0022-538X/08/$08.00
⫹0 doi:10.1128/JVI.01238-08
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Characterization of White Spot Syndrome Virus Envelope Protein
VP51A and Its Interaction with Viral Tegument Protein VP26
䌤
Yun-Shiang Chang,
1* Wang-Jing Liu,
2Tsung-Lu Chou,
1Yuan-Ting Lee,
1Tai-Lin Lee,
1Wei-Tung Huang,
1Guang-Hsiung Kou,
2and Chu-Fang Lo
2*
Department of Molecular Biotechnology, Da-Yeh University, Changhua, Taiwan,
1and Institute of Zoology,
National Taiwan University, Taipei, Taiwan
2Received 14 June 2008/Accepted 17 September 2008
In this study, we characterize a novel white spot syndrome virus (WSSV) structural protein, VP51A
(WSSV-TW open reading frame 294), identified from a previous mass spectrometry study.
Temporal-tran-scription analysis showed that vp51A is expressed in the late stage of WSSV infection. Gene structure analysis
showed that the transcription initiation site of vp51A was 135 bp upstream of the translation start codon. The
poly(A) addition signal overlapped with the translation stop codon, TAA, and the poly(A) tail was 23 bp
downstream of the TAA. Western blot analysis of viral protein fractions and immunoelectron microscopy both
suggested that VP51A is a viral envelope protein. Western blotting of the total proteins extracted from WSSV
virions detected a band that was close to the predicted 51-kDa mass, but the strongest signal was around 72
kDa. We concluded that this 72-kDa band was in fact the full-length VP51A protein. Membrane topology assays
demonstrated that the VP51A 72-kDa protein is a type II transmembrane protein with a highly hydrophobic
transmembrane domain on its N terminus and a C terminus that is exposed on the surface of the virion.
Coimmunoprecipitation, colocalization, and yeast two-hybrid assays revealed that VP51A associated directly
with VP26 and indirectly with VP28, with VP26 acting as a linker protein in the formation of a
VP51A-VP26-VP28 complex.
Viral structural proteins, especially the envelope proteins,
are important, not only because they are involved in virion
morphogenesis, but also because they are the first molecules to
interact with the host. The structural proteins often play vital
roles in cell targeting, virus entry, assembly, and budding (1, 2,
21, 22, 24), as well as triggering host antiviral defenses (26). In
the case of white spot syndrome virus (WSSV) (genus
Whispo-virus, family Nimaviridae) (37), a double-stranded DNA virus
that has caused severe mortality and huge economic losses to
the shrimp farming industry globally for more than a decade
(5, 19), proteomic methods have helped to identify a total of 58
structural proteins, over 30 of which are currently recognized
as envelope proteins (13, 31, 44, 47). Some of the WSSV
envelope proteins involved in shrimp infection have been
iden-tified (12, 14, 34, 36, 41, 43), and these envelope and other
WSSV structural proteins have been used in various studies,
including RNA interference-based gene knockdown to silence
viral structural-protein gene expression (8, 27, 45), DNA and
protein vaccination to elevate host immunity (25, 29, 36, 39),
and antibody neutralization techniques that neutralize the
vi-rus by preventing envelope proteins from interacting with host
cell receptors (12, 34, 41, 43).
In the present paper, we characterize and investigate the
functionality of a WSSV structural protein that was first
re-ported by Tsai et al. (32). This protein, designated VP51A,
corresponds to open reading frame 294 of the WSSV-TW
isolate, and its gene encodes a polypeptide of 486 amino acids
(aa) with a theoretical mass of 51 kDa. A method was recently
established to assign the WSSV structural proteins to one of
three morphologically distinct substructures in the virion: the
nucleocapsid, the intermediate tegument layer that surrounds
the nucleocapsid, and the outer viral envelope (31). We use the
same method here to show that VP51A is an envelope protein,
and we support this conclusion with an in vivo immunogold
assay. Some WSSV structural proteins are known to interact
with other structural proteins. For example, VP28 interacts
with VP24 and VP26 (43, 44), and VP24 interacts with another
WSSV envelope protein, WSV010 (3). Identification of these
relationships among structural proteins is a significant step
toward understanding the function of each protein and is
po-tentially helpful in developing effective anti-WSSV strategies.
We show here that VP51A forms a complex with two other
major WSSV structural proteins, VP26 and VP28.
MATERIALS AND METHODS
Virus.The WSSV-TW strain was isolated from a batch of WSSV-infected
Penaeus monodon shrimp collected in Taiwan in 1994 (18, 38), and it was used as
the template for amplification of the vp51A, vp26, and vp28 coding regions in all of the following experiments.
Temporal-transcription analysis of vp51A by RT-PCR. Adult P. monodon
shrimp (mean weight,⬃20 g) were experimentally infected with WSSV by
in-jection and subsequently collected at 0 (i.e., immediately before infection), 2, 4, 6, 12, 24, 36, 48, and 60 h postinfection (p.i.) according to a procedure described by Tsai et al. (33). Total RNAs were isolated from the gills of the sampled shrimp by using Trizol reagent (Invitrogen Corp.) according to the manufacturer’s in-structions. The isolated RNAs were treated with DNase I (Roche) at 37°C for 1 h and then recovered by phenol-chloroform-isoamyl alcohol extraction and etha-nol precipitation. The RNAs were reverse transcribed with SuperScript II
re-* Corresponding author. Mailing address for Chu-Fang Lo: Institute
of Zoology, National Taiwan University, Taipei 106, Taiwan. Phone:
886-2-33662453. Fax: 886-2-23638179. E-mail: [email protected].
Mailing address for Yun-Shiang Chang: Department of Molecular
Biotechnology, Da-Yeh University, Changhua 515, Taiwan. Phone:
886-4-8511888, ext. 4265. Fax: 886-4-8511326. E-mail: yschang@mail
.dyu.edu.tw.
䌤
Published ahead of print on 1 October 2008.
12555
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
verse transcriptase (RT) (Invitrogen Corp.) and an oligo(dT) anchor primer (Roche). The first-strand cDNA products were subjected to PCR with the vp51A primers WSSV294-F-122 and WSSV294-R-632 (Table 1). For comparison, the WSSV ie1 (an immediate-early gene), dnapol (a DNA polymerase gene), and
vp28 (an envelope protein gene) gene fragments were also amplified from the
same templates using the primer pairs ie1-F/ie1-R, dnapol-F/dnapol-R, and vp28-F/vp28-R, respectively (Table 1). An actin 2 primer pair, P1882-actin F1 and P1883-actin R1, designed by Leu et al. (11) and based on P. monodon actin
2 (AF100987), was used as an internal control for RNA quantity and
amplifica-tion efficiency. The primer sequences are listed in Table 1.
Mapping the 5ⴕ and 3ⴕ termini of vp51A transcripts. The 5⬘ and 3⬘ regions of
the vp51A transcripts were obtained by 5⬘/3⬘ rapid amplification of cDNA ends
(RACE) (6) using a commercial 5⬘/3⬘ RACE kit (Roche) with an avian
myelo-blatosis virus RT. The RNA samples used for 5⬘/3⬘ RACE analysis were isolated
from the gills of WSSV-infected P. monodon at 36 h p.i. and treated with DNase
I as described above. The appropriate gene-specific primers used for 5⬘/3⬘ RACE
are listed in Table 1. The final amplification products were cloned into the pGEM-T Easy vector (Promega) and sequenced. The sequences of the inserts were compared with the WSSV genomic-DNA sequences.
