國 立 交 通 大 學
機 械 工 程 學 系
碩士論文
硫化氫毒化對質子交換膜燃料電池性能
影響之暫態分析
Transient Analysis of Hydrogen Sulfide Poisoning
Effect on Cell Performance of PEM Fuel Cell
研 究 生:林子淵
指導教授:曲新生 教授
硫化氫毒化對質子交換膜燃料電池性能影響
之暫態分析
Transient Analysis of Hydrogen Sulfide Posioning Effect
on Cell Performance of PEM Fuel Cell
研 究 生:林子淵 Student:Tzu-Yuan Lin 指導教授:曲新生 Advisor:Hsin-Sen Chu 國 立 交 通 大 學 機 械 工 程 學 系 碩 士 論 文 A Thesis
Submitted to Department of Mechanical Engineering National Chiao Tung University
In Partial Fulfillment of the Requirements For the Degree of
Master In
Mechanical Engineering June 2006
Hsinchu, Taiwan, Republic of China
硫化氫毒化對質子交換膜燃料電池性能影響
之暫態分析
研究生:林子淵 指導教授:曲新生 國立交通大學機械工程學系摘要
本文旨在發展質子交換模燃料電池在陽極觸媒層受到燃料氣體 中不純物質硫化氫毒化之數學模型,及探討其內部氫氣、硫化氫之濃 度、白金表面覆蓋率、電流密度分佈的暫態行為。在陽極觸媒層內, 由於硫化氫會與白金形成穩定的鍵結,造成氫氣的使用率下降。研究 中主要是尋找各個參數範圍和影響,以及建立起模擬硫化氫毒化現象 的反應項。此數學模型主要是考慮氫氣與硫化氫因吸附、脫附、電化 學氧化行為以及新假設的硫化氫搶奪已吸附氫氣之白金表面項,造成 在白金表面覆蓋率的改變,進而影響到電池的性能。 研究結果顯示,由結果圖形趨勢確定硫化氫毒化模式中必須考慮 本文所假設的硫化氫搶奪已吸附氫氣之白金表面項和硫覆蓋率變化 對硫化氫吸附反應速率常數的影響。用本文所建立之數學模型來預 測,當通入的硫化氫濃度達5ppm 時,電池達穩態時間僅 800 分鐘左 右;硫化氫濃度為 0.2ppm 時,電池達穩態時間約 21 天;硫化氫濃 度降到 0.05ppm 時,電池達穩態時間達 81 天;硫化氫濃度僅剩 0.01ppm 時,電池達穩態時間將有一年的可能性。在硫化氫濃度為 0.2ppm 時,通入純氫性能可回復,但效果不盡理想。Transient Analysis of Hydrogen Sulfide Posioning Effect
on Cell Performance of PEM Fuel Cell
Student:Tzu-Yuan Lin Advisor:Hsin-Sen Chu Department of Mechanical Engineering
National Chiao Tung University
Abstract
A new mathematical model was established to simulate the hydrogen sulfide poisoning effects on the performance of the proton exchange membrane fuel cell. The hydrogen sulfide poisons the anode reaction through preferential adsorbing to the platinum surface and the blocking of active sites of hydrogen. The discussions were focused on finding the range and effects of existing parameters, and on establishing new terms simulating hydrogen sulfide effect. The mechanism was described in terms of adsorption, desorption, electro-oxidation, and terms of hydrogen sulfide on capturing platinum surface from adsorbed hydrogen, by which the performance of PEMFC is affected.
The results showed that it was necessary in the hydrogen sulfide poisoning model to evaluate the effect of hydrogen sulfide on capturing platinum surface, and also the effect of sulfide coverage on the parameter of hydrogen sulfide adsorption. The PEMFC duration was obtained for variable hydrogen sulfide concentration by employing this model. The cell duration was 800 minutes for 5ppm of hydrogen sulfide, 21 days for 0.2ppm, and 81 days for 0.05ppm. For 0.01ppm, the duration could be probably extended to about 1 year.
誌 謝
來讀研究所學習做論文讓我發現到一篇論文的出現,其背後有數 不完的因緣。“得之於人者太多,出之於己者太少",這句國中學過 的名句現在深深的體會到了,看著這即將完稿的論文,回憶起許許多 多應該感謝的人。 首先要感謝我的指導教授 曲老師新生,雖然在group meeting 中 老師一向嚴格謹慎,常常問的問題令我頭腦空白,但若沒有如此精密 嚴格的指導,許多論文上的盲點我可能還難以發現。而老師“絕對精 準"的處世態度更是讓我敬佩。 經此訓練雖不敢說改變成多好,但 在未來行事做人必會憶及師長求學態度與解決問題的方法。再來要感 謝華梵大學校長 顏老師維謀,校長的業務繁重而顏老師除了得顧及 他自己的研究生外還要撥空照顧我們這群外校學生,在和顏老師 group meeting 中,感謝顏老師相當仔細且不厭其煩聽我們報告,並且 提出細節上的建議,使我們得以順利完成論文。 實驗室是每天生活做研究所必須待的地方,在實驗室中學長姐指 導可說是除了老師之外最為重要。要感謝世國學長在工數上的指教, 感謝時明學長時常在軟硬體工具上的經驗分享,感謝建評學長在論文 方向和程式技巧上的指導,感謝純怡學姐幫我們處理實驗室行政業務 以及生活經驗分享,也要感謝聖琰、東洲、清益和為琦學長曾經在學 習上的指導以及論文資訊搜尋的經驗談,還有同學孟熹、智堡和家瑜 在論文格式以及實驗細節的相互討論和指正以及程式上相互切磋,幫 助相當大,另外較少見面的木勝學長也幫過我在理論上的理解,實驗 室的學弟式堯、祥哲和榮祥積極努力學習的精神使我獲得不少激勵。 必須感謝我太極拳老師 江老師弦蒼對我的指導,使我面對研究 生涯能心理安定以及有強健的身體面對耗費精神的研究生涯,更感謝釋迦牟尼佛陀的遺教,使我能超越種種困境和煩惱。在論文上也必須 感謝J.W.Van Zee 等人一系列的研究使我的論文資訊充足,而得以完 成。 以及對中興太極拳社和交大鐘鐸社的全體師生,感謝他們對我 研究生活提供建議。要感謝的人實在太多,無法一一舉出,但是真的 很感謝你們有形無形的幫忙,此論文能完成正是無數因緣的組成,我 感謝這一切因緣。 最後感謝我的父母家人和我女朋友,因為他們的精神上的支持, 使我能夠咬緊牙關撐過難關,直到完成。
目 錄
中文摘要...i
英文摘要... ii
誌 謝... iii
目 錄...v
表目錄... viii
圖目錄...ix
符號說明... xiii
