國
立
交
通
大
學
應用化學研究所系
碩
士
論
文
以自發電化學反應簡易製備
大範圍矽奈米線陣列
A Facile Route to Fabrication of Large Area Silicon Nanowire Arrays via Spontaneous Electrochemical Reactions
研 究 生:劉高翔
指導教授:裘性天 教授
李紫原 教授
以自發電化學反應簡易製備大範圍矽奈米線陣列
學生:劉高翔 指導教授:裘性天 博士 李紫原 博士 國立交通大學應用化學研究所 摘要 矽奈米線以其半導體性質與發展為各式元件的極大潛力,受到學 術界注意。本論文報導一種簡單的方法,解決以固-液-氣法合成矽 奈米線的諸多缺點。可在常溫、常壓、大氣、水相條件下,不需外加 任何形式能量,即可利用自發的電化學反應,以硝酸銀 氫氟酸水溶 液在矽晶片表面大範圍地生成單結晶的矽奈米線陣列。為了進一步縮 小與均化線徑,同時讓陣列排列得更緻密整齊,矽晶片的表面被用濺 鍍和燒結的方式做出一層金奈米粒子。以此做為反應發生的起始點, 可得到線徑更細小、更一致、更緻密的產物。調整金奈米粒子的粒徑, 可間接控制砂奈米線的線徑。將反應液中的硝酸銀更換為硝酸銅或六 氯鉑酸鉀,可利用同樣的原理,在矽奈米線陣列表面修飾上一層銅或 鉑的奈米粒子,更增加其實際應用的可能性。A Facile Route to Fabrication of Large Area Silicon Nanowire Arrays via Spontaneous Electrochemical Reactions
Student: Kao-Hsiang Liu Advisor: Dr. Hsin-Tien Chiu Dr. Chi-Young Lee Institute of Applied Chemistry
National Chiao Tung University
Abstract
Owing to the semi-conductor properties and the great potential for developing various applied devices, there has been growing concern in academics about silicon nanowires (SiNWs) recently. In this thesis, a simple method via electroless metal deposition (EMD) has been reported. This method can circumvent the shortcomings of SiNWs synthesis based on the vapor-liquid-solid (VLS) mechanism. By employing the spontaneous electrochmical reactions with silver nitrate / hydrofluoric acid solution, large areas of single crystal SiNW arrays were produced on silicon wafer in aqueous solution under room temperature, standard pressure, and atmosphere conditions. In order to decrease the SiNWs’ diameter and improve the uniformity, silicon wafers coated with gold nanoparticles (AuNPs), which were prepared by sputtering and annealing, were used as the starting material. When the electrochemical reaction started on AuNPs, the products with smaller, tidier, and more consensus wire diameters were obtained. Replacing silver nitrate with cooper nitrate or potassium hexachloroplatinate in the reaction solution, copper or platinum nanoparticles were coated on SiNW arrays, which
誌謝 念大學部的時候,我認為做實驗首重嚴謹與邏輯。而攻讀碩士的 這兩年,我在裘性天博士與李紫原博士的指導下,學到創造力和想像 力對學術研究的重要性。感謝裘性天博士給學生完全自由的發揮空 間,跟老師天馬行空的討論,將會讓我在未來研究的路上深深懷念。 感謝李紫原博士對學生的細心與耐性,補足學生思緒不夠週延之處。 也感謝陳登銘博士於百忙之中擔任學生的口試委員,提出許多重要建 議使本論文更加完備。 感謝實驗室的伙伴們。宜萱學姊帶我開始進行本論文的實驗,之 後無數次的討論亦激發我許多想法。志豪學長和嘉興學長,好幾次幫 我照電子顯微鏡到深夜。裕煦學長、隆昇學長、焙蓀學姊、曜安學長、 子厚學長、文昭學姊,教我許多實驗的知識。同學亭凱、進興、煌凱 跟我一起努力。學弟暐翰和學妹宇婕打點實驗室諸多雜事,讓我在碩 二時能更專心實驗。清大的正得學長、鈺芬學姊、奕仁、旻橋也都在 儀器方面,給予很大的幫助。 感謝電資研和研二咖啡的朋友們,雖然對論文沒有直接的貢獻, 甚至這兩年間與你們相處的時間也不多,但是擁有這一群我一生中最 重要的摯友們,帶給我很大的精神力量。 感謝我的女朋友蘭蘭,即使分隔台北與新竹,仍然在任何情況下
不斷地支持我、鼓勵我,陪我走過這兩年所有的快樂與不快樂。 感謝榮茂美味的魯肉飯,能讓我忘記一個人在新竹的寂寞。感謝 克立瑪的鍾先生和 8K café 的婉婷,提供有好音樂、好咖啡和好朋友 的愉快的環境。 感謝我的家人,從小到大,這兩年是我第一次離家念書,家人的 關心,卻是不受距離限制的。精神與實質各方面的協助,讓我雖然人 在新竹,卻依然擁有一個跟家一樣舒適的求學環境。 最後,我想將這本論文獻給我的祖父。可惜在病魔纏身多年後, 仍然沒來得及看到我取得碩士學位。同時祝福我的祖母,希望您身體 安健。
目錄