Expression, purification of recombinant VP51A proteins, and antibody pro-duction.PCR fragments representing three different coding regions of vp51A were amplified using the primer sets listed in Table 2, digested with restriction
enzymes, and cloned into pET-28b(⫹) (Novagen). The resulting plasmids,
pET-28b/VP51A139-250, pET-28b/VP51A251-486, and pET-28b/VP51A51-486
(contain-ing the VP51A aa residues 139 to 250, 251 to 486, and 51 to 486, respectively), were transformed into BL21 Codon Plus Escherichia coli cells (Stratagene) and used for protein production. The transformed cells were grown overnight at 37°C
in Luria-Bertani medium supplemented with 50g/ml kanamycin. The cells were
then inoculated into fresh medium at a ratio of 1:50 and grown at 37°C for 1.5 to
2 h. Expression was induced by the addition of 1 mM IPTG (isopropyl--
D-thiogalactopyranoside), and incubation was continued for another 1.5 to 3 h. The
induced bacteria were spun down at 4°C, suspended in ice-cold 1⫻
phosphate-buffered saline (PBS) containing 10% glycerol and a protease inhibitor cocktail tablet (Roche), and sonicated for 3 min on ice. The insoluble debris was collected
by centrifugation, suspended in 1⫻ PBS containing 1.5% sodium lauryl
sar-cosine, and solubilized by shaking at room temperature for 1 h. The supernatant was clarified by centrifugation and mixed with Ni-nitrilotriacetic acid-agarose beads (Qiagen) on a rotating wheel at 4°C for 16 h. The beads were then washed several times with ice-cold wash buffer (1 M NaCl, 10 mM Tris-HCl, pH 7.5) to remove unbound material. The fusion proteins were eluted directly from the beads with sodium dodecyl sulfate (SDS) sample buffer and were then subjected to SDS-polyacrylamide gel electrophoresis (PAGE) analysis. For polyclonal-antibody production from the VP51A midsequence (aa 139 to 250) and C-terminal (aa 251 to 486) fragments, the protein bands containing the fusion proteins were sliced from the gel, minced, mixed with Freund’s adjuvant, and inoculated into rabbits.
TABLE 1. Primer sequences for 5
⬘/3⬘ RACE and RT-PCR
Gene Primer Sequence (5⬘–3⬘) Usage
vp51A
WSSV294-F-122
GGAAGAGATATCGTACCAGTGAG
vp51A RT-PCR
WSSV294-R-632
CTTGGTTTTCCTTGTAGAGTGTC
ie1
ie1-F
GACTCTACAAATCTCTTTGCCA
ie1 RT-PCR
ie1-R
CTACCTTTGCACCAATTGCTAG
dnapol
dnapol-F
TGGGAAGAAAGATGCGAGAG
dnapol RT-PCR
dnapol-R
CCCTCCGAACAACATCTCAG
vp28
vp28-F
CTGCTGTGATTGCTGTATTT
vp28 RT-PCR
vp28-R
CAGTGCCAGAGTAGGTGAC
actin 2
P1882-actin F1
CCGTCATCAGGGTGTGATGGT
actin RT-PCR
P1883-actin R
CCACGCTCAGTCATGATCTTCA
vp51A
vp51A SP1
GTAGGCTCAAAATCGTCTGTGTC
vp51A 5
⬘ RACE
vp51A
vp51A SP2
GAACCATTTCCATTTGTTCCAC
vp51A 5
⬘ RACE
vp51A
vp51A SP3
TTGTTCCAGTTCCTCCGTCTAT
vp51A 5
⬘ RACE
vp51A
vp51A SP4
TTGTGGCCAATAAGAATGACAC
vp51A 3
⬘ RACE
TABLE 2. Primer sequences used for the construction of various expression plasmids
Construct Primera Sequence (5⬘–3⬘)b Tag
pET-28b/VP51A
139-250F
CGCGGATCCGGATACAGACACAGACGATTTTGAG
His
R
CCCAAGCTTTAATATTGTTTGTGCAATTTC
pET-28b/VP51A
251-486F
GGCGAGCTCCAGTCAACTAAGAGAAAAACAC
His
R
CCCAAGCTTTTGGCTGGACAATAAATTTTTG
pET-28b/VP51A
51-486F
CGGGATCCTGACGGCATAGACGGAGGAACT
His
R
CCCAAGCTTTTGGCTGGACAATAAATTTTTG
pcDNA3/VP51A
F
CCCAAGCTTGAAAAAATGTTCGTCATAAGC
None
R
CGCGGATCCTTATTGGCTGGACAATAAATTTTT
pDHsp/VP51A-FLAG-His
F
CCCAAGCTTGAAAAAATGTTCGTCATAAGC
Flag/V5
pDHsp/VP51A-V5-His
R
TCCCCGCGGTTGGCTGGACAATAAATTTTTG
pDHsp/VP26-FLAG-His
F
CCCAAGCTTAGAAAAATGGAATTTGGCAAC
Flag/V5
pDHsp/VP26-V5-His
R
TCCCCGCGGCTTCTTCTTGATTTCGTCCTTG
pDHsp/VP28-V5-His
F
CCCAAGCTTCTCGTCATGGATCTTTCTTTC
V5
R
TCCCCGCGGCTCGGTCTCAGTGCCAGAGTAG
pGBK-VP51A
F
TCCCCCGGGAATGTTCGTCATAAGCATAGC
c-Myc
R
CGGGATCCTTATTGGCTGGACAATAAATT
pGAD-VP26
F
CGGAATTCATGGAATTTGGCAACCTAAC
HA
cR
CCGCTCGAGGCTTCTTCTTGATTTCGTCCT
pGAD-VP28
F
CGGAATTCATGGATCTTTCTTTCACTCTTT
HA
R
CCGCTCGAGGCTCGGTCTCAGTGCCAGAGTA
aF, forward; R, reverse.bThe restriction enzyme cutting sites are underlined.
cHA, hemagglutinin.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
Fractionation of virion proteins by detergent treatment at different NaCl concentrations. Adult crayfish, Procambarus clarkia, were challenged with WSSV, and the virions were purified from the hemolymph of the infected crayfish as described by Tsai et al. (32). The purified virus suspension was treated with 1% Triton X-100 in different concentrations (0, 0.1, 0.5, and 1 M) of NaCl solution, and the soluble and insoluble portions were then fractionated by cen-trifugation as described previously (31). The intact untreated virion suspension served as a control. The proteins in each of the eight resultant fractions and in the intact purified virion control were resolved by Western blot analysis using the anti-VP51A C-terminal-fragment antibody. The bound antibodies were then stripped out of the membrane, and the membrane was reprobed with antibodies to the WSSV envelope protein VP28, to the tegument protein VP26, and to the nucleocapsid protein VP51C (31, 40). An additional Western blot analysis was performed on the intact purified virion proteins using the anti-VP51A midse-quence fragment antibody as the probe.
Western blot analysis.Protein samples were resolved by SDS-PAGE. After separation, the proteins were transferred to polyvinylidene difluoride mem-branes (MSI). The memmem-branes were incubated in blocking buffer (5% skim milk in Tris-buffered saline [50 mM Tris, 500 mM NaCl, pH 7.5]) at 4°C overnight and then incubated with blocking buffer containing primary antibodies for 1 h at room temperature. Next, the membrane was washed three times with 0.5% Tween 20 in Tris-buffered saline and incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody. After three more washes, the proteins were visualized by use of a chemiluminescence reagent (Perkin-Elmer, Inc.).
Localization of VP51A by immunoelectron microscopy (IEM).Following the
method of Tsai et al. (31), aliquots (10l) of purified virion suspension were
adsorbed to Formvar-supported, carbon-coated nickel grids (200 mesh) for 5 min at room temperature, and then the excess solution was removed. The grids were either prefixed for 5 min with 4% paraformaldehyde and 1% Triton X-100 simultaneously in 50 mM Tris buffer (to remove the virus envelope) or were left unfixed and were incubated with incubation buffer only (0.1% Aurion BSA-c, 15
mM NaN3, 10 mM phosphate buffer, 150 mM NaCl, pH 7.4). Other grids were
loaded with purified nucleocapsid samples (5-min absorption at room tempera-ture) prepared by treating purified virions with detergent and NaCl, followed by centrifugal fractionation (31). These nucleocapsid grids were left unfixed. All grids were then blocked with blocking buffer (5% bovine serum albumin, 5% normal goat serum, 0.1% cold-water fish skin gelatin [Aurion], 10 mM phosphate buffer, 150 mM NaCl, pH 7.4) for 15 min and then incubated for 1 h at room temperature with VP51A C-terminal antibody diluted 1:50 in incubation buffer. As a control, an additional grid containing intact virions was incubated using a dilution of preimmune rabbit serum instead of the anti-VP51A C-terminal an-tibody. After several washes with incubation buffer, the grids were incubated for 1 h at room temperature with a goat anti-rabbit secondary antibody conjugated with 15-nm-diameter gold particles (1:40 dilution in incubation buffer). The grids were then washed extensively with incubation buffer, washed twice more with distilled water to remove excess salt, and stained with 2% phosphotungstic acid (pH 7.4) for 30 s. Specimens were examined with a transmission electron micro-scope (Jeol JEM1010).