第一章 緒論...1
1.1
前言
... 1
1.2 燃料電池工作原理:... 2
1.3 燃料電池種類介紹:... 3
1.3.1 鹼性燃料電池 ...4 1.3.2 磷酸燃料電池 ...5 1.3.3 熔融碳酸鹽燃料電池 ...5 1.3.4 質子交換膜燃料電池 ...6 1.3.5 直接甲醇燃料電池 ...71.3.6 固態氧化物燃料電池 ...8
1.4 文獻回顧... 9
1.4.1 燃料電池燃料來源以及不純物質和相關抗毒化方法...9 1.4.2 質子交換膜燃料電池硫化物毒化現象...14 1.4.3 質子交換膜燃料電池其他毒化現象 ...161.5 研究內容... 16
第二章 理論分析 ...23
2.1 硫化氫毒化之數學模式介紹... 23
2.2
基本假設
... 26
2.3
統御方程式
... 26
2.3.1 濃度分佈 ...26 2.3.2 觸媒表面覆蓋率 ...27 2.3.3 電流密度分佈 ...282.4
起始條件
... 29
2.5
邊界條件
... 30
第三章 數值方法 ...34
3.1
有限差分法
... 34
3.2
穩態條件
... 36
3.3
Runge-Kutta方法 ... 37
第四章 結果與討論 ...41
4.1
硫化氫覆蓋率比對來源
... 41
4.2
硫化氫毒化模型結果分析比較與討論
... 41
第五章 結論與建議 ...65
5.1
結論
... 65
5.2
建議以及研究趨勢
... 66
參 考 文 獻...67
表目錄
表1- 1 六種燃料電池資料比較[2] ...18 表2- 1 統御方程式 ...32 表2- 2 邊界條件 ...33 表4- 1 基本操作條件之參數表...48 表4- 2 硫化氫吸附速率常數比對...49圖目錄
圖1- 1 質子交換膜燃料電池工作原理示意圖...19 圖1- 2 各種燃料電池的優點與應用範圍之概括圖[2] ...20 圖1- 3 單一薄膜電極裝置之質子交換膜燃料電池的立體示意圖...21 圖1- 4 蒸氣重組製氫流程圖 ...22 圖2- 1 物理模型示意圖 ...31 圖3- 1 Explicit Method 格點分佈示意圖...39 圖3- 2 Implicit Method 格點示意圖...40 圖4- 1 電池操作溫度50℃時,操作電壓 0.7V,硫化氫濃度 5ppm 下, 時間1 秒時,格點數對氫氣濃度分佈之影響。...50 圖4- 2 電池操作溫度70℃時,操作電壓 0.7V,硫化氫濃度 5ppm下, 硫化物隨時間覆蓋情形,操作參數kfs為 20 A/cm2atm,ksh為 0.56 A/cm2atm。 ...51 圖4- 3 電池操作溫度 50℃、70℃和 90℃時,操作電壓 0.7V,硫化 氫濃度5ppm 下,硫化物隨時間覆蓋情形。...52 圖4- 4 電池操作溫度50℃時,操作電壓 0.7V,硫化氫濃度 5ppm下, 電流密度隨時間變化圖,操作參數kfs為 15 A/cm2atm,ksh為 0.5 A/cm2atm。 ...53圖4- 5 電池操作溫度分別為 50℃、70℃和 90℃時,觸媒層厚度 16μm,操作電壓 0.7V,硫化氫濃度 5ppm 下,電流密度隨時 間之變化圖。...54
圖4- 6 電池操作溫度50℃時,觸媒層厚度 16μm,操作電壓 0.7V,
硫化氫濃度 5ppm下,觸媒層內氫氣覆蓋率分佈隨時間之變
化圖,操作參數kfs為15 A/cm2atm,ksh為0.5 A/cm2atm。...55
圖4- 7 電池操作溫度50℃時,觸媒層厚度 16μm,操作電壓 0.7V,
硫化氫濃度 5ppm下,觸媒層內硫覆蓋率分佈隨時間之變化
圖,操作參數kfs為15 A/cm2atm,ksh為0.5 A/cm2atm。...56
圖4- 8 電池操作溫度50℃時,觸媒層厚度 16μm,操作電壓 0.7V,
硫化氫濃度 5ppm下,觸媒層內氫氣濃度分佈隨時間之變化
圖,操作參數kfs為15 A/cm2atm,ksh為0.5 A/cm2atm。...57
圖4- 9 電池操作溫度50℃時,觸媒層厚度 16μm,操作電壓 0.7V,
硫化氫濃度 5ppm下,觸媒層內硫化氫濃度分佈隨時間之變
化圖,操作參數kfs為15 A/cm2atm,ksh為0.5 A/cm2atm。...58
圖4- 10 電池操作溫度50℃時,觸媒層厚度 16μm,操作電壓 0.7V,
硫化氫濃度 5ppm下,觸媒層內電流密度分佈隨時間之變化
圖,操作參數kfs為15 A/cm2atm,ksh為0.5 A/cm2atm。...59
硫化氫濃度0.2ppm下,觸媒層內電流密度分佈隨時間之變化 圖,操作參數kfs為15 A/cm2atm,ksh為0.5 A/cm2atm。...60
圖4- 12 電池操作溫度50℃時,觸媒層厚度 16μm,操作電壓 0.7V,
硫化氫濃度 0.05ppm下,觸媒層內硫覆蓋率分佈隨時間之變
化圖,操作參數kfs為15 A/cm2atm,ksh為0.5 A/cm2atm。...61
圖4- 13 電池操作溫度50℃時,觸媒層厚度 16μm,操作電壓 0.7V, 硫化氫濃度分別為0.2ppm、0.05 和 0.01ppm 下,電流密度隨 時間之變化圖。...62 圖4- 14 電池操作溫度為50℃時,觸媒層厚度 16μm,操作電壓 0.7V, 硫化氫濃度分別為5ppm、0.2ppm、0.05 和 0.01ppm 下,電池 達穩態時間圖。...63 圖4- 15 電池操作溫度為50℃時,觸媒層厚度 16μm,操作電壓 0.7V, 硫化氫濃度0.2ppm下,電流密度隨時間之變化圖,操作參數
符號說明
a :每單位體積觸媒所含有之表面積(cm2/cm3) bfH :氫氣脫附之速率常數(atm) C :濃度(mole/cm3) D :擴散係數(cm2/s)Δ
E :活化能(J/mol)F :法拉第常數(Faraday constant) (C/mol)
Δ
Gf :吉柏自由能(Gibbs free energy) (J/mol)I :電流密度(A/cm2) I :操作電流密度(A/cm2) keH :氫氣電化學反應之速率常數(A/cm2) kfH :氫氣吸附之速率常數(A/cm2 atm) kfS :硫化氫吸附之速率常數(A/cm2 atm) kSH :硫化氫搶奪已吸附氫氣白金表面之速率常數(A/cm2 atm) Lc :與觸媒層入口處之間的距離(μm) n :電子數 P :壓力(atm) R :理想氣體常數(J/mol K) s :化學計量數 T :溫度(K)
t :時間(s) Uo :開迴路電壓(V) V :電位(V) X :莫耳分率 z :位置座標(μm)
希臘字母
α :傳輸係數 ε :孔隙率 θ :觸媒表面積覆蓋率 ξ :氣體影響觸媒莫耳面積密度乘以法拉第常數 φ :電位(V) η :過電位(V) κ :質子傳導係數(mΩ/cm) σ :電子傳導係數(mΩ/cm) Ω :擴散碰撞積分 λ :每單位硫化氫所影響的範圍上標
0 :起始值 c :觸媒層 in :觸媒層入口(z=0)下標
0 :起始值
anode :陽極
act :活化極化(Activation polarization)
c :觸媒層 cathode :陰極 CO :一氧化碳 cell :電池 eff :有效值 H :氫離子 H2 :氫氣 H2S :硫化氫 oc :開電路(Open circuit)
ohmic :歐姆極化(Ohmic polarization)
s :硫
第一章 緒論
1.1 前言
