回流 兩個小時,直到溶液呈黃色,用二氯甲烷萃取,用正己烷進行 管住層析,並用正己烷再結晶,可得白色片狀結晶 1.53 克,產 率 55%。1 H NMR δ 3.55 (s, 2H), 7.09 (d, 2H), 7.18 (d, 2H)。 E. 4,4-Bis(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene 5
S S R2 R 2 S S 4 RBr, KOH, KI DMSO 5 取化合物 4(0.839g, 4.71mmol)以及將催化劑量的 Potassium iodide(KI) 21.1mg 置於雙頸圓底燒瓶,並於氮氣下注入 Dimethyl sulfoxide(DMSO) 25ml 當作溶劑後,慢慢丟入
Potassium hydroxide(KOH) 0.84 克,再慢慢加入 1-Bromo-octane
(2ml),氮氣下反應溫度控制在 50~600C,反應至隔天,用飽和 食鹽水和二氯甲烷進行萃取,取出有機層,再用飽和 NH4Cl 水溶液萃取第二次,用 MgSO4除水,濃縮後用正己烷當沖提液 進行管柱層析,而得黃色油狀物 1.08 克,產率 57%。1 H NMR δ 0.59 (t, 6H), 0.73 (t, 6H), 0.85~0.89 (m, 18H), 1.84 (m, 4H), 6.90 (d, 2H), 7.07 (d, 2H)。 F. 4,4-Bis(2-ethylhexyl)-2,6-bis-trimethylstannanyl-4H-cyclopenta[ 2,1-b;3,4-b']dithiophene 6 S S R2 R 2 Sn Sn S S R2 R 2 5 6
n-BuLi, Trimethyltin chloride Dry THF
將化合物 5(1g, 2.48mmol)注入至三頸反應瓶,抽乾後,注入
15 毫升無水乙醚,將反應冷卻到 00C,在氮氣下慢慢滴入
反應一個小時,再將溫度控制在 00 C,慢慢滴入 Trimethyltin chloride(9ml, 9mmol),反應 15 分鐘後,將溫度回復到室溫反應 一個小時,用乙醚和水進行萃取,將萃取液濃縮除溶液得棕色 油狀物。 將矽藻土(Celite)填充在抽氣過濾裝置內,擠壓緊密,用三乙 胺(TEA)稀釋油狀物並進行抽氣過濾,再將濾液濃縮得到棕色油 狀物。再一次填充矽藻土,用正己烷稀釋油狀物並進行過濾, 濃縮濾液後得到最後棕色油狀物 1.682 克,產率 95%。1 H NMR δ 0.38 (m, 18H), 0.61 (t, 6H), 0.78 (t, 6H), 0.92 (m, 18H), 1.85 (m, 4H), 6.96 (m, 2H);13C NMR δ 159.71, 142.60, 136.18, 130.23, 52.12, 43.03, 35.11, 34.44, 28.71, 27.58, 22.82, 14.15, 10.77, -8.28。
2-3-2、施體前驅物結構(2)2,5-Bis-trimethylstannanyl-pyrrole-1-carboxylic acid tert-butyl ester
A. 2,5-Dibromo-pyrrole-1-carboxylic acid tert-butyl ester 8
N O O Br Br N O O NBS, THF -75oC →0 oC 7 8 將 N-bromosuccinimide(NBS, 47.1g, 264mmol)加入裝有無 水 THF(750ml)的 2L 三頸瓶當中攪拌至完全溶解後,維持低溫
132.3mmol)並持續低溫攪拌 10 分鐘後,將反應升溫到 0℃反應 至隔天。反應完後,加入 Na2SO3(20g)攪拌 5 分鐘後,減壓迴 旋濃縮得淡黃色固體。再加入 CHCl3 (750ml)進行溶解後抽氣過 濾收集液體。將收集到的液體進行減壓迴旋濃縮得咖啡色油狀 物後立即有晶體析出。將粗產物進行兩次乙醇再結晶,初次再 結晶溫度不可以超過 45℃,產率 55%(23.6g)。1 H NMR δ 1.62 (s, 9H), 6.22 (s, 2H)