Coupled in vitro transcription and translation.A full-length vp51A expression plasmid, pcDNA3/VP51A, was constructed by using PCR to clone the vp51A coding region into the commercial vector pcDNA3 (Invitrogen Corp.). The primers used for the PCR cloning are listed in Table 2. Coupled in vitro tran-scription-translation reactions were conducted in accordance with the manufac-turer’s protocol (TNT System; Promega). One microgram of plasmid pcDNA3/
VP51A DNA and 1l of [35
S]methionine (1,000 Ci/mmol; 10 mCi/ml) were
added to the TNT reaction mixture to make a total volume of 50l, and the
reaction proceeded at 30°C for 90 min. An aliquot of the translation product (5 l) was analyzed by 15% SDS-PAGE. After electrophoresis, the gel was fixed and exposed to Kodak Biomax MS film.
Computer analysis of the VP51A coding region.Based on the deduced amino acid sequence of VP51A, its hydropathic profile was predicted by a computer-assisted procedure that followed the methods of Kyte and Doolittle (10). Topol-ogy predictions were made using the TMHMM (9) program. Both of these programs are accessible on the World Wide Web (http://ca.expasy.org/tools/).
VP51A topology.To investigate the membrane topology of VP51A, the plas-mid pDHsp/VP51A-V5-His was constructed by inserting the full-length WSSV
vp51A gene into a V5-tagged vector containing the heat-inducible Drosophila
heat shock protein 70 promoter (pDHsp/V5-His) (11) by PCR cloning using WSSV genomic DNA as the template. The primers used for this PCR are listed in Table 2. Using Cellfectin reagent (Invitrogen Corp.), this plasmid was trans-fected into Sf9 insect cells that had been seeded onto cover glasses placed in each
well of a four-well microplate (1 ⫻ 105
cells/well) and grown in Sf-900 II serum-free medium (Invitrogen Corp.) overnight at 27°C. After transfection for
16 to 18 h, the cells were heat shocked in a 42°C water bath for 30 min and then returned to 27°C. At 6 h after heat induction, the monolayers were washed with PBS and the cells were fixed in paraformaldehyde (4% in PBS) for 10 min at 4°C. Some cells were then treated with 0.1% Triton X-100 in 4% paraformaldehyde/ PBS solution for 3 min at 4°C, while other cells were left untreated. All the cells were then washed thoroughly two times with PBS. After being blocked in block-ing buffer (5% bovine serum albumin and 2% normal goat serum in PBS) for
16 h at 4°C, the cells were treated with a 1,000⫻ PBS-diluted polyclonal rabbit
anti-V5 antibody (Sigma) (3 h at room temperature). Next, the cells were washed three times (10 min each time) with PBST (0.2% Tween 20 in PBS) and reacted with carboxymethylindocyanine (Cy3) dye-conjugated donkey anti-rabbit immu-noglobulin G (IgG) antibody (1:1,000 in PBS; Jackson ImmunoResearch) for 2 h
at room temperature. Counterstaining of the nucleus was performed with 4
⬘-6⬘-diamidino-2-phenylindole dihydrochloride (DAPI) (Vector Laboratories). After being washed three times (10 min each time) with PBST, the cover glasses were wet mounted with antifade mounting medium (Fluka). During all of the above-mentioned procedures, the cells were kept in darkness. Fluorescence signals were examined using a confocal scanning laser microscope (Leica TCS SP5).
To investigate the topology of VP51A, VP28, and VP26 in the WSSV virion,
we followed the method of Zhu and Yuan (48) and treated aliquots (5g of total
protein) of purified virions with trypsin (5g/ml; Promega) in 100 l of buffer
(50 mM Tris-HCl [pH 7.5], 1 mM CaCl2, 100 mM NaCl) at 37°C for 2 h. Trypsin
digestion was terminated by adding phenylmethylsulfonyl fluoride to a final concentration of 0.5 mM and then adding 1/50 volume of protease inhibitor. In some samples, prior to trypsin digestion, Triton X-100 was added to a final concentration of 1% to dissolve the viral envelope and expose the internal structure to the protease. Samples were analyzed by Western blotting using antibodies to VP51A, to the envelope protein VP28, and to the tegument protein VP26.
Coimmunoprecipitation.Full-length WSSV vp51A, vp26, and vp28 genes were inserted into V5- or FLAG-tagged vectors containing the heat-inducible
Dro-sophila heat shock protein 70 promoter (pDHsp/V5-His and pDHsp/FLAG-His)
(11) by PCR cloning using WSSV genomic DNA as the template. The primers used for PCR are listed in Table 2. For DNA transfection, Sf9 insect cells were
seeded onto a six-well plate (8⫻ 105cells/well) and grown overnight. Plasmids
containing the appropriate genes (including the empty vector) were transfected into the Sf9 cells for 16 to 18 h and then heat shocked (a 42°C water bath for 30 min). At 6 h after the heat shock, the cells were washed with 1⫻ PBS and lysed
in 100l of NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1%
NP-40) supplemented with a protease inhibitor cocktail tablet. The lysis proce-dure was carried out on ice for 10 min with occasional shaking. The lysate was
centrifuged at 12,000⫻ g for 5 min, and an aliquot of the supernatant (10 l) was
reserved for Western blot analysis to confirm the expression of the transfected
genes. The remaining supernatant (90l) was then incubated with 15 l of
anti-FLAG M2 affinity gel (Sigma) at 4°C overnight with rotation. The gel was
then washed five times in 150l of NP-40 lysis buffer. Aliquots of the total cell
lysates and immunoprecipitated complexes were separated by 15% SDS-PAGE and transferred to a polyvinylidene difluoride membrane. V5-tagged fusion pro-teins were detected with rabbit anti-V5 antibody and goat anti-rabbit IgG-HRP conjugate (Sigma). FLAG-tagged VP51A and VP26 proteins were detected with mouse anti-FLAG monoclonal antibody (Sigma) and goat anti-mouse IgG-HRP conjugate (Sigma).
Yeast two-hybrid assay. Protein-protein interaction assays were performed using a commercial yeast two-hybrid system, Matchmaker 3 (Clontech), accord-ing to the manufacturer’s protocol. The bait plasmid, pGBK-VP51A, was con-structed by cloning the PCR-amplified, full-length VP51A gene into the SmaI/ BamHI sites of pGBKT7 (Clontech) in frame with the GAL4 DNA binding domain. The prey plasmids, pGAD-VP26 and pGAD-VP28, were constructed by cloning the PCR-amplified, full-length VP26 and VP28 genes, respectively, into the EcoRI/XhoI sites of pGADT7 (Clontech) in frame with the GAL4 activation domain. The PCR primer sequences are listed in Table 2. For the protein-protein interaction assay, Saccharomyces cerevisiae strain AH109 cells were cotrans-formed with the bait and prey plasmids using the lithium acetate method and plated on selective agar. The proteins were tested for autoactivation by cotrans-forming their respective plasmids with an empty prey or bait plasmid. For the positive control, the AH109 cells were cotransformed with plasmids pGBK-53 (p53) and pGAD-RecT (simian virus 40 large T antigen). Following transforma-tion, the AH109 cells were plated onto synthetic defined (SD) medium lacking leucine (Leu) and tryptophan (Trp) to verify that both of the transformed plasmids were present. To select for yeast that contained interacting proteins, colonies that carried both plasmids were then plated onto SD medium lacking Leu, Trp, and histidine (His) and also onto SD medium lacking Leu, Trp, His,
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
and adenine (Ade) in the presence of 5-bromo-4-chloro-3-indolyl-␣- D-galacto-pyranoside (X-␣-Gal) (Sigma).
Indirect immunofluorescence assays of WSSV-infected shrimp hemocytes.