石油能源存量逐漸趨於用盡是必須嚴格面對的問題,目前處理能 源問題的研究人員正積極尋找其他種類替代能源,從開發利用再生能 源到尋找地球現有的未開發資源。但是大家努力的腳步卻越來越趕不 上科技發展所造成日常生活損耗能量上升的速度。如果大家能在日常 生活節省不必要的能源損耗,而且盡量只用生活所必須的機械,在市 場供需關係下,需求的下降將可成功使得科技發展所造成日常生活能 源損耗量得以緩和。 根據 2004 年世界能源會議的調查顯示,全球目前已知的石油儲 藏量保守估計大約剩餘 1 兆 338 億桶,可能尚有 43 年使用時間,而 燃 燒 化 石 燃 料 排 放 大 量 的 二 氧 化碳,為全球溫室效應的主因,使 得地球暖化之問題日益嚴重。現階段替代化石能源的開發除了要解決 石油用盡的問題,再來就是要減少溫室氣體的產生量。吾人在減少溫 室氣體之前提下,於各種能源方案中略舉一二。 一者為太陽能,其能量來源是太陽,此為最無污染的能源,但是 目前所發展的太陽能發電裝置成本相當高,並且碰上日照不足的日子 根本無法提供有效電力,有時間上和氣候上的限制。而風能、地熱能 以及潮汐能等,因其有地理條件上的限制,降低了普遍發展的可能性。 再者核能源產生的能量可能將會是人類所開發能源中,能量產生 最龐大的發電系統,可是目前使用的核分裂法發電所產生輻射廢棄物 的問題一直難以有效解決,而無輻射問題的核融合發電法卻會產生超 級高溫,目前也還沒有材料能承受核融合所帶來超級高的溫度,而低 溫核融合的發展目前也沒有什麼突破性進展,所以這發展還要很長的一段時間,而且核能燃料的鈾礦儲量約剩 395 萬噸,估計能再用 64 年[1]。 相較於上述幾種能源,氫能源應是最有可能廣泛進入人類日常生 活圈的能源,因為氫能源的使用,同樣無溫室氣體產生,而且較不受 時間氣候和地區限制。燃料電池是氫能源的代表之一,其能源效率 高、原理簡單、低噪音和應用範圍廣泛等優點,而其缺點為大部分種 類燃料電池必須使用白金等貴重金屬來作觸媒,使得燃料電池的價格 目前尚無法降到普遍都能接受的地步。不過研究人員努力成果,使得 今日有數種燃料電池已經進入商業化階段,從太空設備、大型發電 廠、潛水艇到汽車、攝影機和軍事用小型通訊設備等,都已經看得到 燃料電池的蹤跡。並且被美國高技術雜誌< Red Herring >評定為未來 發展的十大趨勢之一。因此燃料電池可能成為未來日常生活主流的能 源裝置。
1.2 燃料電池工作原理:
燃料電池是將化學反應的能量直接轉換成電能,其總反應為電解 水的逆反應,即: O H O 2 1 H2 + 2 ⎯⎯→ 2 。 (1-1) 一般氫氧反應為氫氣和氧氣直接接觸反應生成水,無法得到所需 電能,若欲得電能,則必將反應拆為兩部分,以氫氧電池為例,陽極 (Anode)反應為利用觸媒使氫氣直接離子化(或甲醇離子化),產生氫離 子和電子,氫離子經電解質傳送到陰極;在陰極(Cathode)則為氫離 子、電子和氧氣反應合成水的還原反應,如圖1-1 所示。反應中的電 子外電路循環即為所需之電能,其反應式為:陽極: H ⎯⎯→2H+ +2e−
2 (1-2)
陰極: O2 +4e− +4H+ ⎯⎯→2H2O (1-3)
若人欲得知燃料電池之理論電壓,則須由化學反應之吉柏自由能 (Gibbs free energy)變化和電池電壓的關係來推知:
0 U nF G =− Δ Δ (1-4) 其中n 是反應中電子的數目,F 是法拉第常數(Faraday constant), 是在熱力學平衡下的電壓值。 0 U Δ 燃料電池總反應: O H O 2 1 H2 + 2 ⎯⎯→ 2 , Δ = -237 kj/mol (1-5) G 由(1-4)式,吾人可推出氫氧燃料電池在標準狀態 25℃下的電壓為 1.23V。燃料電池的效率為每單位焓值變化量的有用功,有用功是吉 柏自由能改變量,就是電池釋出淨能量除以生成物和反應物焓值的 差,即: h G Δ Δ = η (1-6)
1.3 燃料電池種類介紹:
燃料電池由19 世紀中葉 William Grove 發明以來,迄今約莫一世 紀半的歲月,燃料電池亦由原先玻璃瓶的簡易設計,發展至今日六種 不同形式的燃料電池。其分類方式主要以電解質材料使用與輸入燃料 不同來作區分。依電解質作區分可分為固態和液態兩類。其中液態電 解質型式的有鹼性燃料電池、磷酸燃料電池和熔融碳酸鹽燃料電池, 而固態電解質型式的有質子交換膜燃料電池、直接甲醇燃料電池和固態氧化物燃料電池。在圖 1-2 [2]表示出各燃料電池應用範圍之概括 圖。 以下簡略介紹上述六種燃料電池。
1.3.1 鹼性燃料電池
此技術於1902 年被提出,1950 年代由劍橋大學 Bacon 驗證完成, 為六種燃料電池中發展最早。1960 年代首度運用於載人太空船阿波 羅號上,但在人類生活圈使用時,遭遇到大氣圈中二氧化碳毒化電解 質的問題,直到 1990 年代後都無法有效解決,因此逐漸停止開發。 不過鹼性燃料電池是所有燃料電池中效率最高的(60~70%),因為在鹼 性介質中氧的還原反應在相同觸媒之反應速率比其他燃料電池高之 故,其多孔性電極則是以鎳或氧化鎳來作為主材料,其電極的反應式 為: 陽極: 2H2 +4OH− ⎯⎯→4H2O +4e− (1-7) 陰極: O2 +4e− + H2O ⎯⎯→4OH− (1-8) 此處和其他燃料電池不同為其水生成於陽極處。 此燃料電池以氫氧化鉀溶液作為電解質,分為動態循環電解質和 靜態非循環電解質兩種方式運作。循環電解質可作為燃料電池的冷卻 系統,也將陽極產生水帶走而省去水管理問題,電解質被水稀釋後, 可於外部直接更換新電解質,但每個單電池必須有獨立的循環系統而 大幅增加空間和重量;非循環電解質雖沒有循環系統佔空間的問題, 但水管理和電解質稀釋的問題卻必須重新面對。真正無法發展鹼性燃 料電池的原因在燃料與氧化劑的使用上必須要用純的氫氣和氧氣,不 然會因為不純的氣體中夾雜的二氧化碳造成電解質的中毒現象,使燃 料電池無法使用。而二氧化碳幾乎必存在一般未純化完全的氫燃料中之氣體,故幾乎斷絕了鹼性燃料電池普遍在日常生活圈使用的可能 性,而只能在不計成本的國家軍事單位使用,如太空和潛艇單位。
1.3.2 磷酸燃料電池
1970 年後,開始研究以酸為電解質的燃料電池,目的是對付燃 料與氧化劑中的二氧化碳,最後選定磷酸為電解質,因為磷酸是同時 有良好熱、化學、及電化學穩定性的無機酸,150℃以上低揮發性且 對二氧化碳容忍度高,其電解質是存放在碳化矽母體(SiC matrix)中的 液態磷酸,但長期運轉會有腐蝕與電解質洩漏等問題。磷酸燃料電池 是最早商業化的燃料電池,故稱第一代燃料電池,主要用在固定式的 發電廠中,工作從分散發電廠的電力負擔到負責啟動發電廠都有。其 反應式為: 陽極: ⎯⎯→ + + − e 2 H 2 H2 (1-9) 陰極: O2 +4e− +4H+ ⎯⎯→2H2O (1-10) 它和質子交換膜燃料電池設計大致相同。兩者差異在中央膜,質子交 換膜燃料電池是固態電解質,磷酸燃料電池是液態磷酸,且磷酸燃料 電池抗水的支撐層設計較質子交換膜燃料電池嚴密,為避免液態磷酸 電解質跑入電極中,產生電極的氣體輸送孔被電解質淹沒的現象,而 造成效率的降低。另一差異在其操作溫度約200℃,比質子交換膜燃 料電池高,對於CO 毒化有較好的容忍力。不過因它的啟動速度長達 數小時,以及操作溫度不夠高,使熱電回收效率不佳,此後其發展趨 緩。1.3.3 熔融碳酸鹽燃料電池
熔融碳酸鹽燃料電池由 1940 年代開始發展,由於它的運轉溫度 較高約600℃~700℃,不僅不需要鉑(Pt)作為觸媒,還可配合發電廠整體系統的設計將廢熱回收利用,其整合效率高達80%,而且可以在 燃料電池內部進行燃料重組過程,所以可直接加入未經處理的天然氣 等化石燃料,可大幅節省成本。此電池是繼磷酸燃料電池後推出的商 用燃料電池,也稱第二代燃料電池。熔融碳酸鹽燃料電池的電解質是 鋰和鈉的熔融混合液,此電解質存放於鋁或陶瓷製的母體(Matrix), 以維持穩定。陽極是鎳和鉻的合金,陰極則是氧化鎳,其反應式為: 陽極: + − ⎯⎯→ + + − (1-11) e CO O H CO H 2 2 2 4 2 2 2 2 3 2 陰極: + + − ⎯⎯→ 2− (1-12) 3 2 2 2CO 4e 2CO O 雖然熔融碳酸鹽燃料電池不需用到昂貴的金屬觸媒,但它運轉時 的溫度高,在燃料電池的材料選擇需要考慮高溫產生的影響,像是熱 膨脹、熱應力等問題。目前使用的材料是鉻、鋁、鎳的合金,造價都 非常昂貴,所以在材料的價格是有待克服的問題。
1.3.4 質子交換膜燃料電池
質子交換膜燃料電池,也稱之為固態聚合物電解質燃料電池 (Protem exchange membrane fuel cell, PEMFC),是以質子交換膜作為電解質。此種燃料電池是一種低溫的燃料電池,通常操作溫度在80℃ ~100℃。1960 年質子交換膜燃料電池首度運用於美國雙子星太空計 劃,而且此種燃料電池在反應後會產生純水,所以也可提供太空人日 常生活的飲用水。但當時電解質所用的薄膜是聚苯乙烯硫磺酸鹽的聚 合物,此種聚合物在操作過程中劣化,無法提供燃料電池持續且穩定 的運轉還污染了生成水,因此在後來的太空計畫中能源的供應,改以 鹼性燃料電池來代替。1966 年對於薄膜主要突破是杜邦公司(Dupont) 發展出的 Nafion 薄膜,此種薄膜主要是以聚四氟乙烯為基礎構造, 不僅比先前的聚苯乙烯硫磺酸鹽聚合物安定,且還有更高的傳導性。