B. 2,5-Bis-trimethylstannanyl-pyrrole-1-carboxylic acid tert-butyl ester 9 N O O Br Br 8 N O O 9 Me3Sn SnMe 3 nBuLi Me3SnCl 將化合物 8(1.5g, 4.62mmol)加入中充滿氮氣並有加入 THF(25ml)的 100ml 三頸瓶中,將溫度控制在-70℃緩緩滴加用
15ml THF 稀釋後之 2.5M nBuLi 的 Hexane 溶液(12mmol,
7.68ml)。反應 75 分鐘後,降溫至-75℃加入 11.1mmol 之
Me3SnCl(1M in Hexane)反應 1 個小時。最後回到室溫攪拌至隔 天。將溶劑移除後,加入乙醚(30ml)和去離子水(30ml)進行萃
機相(30ml)混合後,再用去離子水(30ml)清洗三次。最後收集 有機相使用硫酸鎂乾燥後,迴旋濃縮得黃色晶體。使用乙醇再 結晶。1 H NMR δ 0.28 (s, 18H), 1.62 (s, 9H), 6.48 (s, 2H) 13C NMR δ -6.88, 28.31, 85.24, 123.75 2-3-3、受體結構 2,3-Substituted-thieno[3,4-b]pyrazines A. 2,5-Dibromo-3,4-dinitro-thiophene 10 將濃硫酸(130ml)、發煙硫酸(200ml)以及發煙硝酸(110ml) 於冰浴下逐漸加入至三頸圓底燒瓶,隨後取 2,5-Dibromothiophene(35ml, 75.3g, 311mmol)慢慢滴入反應瓶內, 反應溫度控制在 20-300 C。混合液反應三小時,溶液中會有大 量黃色固體析出後,再加入大量的冰塊去稀釋酸。稀釋後,抽 氣過濾取得黃色固體,並用水清洗,直到濾液無色。將洗過的 黃色固體溶入熱甲醇進行再結晶的動作。抽器過濾取得黃色結 晶狀固體,並在過濾期間用少量甲醇進行清洗動作,產率 55%(56.9 克)。13C NMR δ 113.7, 159.7。 B. 2,5-Dibromo-thiophene-3,4-diamine 11 S Br Br S Br Br O2N NO2 Conc.H2SO4, Fum.H2SO4, Fum.HNO3
10
S Br Br O2N NO2 10 a)Sn, HCl; b)Na2CO3 Br S Br H2N NH2 11 將化合物 1(25g, 75.34mmol)丟入三頸反應瓶,冰浴下加入 濃鹽酸(450ml)。混合半小時後,慢慢加入 7 當量的 Sn(62.6g, 52.74mol),溫度控制在 20~300C,反應至隔天。過濾前,靜置 約兩小時,使得固體逐漸沉澱後,濾掉黃色酸液取得白色固體, 並先用乙醚而後用乙睛清洗白色固體,直到濾液無色為止。此 白色固體能以此形式能保存很久。當要得到化合物 2 時,取出 一部分白色固體溶在去離子水中,在冰浴下加入 4N Na2CO3(aq), 並用乙醚進行萃取,不加熱下濃縮,取得白色固體結晶 4.82 克, 產率 56%。1 H NMR (DMSO-d6): δ 7.31 (s, 2H), 8.44 (s, 6H)。 C. 1,2-Di-thiophen-2-yl-ethane-1,2-dione S S O O Mg a)CuBr, LiBr b) Oxalyl chloride S Br S MgBr 12 用 1N 的鹽酸水溶液清洗鎂粉(4g, 165mmol),並用丙酮將殘餘 的鹽酸水溶液洗去,隨後將鎂丟入雙頸圓底燒瓶後,進行除水抽真 空,隨後再加入催化劑量的碘,將反應瓶充滿氮氣,攪拌約半小時, 加入無水 THF(100ml)後,在 600 C 下回流攪拌一小時,再慢慢滴入 2-Bromo-thiophene(152mmol) , 滴 完 反 應 至 鎂 粉 消 失 , 即 做 成
Grignard 試劑。
取 LiBr(25.5g, 293mmol),以及 CuBr(21.1g, 0.146mol)分別倒入
兩個雙頸圓底燒瓶,抽真空灌氮氣持續三次,再分別加入無水 THF(110ml),攪拌均勻後,將 LiBr 溶液氮氣下注入至 CuBr 溶液 中,此混合液會成綠色透明液。 將綠色透明液冷卻至-780 C,並慢慢滴入 Grignard 試劑,注意 溫度不要超過-700