Hemolymph was collected from healthy P. monodon shrimp and from WSSV-infected shrimp at 72 h p.i. using a syringe that contained cold modified Alsever solution (15). The hemocytes were placed on cover glasses, washed with PBS, and fixed in paraformaldehyde (4% in PBS) for 10 min at 4°C. After acetone treatment (3 min on ice), the hemocytes were incubated with blocking buffer (as
described above) for 16 h at 4°C. The hemocytes were then treated with 500⫻
PBS-diluted polyclonal rabbit anti-VP51A C-terminal antibody, 300⫻ PBS-di-luted polyclonal rat anti-VP26 antibody, or both (16 h at 4°C). Next, the cells were washed with PBST (three washes of 10 min each) and reacted with either
1,000⫻ PBS-diluted Cy3 dye-conjugated donkey anti-rabbit IgG antibody,
fluo-rescein isothiocyanate (FITC)-conjugated donkey anti-rat IgG antibody (1:500 in PBS; Jackson ImmunoResearch), or both for 2 h at room temperature. Coun-terstaining of the nucleus was performed with DAPI. After being washed three times with PBST (10 min each time), the cover glasses were wet mounted and the fluorescent signals were examined as described above.
RESULTS
Temporal-transcription analysis of vp51A.
The expression
profiles of vp51A in the gills of P. monodon at various stages of
WSSV infection were analyzed by RT-PCR (Fig. 1). The vp51A
transcript was first detected at 6 h p.i. By comparison, the
immediate-early gene ie1 and the early gene dnapol were both
transcribed as early as 2 h p.i. and continued to be found to
60 h p.i. The transcript of the WSSV envelope protein gene
vp28 was also detected at 2 h p.i. The actin control confirmed
the quantity of the total RNA templates (Fig. 1E), and another
control using a WSSV genomic-DNA-specific primer pair,
IC-F2/IC-R3 (16), derived from an intergenic region of the WSSV
genome, confirmed that there was no WSSV DNA
contami-nation (data not shown). From these data, we conclude that
vp51A is a gene that is expressed in the late stage of WSSV
infection.
Mapping the 5
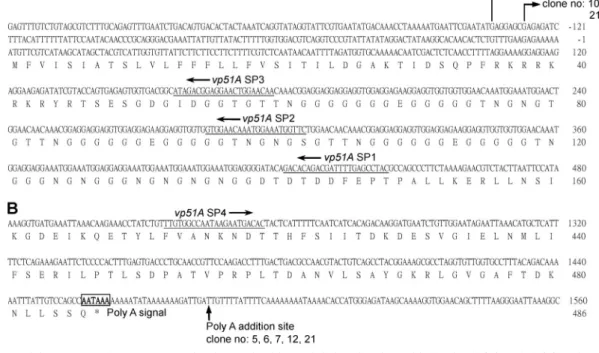
ⴕ and 3ⴕ ends of the vp51A transcript by
RACE.
The 5
⬘ and 3⬘ ends of the vp51A transcripts were
de-termined by the RACE method. The 5
⬘ RACE products were
cloned into the pGEM-T Easy vector, and seven clones were
randomly chosen for sequencing with the results shown in Fig.
2A. Two of the randomly selected 5
⬘ RACE products had their
5
⬘ termini at nucleotide residue ⫺135 (G) and five at ⫺128 (G)
(relative to the A in the translation initiation codon, ATG,
which is defined as
⫹1 [A]) (Fig. 2A). These results suggest
that for WSSV vp51A, there are two candidate transcription
initiation sites, located at nucleotide residues
⫺135 (G) and
⫺128 (G), respectively. The sequence around ⫺128 (G) did
not match any of the known WSSV or baculovirus start site
consensus motifs, and its biological meaning (if any) remains
unclear. Conversely, the sequence around the transcriptional
initiation site at
⫺135 (G) (ATGAG) was a close match to the
WSSV late-gene transcriptional initiation site consensus motif
(ATNAC) (20). We also note that there was an A/T-rich region
upstream of this site. In the 3
⬘ region of vp51A, the
polyaden-ylation signal (AATAAA) overlapped with the stop codon.
Sequence analysis of the cloned 3
⬘ RACE products revealed
that the poly(A) tail addition site was located 22 bp
down-stream of the polyadenylation signal. Like many other WSSV
genes (20), vp51A also had oligo(T) stretches downstream of
the poly(A) tail addition site (Fig. 2B).
Localization of VP51A in the virion.
In this study, we used a
protein fractionation method developed by Tsai et al. (31) to
determine whether VP51A is located in the envelope, the
teg-ument, or the nucleocapsid. Using Western blot analysis of the
eight different fractions of the WSSV virion proteins, VP51A’s
profile was compared to the profiles of known envelope,
teg-ument, and nucleocapsid proteins (VP28, VP26, and VP51C,
respectively) (Fig. 3). The results show that in the 1% Triton
X-100-treated preparations, VP51A was similar to the
enve-lope protein VP28 in that it was almost completely soluble in
both the presence and absence of NaCl. The provisional
con-clusion that VP51A was therefore an envelope protein was
then confirmed using an immunogold assay and IEM
observa-tion. When a gold-labeled secondary antibody was used in
conjunction with an anti-VP51A antiserum derived from a
C-terminal-region fragment (aa 251 to 486), gold particles
were observed on the intact WSSV virions (Fig. 4A) but not on
the tegument layer (Fig. 4B) or on the nucleocapsid surface
(Fig. 4C). Control experiments showed that no gold particles
were found on the envelopes of WSSV virions when normal
rabbit serum was used as the primary antibody (Fig. 4D).
Collectively, these results confirmed that VP51A is a WSSV
envelope protein.
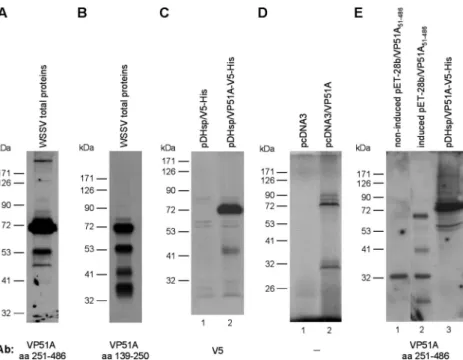
Molecular mass of the VP51A protein.
Western blot analysis
of the WSSV virion proteins using an antibody to a VP51A
C-terminal fragment (aa 251 to 486) detected not only the
theoretically predicted signal around 51 kDa, but also a major
band at
⬃72 kDa (Fig. 5A). This unexpected result was
checked using an antibody against the midsequence region (aa
139 to 250) of the VP51A coding region. The midsequence
antibody detected the major band at
⬃72 kDa, as well as
several other, smaller bands (Fig. 5B) that presumably resulted
from posttranslational processing. The major 72-kDa band was
FIG. 1. Time course analysis of vp51A transcripts by RT-PCR. (A
to E) Total RNAs were extracted from the gills of WSSV-infected
shrimp and subjected to RT-PCR analysis with the indicated primers.
Shrimp actin 2 was included as a template control. The lane headings
show times postinfection (hours). M, 100-bp DNA ladder. The arrows
indicate the size of the amplicon for each gene.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
also detected using both V5 tagging (Fig. 5C, lane 2) and
isotope labeling (Fig. 5D, lane 2). Figure 5E (lane 2) shows
Western blots for a truncated form of vp51A (aa 51 to 486) that
omitted the transmembrane domain in the N-terminal region
and was expressed in E. coli cells. The molecular mass of this
truncated, recombinant VP51A was about 68 kDa, which is a
little lower than the molecular mass of the full-length VP51A
that was overexpressed in Sf9 cells (Fig. 5E, compare lanes 2
and 3). Since no glycosylation can occur in the E. coli BL21
expression system, this result suggests that the 72-kDa band of
VP51A was also not due to glycosylation. Although it is
un-usual for the structural proteins of animal viruses to remain
unglycosylated, this finding for VP51A is consistent with
pre-vious reports that none of the WSSV structural proteins are
glycosylated (35, 44). We therefore tentatively conclude that
the discrepancy between the apparent molecular mass (72
kDa) and the calculated molecular mass (51.5 kDa) of VP51A
must be due to the abundance of negatively charged residues
(14%), which would have the effect of retarding the protein’s
migration through the gel (4, 7).
Membrane topology of VP51A.