質子交換膜燃料電池的兩電極皆是多孔性的氣體擴散電極,以確保燃 料與氧化劑可充分的供應到觸媒層以產生反應,其立體組合示意圖在 圖1-3 表示。在電極與質子交換膜交界有一面鉑觸媒層以促使化學反 應產生,其反應式為: 陽極: H ⎯⎯→2H+ +2e− 2 (1-13) 陰極: O2 +4e− +4H+ ⎯⎯→2H2O (1-14) 質子交換膜燃料電池的優勢為啟動快速,比起需數小時啟動的高 溫燃料電池,它更適合用於緊急用電和家庭用電,而且單位體積小, 在汽車工業未來燃料供應上有雀屏中選之勢。但開發至今有個重大關 卡,就是鉑觸媒遇到不純氫燃料中的一氧化碳(10ppm 以上)會造成嚴 重性能下降的問題,目前解決方案加入釕(Ru)金屬,或通入過氧化氫 (H2O2),還有在氫氣中混入空氣以使一氧化碳氧化成二氧化碳。不過 鉑釕等貴重金屬價錢太高數量不足,而通入過氧化氫研究尚不完全, 通入空氣會造成性能下降。亦有氫氣儲存攜帶安全性和容量的問題。 這些問題必須解決後,質子交換膜燃料電池才有可能真正普及化。
1.3.5 直接甲醇燃料電池
1960 年代開始發展,先是用鹼性電解質,但性能令人不忍卒賭。 1990 年後,因為質子交換膜燃料電池技術快速發展,故開始改用高 分子膜作為電解質,甚至整個系統都改成質子交換膜燃料電池的翻 版,主要區別為甲醇燃料電池用的燃料是甲醇,且用的觸媒不是白 金,而是白金、釕和碳的混合物,其使用釕的原因也是一氧化碳毒化 問題。因甲醇較氫氣容易取得,儲存較容易,燃料攜帶方便,故目前 一直往小型化方便化發展,有可能成為 3C 電子隨身產品的能源來 源。甲醇和白金觸媒的反應與氫氣相類似,其反應式為: 陽極: + ⎯⎯→ + + + − e 6 H 6 CO O H OH CH3 2 2 (1-15)陰極: O2 6H 6e 3H2O 2 3 + + + − → (1-16) 不過甲醇燃料電池是沿用質子交換膜燃料電池結構,薄膜部分無 法阻止甲醇滲透,會產生甲醇從陽極穿透至陰極和陰極觸媒反應的情 形,稱甲醇橫越現象(Methanol crossover) ,此問題使甲醇燃料電池性 能受到相當影響。當前改善想法為增加厚薄膜厚度且將陰極的觸媒換 成不會和甲醇反應的觸媒。若能有效改善,甲醇燃料電池可能成為下 一種商業燃料電池。
1.3.6 固態氧化物燃料電池
這是第三代商用燃料電池,1950 年代末期,先發展出圓管式固態 氧 化 物 燃 料 電 池 , 其 最 大 特 色 為 對 硫 化 物 容 忍 度 很 高 , 在 使 用 300ppmH2S 氫氣燃料下,可運轉 1000 小時以上,再經除硫手續可以 復原,其他種燃料電池只需不到1ppm 的硫化物就使性能嚴重受損, 也無法復原。另有平板式,製程簡單,材料成本比管式低廉,電池內 電阻較低,但因密封不易的問題,所以和圓管式相比各有千秋。固態 氧化物燃料電池的電解質為固態氧化鋯,此電池是以固態與氣態兩相 系統來運作,不會產生觸媒層被水淹沒(沒有液態水的問題)、氧氣還 原反應速度緩慢等問題。其運轉溫度800℃~1000℃,不需昂貴的貴 金屬觸媒,還可用其高溫進行內部燃料重組,可直接使用天然氣等燃 料,而且熱電合併效率高達85%。電極方面,陽極為鎳與陶瓷混合材 料(YSZ-Ni),陰極為 P 型半導體材料,其電極電化學反應式為: 陽極: + − ⎯⎯→ + − (1-17) e CO O CO 2 2 4 2 2 2 陰極: + − ⎯⎯→ 2− (1-18) 2 4e 2O O 相較於熔融碳酸鹽燃料電池,固態氧化物燃料電池供電範圍更廣 (1kw~100mw),小至可攜式電力,大至大型發電設備都是固態氧化物燃料電池的適用範圍,且操作溫度更高,熱電回收效率也更好,可是 其週邊設備多,整個製造成本也相對較高,若能下降製造成本,其普 及化指日可待。
1.4 文獻回顧
1.4.1 燃料電池燃料來源以及不純物質和相關抗毒化方法
燃料電池中不純物質以CO影響最為明顯,當CO濃度超過一定量 時,CO會和氫氣去競爭附著於觸媒層的Pt觸媒上,而CO包覆Pt表面 的量,將影響Pt觸媒催化金屬的表面積,導致於被CO包覆者無法繼 續催化氫氣的電化學反應。在1996年,Tilquin et al. [3]的實驗結果顯 示,陽極燃料氫氣含CO濃度超過10ppm以上時,質子交換膜燃料電池 的性能會受到影響,隨著CO濃度提升,電池性能的下降越嚴重。在 2002年,Bocarsly et al.[4]提到抗CO毒化能力,隨著電池操作溫度上 升而增加。此外,在2003年,Van Zee et al. [5] 實驗發現,電池內部 的溫度和壓力也會對整個毒化現象造成影響。Springer et al. [6]由 Bernardi and Verbrugge [7]燃料電池電化學數學模型,提出一維一氧化 碳毒化模式,模式中描述了當陽極所通入的氣體同時存在氫氣與一氧 化碳時,觸媒表面受到吸附、脫附以及電化學反應之影響,觸媒表面 積因氫氣與一氧化碳覆蓋隨時間改變的情形,而這覆蓋率的改變會影 響到電池所產生的電流量,而其中速率常數的取決,以及速率常數與 覆蓋率之間關係式的發現,都使得理論與實驗的關係性更為準確。 Chan et al.[8]延伸及放寬Bernardi and Verbrugge [7]將觸媒層視為邊界 條件的限制,提出一完整質子交換膜燃料電池電化學反應模式。並且 加入一氧化碳毒化模式和實驗比對,結果比之前文獻更精確。Wang and Bhatia [9]改良Springer et al. [6]的數學模式,成為一個暫態的數學 模式,以及討論氫濃度變化對毒化後對電池在陽極處性能的影響,研究結果發現,一氧化碳濃度越高,電池達穩態所需的時間越短。 在面對CO毒化問題前,要先了解燃料中為何會有CO存在。目前 氫氣燃料的來源主要有電解水,和由化石燃料重組兩種主要方法,而 電解水所損耗的能源和成本過高,而且電解出的氫氣又拿去發電,不 符合經濟效益,並不適合作為燃料電池氫氣來源,所以當前氫氣主要 來源是化石燃料重組。化石燃料重組約分為三種:蒸氣重組法(Steam reforming) 、 部 分 氧 化 法 (Partial oxidation) 、 水 煤 氣 法 (Coal gasification) 。在此只舉出蒸氣重組法[10],此法是最有效率且最廣泛 被用來製造氫氣之製程,其係利用蒸氣與低碳素的石化原料(常用的 是甲烷)反應產生氫氣及二氧化碳的程序,其反應可分為以下兩個主 反應:
合成氣(Synthesis gas generation)生成反應:
CH4+H2O →3H2+CO (1-19)
水-氣轉化反應:
CO+H2O→H2+CO2 (1-20)
總反應: CH4+2H2O+熱→4H2+CO2 (1-21) 此反應所使用的鎳觸媒會遭到硫化物的毒化,而且燃料電池中 Pt 觸媒若遭硫化物污染,連復原的機會都沒有[11],甲烷中含硫化物 約24.5ppm,故甲烷在進入重組器之前要先進入脫硫器脫硫,使硫含 量降至 0.5ppm 以下,以免毒害觸媒,此為高度吸熱反應因此重組器 需要燃料燃燒加熱,藉由空氣與燃料反應產生蒸氣與熱,使反應溫度 維持在 650~700℃左右,壓力依操作條件不同可操作於 7~48 大氣壓 之間。重組器出口氣體溫度在870~885℃之間,壓力於 22~24 大氣壓
之間,甲烷轉化率可達 95%,組成為 76%H2,12%CO,10%CO2, 1.3%CH4與少量水蒸氣,重組器出口氣體經由熱交換器將一部份的熱 與蒸氣回流,再進一步於轉化器內用鋁為擔體的銅鋅觸媒溫度於 200~300℃ 之 間 進 行 水 - 氣 轉 化 反 應 , 生 成 86%H2,22%CO2, 0.25%CO,1.3%CH4,轉化器出來的氣體進一步進入二氧化碳回收系 統,於此系統中將氣體重新加熱至315℃,藉由氧化鎳觸媒將二氧化 碳轉化成甲烷,並回收作為進料,移除二氧化碳之後再進一步純化得 到所要的產品氫氣(98%)。故此製程並無二氧化碳排放問題。如圖 1-1 說明甲烷製氫重組的流程。 但水氣轉換反應會有逆反應出現[12],因此一氧化碳的消除並不 完全。整個產氫過程最後仍有 20ppm 左右的 CO,若以此不純燃料當 質子交換膜燃料電池的燃料,使用一小段時間後,性能將受損至電池 無法輸出所需功率的程度,所以燃料電池本身必須有抗CO 毒化設計。 當前的燃料電池抗毒化設計有(1)純化燃料氣體, (2)發展抗毒化 的觸媒層,以及(3)通入氧化劑進入燃料氣體中[13] 。 純化燃料氣體方法主要有兩目的[10,11],第一就是除去硫化物到 濃度0.1ppm 以下,因為硫化物毒化觸媒 PtRu 的力量更強,而且無法 回復性能;第二就是水-氣轉換反應(Water-gas-shift): 2 2 2O CO H H CO+ → + (1-22) 此反應目的是將一氧化碳大量轉換成二氧化碳,但經兩次水-氣轉換 反應後,剩餘的一氧化碳量仍高於 20ppm 以上,此濃度對未經抗 CO 毒化設計的質子交換膜燃料電池而言,還是過高的濃度。在 2004 年, Rosso et al. [10]提出沸石輔助貴金屬設計預先將氫燃料中一氧化碳反 應 掉 的 方 法 , 使 製 氫 設 備 所 產 出 的 氫 燃 料 中 一 氧 化 碳 濃 度 小 於 10ppm,而圖 1-4 就是製氫去硫去 CO 以及水氣轉換的流程圖。