C,滴完後,再將 Oxalyl chloride(7.77g, 61mmol)
於-780 C 下慢慢滴入至反應瓶,反應溫度不能超過-700C,反應三小 時。 反應完後,讓其溫度逐漸回到室溫,並用飽和 NH4Cl 水溶液 做終止反應。此混合液用乙酸乙酯進行萃取,用 MgSO4將萃取液 除水,濃縮後,用正己烷當沖提液進行管柱層析,取得黃色化合物, 產率 93%。1 H NMR δ 7.00 (dd, 2H), 7.21 (dd, 2H), 7.50 (dd, 2H)。 D. Thieno[3,4-b]pyrazine 13 S Br Br H2N NH2 11 H H O O H2O, Na2CO3(s) S N N 13 將 化 合 物 11(0.852g, 7.47mmol) 置 入 反 應 瓶 內 , 並 將
5%Na2CO3(aq)約 40ml 倒入與其混合均勻。取 40% Glyoxal solution 約 0.6 克,用去離子水稀釋到 20ml 後,加入至反應瓶。室溫暗室
下反應兩小時。反應結束後,加入 100ml 的水並用乙醚去萃取,收 集萃取後的有機層,再用水清洗。洗完後,用 MgSO4除水 並濃縮 得褐色油狀物,用乙醚當沖提液進行管柱層析而得到棕褐色固體 (0.627g, 產率 61.15%)。1H NMR δ 8.01 (s, 2H), 8.46 (s, 2H)。 E. 2,3-Dithiophen-2-yl-thieno[3,4-b]pyrazine 13 S Br Br NH2 NH2 + S S O O 12 11 S N N S S 14 EtOH 將化合物 11(1.38g, 12.09mmol)和 1.1 當量的化合物 12 都置入 於反應瓶內,用純度 99.5%的酒精當溶劑而形成紅橙色溶液,反應 約三個小時後,用濃縮機將酒精除去,得到的固體再用石油醚進行 清洗固體,取得濾液再將其濃縮而的棕褐色固體。用二氯甲烷 : 正 己烷等於 1 :1 的比例當沖提液進行管住層析得到黃色固體,產率 60%。1H NMR δ 7.00 (dd, 2H), 7.21 (dd, 2H), 7.50 (dd, 2H)。 F. 2,3-Diphenylthieno[3,4-b]pyrazine 14
S Br Br NH2 NH2 + O O 11 S N N EtOH 15 將 化 合 物 11(1.38g, 12.09mmol) 和 1.1 當 量 的 化 合 物 1,2-Diphenyl-ethane-1,2-dione(Benzil)都置入於反應瓶內,用純度 99.5%的酒精當溶劑而形成紅橙色溶液,反應約三個小時後,用濃 縮機將酒精除去,得到的固體再用石油醚進行清洗固體,取得濾液 再將其濃縮而的棕褐色固體。用二氯甲烷 : 正己烷等於 1:6 的比例 當沖提液進行管柱層析而得黃褐色固體,產率 85%。1H NMR δ 7.30-7.45 (m, 10H), 8.05 (s, 2H)。 G. 5,7-Dibromo-2,3-substituted thieno[3,4-b]pyrazines 16-18 S N N R1 R1 S N N R1 R1 Br Br NBS, DMF S N N S S Br Br S N N Br Br S N N Br Br 16 18 17 取 0.1 克 2,3-Substituted thieno[3,4-b]pyrazines 置入反應瓶,在 氮氣下注入足量的 Dimethylformamide(DMF)當溶劑。將 2.1 當量的