A hydrophobicity profile
gen-erated using Kyte-Doolittle hydropathy plot analysis showed a
FIG. 2. Partial sequences of WSSV vp51A showing nucleotides and deduced amino acids for the 5
⬘ (A) and 3⬘ (B) regions. The primers used
for 5
⬘ RACE and 3⬘ RACE (vp51A SP1, vp51A SP2, vp51A SP3, and vp51A SP4) are underlined. The bent arrows indicate the transcriptional start
sites as revealed by sequencing seven randomly chosen 5
⬘ RACE clones. The polyadenylation signal (AATAAA) is boxed and in boldface. The
poly(A) addition site occurs 22 bp downstream of the polyadenylation signal and is indicated by a vertical arrow.
FIG. 3. Determination of VP51A’s location in the WSSV virion.
Intact WSSV virions were subjected to detergent and NaCl treatment
as indicated. After fractionation, the pellet (P) and supernatant
(S) fractions were separated on SDS-PAGE and detected by Western
blotting to produce profiles that are characteristic of envelope,
tegu-ment, and nucleocapsid proteins (31). Three representative WSSV
structural proteins are shown for comparison. Lane V is the untreated
purified virus.
FIG. 4. Localization of VP51A in the WSSV virion by immunogold
assay using a rabbit anti-VP51A C-terminal antibody probe followed
by a gold-labeled secondary antibody. (A to C) IEM images of purified
WSSV virions (A), unenveloped virions (i.e., the
tegument-nucleocap-sid structure) (B), and the viral nucleocaptegument-nucleocap-sids (C). (D) IEM of WSSV
virions, with preimmune rabbit serum used as the probe instead of the
primary antibodies. The gold particle signals (arrows) were detected
only on the enveloped virion. Scale bars, 100 nm.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
high hydrophobicity in the N-terminal region of VP51A (data not
shown), while sequence analysis of VP51A using the TMHMM
program predicted that VP51A encodes a transmembrane helix
between aa 2 and 24 (data not shown). To confirm these
pre-dictions, a recombinant VP51A fusion protein (rVP51A-V5)
with a V5 tag on its C terminus was subjected to indirect
immunofluorescence assays in transfected Sf9 cells. In cells
that were treated with Triton X-100 to render them permeable
to the anti-V5 antibody, the full-length rVP51A-V5 was
de-tected both in the plasma membrane region and in the
cyto-plasm (Fig. 6A, top). In the nonpermeabilized cells, however,
even though the rVP51A-V5 could no longer be detected in
the cytoplasm, it was still detected on the outside surface of the
plasma membrane (Fig. 6A, bottom). We conclude that the
C-terminal region of the protein must therefore be located
outside the cell membrane, because otherwise the V5 tag
would not have been accessible to the anti-V5 antibody. This
conclusion was further confirmed by conducting a similar assay
for PmSTAT, a shrimp protein that is expressed in both the
nucleus and cytoplasm of Sf9 cells (17). We found that in the
absence of Triton X-100, no positive PmSTAT signals could be
detected (data not shown). Taken together, these data suggest
that VP51A is a type II transmembrane protein. A schematic
of the proposed transmembrane topology of the V5-tagged
VP51A is shown in Fig. 6B.
Virion topology of VP51A, VP26, and VP28.
The topology of
these three structural proteins in the WSSV virion was
inves-tigated by using trypsin to distinguish between proteins that
were accessible to proteolysis and those that were protected
from digestion by the lipid bilayer. WSSV virions were either
left untreated or treated with trypsin in the absence or
pres-FIG. 5. Molecular masses of the VP51A protein. (A and B) Total proteins from WSSV virions were probed with antibodies to either the
C-terminal (aa 251 to 486) (A) or midsequence (aa 139 to 250) (B) region of VP51A. (C) Western blotting using anti-V5 antibody to detect
transiently expressed V5-tagged VP51A in Sf9 cells (lane 2). Lane 1 is the empty vector. (D) Coupled in vitro transcription and translation products
of the empty vector (lane 1) and plasmids containing the VP51A gene (lane 2). (E) Total proteins of IPTG-induced (lane 2) and noninduced (lane
1) pET-28b/VP51A
51-486-transformed E. coli BL21 and pDHsp/VP51A-V5-His-transfected Sf9 cells (lane 3) probed with anti-VP51A C-terminal
antibody.
FIG. 6. Membrane topology of WSSV VP51A. (A) V5-tagged
recombinant VP51A was transiently expressed in Sf9 cells that were
fixed with paraformaldehyde and permeabilized with Triton X-100
(a to d) or not permeabilized (e to h). rVP51A-V5 was visualized
with rabbit anti-V5 antibody and Cy3-conjugated donkey anti-rabbit
IgG antibody (a and e). Nuclei were visualized by counterstaining
them with DAPI (b and f). Images c and g show the merged Cy3 and
DAPI signals. The last column (d and h) shows the merged signal
overlaid with the bright-field images of the corresponding cells.
Scale bar, 10
m. (B) Schematic of the proposed transmembrane
topology of VP51A.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
ence of the detergent Triton X-100, and the digested products
were analyzed by Western immunoblotting using antibodies
against the VP51A C terminus (aa 251 to 486) or against the
full length of VP26 or VP28. As expected, the VP51A antibody
recognized the VP51A 72- and 51-kDa proteins in the
un-treated virions (Fig. 7, lane 1). However, after digestion with
trypsin in the absence or presence of Triton X-100, the 72-kDa
protein was no longer detected, even though the 51-kDa
pro-tein was still present (Fig. 7, lanes 2 and 3). We note that the
amount of the 51-kDa protein was only slightly decreased in
the presence of Triton X-100 (Fig. 7, lane 3), which suggests
that the 51-kDa VP51A protein was still protected from trypsin
digestion even after the envelope was removed. Similar
immu-noblotting results were obtained with the antibody against the
VP51A midsequence (data not shown). It is not clear why
the 51-kDa VP51A was not degraded by trypsin digestion. The
treated virions were also subjected to Western blotting to
de-tect the envelope protein VP28 and the tegument protein
VP26. VP28 was digested into two bands in the absence of
Triton X-100 and completely digested in the presence of
Tri-ton X-100 (Fig. 7, lanes 4 to 6). VP26 was digested only in the
presence of Triton X-100 (Fig. 7, lanes 7 to 9). Similar results
for VP26 and VP28 were also demonstrated by Tsai et al. (31).
VP51A interacts with VP26.
Interactions between VP51A
and two major WSSV structural proteins, VP26 and VP28,
were investigated using a coimmunoprecipitation assay in
which FLAG-tagged VP51A (VP51A-FLAG)/V5-tagged VP26
(VP26-V5), or VP51A-FLAG/V5-tagged VP28 (VP28-V5)
was coexpressed in Sf9 insect cells. As shown in Fig. 8A, a, all
three of these inputs, VP26-V5, VP28-V5, and VP51A-FLAG,
were successfully expressed in the Sf9 cells, and a pilot
exper-iment confirmed that the VP51A-FLAG proteins could be
efficiently precipitated by the anti-FLAG antibody (data not
shown). In the coimmunoprecipitation assays, VP26-V5 was
coimmunoprecipitated with VP51A-FLAG while VP28-V5
was not (Fig. 8A, b, lanes 3 and 5). A reverse experiment using
FLAG-tagged VP26 and V5-tagged VP51A produced the
same results (data not shown). The binding specificity of
VP51A-FLAG with anti-FLAG M2 affinity gel was
recon-firmed by subjecting VP51A-FLAG protein to reaction with
anti-hemagglutinin antibody-conjugated beads (data not shown).
From these results, we conclude that the interaction between
VP51A and VP26 is specific and independent of the tags. The
interaction between VP51A and VP26 was further confirmed
by a yeast two-hybrid assay. Image a in Fig. 8B shows that all
of the cotransformants were able to grow on the SD/
⫺Leu/
⫺Trp plate, and all of these constructs were also able to be
expressed successfully in yeast cells (data not shown). The
FIG. 7. Western blot analysis showing trypsin digestion of VP51A
and two representative proteins, VP28 (envelope) and VP26
(tegu-ment), in intact and detergent-treated virions. VP51A was detected
using the antibody to its C terminus (aa 251 to 486).