此法使燃料一氧化碳濃度直接降到 10ppm 以下是根本的做法, 但整體製氫成本上升許多,所以還只是選擇之一的做法。 發展抗毒化觸媒層,目前針對 CO 毒化而設計的觸媒層有 PtRu 設計[14,15] ,此種設計目前大量應用在甲醇燃料電池,而質子交換 膜燃料電池尚在實驗室使用此設計。 會運用 PtRu 設計的原因是,當一氧化碳開始接觸鉑觸媒後,將 開始佔據鉑表面,將影響氫氣接觸鉑表面的機會,而附著於鉑表面的 一氧化碳並不會脫離,因此長時間大大影響電池性能。加入釕(Ru)的 原因正是為了處理一氧化碳並不會脫離鉑表面的問題,而Pt:Ru 以 1:1 的比例效果最好。釕金屬和水接觸後可產生氫氧根,而附著在釕表面 的氫氧根可和鉑表面的一氧化碳反應成二氧化碳和水,藉此剔除附著 於鉑表面的一氧化碳,使電池性能不再受到影響,其反應化學式為: − + + + + + → − + −CO Ru OH Pt Ru CO H e Pt 2 (1-23) 此設計雖可使質子交換膜燃料電池的氫氣來源不用太嚴格要 求,但此設計卻有幾個難題而使其難以實施。首先,釕(Ru)太貴了, 這是最主要的原因,而且釕遭遇到硫化物毒化後完全無法再使用 [14] ,其次因為使用釕,使鉑在觸媒層中比例下降而降低了電池原有 性能[11],若通入純氫則幾乎是一種浪費。
另一種為PtMo 設計[17,18] ,其抗毒化原理和 PtRu 相同,Pt:Mo
以5:1 的比例效果最好,和 PtRu 觸媒層相比,PtMo 觸媒層的 CO 容 忍力更好。不過面對二氧化碳 PtMo 觸媒層性能就會受影響。不巧的 是,當一氧化碳被氧化處理掉之後就是變成二氧化碳,如此一來PtMo 觸媒層的設計就有待研究。 而在2002 年 Yu et al. [13] 實驗複合型觸媒層的抗毒化設計,他 們將PtRu 型觸媒和 Pt/C 觸媒層作為三明治架構,因為 Pt/C 觸媒層對
純氫氣有較高的反應效率,所以將 PtRu 型觸媒放在靠氣體擴散層去 處理剛進來的一氧化碳,剩餘純氫氣再和內層 Pt/C 觸媒層反應得到 高效率反應,此實驗的設計確實將抗毒化設計和維持反應效率的目標 達成。 Ishikawa et al. [19]提出一氧化碳吸附於Pt和其他貴金屬的電化學 理論之理論模式。Norskov et al. [20]進一步將氫氣和一氧化碳在Pt、 Ru、PtRu、Pt3Sn作為觸媒的電化學模式建立出來,比較結果發現, Pt3Sn無論在有無一氧化碳毒化的情況下,其效能都比其他合金作為觸
媒要來得高。2005 年Sofia Enback and Goran Lindbergh [21]以實驗及 分析方式探討建立一氧化碳在PtRu/C觸媒上影響的模式,理論與測量 結果相當吻合。 通入氧化劑進入燃料氣體中的方法是這三種方法中最便宜的方 法。當氧化劑進入電池後,將會產生氫氧根,把附著在鉑表面的一氧 化碳氧化成二氧化碳,達到解除毒化的目的。目前通入的氧化劑有, 通入氧氣、通入空氣(Air bleed)以及通入過氧化氫。
Rohland and Plzak[22]發現通入氧氣的方法能將一氧化碳大量氧 化掉,解除毒化現象,但是使用氧氣當氧化劑有非常嚴重的問題,就 是氧氣將會在鉑表面和氫氣反應產生水,如此將會導致電池性能下降 [13]。而通入空氣,空氣中氧氣濃度只有五分之一左右, Van Zee et al. [23]實驗結果顯示,通入空氣也有良好抗 CO 毒化能力,而且對電池 性能影響比氧氣小,更重要的是空氣是免費的。此外,Van Zee 和其 研究團隊[4]亦發現,溫度和壓力也對通入空氣抗毒化能力有所影響。 再來是通入過氧化氫, Schmidt et al. [24-26] 和 Bellows et al. [27]的
實驗發現,就算濃度高達100ppm 一氧化碳的不純燃料,若使用過氧
H2O2解除CO 毒化的機制有下列兩種, 第一種方式: H2O2+M →H2O+M
( )
O ad (1-24) 2M( )
O ad →2M +O2 (1-25a)( )
O M( )
CO 2M CO2 M ad + ad → + (1-25b) 第二種方式:(
M OH)
O H M + → 2 − 2 2 2 (1-26)( )
+( )
→ + + + + − e H CO M OH M CO M ad ad 2 2 (1-27) 此兩種方式同時在進行,所以效果不錯。 但Furuya et al. [28]研究,在沒有一氧化碳時使用過氧化氫則可能 使電池性能減弱,長期使用有可能對質子交換模造成損傷,雖然過氧 化氫抗毒化能力強又便宜,但是因為這些因素的影響,所以此方法尚 待深入的實驗以了解其實際運用的可行性。1.4.2 質子交換膜燃料電池硫化物毒化現象
除了一氧化碳是質子交換模燃料電池頭號殺手外,另外硫化物也 會影響電池性能的物質。硫化氫是由厭氧菌分解有機物質或硫酸鹽類 之代謝產物,而化石燃料是由古代生物屍體經高溫高壓且無氧的狀態 下逐漸變化而成,所以由重組而來的燃料氣體中必有硫化物存在。Miller and Koningsberger [29]所發表的文章中敘述到,硫化物毒 化貴金屬觸媒的毒化機制,硫化物化學吸附至白金表面後,將白金原 子表面遮蓋住,使白金表面無法和氫氣反應,造成毒化狀態。此毒化 機制與一氧化碳毒化機制相同。
Van Zee et al. [11]的實驗結果顯示硫化物對燃料電池的影響為最
可使電池觸媒完全失效。二氧化硫濃度 0.05ppm 和硫化氫濃度 1ppb 就可以經可以毒化電池。 Yan et al. [30]在 2001 年發表的論文中,證 實了硫化物毒化在一般操作溫度下性能無法還原,因為吸附於白金上 的硫難以脫附。尤其硫化物對PtRu 觸媒的性能影響更為嚴重[16],其 毒化所需濃度更低,而且反應毒化影響的速度更快,所以燃料製備過 程必有除硫手續。 硫化物吸附於白金的化學反應式分為, H2S 的反應式: 2 2S Pt (Pt S) H H + → − + (1-28) (1-29) − + − + + → + H O SO H e S H2 4 2 42 10 8 (1-30) Pt e H SO O H S Pt− )+3 ↔ +6 + +6 − + ( 2 3 (1-31) Pt e H SO O H S Pt− )+4 ↔ − +8 + +6 − + ( 2 4 2
(
)
2 2 2 3 ) (Pt S H H Pt S H + − ↔ − + (1-32) SO2的反應式: (1-33) O H SO Pt e H SO2 +2 + +2 − ⎯⎯→Pt ( − )+ 2 (1-34) O H S Pt e H SO Pt ) 2 2 Pt ( ) 2 ( − + + + − ⎯⎯→ − +Van Zee et al. [11]以實驗驗證他們所提硫化物於觸媒上覆蓋的數 學模式,研究結果發現,理論結果與實驗結果相近。 面對硫化物毒化,目前是以強化重組器脫硫程序為主,在Ahmed et al. [31]整理的資料中,了解目前重組器脫硫後硫化氫的殘留量在 2003 年尚有 200ppb 的硫化氫,而在 2005 年進步到剩 50ppb 的含量, 雖然 50ppb 對電池影響已經小很多,但比起 1ppb 的含量依舊是偏高。 目前預期在 2010 年硫化氫的殘留量能低於 10ppb。在 Fukunaga and Ponec [32]研究中發現,使用氧化鉀來去除已吸附於白金上的硫有相