N-bromosuccinimide(NBS)溶 DMF(2ml)中,暗室氮氣下慢慢滴入反 應瓶內,溫度控制在-150 C,反應 2 小時,逐漸回到室溫。到入足 量的水,並用 CH2Cl2進行萃取,用 MgSO4除水,濃縮取得固體, 進行管柱層析而得暗色固體。 5,7-Dibromo-thieno[3,4-b]pyrazine 16 得到黃褐色固體,並用 熱甲醇進行再結晶,得土黃色針狀結晶固體 97.15 毫克,產率 45%。 1H NMR δ 8.51(s, 2H)。 5,7-Dibromo-2,3-dithiophen-2-yl-thieno[3,4-b]pyrazine 17 得 到黃褐色固體,並用熱正己烷進行再結晶,得到黃褐色顆粒狀固體 75.7 毫克,產率 50%。1H NMR δ 7.00 (dd, 2H), 7.21 (dd, 2H), 7.50 (dd, 2H)。 5,7-Dibromo-2,3-diphenyl-thieno[3,4-b]pyrazine 18 得到黃綠 色固體,並用熱正己烷進行再結晶,得黃綠色細針狀結晶固體 85.1 毫克,產率 55%。1H NMR δ 7.30~7.37(m, 6H), 7.44~7.48(m, 4H)。 2-3-4、高分子聚合
我們採用的聚合方法是Stille coupling,而不用Suzuki coupling的最主要
原因是其硼試劑的單體不易純化,經過管柱層析後就有局部的化合物變成
雜質,會影響到聚合後所得到的高分子分子量。Stille coupling所用的單體
合的單體,再利用簡單的清洗過濾,就能得到高純度的單體。以下就簡單 說明本實驗高分子聚合的步驟: 2-3-4-1、針對P1、 P2、 P3聚合步驟: S S R R R=C8H17 S N N * * n S S R R R=C8H17 S N N * * n S S R R R=C8H17 S N N * * n S S P1 P2 P3 將 1mmol 的 受 體 單 體 和 1mmol 的 施 體 單 體 (4,4-Bis-(octyl)-4H-cyclopenta[2,1-b;3,4-b']dithiophene)丟入反應瓶內,抽灌 氮氣使反應瓶充滿乾燥後的氮氣,再注入適當量的溶劑將反應物溶解,並 丟入80毫克的Pd(PPh4)4後,隨即封住注入口,再一次抽灌氮氣確保反應瓶 內充滿氮氣,在室溫下攪拌10分鐘後,將溫度加熱到800 C,反應五天。 收反應時,將濃稠的高分子混合液慢慢滴入甲醇裡,讓其在甲醇裡沉 澱,過濾後再用適當的溶劑去進行清洗動作,抽乾溶劑即可得到純的高分 子固體。 2-3-4-2、針對IP4、IP5、IP6聚合步驟:
S N N * H H N BOC * n S N N * N BOC * n S N N * N BOC * n S S
IP4 IP5 IP6
BOC= R O
O
由於IP4、IP5、IP6使用Pd(PPh4)4 進行聚合反應產率過低,無法產出高 分子量產物。故我們嘗試多種不同Stille coupling催化劑,如: Pd2dba3、 (Ph3P)2PdCl2、PdCl2(AsPh3)2進行聚合反應,其中發現以使用(Ph3P)2PdCl2 做 為IP4、IP5、IP6聚合最為適當。其步驟和P1、P2、P3實驗步驟類似,不同 點在於我們還加入了AsPh3來和催化劑(Ph3P)2PdCl2形成錯合物,穩定催化 劑持續漫長五天的反應中存在。反應溫度也只控制在750 C下進行反應,盡 量確保在反應進行的同時,施體單體上的酯類官能基不會同時離去。 2-4
、有機熱電元件量測步驟
2-4-1、電阻量測 1. 在壓克力片上等距離 2.66mm 刻出 4 條小凹槽。 2. 同時量取適當長度的白金線固定在 4 條凹槽上,此時所刻出的四條 凹槽不能大於白金線。 3. 利用絕緣膠帶將 4 條白金線固定。 4. 量測前將壓克力鎖在裝置上,再將埋有熱電偶的銅塊鎖上,此時必頇 拿 捏 力 道 避 免 將 樣 本 壓 壞 或 將 壓 克 力 片 鎖 斷 。 打 開NANOVOLTMETER 和 DC AND AC CURRENT SOURCE 的電源,
等 待 DC AND AC CURRENT SOURCE 螢 幕 上 出 現 DHCP
CONNECT 後按下 EXIT 鍵,如圖 2-4-1-1 所示,再按 EDIT/LOCAL
鍵將 Compliance 調整至 100.00V 及調整所需要加的電流,如圖 2-4-1-2、2-4-1-3 所示。 圖 2-4-1-1 電阻儀器量測圖示一 圖 2-4-1-2 電阻儀器量測圖示二 螢幕上出現 DHCP CONNECT 後按下 EXIT 鍵 EDIT/LOCAL 鍵 POWER
圖 2-4-1-3 電阻儀器量測圖示三
5.將最外測白金線連接至 DC AND AC CURRENT SOURCE 儀器上,而