第一章 緒論 ...1 第二章 實驗 ...6 2.1 試藥 ...6 2.2 實驗步驟 ...6 2.2.1 常溫常壓大氣水相下大範圍製備矽奈米線陣列 ...6 2.2.2 以金奈米粒子輔助控制矽奈米線生成 ...7 2.2.3 以自發電化學反應將金屬奈米粒子修飾於矽奈米線上 ...8 2.3 分析儀器及樣品製備 ...8 2.3.1 掃描式電子顯微鏡...8 2.3.2 X-光射線能量散佈分析儀 ...9 2.3.3 穿透式電子顯微鏡...9 2.3.4 高解析穿透式電子顯微鏡 ...9 2.3.5 拉曼光譜儀...9 第三章 結果與討論 ...11 3.1 常溫常壓大氣水相下大範圍製備矽奈米線陣列 ...11 3.1.1 產物形貌之 SEM 及 EDS 分析...113.1.2 產物形貌與晶型之 TEM、SAED 及 HRTEM 分析...13 3.1.3 反應機制的研究...14 3.1.4 討論...16 3.2 以金奈米粒子輔助控制矽奈米線生成 ...18 3.2.1 產物形貌之 SEM 及 EDS 分析...18 3.2.2 產物形貌與晶型之 TEM 及 HRTEM 分析 ...20 3.2.3 拉曼光譜分析...21 3.2.4 反應機制的研究...22 3.2.5 討論...23 3.3 以自發電化學反應將金屬奈米粒子修飾於矽奈米線上 ...25 3.3.1 產物形貌之 SEM 及 EDS 分析...25 3.3.2 產物形貌之 TEM 分析 ...27 3.3.3 反應機制的研究...28 3.3.4 討論...28 第四章 結論 ...30 第五章 參考文獻 ...32
表目錄
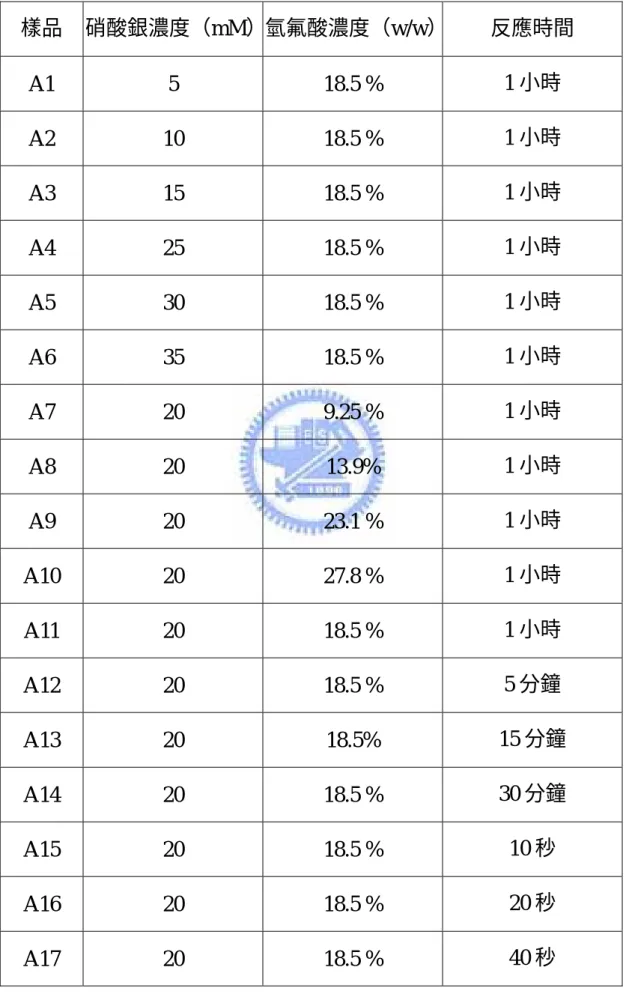
表 一 A 類樣品詳細製備條件 ...35
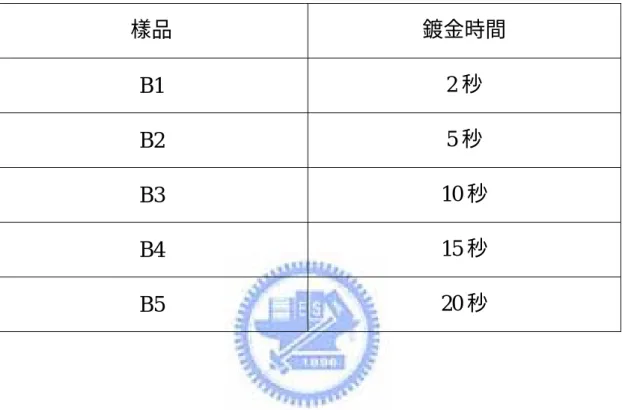
表 二 B 類樣品詳細製備條件...36
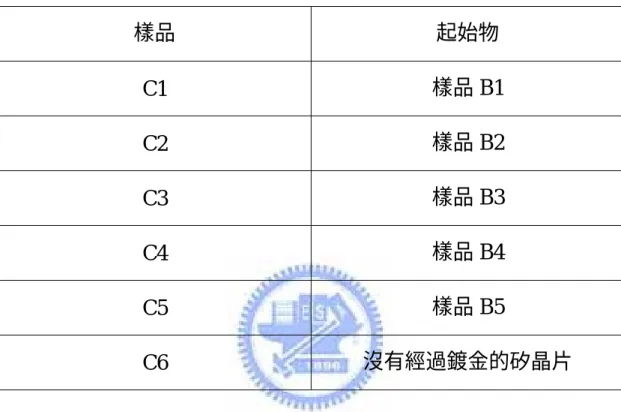
表 三 C 類樣品詳細製備條件...37
圖目錄
圖 一 (a)樣品 A1 的 SEM 影像 (b)樣品 A2 的 SEM 影像 ...39 圖 二 樣品 A6 的(a)側視 SEM 影像 (b)俯視 SEM 影像 ...40 圖 三 樣品 A8 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像.41 圖 四 樣品 A10 的 SEM 影像 ...42 圖 五 樣品 A11 的(a)低倍率 SEM 影像 (b)中倍率 SEM 影像 (c)
高倍率 SEM 影像 (d)EDS 圖譜 ...43 圖 六 反應時間對矽奈米線長度作圖 ...45 圖 七 銀沈積物的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像46 圖 八 樣品 A11 的(a)TEM 影像 (b)SAED 影像 (c)HRTEM
影像...47 圖 九 樣品 A15 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像
...48 圖 十 樣品 A16 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像 (c)
EDS 圖譜 ...49 圖 十一 (a)樣品 A17 的 SEM 影像 (b)矽奈米線頭部的 TEM 影
像...50 圖 十二 以自發電化學反應製備矽奈米線陣列之機制示意圖 ...51 圖 十三 (a)樣品 B1 (b)樣品 B2 (c)樣品 B3 (d)樣品 B4 (e)
樣品 B5 的 SEM 影像 (f)樣品 B5 的 EDS 圖譜...52 圖 十四 (a)樣品 C1 (b)樣品 C2 (c)樣品 C3 (d)樣品 C4 (e) 樣品 C5 (f)樣品 C6 的低倍率 SEM 影像 ...54 圖 十五 (a)樣品 C1 (b)樣品 C6 的高倍率 SEM 影像 (c)C 類樣品的 EDS 圖譜...56 圖 十六 (a)樣品 C1 (b)樣品 C2 (c)樣品 C3(d)樣品 C4 (e) 樣品 C5 的 TEM 影像 (f)樣品 C2 的 HRTEM 影像 ...57 圖 十七 鍍金時間對金奈米粒子粒徑與矽奈米線線徑作圖 ...59 圖 十八 C 類樣品的拉曼光譜圖...60 圖 十九 以金奈米粒子輔助控制矽奈米線生長機制示意圖 ...61 圖 二十 D 類與 E 類樣品的共同起始物 SEM 影像...62
圖 二十一 樣品 D1 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影 像...63
圖 二十二 樣品 D2 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影 像 (c)EDS 圖譜 ...64
圖 二十三 樣品 E1 的 SEM 影像...65
圖 二十四 樣品 E2 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影 像 (c)EDS 圖譜 ...66
圖 二十六 將矽晶片以樣品 D1 的條件進行反應後的 SEM 影像 ...68 圖 二十七 在矽奈米線的表面修飾銅或鉑的金屬奈米粒子的示意圖
...69 圖 二十八 總反應示意圖 ...70
第一章 緒論
近十餘年來,學術界專注於奈米科學的研究。主要的原因就是當 材料被縮小至奈米尺度時,其所展現出與塊材截然不同的特殊性質。 其中,特別是低維度的奈米材料吸引了最多的注意。在零維的奈米粒 子(nano particles)或量子點(quantum dots)在催化觸媒方面展現極 高的成就時[1,2],一維的奈米柱(nanorods)[3]、奈米線(nanowires) [4,5] 、奈米管(nanotubes)[4,6]、奈米帶(nanobelts)[7]等等愈來愈多樣 性的材料,也在各方面發展出實際應用的潛能。 一維奈米材料[8]在物理、化學、機械、電性、磁性、光學、量子、 傳導、運輸等特性,使它們有很大的機會做為奈米微機電元件的基本 組成。目前已經有多種金屬、碳材、氧化物、或其他材料的一維奈米 材料被製造及鑑定,其中亦有許多已經被實際製成場效應電晶體或其 他應用元件的例子[9-12]。 在各式各樣的材料中,矽的半導體性質佔了很大的優勢[13]。透過 熟知的製程,包括氧化、摻雜、蝕刻等等,可輕易讓矽材表現出所需 的性質,這也的確讓矽在半導體產業上舉足輕重,但是目前的微影技 術已逼近解析度的極限,若想要進一步縮小元件的尺度,勢必發展全 新的技術。 綜合以上兩者,製備一維矽奈米材料是極有學術及應用上的價值
的,特別是擁有極高長寬比的矽奈米線(silicon nanowires)[14]。目前 製備矽奈米線最主要都是透過 1960 年代由 Wagner 提出的固-液-氣 相法(vapor-liquid-solid method, VLS)[15,16]。氣態的矽擴散進入由矽 和金屬催化物的合金液滴,當矽的濃度過飽和時,就會從液滴析出, 當氣態矽持續加入反應,固態的矽也就不斷地在液-固界面間成長, 而矽奈米線的直徑,即取決於合金液滴的大小。從此之後,許多基於 此生長機構的方法都被發展出來。包括化學氣相沈積(chemical vapor deposition, CVD)[17]、熱揮發(thermal evaporation)[18]、雷射剝離(laser ablation)[19]等等。但是,高溫、高能量、高真空、複雜的製程、昂 貴的設備、嚴苛的條件、危險的起始物、緩慢的生長速度、不易量產, 是這些方法的共同缺點。過高的成本及技術門檻,使得這些技術只有 少數先進的實驗室能夠掌握。 另一方面,為了能夠更方便對矽奈米線做檢測,或進一步應用於 元件之中,矽奈米線的排列方向性也很重要。因此,許多研究開始針 對發展出製備矽奈米線陣列(silicon nanowire arrays)的方法。其中, 最主要的是固-液-氣相磊晶法(vapor-liquid-solid epitaxy technique, VLSE)[20],或以陽極處理氧化鋁(anodic aluminum oxide, AAO)[21] 之類的奈米通道模版輔助。但是這樣的製程,除了有固-液-氣相法 的缺點之外,對於模板、基材、以及催化物,都有更高的要求。