FIG. 8. VP51A interacts with VP26, but not with VP28. (A)
Coim-munoprecipitation of V5-tagged VP26 or VP28 with FLAG-tagged
VP51A from transfected cells. Sf9 cells were transfected with plasmids
expressing V5-tagged VP26, V5-tagged VP28, FLAG-tagged VP51A,
or empty plasmid (vector) as indicated. (a) At 6 h after heat shock, the
cell lysates were harvested and separated by SDS-PAGE, and input
expression was confirmed by Western blotting (blot) using either
anti-V5 antibody or anti-FLAG antibody as a probe. The arrows
indi-cate the expressed V5-tagged VP26, V5-tagged VP28, and
FLAG-tagged VP51A. (b) The cell lysates were immunoprecipitated (I.P.)
with anti-FLAG M2 affinity resins, and then the immunoprecipitated
complexes were subjected to Western blot analysis with an anti-V5
antibody probe. (B) The yeast two-hybrid results confirmed that
VP51A specifically interacted with VP26, but not with VP28. (a) Yeast
growth on medium lacking both Leu and Trp indicated the presence of
each respective pair of plasmids. (b and c) Yeast growth on
low-stringency (
⫺Leu/⫺Trp/⫺His) and high-stringency (⫺Leu/⫺Trp/
⫺His/⫺Ade) media, respectively. The blue signal in image c is due to
the presence of X-
␣-Gal. The positive signals represent
protein-pro-tein interactions. (C) Intracellular localizations of VP51A and VP26 in
WSSV-infected P. monodon hemocytes by confocal microscopy. (a)
VP51A was visualized using rabbit anti-VP51A antibody and
Cy3-conjugated donkey anti-rabbit IgG antibody. (b) VP26 was visualized
with rat anti-VP26 antibody and FITC-conjugated donkey anti-rat IgG
antibody. (c) Nuclei were visualized by counterstaining them with
DAPI. (d) Merged Cy3, FITC, and DAPI signals. Scale bar, 10
m.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
VP51A protein and the empty vector (pGBK-VP51A/
pGADT7) did not induce reporter gene activation (Fig. 8B, b
and c), from which we conclude that VP51A does not have
autonomous activation ability. Growth on the low-stringency
(SD/
⫺Leu/⫺Trp/⫺His) and high-stringency (SD/⫺Leu/⫺Trp/
⫺His/⫺Ade/X-␣-Gal) plates was observed only when the yeast
was transformed with pGBK-VP51A/pGAD-VP26 or with the
positive control (Fig. 8B, b and c). Again, these results showed
that VP51A interacts with VP26 but does not interact with
VP28.
In order to assess the intracellular distribution of VP51A
and VP26 in WSSV-infected P. monodon cells, hemocytes were
collected at a late stage of infection (72 h p.i.). The
virus-infected cells were fixed, permeabilized, stained for rabbit
anti-VP51A and rat anti-VP26 antibodies, and analyzed by confocal
microscopy. As shown in Fig. 8C, strong punctate signals from
both VP51A (image a) and from VP26 (image b) were
ob-served in the plasma membrane and cytoplasm, and the
merged image shows that these signals were almost completely
superimposed (image d). No signals were detected in the
un-infected P. monodon hemocyte controls (data not shown).
Clearly, this suggests that both VP51A and VP26 colocalized
to the same subcellular locations.
VP26 links VP51A and VP28.
A previous study showed that
VP26 interacts with the most abundant envelope protein,
VP28 (44), and this result was reconfirmed here by a
coimmu-noprecipitation assay with V5-tagged VP28 and FLAG-tagged
VP26 expressed in Sf9 insect cells. The two images in Fig. 9A,
a, show that both VP26-FLAG and VP28-V5 were successfully
expressed in Sf9 cells. Complexes consisting of VP28-V5 plus
VP26-FLAG were coimmunoprecipitated by anti-FLAG M2
affinity gel and detected by Western blotting with anti-V5
an-tibody (Fig. 9A, b, lane 2). As discussed above, we had already
established that VP51A interacts with VP26 but not with VP28
(Fig. 8A and B), so we next designed a coimmunoprecipitation
experiment to investigate the interactions among all three of
these proteins. Expression of VP51A-FLAG, VP26-V5, and
VP28-V5 in the cotransfected Sf9 cells was confirmed by
West-ern blot analysis with anti-FLAG and anti-V5 antibodies (Fig.
9B, a). The putative VP51A-VP26-VP28 complex was
immu-noprecipitated by using an anti-FLAG M2 affinity gel against
the FLAG epitope on VP51A, and the presence of both VP26
and VP28 in the complex was demonstrated by Western
blot-ting using anti-V5 antibody (Fig. 9B, b). For the control, cells
were cotransfected with an empty V5 vector instead of the
VP26-V5 plasmid, with the result that neither VP26 nor VP28
was observed in the Western blot analysis (Fig. 9B, b, lane 1).
We therefore conclude that VP26 plays a key role in the
for-mation of a VP51A-VP26-VP28 ternary complex.
DISCUSSION
Our results suggest that the major VP51A protein species is
unglycosylated and that its apparent mass of 72 kDa is in fact
due to a high proportion of charged residues that retard the
protein’s migration in the gel. We note that two earlier studies
identified only a single VP51A (or VP52A) band in the region
of
⬃51 kDa, and neither study reported the major 72-kDa
band. In one study (32), the 72-kDa region of the gel was
dominated by a very high-intensity band of crayfish
hemocya-nin that obscured all of the neighboring bands. In the other
study (44), the 72-kDa VP51A protein was not reported, even
though the entire crayfish was used as the source material and
the problem of hemocyanin contamination was solved by using
a different purification method. We found, however, that by
using the same source tissues and protocols as Xie et al. (44),
FIG. 9. VP51A, VP26, and VP28 form a ternary complex. (A) (a)
V5-tagged VP28 was transiently coexpressed in Sf9 cells with either the
empty plasmid (vector) or a plasmid that expressed FLAG-tagged
VP26. At 6 h after heat shock, the cell lysates were harvested and input
expression was monitored using Western blot (blot) analysis with
ei-ther anti-V5 or anti-FLAG antibody as the probe. (b) Lysates from
cells cotransfected with V5-tagged VP28 and vector or with V5-tagged
VP28 and FLAG-tagged VP26 were immunoprecipitated (I.P.) with
anti-FLAG M2 affinity resins. The immunoprecipitated complexes
were then subjected to Western blot analysis with an anti-V5 antibody
probe. Bands corresponding to the V5-tagged VP28 and nonspecific
bound IgG are indicated. (B) (a) Sf9 cells were cotransfected with
plasmids expressing FLAG-tagged VP51A, tagged VP28, and
V5-tagged VP26. For the control, the VP26 plasmid was replaced by empty
plasmid (vector). Expression of each input was confirmed by Western
blot analysis of the cell lysates with the indicated antibodies. (b) At 6 h
after heat shock, the cell lysates were harvested and
immunoprecipi-tated with anti-FLAG M2 affinity resins. The immunoprecipitates were
then subjected to Western blot analysis with an anti-V5 antibody
probe, and both V5-tagged VP26 and VP28 and the nonspecific bound
IgG were detected.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
we were able to detect the 72-kDa VP51A in the purified
virions, and in fact, Western blotting showed that the smaller
species of VP51A (i.e., 51 kDa and others) (Fig. 5A and B)
were consistently present as well (data not shown). Since the
51-kDa protein could be detected by both the anti-VP51A
midsequence and C-terminal antibodies (Fig. 5A and B) and
since the peptide sequences deduced from the tandem mass
spectrometry data of Tsai et al. (32) matched only the
C-terminal region of VP51A (J.-M. Tsai, personal
communica-tion), we hypothesize that the 51-kDa band was in fact
pro-duced by N-terminal truncation of the full-length protein.
Further, since Western blotting consistently produced the
same protein profiles regardless of the purification method
that was used, we infer that VP51A is sensitive only to
site-specific proteases and is otherwise not easily degraded. Lastly,
we note that since the 72-kDa, 51-kDa, and some other,
smaller VP51A proteins are all readily detected in the WSSV
virion (Fig. 5A and B), it seems likely that all of these forms are
essential components of the viral particle, and they may all
participate at various stages of virus morphogenesis.