內側兩條則連接至 NANOVOLTMETER 儀器上。 6. 紀錄 0 點的電壓值後,依序從 0.01mA~0.1mA 或不同的電流範圍量 測。 7. 量測結束後將壓克力片拆下,是為了排除不必要因素而影響 seebec 係數量測。 2-4-2、Seebeck 係數量測 1. 先將熱電偶連接到觸控式溫度蒐集器上,熱電偶的 Cu 線接在正極而 負極則連接 Cu/Ni 線,如圖 2-4-2-1 所示。 調整電流大小 控制游標 OUTPUT ON/OFF 鍵, 量測開始鍵
圖 2-4-2-1 Seebeck 係數量測儀器之圖示一 2. 打開觸控式溫度蒐集器開關,如圖 2-4-2-1 所示。 圖 2-4-2-2 Seebeck 係數量測儀器之圖示二 3. 將銅塊上加熱棒以串連方式連接,因為只有量測室溫的 seebeck 係數, 只需要串連單邊銅塊的加熱棒。 4. 將絕緣膠帶貼在銅塊和熱電偶上,如圖 2-9 所示。避免銅塊和熱電偶 影響樣本的量測。此時再將白金線在銅塊和樣本之間,當作量測電位 電源開關 Cu/Ni 線連接處(負極) Cu 線連接處(正極)
差導線用。 圖 2-4-2-3 Seebeck 係數量測示意圖 5. 溫度平穩後即可開始測量,同樣的先紀錄原點的電位差再進行加熱 動作,加熱時給予加熱棒一定的電壓,等待溫度穩定後即可紀錄電 位差變化。 熱電偶 銅塊 樣本 絕緣膠帶
參、結果與討論
3-1、高分子的基礎性質(GPC、TGA、DSC)
Mn
Mw
PDI
Td(℃)
P1
10173
12183
1.14
369
P2
9028
13343
1.36
358
P3
11943
14643
1.36
374
IP4
2781
4028
1.44
151(475)
IP5
2393
3113
1.30
186(512)
IP6
4934
6694
1.42
156(490)
表 3-1-1 高分子分子量、裂解溫度 由於高分子施體的不同,導致了高分子在基礎性質方面有極端的不 同之處,所以分成兩組來討論。P1、P2、P3 三個高分子的分子量大小 大都在 10000 左右,可得知在各高分子聚合的條件上控制良好;在單體的純度方面也無太大的差異。此外 PDI(The Polydispersity Index)值皆
小於 1.5 以下,顯示出單體(雙三甲基錫雙噻吩環戊烷)的聚合官能基(三
甲基三甲基錫)在聚合的過程當中,可以穩定的存在,亦表示了我們選
用了適當的催化劑和催化條件。從下圖 3-1-1 中可以知道三個高分子在
裂解溫度上面並沒有太大的差異且達到 350 度以上,代表在高溫的環
圖 3-1-1 高分子 P1、P2、P3 熱重分析圖
IP4、IP5、IP6 的分子量均偏低,推測其原因有兩個:一是因為本
身高分子僅可溶於熱的 Tetrahydrofuran(THF)之部分分子量,所以實際
的分子量應更大。第二點可能是因為施體結構(Pyrrole-1-carboxylic acid
tert-butyl ester)雖然有 tert-butyl 碳鏈的酯基官能基來增加溶解度,但其
增加溶解度依然不足以讓分子聚合成高分子後維持其溶解特性,導致
高分子在分子量不高時即從溶劑中沈澱下來,導致分子量無法達到高
分子的水準。雖然分子量無法達到高分子的水準,但比起文獻中在聚
合相似的高分子而言,將兩步的聚合反應簡化成一步反應且文獻能達





![圖 1-5-1 單體結構作用力示意圖 除了施體受體結構在得電子能力強弱的考量之外,我們也導入了併雜環 (Fused ring)的構想 46 ,其效益在於若有堅硬的結構結構主幹,會更有益於 高分子堆疊、提高載子的移動率和熱穩定性。 實 驗 第 一 部 分 所 中 採 用 的 受 體 單 體 是 2,3-Substituted-thieno[3,4-b]pyrazines ; 施 體 單 體 是 4,4-Dioctyl-4H-cyclopenta[2,1-b;3,4-b']dithiophene。高分子](https://thumb-ap.123doks.com/thumbv2/9libinfo/8515914.186145/42.892.180.804.135.227/結構結構主幹會更有益高分子堆疊載子移動率和熱穩定性第高分子.webp)