當好的效果,因為氧化鉀對硫化氫的反應力比硫化氫對白金的吸附力 更強所導致。 Apesteguia et al. [33]研究發現,一氧化碳和硫化氫同時存在於燃 料氣體環境時,對於白金吸附情形,同一白金可同時吸附一氧化碳和 硫化氫,如此較不會有白金觸媒遭到其中一種毒化物毒化,而另一種 毒化物質去覆蓋其餘空白金表面。 以上數種硫化氫和白金反應的研究,雖然針對燃料電池的部份數 量並不多,但足以幫助對硫化氫毒化數學模式的建立。
1.4.3 質子交換膜燃料電池其他毒化現象
在日本汽車工業Akai et al. [16]研究氫燃料中不純物質對質子交 換膜燃料電池性能之影響,發現不只一氧化碳和硫化物會影響電池性 能其他碳氫化合物幾乎都會降低電池性能。在2004 年, Van Zee et al.[34]開始實驗分析由陰極進入的空氣中污染物所造成的影響,發現 除了硫化物外,還有臭氧和二氧化氮也會影響電池性能。 另外在海島型國家,空氣中含有氯化鹽類,劉又綸[35]的實驗證 明,當氯化鈣和氯化鈉伴隨空氣由陰極進入燃料電池,確實造成燃料 電池性能下降,此乃氯化鈉造成陰極集電板的腐蝕,使陰極過電為增 加。而氯化鈣的入侵則是因鈣離子影響膜的結構和水傳輸現象,使得 陽極缺水造成電池性能下降。Okada [36,37]當中提到,當燃料電池在 長時間運轉的過當中,這些污染離子進入電池內部的含量小,事實上 對於電池的影響不大,一但真的發生影響,這些陽離子對觸媒層所造 成的影響將遠超過薄膜。1.5 研究內容
隨著國際原油價錢高速攀升而毫無回跌的趨勢,運輸成本也隨之上升,生活物資價格呈現上揚趨勢,直到原油趨於用盡的一天。燃料 電池開發到現在,技術上相當成熟,也已經有燃料電池產品上市,不 過都是高等價位,而且在其燃料上還有問題需要克服,就是氫氣和空 氣中的少量不純物質。 由重組器所產出的氫氣中有約 25ppm 的硫化氫,對質子交換膜 燃料電池而言,25ppm 的一氧化碳足以造成電池性能下降,而如果除 硫未完善,只要有超過 1ppb 的硫化氫,燃料電池性能開始下降而且 也難以復原。硫化物除了由燃料氣體處理不良而進入燃料電池外,若 空氣中含硫化物的量過高的話,電池陰極觸媒也將受到毒化破壞。由 空氣品質監測報告可知,例如像溫泉或火山地帶以及空氣品質較差的 工業區,甚至空氣污染較嚴重的都會區,空氣中含硫化物的量可能到 達足以影響燃料電池的邊緣。 正因為一氧化碳和硫化物是不論在燃料氣體或空氣中都會存在 的物質,因此對於燃料電池受到一氧化碳和硫化物毒化的現象,尤其 是在硫化物毒化的部分,理論模式缺乏。硫化物中以硫化氫影響最為 明顯,只需極少量就造成影響,所以硫化物的部分以硫化氫為代表參 與毒化反應。因此,本文所要探討的方向,是提出和發展硫化氫毒化 的理論模式,先利用數值模擬的方式比對實驗,進而探討當硫化氫進 入電池後,對電池性能影響之暫態影響,希望能更進一步了解電池受 到硫化氫毒化所發生的現象。
表
1- 1 六種燃料電池資料比較[2]
AFC PAFC MCFC PEMFC DMFC SOFC
操作溫度 溫度範圍 100℃ 80-200℃ 205℃ 180-210℃ 650℃ 600-800℃ 80℃ 80-100℃ 90℃ 60-90℃ 1000℃ 800-1000℃ 理想電壓 1.15V 1.14V 1.03V 1.18V 1.183V 0.91V 操作電壓 0.8-0.95V - - 0.6-0.8V 0.4-0.5V - 發電效率 60-70% 37-42% >50% 43-58% ~40% 50-60% 熱電回收 - 60-70% 80% - - 85%
e -e- H+ e -e -O2 H2 e -e -H2O H2 O2 H+ e- e -e- e -圖1- 1 質子交換膜燃料電池工作原理示意圖
applications Portable electronics equipment Cars,boats, and domestic CHP Distributed power generation CHP, also buses POWER in W Typical atts 1 10 100 1k 10k 100k 1M 10M Main Advantages Higher energy density than batteries.
Faster recharging
Potential for zero emissions, higher efficiency Higher efficiency less pollution quiet Range of application of the diff rent
types of fuel cell MCFC AFC SOFC e PEMFC PAFC 圖1- 2 各種燃料電池的優點與應用範圍之概括圖[2]
陽極平板 陽極氣體擴散層 陽極觸媒層 陰極氣體擴散層 陰極觸媒層 質子交換膜 陰極平板 圖1- 3 單一薄膜電極裝置之質子交換膜燃料電池的立體示意圖
空氣和燃料 熱交換器 水-氣轉換 二氧化碳回收系統 純化器 98%氫氣和約 0.2ppm 硫化氫 以及20ppm 一氧化碳 甲烷 脫硫器 硫 重組器 圖1- 4 蒸氣重組製氫流程圖
第二章 理論分析
2.1 硫化氫毒化之數學模式介紹
在1988 年,Gottesfeld and Pafford [38]就已做出實驗證實,一氧 化碳影響電池性能衰退的機制,在於毒化陽極觸媒層內之白金,一氧 化碳會與白金形成穩定之鍵結,造成氫氣可以利用之空間減小,由於 一氧化碳幾乎不會產生電流密度,因此一但氫氣的的使用率下降,直 接會影響到電池的發電量。 2001 年,Springer et al. [6],由化學反應的角度,從氫氣與一氧 化碳在白金表面的化學反應式中,找到對應之數學關係,其化學反應 式如下: ) ( 2 2 2 Pt H Pt H + ←⎯→ − (2-1) ) (2-2) (CO Pt Pt CO + ←⎯→ − − + + + ⎯→ ⎯ −Pt Pt H e H (2-3) − + + + + ⎯→ ⎯ + −Pt H O Pt CO H e CO ) 2 2 ( 2 2 (2-4) (2-1)式代表氫氣在白金表面所造成的吸附與脫附之現象,(2-2) 式則代表一氧化碳在白金表面的吸附與脫附現象,前二式只是單純的 代表與白金鍵結與脫離現象;而的(2-3)式則是氫氣電化學反應式,代 表氫氣消耗產生電流,同樣的 (2-4)式代表一氧化碳消耗產生電流。 一氧化碳毒化數學模式即在利用氫氣與一氧化碳在白金表面的化學 反應,來描述氫氣與一氧化碳的質量守恆現象,其數學方程式表示如 下:
(
)
⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − − − = RT F n k k b P X k dt d H anode H eH H fH fH CO H H fH H 2 sinh 2 1 2 2 η θ θ θ θ θ ξ (2-5)(
)
⎟ ⎠ ⎞ ⎜ ⎝ ⎛ − − − − = RT F n k k b P X k dt d CO anode CO eCO CO fCO fCO CO H CO fCO CO 2 sinh 2 1 θ θ θ θ η θ ξ (2-6) (2-5)式中左邊第一項代表氫氣在白金表面的覆蓋率隨著時間的 改變量,右邊第一項代表氫氣吸附的增加量,第二項代表氫氣脫附的 減少量,第三項則代表電化學反應之消耗量。而(2-6)式,則是代表一 氧化碳在白金表面所牽涉到的化學反應,與(2-5)式意義相似。 在一氧化碳毒化數學模式發展出來之後,Springer et al.[6]開始尋 找適當之反應速率常數,為的是要與實驗現象相符合,在過程中, Springer et al.[6]又更進一步發現,氫氣吸附的速率常數與一氧化碳脫 附的速率常數與一氧化碳在觸媒表面的覆蓋率有關,因此更準確的與 實驗值相符合,在往後的研究當中成為了一氧化碳毒化的理論模式基 礎,如2004 年,Wang et al. [5],所解出之暫態現象即是引用 Springer et al. [6],所發展出來的數學模式,不過 Wang 等人所求解的方式,僅 是單純的零維度模式(Zero dimensional),也就是只解陽極觸媒層部 分,由於不解厚度方向的分佈情形,電池的電流密度即是(2-5)及(2-6) 式的電化學反應項之總和。由於不考慮觸媒層的厚度,以及其他各層 的效應,因此所得到的結果並不是那麼的準確。 Akai et al. [16]研究發現氫燃料中不純物質影響,硫化物對燃料電 池的影響是最為嚴重的。Van Zee et al. [11]由化學反應的角度,從硫 化氫在白金表面的化學反應式中,找到對應之數學關係,其化學反應 式如下:H