最近幾年發展出的無電金屬沈積法(electroless metal deposition, EMD)[22-24]很可能是對以上問題的一個解決方案。與傳統的電鍍法不 同,電子不由外加電路供給,僅由自發性的電化學反應進行。電解液 中的還原劑直接把電子傳遞給金屬離子,而生成被還原的金屬沈積。 因為此法的高純度、高選擇性、簡易性、較低的反應溫度、以及低成 本,無電金屬沈積法已被廣泛應用於微機電工業中。 從 2002 年開始,Zhu et al.發展出一系列無電金屬沈積法製備大 面積矽奈米線陣列的方法[25-29]。將矽晶片(silicon wafer)浸入硝酸銀 (silver nitrate)與氫氟酸(hydrofluoric acid)的水溶液,再將此系統 置入高壓彈體中,加熱至 50℃反應,即可以在矽晶片表面蝕刻出大 面積矽奈米線陣列。此法不需要外加模板,僅透過在矽基材表面自發 的電化學氧化還原反應即可完成。可說是一舉克服了以固-液-氣相 磊晶法製備矽奈米線陣列的所有缺點。從另一方面來說,此法利用矽 晶片為起始物做由大而小的反應(top-down reaction),也遠比固-液 -氣相磊晶法的由小而大反應(bottom-up reaction)更容易得到高純 度和高結晶性的產物。 然而,這種新的方法也不是全無缺點,本篇論文即基於此一方 法,對其做進一步的調整與改良。主要分為三個部份。 第一部份,嘗試在更加溫和的條件下進行反應,以解決將反應系
統加溫加壓所可能造成的危險。在這一部份中,嘗試不使用高壓彈 體,而直接在常溫常壓下,僅藉由調整反應物濃度來進行實驗。希望 能得到近似,或甚至更高品質的矽奈米線陣列。同時,觀察極短反應 時間的樣品,透過研究副產物對主產物的影響,期望更深入瞭解此反 應的機制。 第二部份,嘗試解決 Zhu et al.實驗中最大的一個問題-產物線徑 的一致性與控制性。截至本論文之前,無電金屬沈積法製備矽奈米線 的線徑,完全取決於反應之初,銀離子在矽晶片表面沈積出的銀金屬 成核點大小。但是,這並不能由反應物濃度來簡單控制,因為改變濃 度的同時,很可能連帶影響產物的品質。而且成核點大小似乎是取決 於反應時微區中的銀離子濃度,此成核點大小又將影響線徑粗細。在 Zhu et al.的實驗之中,最粗與最細的矽奈米線,其線徑相差幾達十 倍,且完全無法控制,排列整齊緻密程度也不理想。為了讓矽奈米線 陣列能真正做為奈米微機電元件的一部份,製備更細小、可控制、更 一致、且更整齊的矽奈米線陣列是必要的。在這一部份中,嘗試先在 矽晶片表面做一層金奈米粒子,以此做為起始點,讓銀沈積於其上來 進行反應。因為金奈米粒子的大小可以控制,希望能間接控制矽奈米 線的線徑,也將線徑限制在一較小的範圍中,同時增進整齊程度。 第三部份,嘗試利用同樣的簡單電化學反應,在矽線的表面做一
層金屬的奈米顆粒修飾。雖然矽有良好的半導體性質,但是長久置於 大氣之下,表面極易產生一層無結晶性的氧化絕緣層,將大幅影響矽 在電性方面的表現。此一情形在奈米尺度的矽線上更為嚴重,因此希 望能以一層金屬保護。此反應亦透過無電金屬沈積法進行,僅利用不 同金屬離子在沈積時的性質不同,對矽線表面進行修飾。同時又保有 無電金屬沈積法簡單、低成本等優點。在應用方面,最直接的就是發 展成半導體或其他電性元件,如蕭基二極體(Schottky-barrier diodes) [30] 。此外,矽奈米線陣列的表面積極大,在其表面所修飾的金屬奈米 粒子,很有潛力發展成為觸媒,且其安定性將比溶液態時更好,也更 容易達成異相催化(heterogeneous catalysis)[31]的理想。或者,以其 高長寬比,容易吸附物質的特性,發展成為偵檢器[32]。在這一部份中, 嘗試在矽奈米線陣列的表面修飾上銅或鉑的金屬奈米粒子。
第二章 實驗
2.1 試藥
矽晶片 Silicon wafer n-type (100)
硝酸銀 AgNO3 Showa 氫氟酸 HF Arcos (48-51%) 硝酸 HNO3 Showa 硝酸銅 Cu(NO3)2 Showa 六氯鉑酸鉀 K2PtCl6 Showa 氮氣 N2 洽隆 (99.99%) 正己烷 n-Hexane, C6H14 Merck
2.2 實驗步驟
2.2.1 常溫常壓大氣水相下大範圍製備矽奈米線陣列 矽晶片切成 0.5 cm2大小,以標準 RCA 程序清洗[13]。在塑膠樣品 瓶中,配製 AgNO3 5mM,HF 18.5%水溶液為反應液。在常溫常壓下, 將矽晶片浸入反應液中 1 小時。反應結束後,取出矽晶片,可得到大 量銀灰色物質沉積於矽晶片表面。先以 D.I. water 洗去殘留反應液, 之後以硝酸洗去表面沉積物,再以 D.I. water 洗去殘留硝酸,最後以氮氣槍吹乾矽晶片。此時矽晶片表面為深褐色至黑色。此為樣品 A1。 改變不同的反應條件,包括反應時間、AgNO3及 HF 濃度,製備其他 的 A 類樣品。(最佳反應條件為樣品 A11,AgNO3 20mM,HF 18.5%。) 詳細反應條件列於表一。 2.2.2 以金奈米粒子輔助控制矽奈米線生成 矽晶片切成 0.5 cm2大小,以標準 RCA 程序清洗[13]。以濺鍍機在 矽晶片表面濺鍍一層金膜,濺鍍時間為 2 秒,靶材為純金。將矽晶片 於低壓下封入 Pyrex 玻璃管中。置於高溫爐中熱處理,熱處理條件為 昇溫速率每分鐘 1.6 K,熱處理溫度為 673 K,持溫 3 小時,自然降 溫。破管後可得到金奈米粒子附著於矽晶片表面。此為樣品 B1。改 變不同濺鍍時間,製備其他的 B 類樣品,以得到不同大小的金奈米 粒子。詳細反應條件列於表二。 以樣品 B1 為起始物。在塑膠樣品瓶中,配製 AgNO3 20mM,HF 25%水溶液為反應液。在常溫常壓下,將矽晶片浸入反應液中 30 分 鐘。反應結束後,取出矽晶片,可得到大量銀灰色物質沉積於矽晶片 表面。先以 D.I. water 洗去殘留反應液,之後以硝酸洗去表面沉積物, 再以 D.I. water 洗去殘留硝酸,最後以氮氣槍吹乾矽晶片。此時矽晶 片表面為深褐色至黑色。此為樣品 C1。以表二中其他樣品為起始物, 製備其他的 C 類樣品。詳細反應條件列於表三。
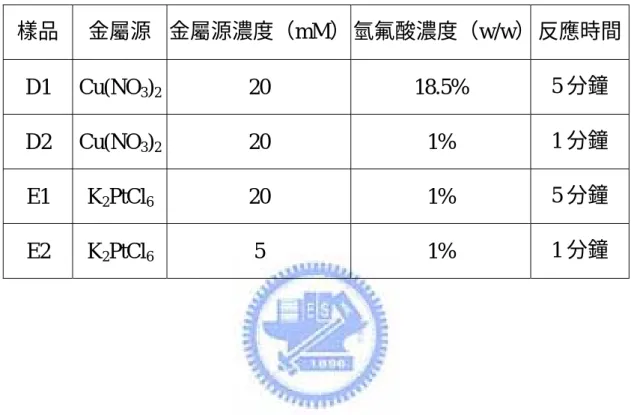
2.2.3 以自發電化學反應將金屬奈米粒子修飾於矽奈米線上 以樣品 A13 為起始物。在塑膠樣品瓶中,配製 Cu(NO3)2 20mM, HF 18.5%水溶液為反應液。在常溫常壓下,將矽晶片浸入反應液中 5 分鐘。反應結束後,取出矽晶片,可得到帶有帶有金屬光澤的紅褐色 薄膜生成於矽晶片表面。以 D.I. water 洗去殘留反應液,再以氮氣槍 吹乾矽晶片。此為樣品 D1。改變不同的反應條件,包括反應時間、 Cu(NO3)2及 HF 濃度,製備其他的 D 類樣品。(最佳反應條件為樣品 D2,Cu(NO3)2 20mM,HF 1%,反應時間 5 分鐘。)亦嘗試將 Cu(NO3)2 更換為 K2PtCl6,製備其他的 E 類樣品。(最佳反應條件為樣品 E2, K2PtCl6 5mM,HF 1%,反應時間 1 分鐘。)詳細反應條件列於表四。