We note that there is similar evidence for the proteolytic
processing of at least one other WSSV structural protein. Xie
et al. (44) identified five different species of VP150 in purified
WSSV virions, and the smallest of these proteins had the same
apparent mass as VP53A of Tsai et al. (32). Since these two
studies used different purification methods, and since VP53A
and the 53-kDa species of VP150 were both identified as
prod-ucts of the same open reading frame (WSV011 in the China
isolate; WSSV067 in the Taiwan isolate), we hypothesize that
WSSV VP150 may also be subjected to proteolytic processing
by a specific virus or host protease.
The ability of WSSV to replicate successfully in a wide range
of crustacean hosts suggests that a common crustacean
pro-tease might be involved in VP51A proteolysis. Furin is one
possible candidate. This extensively studied cellular proprotein
convertase is used by many cleavable viral envelope proteins
(23, 28), and in WSSV VP51A, we found two overlapping
instances of the furin recognition motif RXK/RR near the N
terminus (
36RKRRKR
41). However, our pilot experiments
suggested that furin is not in fact involved in the proteolytic
processing of the VP51A 72-kDa protein molecule (data not
shown). Thus, for the moment, the cellular or viral factors
involved in VP51A cleavage and the events governing the
traf-ficking of the precursors remain unknown.
The protein domains exposed on the surfaces of viruses play
fundamental roles in infection by binding to cell receptors,
promoting cell fusion processes, or interacting with elements
of the host immune system (1, 24, 26). Thus, the determination
of a protein’s membrane topology is an important first step
toward understanding its function. Topological predictions
made by the program TMHMM suggested that VP51A
en-codes a transmembrane helix near its N terminus and that the
entire C-terminal region of the protein is on the outside
sur-face of the plasma membrane. Other membrane topology and
hydropathic prediction programs yielded similar results (data
not shown). Immunofluorescence assays performed on
recom-binant VP51A fusion protein expressed in Sf9 cells confirmed
these predictions; that is, in the intact Sf9 cells, the C-terminal
V5 epitopes of the recombinant VP51A could be detected only
on the outer surfaces of the cell membranes (Fig. 6A). In the
WSSV virions, VP51A topology was investigated using two
experimental approaches. First, an antibody to the VP51A C
terminus directly recognized VP51A on the outer surfaces of
the envelopes of purified intact virions (Fig. 4A), and second,
trypsin digestion of the VP51A 72-kDa protein in the absence
of detergent (Fig. 7, lane 2) also confirmed that the C terminus
of VP51A was exposed on the outside of the virion.
Interactions between structural proteins are common in
en-veloped viruses, but to date, protein-protein interactions have
been reported only in four major WSSV virion proteins, VP24,
VP26, VP28, and WSV010 (3, 43, 44). In the present study, we
found that VP51A formed a ternary complex with VP26 and
VP28, with VP26 acting as the linker protein (Fig. 8 and 9).
The functional significance of this complex is unknown, but we
note that VP26 and VP28 are both major WSSV structural
proteins. VP28 is an envelope protein that is implicated in cell
attachment during infection (12, 34, 46), and although the
location of VP26 is still disputed (e.g., by Tang et al. [30]), most
of the recent evidence (31, 42, 44) (Fig. 7) suggests that it is in
fact a tegument or linker protein. In the virion, the
hydropho-bic N-terminal region of VP26 may be anchored in the
enve-lope, while its C terminus is bound to the nucleocapsid (42).
Based on the fact that VP26 binds with actin, Xie and Yang
(42) further hypothesized that it may be instrumental in
traf-ficking the WSSV nucleocapsid into the host nucleus via the
cytoskeleton. Given the propensity of VP51A to form a
com-plex with VP26 and VP28, it will be interesting to investigate
the extent to which VP51A may contribute to the functionality
of either or both of these two major WSSV structural proteins.
ACKNOWLEDGMENTS
This investigation was supported financially by National Science
Council grants (NSC95-2313-B-212-006-MY2,
NSC96-2317-B-002-015, and NSC96-2317-B-002-020).
We are indebted to Paul Barlow for his helpful criticism.
REFERENCES1. Campadelli-Fiume, G., M. Amasio, E. Avitabile, A. Cerretani, C. Forghieri,
T. Gianni, and L. Menotti.2007. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol. 17:313–326. 2. Chazal, N., and D. Gerlier. 2003. Virus entry, assembly, budding, and
mem-brane rafts. Microbiol. Mol. Biol. Rev. 67:226–237.
3. Chen, J., Z. Li, and C.-L. Hew. 2007. Characterization of a novel envelope protein WSV010 of shrimp white spot syndrome virus and its interaction with a major viral structural protein VP24. Virology 364:208–213.
4. Dunker, A. K., and R. R. Rueckert. 1969. Observations on molecular weight determinations on polyacrylamide gel. J. Biol. Chem. 244:5074–5080. 5. Escobedo-Bonilla, C. M., V. Alday-Sanz, M. Wille, P. Sorgeloos, M. B.
Pensaert, and H. J. Nauwynck.2008. A review on the morphology, molecular characterization, morphogenesis and pathogenesis of white spot syndrome virus. J. Fish Dis. 31:1–18.
6. Frohman, M. A., M. K. Dush, and G. R. Martin. 1988. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA 85:8998–9002. 7. Graceffa, P., A. Jancso, and K. Mabuchi. 1992. Modification of acidic
resi-dues normalizes sodium dodecyl sulfate-polyacrylamide gel electrophoresis of caldesmon and other proteins that migrate anomalously. Arch. Biochem. Biophys. 297:46–51.
8. Kim, C. S., Z. Kosuke, Y. K. Nam, S. K. Kim, and K. H. Kim. 2007. Protection of shrimp (Penaeus chinensis) against white spot syndrome virus (WSSV) challenge by double-stranded RNA. Fish Shellfish Immunol. 23: 242–246.
9. Krogh, A., B. Larsson, G. von Heijne, and E. L. Sonnhammer. 2001. Pre-dicting transmembrane protein topology with a hidden Markov model: ap-plication to complete genomes. J. Mol. Biol. 305:567–580.
10. Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132.
11. Leu, J.-H., Y.-C. Kuo, G.-H. Kou, and C.-F. Lo. 2008. Molecular cloning and characterization of an inhibitor of apoptosis protein (IAP) from the tiger shrimp, Penaeus monodon. Dev. Comp. Immunol. 32:121–133.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
12. Li, L. J., J. F. Yuan, C. A. Cai, W. G. Gu, and Z. L. Shi. 2006. Multiple envelope proteins are involved in white spot syndrome virus (WSSV) infec-tion in crayfish. Arch. Virol. 151:1309–1317.
13. Li, Z., Q. Lin, J. Chen, J. L. Wu, T. K. Lim, S. S. Loh, X. Tang, and C.-L.
Hew.2007. Shotgun identification of the structural proteome of shrimp white
spot syndrome virus and iTRAQ differentiation of envelope and nucleocap-sid subproteomes. Mol. Cell Proteomics. 6:1609–1620.
14. Liang, Y., J. Huang, X.-L. Song, P.-J. Zhang, and H.-S. Xu. 2005. Four viral proteins of white spot syndrome virus (WSSV) that attach to shrimp cell membranes. Dis. Aquat. Organ. 66:81–85.
15. Lin, S.-T., Y.-S. Chang, H.-C. Wang, H.-F. Tzeng, T.-Z. Chang, J.-Y. Lin,
C.-H. Wang, C.-F. Lo, and G.-H. Kou.2002. Ribonucleotide reductase of shrimp white spot syndrome virus (WSSV): expression and enzymatic activity in a baculovirus/insect cell system and WSSV-infected shrimp. Virology
304:282–290.
16. Liu, W.-J., H.-T. Yu, S.-E. Peng, Y.-S. Chang, H.-W. Pien, C.-J. Lin, C.-J.
Huang, M.-F. Tsai, C.-J. Huang, C.-H. Wang, J.-Y. Lin, C.-F. Lo, and G.-H.
Kou.2001. Cloning, characterization and phylogenetic analysis of a shrimp
white spot syndrome virus (WSSV) gene that encodes a protein kinase. Virology 289:362–377.