2S
+
Pt
⎯
⎯→
kfs(
Pt
−
S
)
+
H
2 (2-7) H2S +4H2O → SO42− +10H+ +8e− (2-8)(
Pt
−
S
)
+
3
H
2O
↔
SO
3+
6
H
++
6
e
−+
Pt
(2-9) (2-10)(
Pt −S)
+4H2O ↔ SO42− +8H+ +6e− + Pt2
(
)
2 2 3 ) (Pt S H H Pt S H + − ↔ − + (2-11) (2-7)式代表硫化氫在白金表面所造成的吸附現象, (2-8)式則代 表硫化氫和水的反應,而 (2-9)式和(2-10)式則是硫化氫電化學反應 式,代表硫化氫消耗產生電流,但和氫氣以及一氧化碳比較之下,(2-9) 式和(2-10)式的反應幾乎可視為不存在,因為相當微弱。而(2-11)式是 硫化氫能將已吸附的氫拔除下來自己取而代之的特別反應[34]。數學 模式的建立,就在利用硫化氫在白金表面的化學反應,來描述硫化氫 覆蓋觸媒現象,其數學方程式表示如下[11]:(
s H S H fs s P k dt dθ θ θ)
− − = 1 2 (2-12) 上式中左邊第一項代表硫化氫在白金表面的覆蓋率隨著時間的 改變量,右邊第一項代表硫化氫吸附的增加量。氫覆蓋率被假設為比 硫覆蓋率小很多,所以可將θH忽略。方程式即變為:(
s S H fs s k P dt dθ θ − = 1 2)
(2-13) 經初始條件t=0 時 θs=0,推出 t P k s S H fs e 2 1− − = θ (2-14) 此式繪出圖形和實驗比對,在 300 分鐘內預測還算準確,但超過 300 分鐘後,誤差開始加大。 因為硫化物模式的建立算是相當近期才開始開發的方向,目前針 對硫化物毒化模式探討的文章相當少量,所以在跟隨探討一氧化碳毒 化模式之下,會希望增加硫化氫毒化的討論,甚至進一步比較一氧化 碳和硫化物毒化的差異,以及同時遭毒化的狀況探討。2.2 基本假設
本文所要探討的是當電池受到硫化氫毒化後,陽極觸媒層內氫氣 與硫化氫的觸媒表面覆蓋率以及到達穩態所需要的時間,因此,主要 考慮的範圍暫定在陽極觸媒層部分(0≤ z≤ Lc),主要的假設如下: 1. 一維之直角座標系統。 2. 電池處於恆溫狀態。 3. 氣體為理想氣體。 4. 多孔性材質之孔隙為均勻分布。 5. 擴散為唯一的傳輸模式。 6. 陽極觸媒層內考慮氫氣與硫化氫之傳輸行為。 7. 忽略硫化氫的電化學反應。2.3 統御方程式
文章所使用之物理模型如圖2-1 所示,由於主要探討硫化氫毒化 影響的是陽極觸媒層,所以將討論區間定在陽極觸媒層部分,因此將 原點定在陽極氣體擴散層與陽極觸媒層之交界處,A 點定為陽極觸媒 層與薄膜交界點。2.3.1 濃度分佈
在陽極觸媒層內,氫氣與一氧化碳的濃度分佈,主要受到擴散與 電化學反應的影響,在隨著位置與時間的分佈可表示如下[6]: H2: ⎟⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ − ∂ ∂ = ∂ ∂ F n s dz di z C D t C H H H H H c H c 2 2 2 2 2 2 2 2 ε ε c L z≤ ≤ 0 (2-15) H2S: ⎟⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ − ∂ ∂ = ∂ ∂ F n s dz di z C D t C S H S H S H S H S H c S H c 2 2 2 2 2 2 2 2 ε ε c L z≤ ≤ 0 (2-16)其中εc代表孔隙率、D為擴散係數、i是所產生之電流密度、s是化學 計量數、n代表電量、 則是法拉第常數,第一項代表控制體積內濃 度隨時間變化的增加量,第二項代表經由擴散作用進出控制體積濃度 的變化量,第三項則是代表參與電化學反應所造成之濃度消耗量。 F 硫化氫擴散係數DH2S的來源可由文獻[39]而來,參數經驗公式表 示如下: D 2 S/H H 2 1 S/H H 2 3 S/H H 2 2 2 2 2 2
PM
0.00266T
D
Ω
=
σ
(2-17) 其中P(bar)代表壓力、T(K)為絕對溫度、σ(Αo )是特徵長度、 是 擴散碰撞積分、M 代表分子量相互影響關係。研究中的硫化氫相對於 氫氣的擴散速率 為0.27684。 D Ω 2 2S/H H D2.3.2 觸媒表面覆蓋率
當陽極觸媒層同時存在氫氣、硫化氫和一氧化碳時,氫氣、硫化 氫和一氧化碳為了進行化學反應,會與空出來的白金表面進行吸附、 脫附以及電化學反應產生電流,影響到白金表面氫氣、硫化氫和一氧 化碳的覆蓋率,而進行這三種程序的難易程度,取決於各個過程所中 所涉及到的速率常數,其表示式如下[11]:(
)
h H h anode H H eH H fH fH H s H fH H k P RT F n k k b P X k dt dθ θ θ θ θ η θ ξ 2 s 2S 2 1 2 sinh 2 ⎟⎟− ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − − − = c L z≤ ≤ 0 (2-18)(
s H)
fs fs s h H h S H fs s P k k b P X k dt dθ θ θ θ θ ξ s S 2 2 1− − − + = 0≤ z≤ Lc (2-19)ξ為觸媒表面的面積密度與法拉第常數的乘積,k、b代表速率常 數,X 為莫耳分率, 為壓力,P ηanode為陽極過電位,R為萬用氣體常 數,T代表電池溫度。等式左邊代表覆蓋率隨時間的變化,等式右邊 由左至右分別代表氫氣的吸附項、脫附項與電化學反應造成之消耗 項,以及白金表面積搶奪項。其中莫耳分率是覆蓋率方程式和濃度方 程式相互影響的關鍵參數。Van Zee 等人[40]發現陽極過電位超過 0.8V 時,才出現硫脫附反應。 另外,氫氣吸附的速率常數與硫化氫在觸媒表面的覆蓋率有關, 因此可表示如下[5]:
(
)
⎥ ⎥ ⎦ ⎤ ⎢ ⎢ ⎣ ⎡ ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − Δ − ⋅ = 1 exp 1 exp 0 S S H fH fH RT E k k θ λθ δ 0≤ z ≤ Lc (2-20) 此研究中假設硫化氫吸附的速率常數與硫化氫在觸媒表面的覆蓋率 有關,因此可表示如下:( )
⎥
⎥
⎦
⎤
⎢
⎢
⎣
⎡
⎟
⎟
⎠
⎞
⎜
⎜
⎝
⎛
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛
−
−
Δ
−
⋅
=
1
exp
1
exp
S 0 S S S S f fRT
E
k
k
θ
λθ
δ
0≤ z ≤ Lc (2-21) 其中 ΔES 代表硫化氫吸附所需之活化能。2.3.3 電流密度分佈
由於受到硫化氫毒化的影響,觸媒的表面覆蓋有氫氣與硫化氫兩 種分子,但一般情形硫化氫附著後相當安定,因此電化學反應所產生 之傳輸電流密度,主要由氫氣產生,因此,傳統的 Bulter-Volmer 方 程式必須對白金表面覆蓋率進行修正[6]:⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ = = RT F n ak dz di dz di H anode H eH H 2 sinh 2 2 2 θ η 0≤ z≤ Lc (2-22) 其中 代表每單位體積觸媒所含有的表面積,右式代表氫氣反應產生 子,所以在產生電流密度式子中不加入硫化物產生的電流密度項。 a
在電壓電流密度圖的繪製上,以Wang and Bhatia [9]計算電壓電
流密度圖的方程式來使用: ohmic c a o cell
V
V
=
−
η
−
η
−
η
(2-23) 其中: ohmic ohmic oc c h eh aiR
i
i
F
RT
k
i
F
RT
=
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛
=
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛
=
−η
α
η
θ
α
η
ln
2
sinh
1 (2-24) ηa為陽極活化過電位,ηc為濃度過電位,ηohmic為歐姆過電位,V0為初始電壓,Vcell為電池實際電壓,ioc為開迴路電流,Rohmic為電池內電
阻。以上統馭方程式整理於表4-1。
2.4
起始條件
在本文中所使用到的起始條件,是假定燃料電池系統處在一開始 未啟動的狀態,也就是t = 0 時,電池內部沒有任何氣體,當 t > 0 時, 才開始有氣體通入,其數學式表示如下:( )
,0 0( )
,0 2 2 z C z CH = H t = 0 (2-25)( )
,0 0(
,0 2 2 z C z CHS = HS)
t = 0 (2-26)0 ) 0 , ( H H z
θ
θ
= t = 0 (2-27) 0 2 2S( ,0) H S H zθ
θ
= t = 0 (2-28)2.5
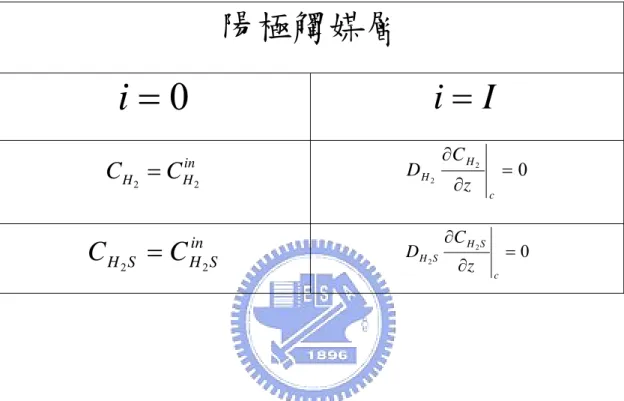
邊界條件
在濃度的邊界條件部份,是給予陽極觸媒層入口處( z = 0 ) ,固定 之濃度值,在觸媒層與薄膜交界處( z = Lc ),由於薄膜有阻隔氣體的 作用,因此給予通量為零的邊界條件,整理於表4-2 並如下所示: in , z = 0 (2-29) H H C C 2 = 2 in S H S H C C 2 2 = 2 0 2 ∂ = ∂ z C DH H , 2 0 2 ∂ = ∂ z C DHS HS z = Lc (2-30) 在電流密度部分,由於在陽極觸媒層入口處( z = 0 ),是電化學反應最 先發生的地方,因此電流也由此處開始產生,在這裡是將電流密度定 為零作為邊界條件: z = 0 (2-31) 0 = iLac Lm Membrane CL GDL Anode z 圖2- 1 物理模型示意圖
表
2- 1 統御方程式
陽極觸媒層
(
)
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛
−
=
=
RT
F
n
ak
dz
di
dz
di
Ha solid H eH H2
sinh
2
θ
φ
φ
⎟
⎟
⎠
⎞
⎜
⎜
⎝
⎛
−
∂
∂
=
∂
∂
F
n
s
dz
di
z
C
D
t
C
H H H H eff c H c m H c m 2 2 2 2 2 2 2 ,ε
ε
2 2 , 2 2 2z
C
D
t
C
eff H S c S H c m S H c m∂
∂
=
∂
∂
ε
ε
(
)
h H h anode H H eH H fH fH H s H fH Hk
P
RT
F
n
k
k
b
P
X
k
dt
d
θ
θ
θ
θ
θ
η
θ
ξ
2 2 s 2S2
sinh
2
1
⎟⎟
−
⎠
⎞
⎜⎜
⎝
⎛
−
−
−
−
=
(
s H)
fs fs s h H h S H fs sk
X
P
b
k
k
P
dt
d
θ
θ
θ
θ
θ
ξ
=
21
−
−
−
+
s 2S表
2- 2 邊界條件
陽極觸媒層
0
=
i
i
=
I
0 2 2 ∂ = ∂ c H H z C D in H HC
C
2=
2 in S H S HC
C
2 2=
0 2 2 ∂ = ∂ c S H S H z C D第三章 數值方法
一般在處理複雜的偏微分方程式時,為了增加計算效率,通常都 會借助電腦來幫我們處理,而為了要將方程式化為電腦可以計算的語 言,首先必需將微分方程式加以離散化,變為線性代數聯立方程組, 使成為電腦可以處理的形式,而在本文中所選用的方法為有限差分法 ( Finite difference method )。
3.1 有限差分法
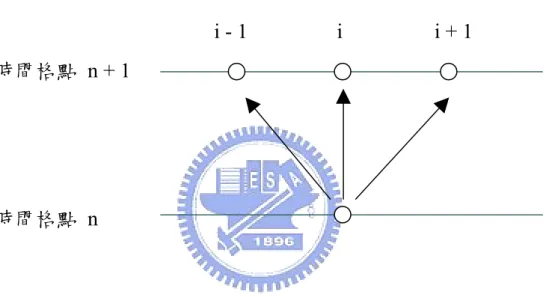
對於利用有限差分法來離散方程式的方法,通常有三種,前差分 ( Forward difference )、後差分( Backward difference )以及中央差分法 ( Central difference ): 對於一個函數 f x( ),可以對其作兩種不同方向的泰勒展開(Taylor series expansion),其結果分別為: 向前展開: ( ) ( ) ( )2 22 ( )3 33 2! 3! f x f x f f x x f x x x x x ∂ Δ ∂ Δ ∂ + Δ = + Δ + + + ⋅⋅⋅ ∂ ∂ ∂ (3-1) 向後展開: ( ) ( ) ( )2 22 ( )3 33 2! 3! f x f x f f x x f x x x x x ∂ Δ ∂ Δ ∂ − Δ = − Δ + − + ⋅⋅⋅ ∂ ∂ ∂ (3-2) 將上述兩式相減,可以得到: 3 3 3 ( ) ( ) ( ) 2 2 3! f x f f x x f x x x x x ∂ Δ ∂ + Δ − − Δ = Δ + + ⋅⋅⋅ ∂ ∂ (3-3) 經過整理,可以得到函數 f 對變數x一次微分的表示式為: 2 ( ) ( ) O( ) 2 f f x x f x x x x x ∂ = + Δ − − Δ + Δ ∂ Δ (3-4) 其中O(Δx)為誤差項的總和,經由如圖3-1 的格點劃分後,可以將(3-1) 以另一種形式表示成: 2 1 1 O( ) 2 i i i f f f x x x + − ∂ = − + ∂ Δ Δ (3-5)
這個用來將微分式轉換成代數式的方法稱為中央差分法。相同的將 (3-1)式和(3-2)式相加,並經過整理後可以得到: 2 2 2 2 ( ) 2 ( ) ( ) O( ) ( ) f f x x f x f x x x x x ∂ + Δ − + − Δ = ∂ Δ + Δ (3-6) 或是: 2 2 1 1 2 2 2 O( ) ( ) i i i i f f f f x x x + − ∂ = − + ∂ Δ + (3-7) 以上用了中央差分法來處理函數 f 對變數x一階微分和二階微分 的項,則欲求得於格點i的微分項,可以經由格點i+1和 所構成的 代數式而得。所以我們對於統御方程式中濃度對位置變數 1 i− x的微分 項,用誤差型態為(Δx)的中央差分處理之。 差分式又分兩種方法處理,一種是顯函數法(Explicit method),此 方法會受限於時間格點與位置格點的大小,原因在於求解下一個時間 點的變數時,需要這個時間點所對應相鄰三個點的變數值進行計算, 如圖 3-1 所示,如果時間與位置格點取的值不恰當,將會造成 i 格點 有低溫往高溫這種違反熱力學第二定律的現象,有穩定性的問題存 在。而隱函數法(Implicit method)則不受這個條件的限制。 文中所遭遇的偏微分方程屬於拋物線型(Parabolic type),就是熱 傳方程式或擴散方程式的型式: 2 2 z C D t C ∂ ∂ = ∂ ∂ (3-8) 若使用顯函數法(Explicit method)來解此偏微分方程式,要注意必 須滿足 2 1 ( ) 2 D t x Δ ≤ Δ 這個限制條件,這樣所算出來的解才會穩定(stable), 所以時間區間受到限制,必須取很小,造成運算時間過長。所以在此
![表 1- 1 六種燃料電池資料比較[2]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8624638.191805/35.892.128.771.345.700/表11六種燃料電池資料比較2.webp)

![表 4- 1. 基本操作條件之參數表 參數 符號 值 溫度 T 323 K 壓力 P 101325 Pa 觸媒層厚度 L c 1.6×10 -3 cm 觸媒層之有效孔隙度 ε c 0.4 [8] 陽極壓力 P 1 atm [8] 開迴路電壓 U o 1.23 V [8] 傳輸係數 α 0.5 [6] 氫氣化學計量數 S H 2 0.5 [6] 氫氣反應之電子數 H 2n 1 [6] 氫氣擴散係數 H 2D 2×10 -](https://thumb-ap.123doks.com/thumbv2/9libinfo/8624638.191805/65.892.127.770.128.1164/層厚觸媒層之有效孔隙εc陽極壓力V化學計量S氫氣反應之電子數.webp)