2.3 分析儀器及樣品製備
2.3.1 掃描式電子顯微鏡(Scanning Electron Microscopy, SEM) FE-SEM JEOL JSM-6330F
樣品切成小片,以碳膠固定於金屬載具。觀察矽奈米線陣列時, 樣品傾斜約 45∼60 度,以顯現其一維結構。電子源為冷陰極電子槍, 加速電壓為 0.5∼30 kev,放大倍率為 10∼5×105倍,解析度為 1.5 奈 米(15 kev),5.0 奈米(1 kev)。
2.3.2 X-光射線能量散佈分析儀
(Energy Dispersive X-ray Spectroscopy, EDS) EDX OXFORD Link Pentafet
偵測窗口為鈹窗,厚度為 7.8∼8 微米。
2.3.3 穿透式電子顯微鏡
(Transmission Electron Microscopy, TEM) JOEL JEM-2010 樣品切成小片,浸入正己烷中,以超音波振盪 30-60 分鐘。將 2-3 滴懸浮液滴在 TEM 專用銅網上,抽乾溶劑。儀器電子源為 LaB6,加 速電壓為 80∼200 kev,其放大倍率為 50∼1.2×106倍,解析度為 0.25 奈米。 2.3.4 高解析穿透式電子顯微鏡
(High Resolution Transmission Electron Microscopy, HRTEM) JOEL JEM-4000EX
同 TEM 樣品製備。儀器電子源為 LaB6,加速電壓為 400 kev,
其放大倍率為 50∼2×106倍,解析度為 0.18 奈米。
2.3.5 拉曼光譜儀
(Raman Spectroscopy) Raman JABIN-YVON T-64000
樣品切成小片,固定於載玻片上。儀器入射波長為 632.8 奈米, 掃描範圍為 200∼1000 cm-1,顯微鏡放大倍率為 500 倍。
第三章 結果與討論
3.1 常溫常壓大氣水相下大範圍製備矽奈米線陣列
本實驗希望能克服在之前的文獻報導中,必需以高壓彈體反應的 問題,而僅由調控反應液濃度和比例進行。試圖以更加溫和的反應條 件,降低實驗進行時的危險性和難度。 3.1.1 產物形貌之 SEM 及 EDS 分析 首先透過樣品 A1∼A6 探討硝酸銀濃度與反應的關係。圖一(a) 為樣品 A1 的照片,此時硝酸銀濃度為 5mM。可見矽晶片的表面僅有 凹凸不平的被侵蝕痕跡,完全沒有形成矽奈米線的產物。圖一(b) 為樣品 A2 的照片,將硝酸銀濃度提高為 10mM。開始可以在矽晶片 的局部區域,生成矽奈米線,但並不均勻,也無法大範圍地得到產物。 硝酸銀濃度提高為 15mM 的樣品 A3 與樣品 A2 類似,惟反應區域略 廣。樣品 A4∼A6 繼續提高硝酸銀濃度。圖二(a)及圖二(b)分別 為樣品 A6 的側視與俯視照片,此樣品中硝酸銀濃度提高到 35mM。 此時雖然矽晶片的全部區域都已生成矽奈米線,但由圖二(a)可見, 矽奈米線混亂不均,亦多有斷折倒塌;由圖二(b)可更清楚看到, 矽奈米線混亂及斷折的情形。樣品 A4 與樣品 A5 亦有類似情形,惟 斷折情形稍緩。由此推論,當硝酸銀濃度太低時,反應將不進行或僅有部份進行;而硝酸銀濃度太高時,則會讓反應過於劇烈,無法得到 整齊排列的矽奈米線陣列。 接著透過樣品 A7∼A10 探討氫氟酸濃度與反應的關係。圖三(a) 與圖三(b)分別為樣品 A8 的低倍率及高倍率照片,此時氫氟酸濃 度為 13.9%。在圖三(a)中可見,矽奈米線陣列已全面性地生成。 但是圖三(b)放大觀察矽奈米線的頂端時,可見矽奈米線並沒有根 根獨立,而是與附近的數根相連,形成片狀或柱狀。此情形在氫氟酸 濃度更低的樣品 A7 中更為嚴重。圖四為樣品 A10 的照片,氫氟酸濃 度為 27.8%。類似樣品 A6 的情形,矽奈米線傾倒斷折情形嚴重。氫 氟酸濃度為 23.1%的樣品 A9 亦同,惟斷折情形稍緩。由此推論,當 氫氟酸濃度太低時,矽奈米線無法被完全蝕刻分離;而氫氟酸濃度太 高時,則會過度地侵蝕產物,亦無法得到整齊排列的矽奈米線陣列。 圖五為樣品 A11 的照片,此樣品硝酸銀濃度為 20mM,氫氟酸濃 度為 18.5%。由圖五(a)的低倍率照片中,可見大範圍的矽晶片, 皆已生成矽奈米線陣列產物,矽奈米線長度約為 35 微米;在圖五(b) 的中倍率照片中,可見幾乎沒有斷折的情形發生;在圖五(c)的高 倍率照片中,放大矽奈米線的頂端,可在相鄰兩根矽奈米線間,看到 清楚的界面,證實在這個樣品中的矽奈米線是根根獨立的。而線徑大 約分佈在 80∼180 奈米之間。圖五(d)為產物的 EDS 圖譜。可見到
只有矽與極微弱的氧訊號,以此判斷產物除表面輕微氧化之外,其餘 部份保持為純矽。根據以上結果認定樣品 A11 為 A 類樣品中的最佳 條件,後續分析將以此樣品為主。 透過觀察其他以同樣反應液濃度,但反應時間不同的樣品 A12∼ A14 中,試圖分析此反應生成矽奈米線的速度。圖六為硝酸銀濃度 20mM,氫氟酸濃度 18.5%下,不同反應時間對矽奈米線長度作圖。 發現大略呈線性關係,每分鐘平均生成線長在 0.5∼0.6 微米之間。若 持續延長反應時間,矽奈米線長度可達起始物矽晶片的厚度。 圖七是此反應進行時,所產生銀沈積副產物的照片。此產物外觀 為銀白色至銀灰色,於反應時生成於矽晶片表面,並包覆於其上。在 反應結束後,可用硝酸將其完全洗去。圖七(a)和圖七(b)分別是 此副產物的低倍率與高倍率照片。可清楚看見到它長條形樹枝狀的結 構。 3.1.2 產物形貌與晶型之 TEM、SAED 及 HRTEM 分析
圖八(a)為樣品 A11 的 TEM 照片。在這單支矽線的影像中,可 見其線徑為 100 奈米,除末端外,線徑大略保持一致,乃是一實心的 結構。表面有大約 2 奈米厚度的無晶相氧化層,應是樣品處於大氣下 自然生成的氧化薄層。圖八(b)為同一矽線的 SAED 影像。由它清 晰的點狀影像,可推論產物為單晶的組成。此圖符合文獻中單結晶矽
[011]的電子繞射圖。圖八(c)為同一根矽線的 HRTEM 原子影像。 其層間距為 0.313 奈米,符合文獻中矽(111)面的層間距。此矽線 的軸向為[100]方向,符合以(100)矽晶片為起始物,垂直於基材表 面的矽線應有的方向性。 3.1.3 反應機制的研究 透過 SEM 及 TEM 對短時間反應的樣品做觀察,試圖了解此反應 進行的機制。 圖九為僅反應 10 秒的樣品 A15 之照片,不以硝酸洗去其表面的 副產物,以觀察副產物與主產物之間的關係。圖九(a)為低倍率的 照片,可見矽晶片表面佈滿顆粒狀的沈積物。圖九(b)為高倍率的 照片,可見這些顆粒的粒徑大小與排列情形,大致與矽奈米線產物的 線徑與排列情形相符。 圖十為反應 20 秒的樣品 A16 之照片,同樣不洗去表面的副產物。 圖十(a)為低倍率的照片,可見矽晶片表面所佈滿的顆粒狀沈積物, 並沒有因為反應時間增長而變大,也沒有互相融合的情形。圖十(b) 為高倍率的照片,更可以確認銀顆粒並不是長大,而是以垂直基材的 方向變長,並且開始出現分枝。另外,可以在樣品表面看到少量長條 樹枝狀沈積物橫陳於樣品表面,這應是沈積物繼續生成的結果。隨著