17. Liu, W.-J., Y.-S. Chang, A. H.-J. Wang, G.-H. Kou, and C.-F. Lo. 2007. White spot syndrome virus annexes a shrimp STAT to enhance expression of the immediate-early gene ie1. J. Virol. 81:1461–1471.
18. Lo, C.-F., H.-C. Hsu, M.-F. Tsai, C.-H. Ho, S.-E. Peng, G.-H. Kou, and D. V.
Lightner.1999. Specific genomic DNA fragments analysis of different geo-graphical clinical samples of shrimp white spot syndrome virus. Dis. Aquat. Organ. 35:175–185.
19. Lo, C.-F., S.-E. Peng, Y.-S. Chang, and G.-H. Kou. 2005. White spot syn-drome—what we have learned about the virus and the disease, p. 421–433.
In P. J. Walker, R. G. Lester, and M. G. Bondad-Reantaso (ed.), Diseases in
Asian aquaculture V. Proceedings of the 5th Symposium on Diseases in Asian Aquaculture. Fish Health Section, Asian Fisheries Society, Manila, Philippines.
20. Marks, H., X.-Y. Ren, H. Sandbrink, M. C. W. van Hulten, and J. M. Vlak. 2006. In silico identification of putative promoter motifs of white spot syn-drome virus. BMC Bioinformatics 7:309. http://www.biomedcentral.com /1471–2105/7/309.
21. Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167–180.
22. Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2006. Herpesvirus assem-bly: a tale of two membranes. Curr. Opin. Microbiol. 9:423–429. 23. Nakayama, K. 1997. Furin: a mammalian subtilisin/Kex2p-like endoprotease
involved in processing of a wide variety of precursor proteins. Biochem. J.
327:625–635.
24. Rajca´ni, J.2003. Molecular mechanisms of virus spread and virion
compo-nents as tools of virulence. A review. Acta Microbiol. Immunol. Hung.
50:407–431.
25. Rajesh Kumar, S., V. P. Ishaq Ahamed, M. Sarathi, A. Nazeer Basha, and
A. S. Sahul Hameed.2008. Immunological responses of Penaeus monodon to DNA vaccine and its efficacy to protect shrimp against white spot syndrome virus (WSSV). Fish Shellfish Immunol. 24:467–478.
26. Reske, A., G. Pollara, C. Krummenacher, B. M. Chain, and D. R. Katz. 2007. Understanding HSV-1 entry glycoproteins. Rev. Med. Virol. 17:205–215. 27. Robalino, J., T. Bartlett, E. Shepard, S. Prior, G. Jaramillo, E. Scura, R. W.
Chapman, P. S. Gross, C. L. Browdy, and G. W. Warr. 2005. Double-stranded RNA induces sequence-specific antiviral silencing in addition to nonspecific immunity in a marine shrimp: convergence of RNA interference and innate immunity in the invertebrate antiviral response? J. Virol. 79: 13561–13571.
28. Rockwell, N. C., and J. W. Thorner. 2004. The kindest cuts of all: crystal structures of Kex2 and furin reveal secrets of precursor processing. Trends Biochem. Sci. 29:80–87.
29. Rout, N., S. Kumar, S. Jaganmohan, and V. Murugan. 2007. DNA vaccines
encoding viral envelope proteins confer protective immunity against WSSV in black tiger shrimp. Vaccine 25:2778–2786.
30. Tang, X., J. Wu, J. Sivaraman, and C.-L. Hew. 2007. Crystal structures of major envelope proteins VP26 and VP28 from white spot syndrome virus shed light on their evolutionary relationship. J. Virol. 81:6709–6717. 31. Tsai, J.-M., H.-C. Wang, J.-H. Leu, A. H.-J. Wang, Y. Zhuang, P. J. Walker,
G.-H. Kou, and C.-F. Lo.2006. Identification of the nucleocapsid, tegument, and envelope proteins of the shrimp white spot syndrome virus virion. J. Vi-rol. 80:3021–3029.
32. Tsai, J.-M., H.-C. Wang, J.-H. Leu, H.-H. Hsiao, A. H.-J. Wang, G.-H. Kou,
and C.-F. Lo.2004. Genomic and proteomic analysis of thirty-nine structural proteins of shrimp white spot syndrome virus. J. Virol. 78:11360–11370. 33. Tsai, M.-F., G.-H. Kou, H.-C. Liu, K.-F. Liu, C.-F. Chang, S.-E. Peng, H.-C.
Hsu, C.-H. Wang, and C.-F. Lo.1999. Long-term presence of white spot syndrome virus (WSSV) in a cultured shrimp population without disease outbreaks. Dis. Aquat. Organ. 38:107–114.
34. van Hulten, M. C. W., J. Witteveldt, M. Snippe, and J. M. Vlak. 2001. White spot syndrome virus envelope protein VP28 is involved in the systemic infection of shrimp. Virology 285:228–233.
35. van Hulten, M. C. W., M. Reijns, A. M. G. Vermeesch, F. Zandbergen, and
J. M. Vlak.2002. Identification of VP19 and VP15 of white spot syndrome virus (WSSV) and glycosylation status of the WSSV major structural pro-teins. J. Gen. Virol. 83:257–265.
36. Vaseeharan, B., T. Prem Anand, T. Murugan, and J. C. Chen. 2006. Shrimp vaccination trials with the VP292 protein of white spot syndrome virus. Lett. Appl. Microbiol. 43:137–142.
37. Vlak, J. M., J. R. Bonami, T. W. Flegel, G.-H. Kou, D. V. Lightner, C.-F. Lo,
P. C. Loh, and P. J. Walker.2004. Nimaviridae, p. 187–192. In C. M. Fauquet, M. A. Mayo, J. Maniloff, U. Desselberger, and L. A. Ball (ed.), Virus taxonomy. Eighth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA.
38. Wang, C.-H., C.-F. Lo, J.-H. Leu, C.-M. Chou, P.-Y. Yeh, H.-Y. Chou, M.-C.
Tung, C.-F. Chang, M.-S. Su, and G.-H. Kou.1995. Purification and genomic analysis of baculovirus associated with white spot syndrome (WSBV) of
Penaeus monodon. Dis. Aquat. Organ. 23:239–242.
39. Witteveldt, J., J. M. Vlak, and M. C. W. van Hulten. 2004. Protection of
Penaeus monodon against white spot syndrome virus using a WSSV subunit
vaccine. Fish Shellfish Immunol. 16:571–579.
40. Wu, C., and F. Yang. 2006. Localization studies of two white spot syndrome virus structural proteins VP51 and VP76. Virol. J. 3:76. http://www.virologyj .com/content/3/1/76.
41. Wu, W., L. Wang, and X. Zhang. 2005. Identification of white spot syndrome virus (WSSV) envelope proteins involved in shrimp infection. Virology 332: 578–583.
42. Xie, X., and F. Yang. 2005. Interaction of white spot syndrome virus VP26 protein with actin. Virology 336:93–99.
43. Xie, X., and F. Yang. 2006. White spot syndrome virus VP24 interacts with VP28 and is involved in virus infection. J. Gen. Virol. 87:1903–1908. 44. Xie, X., L. Xu, and F. Yang. 2006. Proteomic analysis of the major envelope
and nucleocapsid proteins of white spot syndrome virus. J. Virol. 80:10615– 10623.
45. Xu, J., F. Han, and X. Zhang. 2007. Silencing shrimp white spot syndrome virus (WSSV) genes by siRNA. Antivir. Res. 73:126–131.
46. Yi, G., Z. Wang, Y. Qi, L. Yao, J. Qian, and L. Hu. 2004. VP28 of white spot syndrome virus is involved in the attachment and penetration into shrimp cells. J. Biochem. Mol. Biol. 37:726–734.
47. Zhang, X., C. Huang, X. Tang, Y. Zhuang, and C.-L. Hew. 2004. Identifica-tion of structural proteins from shrimp white spot syndrome virus (WSSV) by 2DE-MS. Proteins 55:229–235.
48. Zhu, F. X., and Y. Yuan. 2003. The ORF45 protein of Kaposi’s sarcoma-associated herpesvirus is sarcoma-associated with purified virions. J. Virol. 77:4221– 4230.