分緻密,只是鬆散地繼續堆疊。因此,反應液始終可以透過沈積物間 的縫隙,與矽基材表面接觸而持續反應。圖十(c)的 EDS 圖譜,僅 出現銀與矽的訊號。矽的訊號來自基材矽晶片,可推斷此沈積物為銀 的顆粒。 圖十一為反應 40 秒的樣品 A17 之照片,圖十一(a)已經以硝酸 洗去表面的副產物。可見表面布滿坑洞,這些坑洞的大小與排列情 形,大致與附著於其上的銀顆粒相符,亦與矽奈米線產物的線徑與排 列情形相符。由此推論這些坑洞是銀顆粒沈積於矽基材表面時所造成 的。但是坑洞的深度,卻不隨著反應時間增長而增加,反而是坑洞與 坑洞之間的縫隙逐漸被往下蝕刻。可能是因為坑洞的部份已經被銀顆 粒附著而保護住,只有裸露於反應液中的縫隙處,才能讓蝕刻繼續進 行。圖十一(b)是矽奈米線頭部的 TEM 照片,由此可以更清楚看到 銀顆粒在矽奈米線頭部所造成的坑洞凹陷,亦證明蝕刻不是由銀顆粒 正下方的坑洞一路往下進行,而是蝕刻坑洞週圍的隙縫處。如圖八(a) 所見表面凸起,而非凹陷的矽奈米線末端,並不是矽奈米線的頭部, 而是製備 TEM 樣品時,以超音波振斷的矽奈米線根部(原本與矽基 材相連的部份)。 綜合以上所述,推論其反應機制應如同圖十二所示。由矽提供電 子,讓反應液中的銀離子被還原,而以銀金屬的形式,於矽表面析出。
此時,反應液中的氫氟酸將銀顆粒週圍被氧化的矽溶解帶走。分別計 算氧化和還原兩個半反應,發現這個反應的電位為正,得在不外加能 量下,自發進行。再加上銀於此反應中持續析出時的獨特性質-形成 長條樹枝狀的結構,而非一般金屬析出時常見的形貌-由顆粒聚集而 成的緻密薄膜。使得此反應不會因為緻密的沈積物完全覆蓋矽表面, 而使反應被迫中止。於是,每一顆沈積於矽表面的銀顆粒與其週圍的 部份,就獨立形成了一個奈米級的氧化還原反應區。銀繼續析出,週 圍被氧化的矽也持續被氫氟酸帶走。而且,這樣的反應在(100)晶 面的矽晶片上顯然是非等向性的,垂直於基材表面是最偏好的方向。 最後,當週圍的矽被垂直往下蝕刻到一定程度時,被留下來的部份便 成為一根獨立的矽奈米線。而在整片矽晶片上,這樣的微反應區大致 均勻地分佈於其上,所得到的產物變是整齊排列,根根獨立的矽奈米 線陣列。倘若更換其他晶面的矽晶片為起始物,則蝕刻方向仍將保持 為延著[100]方向進行,而生成非垂直基材表面的產物。 3.1.4 討論 在這一部份的實驗中,由以上數據與文獻[25,26]比較可知,所得到 的產物與文獻報導中的極為相似。證明此反應未必需要以高壓彈體加 溫加壓來進行,於常溫常壓的反應條件下亦可達成。
仿,所以其品質大致相同。若比較使用高壓彈體與否時的生成速度 [28] ,雖然常理上較和緩的反應條件生成速度應該較慢,但是因為反應 液的組成亦有調整,所以實際上兩者的生成速度相當接近。最後比較 線徑的粗細與分佈範圍,則有一些差距。使用高壓彈體時[25,26],線徑 粗細之間,可相差至十倍;然而,於常溫常壓下的反應,因為反應條 件更趨溫和,產物的線徑亦更趨於一致,粗細之間僅有二∼三倍左右 的差距。
3.2 以金奈米粒子輔助控制矽奈米線生成
目前文獻所報導過以自發電化學反應製備矽奈米線的方法中,對 於線徑大小無法有效控制,且極不一致。在本論文 3.1 節中以常溫常 壓進行反應,雖然已將線徑的差異縮小,仍難以滿足實際應用時的希 冀。透過上一節中對反應機制的了解,在本節中,試圖利用可控制大 小的金奈米粒子,對線徑大小及分佈,做一間接的限制。 3.2.1 產物形貌之 SEM 及 EDS 分析 將矽晶片濺鍍上一層金膜,再以高溫燒結成金奈米顆粒,因為顆 粒極小,以 SEM 的低倍率影像,幾乎觀察不到金奈米粒子,只有當 放大倍率達到十萬倍以上時,才能清楚看到。圖十三(a)∼圖十三 (e)分別是樣品 B1∼B5 的高倍率 SEM 影像。 圖十三(a)中可見,鍍金時間為 2 秒的樣品 B1,燒結後其金奈 米粒子的粒徑分佈於 10∼13 奈米左右,且十分均勻地分佈在矽晶片 表面。圖十三(b)中可見,鍍金時間為 5 秒的樣品 B2,燒結後其金 奈米粒子的粒徑分佈於 18∼22 奈米左右,分佈於矽晶片表面的情形 亦頗均勻。圖十三(c)中可見,鍍金時間為 10 秒的樣品 B3,燒結 後其金奈米粒子的粒徑分佈於 26∼31 奈米左右,其分佈均勻程度, 略遜於前兩個樣品,但仍大致均勻。圖十三(d)中可見,鍍金時間為 15 秒的樣品 B4,燒結後其金奈米粒子的粒徑分佈於 33∼44 奈米 左右,粒徑大小開始有較大的差異,其分佈也比之前的樣品較不均 勻。圖十三(e)中可見,鍍金時間為 20 秒的樣品 B5,燒結後其金 奈米粒子的粒徑分佈於 35∼58 奈米左右,粒徑大小差異更大,甚至 開始出現粒子間互相聚集的現象,也造成分佈更不均勻。圖十三(f) 為樣品 B5 的 EDS 圖譜,僅有金與矽的訊號。金的訊號來自表面的金 奈米粒子,矽的訊號來自矽晶片。其他 B 類樣品的 EDS 圖譜亦與圖 十三(f)相似。 樣品 C1∼C5 分別是以樣品 B1∼B5 為起始物,而 C6 是以沒有 經過鍍金燒結處理的矽晶片為起始物,當作本系列實驗的對照組。 圖十四(a)∼圖十四(f)分別是樣品 C1∼C6 低倍率下的 SEM 影像。這六張照片拍攝於同一放大倍率,以此比較不同大小的金奈米 粒子,對於產物線徑大小、線徑分佈、與排列情形之影響。首先就線 徑大小分析,從圖十四(a)∼圖十四(f)呈現一明顯遞增的趨勢。 顯示金奈米粒子粒徑與矽奈米線線徑的確有相關性。再就線徑分佈討 論,其線徑一致性從圖十四(a)∼圖十四(f)呈現遞減趨勢。符合 其起始物表面金奈米粒子粒徑一致性的趨勢。最後探討產物排列情 形,從圖十四(a)∼圖十四(f)中可發現產物陣列從極為緻密愈趨 鬆散混亂。這樣的趨勢亦符合其起始物表面金奈米粒子分佈的整齊程
度。 圖十五(a)與圖十五(b)分別是樣品 C1 和 C6 在同樣放大倍 率下的 SEM 照片。在較高的倍率下,可以更清楚比較出有沒有以金 奈米粒子輔助控制矽奈米線生成,產物在線徑上的差異。圖十五(c) 是 C 類樣品普遍的 EDS 圖譜,僅有矽與極弱的氧訊號。矽來自於產 物矽奈米線,氧來自於產物處於大氣下發生的自然氧化;沒有任何金 的訊號,說明反應結束後的清洗流程中,可將銀沈積物與金奈米粒子 一起帶走,不至對產物純度造成影響。 3.2.2 產物形貌與晶型之 TEM 及 HRTEM 分析 圖十六(a)∼圖十六(e)分別是樣品 C1∼C5 的 TEM 影像。 每張照片僅顯示單根矽奈米線,但每個樣品的照片皆是從多張照片中 挑出具有代表性且清楚呈現形貌與線徑者。所有矽奈米線皆為實心結 構,表面包覆無晶相氧化薄層,厚度約為數奈米,乃置於大氣下自然 氧化所產生。樣品 C1 線徑大約分佈於 16∼24 奈米左右。樣品 C2 線 徑大約分佈於 27∼35 奈米左右。樣品 C3 線徑大約分佈於 33∼41 奈 米左右。樣品 C4 線徑大約分佈於 46∼59 奈米左右。樣品 C5 線徑大 約分佈於 47∼72 奈米左右。而對照組的線徑分佈於 80∼180 奈米。 圖十六(f)是樣品 C2 的 HRTEM 照片,清晰的原子影像說明樣品具
面的層間距。此矽線的軸向為[100]方向,符合以(100)矽晶片為起 始物,垂直於基材表面的矽線應有的方向性。其他 C 類樣品的 HRTEM 照片,亦與圖十六(f)表現出相同的訊息。 透過 TEM 對線徑的分析,可以更清楚看出金奈米粒子粒徑與矽 奈米線線徑的關係,同一樣品中線徑的均勻亦程度大幅提昇,若再跟 以高壓彈體進行反應的樣品比較,產物的品質更是有長足地進步。圖 十七是以鍍金時間對金奈米粒子粒徑與矽奈米線線徑作圖。 3.2.3 拉曼光譜分析 圖十八為樣品 C1 的拉曼光譜,其他矽奈米線陣列產物的拉曼光 譜亦與圖十八相似。具有結晶性的矽塊材,拉曼特徵鋒位於 520 cm-1, 且半高寬(FWHM)僅有 2.8 cm-1,此為其 the first-order optical phonon mode(TO)[33]。與圖十八比較,可發現樣品的 TO mode 略微向低波 數位移至 517 cm-1,半高寬增加至大約 15 cm-1,且出現一明顯的肩 部。根據文獻報導,這些現象普遍出現在矽奈米線樣品的拉曼光譜 中 , 主 要 源 自 樣 品 細 小 的 線 徑 與 高 長 寬 比 造 成 的 量 子 限 制 效 應 (quantum confinement effect)[33-36]。此外,接近 297 cm-1出現的寬廣 訊號乃是 the second-order transverse acoustic phonon mode (2TA), 而接近 940 cm-1出現的寬廣訊號乃是 the second-order optical phonon mode(2TO)。與具有結晶性的矽塊材比較,這兩處的訊號,都明顯
向低波數處位移,相對強度亦增加,這也都是具有結晶相矽奈米線的 拉曼光譜特徵[33]。 3.2.4 反應機制的研究 綜合以上數據,推論此部份反應機制應如同圖十九所示。經過濺 鍍與燒結生成於矽晶片表面的金奈米粒子,一般認為是比矽晶片本身 更容易聚集負電荷。因此,銀離子析出成金屬沈積會以金奈米粒子為 起始點。當銀在金奈米粒子上被還原時,金扮演著傳遞矽所提供電子 的角色。隨著銀繼續析出,其本身的特性讓沈積物發展成為長條樹枝 狀,而銀週圍的矽在失去電子後被氫氟酸帶走,這一部份的反應機制 與 3.1.3 節中所提出者相同。所以,金奈米粒子的粒徑大小、粒徑分 佈、和粒子分佈,就間接影響了矽奈米線的生成。當金奈米粒子的粒 徑愈小、愈一致、散布愈緻密平均,將使得矽奈米線陣列的線徑愈細、 愈一致、排列愈緻密整齊。 關於這一類以自發電化學反應製備矽奈米線陣列的機制,最早投 入此一領域研究的 Zhu et al.於 2002∼2004 年有多篇文獻討論[25-28]。 其所提出的機制,大致與本文 3.1.3 節中所提出的類似。然則,Zhu et al.於 2005 年有一篇最新的文獻提出了一個截然不同的機制[29]。茲簡 述如下:其實驗步驟分為兩部份,先將矽晶片以硝酸銀 氫氟酸反應
酸反應液進行蝕刻。由氫氟酸將被氧化的矽帶走,而鐵(III)被還原 成鐵(II),因此不會有鐵金屬沈積。但是,在這一篇報導中,Zhu et al.認為,被氧化的矽並不是在銀沈積物的週圍,而是其正下方。當氫 氟酸將銀正下方的氧化物帶走,銀沈積物就往下沈降,而鐵(III)繼 續透過銀沈積物被還原,將銀正下方的矽氧化。如此持續進行,當蝕 刻進行到一個程度,剩下在反應之初沒有被銀沈積物附著之處,就被 留下來成為矽奈米線,而銀顆粒最後也沈降到矽奈米線與矽晶片基材 的交界處。 但是,根據本實驗的觀察,以金奈米粒子粒徑與矽奈米線線徑的 關係為最主要證據,輔以圖十一對樣品 A17 的觀察,本實驗的反應 機制應如同本節之初所述,以本論文 3.1.3 節所提出機制為基礎,亦 與 Zhu et al.於 2002∼2004 年間提出的機制觀點類似,而不採用 Zhu et al.於 2005 年所提出的新機制。主要的原因或許是反應液不同所造成 的差異。 3.2.5 討論 在這一部份的實驗中,成功地用金奈米粒子間接控制矽奈米線生 成。對於產物的線徑大小、線徑分佈、與排列整齊程度,可以做到大 略的控制。以不同的濺鍍時間控制金奈米粒子的粒徑,可以在 20∼ 60 奈米間調整產物線徑。產物的線徑一致度與排列整齊度,亦大幅
提昇。若想以這樣的想法對產物有更大的控制力,則應該思考金奈米 粒子的品質。透過濺鍍與燒結的方式產生的金奈米粒子有四個最主要 的缺點:首先,其粒徑本身已經有某種程度的差異,而以這樣的起始 物進行反應,產物線徑的差異只會比金奈米粒子粒徑的差異更大。二 者,粒徑所能控制的範圍太小,如果將金奈米粒子做得更密更小,銀 析出時可能同時以多顆粒子為起始點,而失去效果;反之,過大的顆 粒,其粒徑不均、互相聚集的情形會更嚴重,亦效果不彰。三者,其 顆粒對矽晶片並沒有很強的附著力,在操作實驗時稍一不慎,非常容 易讓金奈米粒子從矽晶片表面剝落,如此便失去了對產物的限制。最 後,金奈米粒子只能隨機排列於矽晶片表面,無法對其間距或排列做 控制。若能以更嚴謹的製程製備起始點,相信對產物的控制亦將有長 足的進步。 另外,在這一部份的實驗中,氫氟酸的濃度需要適度提高。因為 當產物的線徑大幅縮小後,需要氫氟酸蝕刻的部份也變多。若維持前 節中的濃度,產物的形貌將與樣品 A8 類似,無法根根獨立。
3.3 以自發電化學反應將金屬奈米粒子修飾於矽奈米線上
矽材料置於大氣之下,表面將難以避免地生成無晶相的絕緣氧化 層,此一現象在奈米尺度更加嚴重影響其電性方面的表現。本節嘗試 將金屬奈米粒子修飾於矽奈米線上,保持產物表面良好的導電性,增 加其多方應用的可能性。 3.3.1 產物形貌之 SEM 及 EDS 分析 圖二十為本節反應共同的起始物照片,其反應條件如樣品 A13, 反應時間縮短為 15 分鐘,因此矽奈米線長僅有約 7.5 微米。 圖二十一(a)及圖二十一(b)分別是樣品 D1 的低倍率和高倍 率 SEM 照片。在圖二十一(a)的大範圍影像中,可見矽晶片表面的 線狀產物幾乎全部斷折,僅有少數傾倒散布於基材表面,應該是反應 條件太過劇烈造成。圖二十一(b)放大觀察殘留的矽奈米線,可見 表面佈滿顆粒狀的產物。說明以自發電化學反應將金屬奈米粒子修飾 於矽奈米線上的構想是可以成功的,惟反應條件應更小心控制。 圖二十二(a)及圖二十二(b)分別是樣品 D2 的低倍率和高倍 率 SEM 照片。在大幅降低氫氟酸濃度及反應時間後,從圖二十二(a) 的大範圍影像中可見幾乎所有的矽奈米線都仍然保持完好。圖二十二 (b)放大觀察矽奈米線的尖端部份,可見矽奈米線的表面已被包覆一層奈米粒子,其粒徑約為數奈米至數十奈米。而且,從圖中可看出, 產物多半具有細小的尖端,最頂端的部份亦有奈米粒子附著。圖二十 二(c)為樣品 D2 的 EDS 圖譜,僅有矽與銅的訊號。其中矽來自矽 奈米線,而銅的訊號確認了包覆在矽奈米線表面的顆粒是銅。 在成功將銅奈米粒子修飾於矽奈米線上之後,嘗試修飾熔點更 高、活性更低的鉑。 圖二十三是樣品 E1 的 SEM 照片,可見有大量的奈米粒子附著於 表面。但是顆粒的量太過龐大,幾乎已經失去矽奈米線的一維結構, 因此需嘗試更溫和的反應條件。圖中偶然可見深色巨大顆粒,經 EDS 分析是在反應過程中產生的鹽類,稍微長時間的水洗,可以將之完全 去除。 圖二十四(a)及圖二十四(b)分別是樣品 E2 的低倍率和高倍 率 SEM 照片。在更溫和的反應條件下,從圖二十四(a)的大範圍影 像中可見幾乎所有的矽奈米線都仍然保持完好。圖二十四(b)放大 觀察矽奈米線的頂端部份,可見矽奈米線的表面已被包覆一層奈米粒 子,其粒徑約為數十奈米。圖二十四(c)為樣品 E2 的 EDS 圖譜, 僅有矽與鉑的訊號。其中矽來自矽奈米線,而鉑的訊號確認了包覆在 矽奈米線表面的顆粒是鉑。
3.3.2 產物形貌之 TEM 分析 圖二十五(a)為樣品 D2 的 TEM 照片,可見圖中的矽奈米線表 面,大致均勻地包覆著銅奈米粒子,其粒徑大小介於數奈米至數十奈 米之間。圖中所見到的三組矽奈米線,正好展現出矽奈米線的頂端(從 畫面左方向右上延伸)、中段(從左上方向右方橫跨畫面)、以及根部 (畫面左下角)。從這三組矽奈米線,可以更清楚看出當銅奈米粒子 附著於矽奈米線時,是均勻地分佈於矽奈米線的各個部位,其粒徑、 數量、密度都是大致相同的。再者,根據這張照片,可以發現銅奈米 粒子對矽奈米線的附著力是夠強的,經過將近一小時的超音波振盪, 即使矽奈米線已經從矽晶片基材上斷折,銅奈米粒子仍然附著於矽奈 米線的表面,說明若透過這樣的方式製備實際應用元件,其機械強度 是完全沒有問題的。 圖二十五(b)為樣品 E2 的 TEM 照片。透過 TEM 照片,可以 清楚看到,在 SEM 照片中看似粒徑數十奈米大小的鉑顆粒,其實是 由非常多顆粒極小的鉑奈米粒子聚集而成。推測可能是鉑金屬在反應 析出時的特性使然。圖中顯示數根矽奈米線,鉑奈米粒子散布成數個 集團附著於分佈於上半部的數根矽奈米線的頂端或頂端附近;而畫面 中最下面一根矽奈米線,展現的是矽奈米線的中段,這個部份完全沒 有鉑奈米粒子附著。另外,與樣品 D2 相同,經過將近一小時的超音
波振盪,鉑奈米粒子仍穩固地附著於矽奈米線表面,足以應付實際應 用所需的機械強度。 3.3.3 反應機制的研究 這一部份實驗的反應機制,應可以直接套用本論文 3.1.3 節所提 出的反應機制。但是,不論是銅或鉑,於反應中沈積時,其析出的形 態與銀有很大的不同。銀會形成長條樹枝狀,使矽晶片表面始終有部 份裸露於反應中;銅或鉑傾向形成緻密的薄膜,當這一層金屬膜完全 蓋住矽晶片表面,反應也隨之中止[26]。圖二十六是將矽晶片以樣品 D1 的條件進行反應後所得產物的照片。可見在大面積下所形成連續 的銅膜。本節的反應即利用這樣的特性,在矽奈米線的表面修飾銅或 鉑的金屬奈米粒子。如圖二十七的示意圖。 根據這樣的反應機制及電位計算,只要在金屬活性表上,銅以下 的金屬皆可進行此反應,包括銅、汞、銀、鉑、金等。但是汞於常溫 下為液態,又具有高毒性,不予採用。銀和金因為沈積時,其析出形 狀特殊,並不適合[26,27]。故本實驗僅進行銅與鉑的部份。[26] 3.3.4 討論 在這一部份的實驗中,需要比之前更嚴格地調整反應條件。因為 當反應的起始物是塊材的矽晶片時,稍有誤差的條件,並不會造成明
顯的差異;若以矽奈米線陣列為起始物,反應只要稍微劇烈,很容易 就會把矽奈米線給弄斷。或者即使矽奈米線仍完好,但過多的金屬沈 積物將矽一維材料的特性完全遮蔽。都將難以保持產物的良好品質。 另外,比較修飾銅的 D 類反應與修飾鉑的 E 類反應,可發現雖 然同樣成功使金屬奈米粒子附著於矽奈米線表面,但其分佈的型態並 不相同。銅奈米粒子傾向平均包覆於整根矽奈米線;而鉑奈米粒子則 傾向於集中在矽奈米線的頂端,而在矽奈米線的中段與根部,都只有 零星的分佈。推測除了不同金屬析出特性之間的差異,尚有一重要原 因:以硝酸銅進行反應時,除了會有銅沈積之外,硝酸根離子始終處 於溶液態;然而以六氯鉑酸鉀進行反應時,除了鉑的沈積,還會伴隨 鹽類的析出。這些鹽類會沈積於矽奈米線與矽晶片的交界處,進而對 鉑的沈積造成一定程度的阻礙,才造成兩種金屬在類似反應中不同的 形貌。
第四章 結論
本實驗分為三部份,圖二十八為總示意圖。皆以自發電化學反應 為主要反應機制,在矽晶片表面大範圍地製備整齊排列的矽奈米線陣 列。克服以固-液-氣相法製備矽奈米線陣列時的諸多缺點。 在第一部份中,成功地在常溫、常壓、大氣、水相的反應條件下 生成矽奈米線陣列。將矽晶片浸入適當比例的硝酸銀 氫氟酸水溶液 中,即可在很短的時間內簡單地得到產物。透過電子顯微鏡的鑑定, 發現其線徑約介於 80∼180 奈米之間;長度可由反應時間控制,生成 速度約為每分鐘 0.5∼0.6 微米,最長可穿透整片矽晶片。除表面極薄 的氧化層之外,仍保持極高的純度與良好結晶性。反應範圍僅受限於 矽晶片與反應容器大小。 在第二部份中,以濺鍍和燒結的方式,先在矽晶片表面生成一層 金奈米粒子,以此做為起始點,再進行反應。成功地將產物的線徑均 勻程度大幅提昇;且矽奈米線線徑可藉由調整金奈米粒子的大小,於 20∼60 奈米之間大致選擇;矽奈米線陣列的排列亦更加緻密整齊。 而反應結束後,金奈米粒子可以很簡單地被移除,不至於對產物純度 造成影響。 第三部份中,將反應液中的硝酸銀換成硝酸銅或六氯鉑酸鉀,即 可透過同樣簡單的自發電化學反應,利用不同金屬離子還原成金屬時不同的生成形態,成功地在矽奈米線陣列表面修飾上銅或鉑的金屬奈 米粒子。其顆粒大小約在數奈米至數十奈米之間,能保護矽奈米線不 在大氣環境下被氧化,增進其電性表現,亦增加其做為應用元件的機 會。
第五章 參考文獻
1. M.-C. Daniel, D. Astruc, Chem. Rev. 2004, 104(1), 293-346.
2. R. M. Crooks, M. Zhao, L. Sun, V. Chechik, L. K. Yeung, Acc. Chem.
Res. 2001, 34(3), 181-190.
3. L.-S. Li, J. Walda, L. Manna, A. P. Alivisatos, Nano Lett. 2002, 2(6), 557-560.
4. J. Hu, T. W. Odom, C. M. Lieber, Acc. Chem. Res. 1999, 32(5), 435-445.
5. David C. Bell, Yue Wu, Carl J. Barrelet, Silvija Grade ak, Jie Xiang, Brian P. Timko, Charles M. Lieber, Microscopy Research and
Technique, 2004, 64(5-6); 373-389.
6. P. M. Ajayan, Chem. Rev. 1999, 99(7), 1787-1800.
7. M. S. Arnold, P. Avouris, Z. W. Pan, Z. L. Wang, J. Phys. Chem. B.
2003, 107(3), 659-663.
8. Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim, H. Yan, Adv. Mater. 2003, 15(5), 353-389.
9. E. C. Walter, R. M. Penner, H. Liu, K. H. Ng, M. P. Zach, F. Favier,
Surface and Interface Analysis, 2002, 34(1), 409-412.
10. Z. L. Wang, Adv. Mater. 2003, 15(5), 432-436.
11. Y. Cui, Z. Zhong, D. Wang, W. U. Wang, C. M. Lieber, Nano Lett.
2003, 3(2), 149-152.
12. Z. Zhong, F. Qian, D. Wang, C. M. Lieber, Nano Lett. 2003, 3(3), 343-346.
發展協進會,台北市,1997.
14. B. Zheng, Y. Wu, P. Yang, J. Liu, Adv. Mater. 2002, 14(2), 122-124. 15. R. S. Wagner, W. C. Ellis, Appl. Phys. Lett. 1964, 4, 89-91.
16. Y. Wu, P. Yang, J. Am. Chem. Soc. 2001, 123(13), 3165-3166.
17. T. I. Kamins, R. S. Williams, Y. Chen, Y. Chang, Y. A. Chang, Appl.
Phys. Lett. 2000, 76(5), 562-654.
18. Z. W. Pan, Z. R. Dai, L. Xu, S. T. Lee, Z. L. Wang, J. Phys. Chem. B.
2001, 105(13), 2507-2514.
19. A. M. Morales, C. M. Lieber, Science, 1998, 279(5348), 208-211. 20. S. Ge, K. Jiang, X. Lu, Y. Chen, R. Wang, S. Fan, Adv. Mater. 2005,
17(1), 56-61.
21. X.-Y. Zhang, L.-D. Zhang, G.-W. Meng, G.-H. Li, N.-Y. Jin-Phillipp, F. Phillipp, Adv. Mater. 2001, 13(16), 1238-1241.
22. G. V. Kuzentsov, V. A. Skryshevsky, T. A. Vdovenkova, A. I. Tsyganova, P. Gorostiza, F. G. Sanz, J. Electrochem. Soc. 2001, 148, C528.
23. P. Gorostiza, R. Diaz, J. Servat, F. Sanz, J. R. Morante; J.
Electrochem. Soc. 1997, 144(3), 909-914.
24. P. Gorostiza, M. A. Kulandainathan, R. Diaz, F. Sanz, P. Allongue, J. R. Morante; J. Electrochem. Soc. 2000, 147(3), 1026-1030.
25. K.-Q. Peng, Y.-J. Yang, S.-P. Gao, J. Zhu; Adv. Mater. 2002, 14(16), 1164-1167.
26. K.-Q. Peng, Y.-J. Yang, S.-P. Gao, J. Zhu; Adv. Func. Mater. 2003,
13(2), 127-132.
558 35-39.
28. K.-Q. Peng, Z.-P. Huang, J. Zhu; Adv. Mater. 2003, 16(1), 73-76. 29. K.-Q. Peng, Y. Wu, H. Fang, X.-Y. Zhong, Y. Xu, J. Zhu, Angew.
Chem. 2005, 117, 2797-2802.
30. X. Ji, A. Zuppero, J. M. Gidwani, G. A. Somorjai, Nano Lett. 2005,
5(4), 753-756.
31. R. B. Bedford, U. G. Singh, R. I. Walton, R. T. Williams, S. A. Davis,
Chem. Mater. 2005, 17(4), 701-707.
32. M. Su, S. Li, V. P. David, J. Am. Chem. Soc. 2003, 125(33), 9930-9931.
33. M. Lu, M-K. Li, L-B. Kong, X-Y. Guo, H-L. Li, Composites B. 2004, 179-184.
34. B. Li, D. Yu, S-L. Zhang, Phys. Rev. B, 1999, 59(3), 1645-1648. 35. J. Qi, J. M. White, A. M. Belcher, Y. Masumoto, Chem. Phys. Lett.
2003, 372, 763-766.
36. K. W. Adu, H. R. Gutierrez, U. J. Kim, G. U. Sumanasekera, P. C. Eklund, Nano Lett. 2005, 5(3), 409-414.
表 一 A 類樣品詳細製備條件 樣品 硝酸銀濃度(mM)氫氟酸濃度(w/w) 反應時間 A1 5 18.5 % 1 小時 A2 10 18.5 % 1 小時 A3 15 18.5 % 1 小時 A4 25 18.5 % 1 小時 A5 30 18.5 % 1 小時 A6 35 18.5 % 1 小時 A7 20 9.25 % 1 小時 A8 20 13.9% 1 小時 A9 20 23.1 % 1 小時 A10 20 27.8 % 1 小時 A11 20 18.5 % 1 小時 A12 20 18.5 % 5 分鐘 A13 20 18.5% 15 分鐘 A14 20 18.5 % 30 分鐘 A15 20 18.5 % 10 秒 A16 20 18.5 % 20 秒 A17 20 18.5 % 40 秒
表 二 B 類樣品詳細製備條件 樣品 鍍金時間 B1 2 秒 B2 5 秒 B3 10 秒 B4 15 秒 B5 20 秒
表 三 C 類樣品詳細製備條件 樣品 起始物 C1 樣品 B1 C2 樣品 B2 C3 樣品 B3 C4 樣品 B4 C5 樣品 B5 C6 沒有經過鍍金的矽晶片
表 四 D 類及 E 類樣品詳細製備條件 樣品 金屬源 金屬源濃度(mM) 氫氟酸濃度(w/w) 反應時間 D1 Cu(NO3)2 20 18.5% 5 分鐘 D2 Cu(NO3)2 20 1% 1 分鐘 E1 K2PtCl6 20 1% 5 分鐘 E2 K2PtCl6 5 1% 1 分鐘
圖 一
(a)樣品 A1 的 SEM 影像 (b)樣品 A2 的 SEM 影像
(a)
圖 二
樣品 A6 的(a)側視 SEM 影像 (b)俯視 SEM 影像
(a)
圖 三
樣品 A8 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像
(a)
圖 四
圖 五
樣品 A11 的(a)低倍率 SEM 影像 (b)中倍率 SEM 影像 (c)高倍率 SEM 影像 (d)EDS 圖譜
(b) (a)
圖 五 (承上頁)
(c)
0 10 20 30 40 0 15 30 45 60 圖 六 反應時間對矽奈米線長度作圖 反應時間(分鐘) 矽奈米線長度(微米)
圖 七
銀沈積物的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像
(a)
圖 八
樣品 A11 的(a)TEM 影像 (b)SAED 影像 (c)HRTEM 影像
(a)
圖 九
樣品 A15 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像
(a)
圖 十
樣品 A16 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像 (c)EDS 圖譜 Si Ag (a) (b) (c)
圖 十一
(a)樣品 A17 的 SEM 影像 (b)矽奈米線頭部的 TEM 影像
(a)
圖 十二
圖 十三
(a)樣品 B1 (b)樣品 B2 (c)樣品 B3 (d)樣品 B4 (e)樣品 B5 的 SEM 影像 (f)樣品 B5 的 EDS 圖譜
(a) (b) (c) 鍍金 2 秒 鍍金 5 秒 鍍金 10 秒
圖 十三 (承上頁) (d) (e) (F) 鍍金 15 秒 鍍金 20 秒 Si Au
圖 十四
(a)樣品 C1 (b)樣品 C2 (c)樣品 C3 (d)樣品 C4
(a)
(b)
圖 十四 (承上頁)
(d)
(e)
圖 十五 (a)樣品 C1 (b)樣品 C6 的高倍率 SEM 影像 (a) (b) (c) Si O
圖 十六
(a)樣品 C1 (b)樣品 C2 (c)樣品 C3(d)樣品 C4 (e)樣品 C5 的 TEM 影像 (f)樣品 C2 的 HRTEM 影像
(a) (b)
圖 十六 (承上頁)
0 20 40 60 80 100 120 140 160 180 200 0 5 10 15 20 25 30 金奈米粒子粒徑(nm) 矽奈米線線徑(nm) 圖 十七 鍍金時間對金奈米粒子粒徑與矽奈米線線徑作圖 對照組 鍍金時間(秒)
圖 十八
C 類樣品的拉曼光譜圖 TO
2TO 2TA
圖 十九
圖 二十
圖 二十一
樣品 D1 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像
(a)
圖 二十二
樣品 D2 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像
(a) (b) (c) Si Cu Cu
圖 二十三 樣品 E1 的 SEM 影像
圖 二十四
樣品 E2 的(a)低倍率 SEM 影像 (b)高倍率 SEM 影像
(a) (b) (c) Si Pt Pt
圖 二十五
(a)樣品 D2 (b)樣品 E2 的 TEM 影像
(a)
圖 二十六
圖 二十七
圖 二十八 總反應示意圖