台大農藝系 遺傳學 601 20000 Chapter 9 slide 1
CHAPTER 9
Genome Analysis
Peter J. Russell
edited by Yue-Wen Wang Ph. D. Dept. of Agronomy, NTU
台大農藝系 遺傳學 601 20000 Chapter 9 slide 2
Structural Genomics
1. The advent of DNA sequencing techniques changed experimental biology, and automation has enhanced the rate of change.
2. Genomics is the development and application of techniques for:
a. Mapping chromosomes. b. Sequencing genomes.
c. Computational analysis of entire genomes.
3. Subfields of genomics are:
a. Structural genomics, the genetic and physical mapping and sequencing of chromosomes.
b. Functional genomics, comprehensive analysis of gene functions and of non-gene sequences in entire genomes.
c. Comparative genomics, comparison of entire genomes across species, looking at functions and evolutionary relationships.
4. This section focuses on structural genetics, specifically genome sequencing.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 3
Genomic Sequencing Using a Mapping
Approach
1. Genome projects use two general approaches:
a. The mapping approach divides the genome into
segments with genetic and physical mapping, refines
the map of each segment, and finally sequences the
DNA.
b. A “shotgun” approach breaks the genome into
random, overlapping fragments, and sequences each
fragment. Based on overlaps, the sequences are
assembled by computer. An advantage is that physical
mapping is not required.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 4
Genetic Mapping of a Genome
1. A genetic map derives from the frequency of recombination between genes. Genetic mapping involves determining, the location of genes on a chromosome relative to other genes, using genetic crosses and
pedigree analysis.
2. A genetic map of the human genome has 24 different maps, one for each autosomal pair, plus X and Y.
3. Marker alleles in genetic crosses help determine the crossover rate between linked genes:
a. Individuals with different alleles at two or more loci are crossed, and their offspring examined.
b. Most of the offspring will have phenotypes corresponding to the linked alleles. A few progeny will be recombinant.
c. The frequency of the recombinant phenotype is calculated as a
percentage of the total offspring, giving the recombination frequency or genetic distance. Units are map units (mu) or centiMorgans (cM).
台大農藝系 遺傳學 601 20000 Chapter 9 slide 5
4. Experimental crosses are not done in humans and so genetic mapping relies on pedigree analysis, and is limited by rarity of large,
multigenerational pedigrees showing segregation of defined linked traits.
5. Usually, the lod (logarithm of odds) score method is used for statistical analysis of pedigree data.
a. A lod score compares the expected distributions of traits if they are linked, and if they are not linked.
b. The lod score is the log10 of the ratio of the two probabilities. The higher the lod score, the closer the two genes.
6. The map distance for linked markers is computed from the
recombination frequency given by the highest lod score, by solving lod scores for a range of proposed map units. For the human genome, 1 mu is approximately 1 megabase (Mb).
台大農藝系 遺傳學 601 20000 Chapter 9 slide 6
Genetic Markers for Genetic Mapping
Experiments
1. Genes have historically been used as markers for genetic mapping experiments, but in humans only a few allelic forms are easily studied, and the genes are spaced too widely on the chromosome for high- density mapping.
2. DNA markers are used in association with gene markers for genetic and physical mapping of chromosomes. DNA markers are distinguishable polymorphic alleles that do not
encode proteins, and therefore are neither dominant nor recessive. Four major types are used for humans:
a. Restriction fragment length polymorphisms (RFLPs) result from mutations that create or abolish restriction sites, or from insertions or deletions of DNA between sites under study (Figure 8.5). The procedure to detect polymorphisms is:
i. Isolate genornic DNA and digest with a restriction enzyme. ii. Electrophorese and transfer DNA to a membrane filter. iii. Probe with labeled DNA from the polymorphism region. iv. Monozygotes show one band, heterozygotes two.
v. PCR amplification is an alternative method.
vi. RFLP probes are discovered by chance when random DNA fragments are used in Southern blots.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 7
b. Variable Number of Tandem Repeats (VNTRs), also called minisatellites, are short sequences (5-lOs) repeated 10s-to-l000s of times (Figure 9.1).
i. DNA is digested with a restriction enzyme that cuts flanking the VNTR. ii. Fragments are electrophoresed, and blotted to a filter.
iii. The blot is probed with the VNTR repeating sequence.
iv. Some VNTR sequences are in only one genomic locus, corresponding to a monolocus probe.
v. Other VNTR sequences map to a number of genomic loci, corresponding to a multilocus probe.
vi. PCR can also find VNTR lengths, if the flanking sequences are known.
c. Short Tandem Repeats (STRs), or microsatellite sequences, contain very short (1-4) tandem repeats, and are highly polymorphic.
i. STR alleles are more useful than VNTRs for human genetic mapping. ii. STRs are typed by probing or by PCR, to which they are more suited than
VNTRs.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 8
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
Fig. 9.1 Variable number of tandem repeats (VNTRs), also known as minisatellite
台大農藝系 遺傳學 601 20000 Chapter 9 slide 9
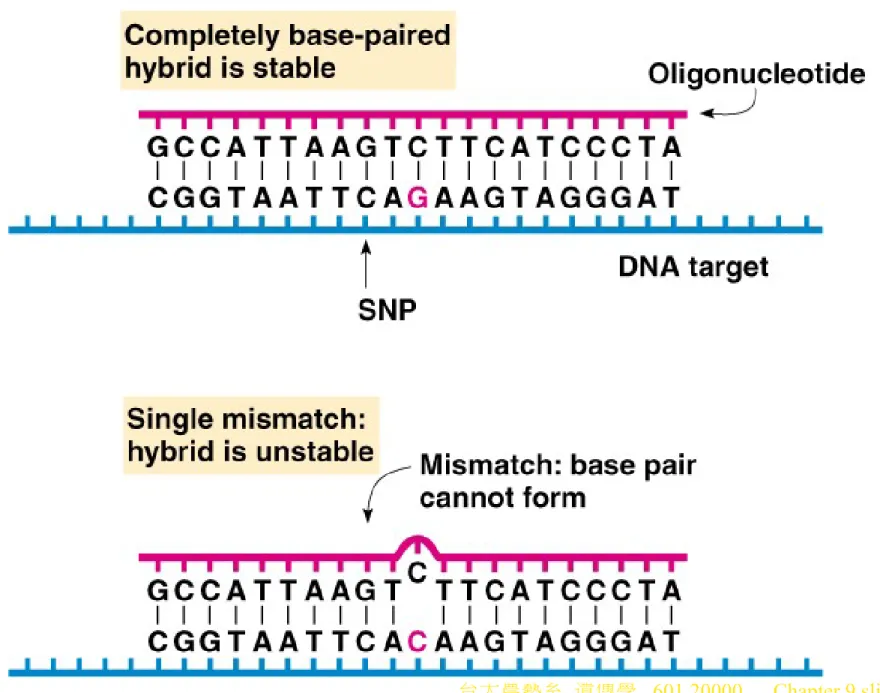
d. Single Nucleotide Polymorphisms (SNPs, “snips”) are base-pair differences between individuals. If at least 1% of the population has an altered base pair at a particular site, it is an SNP. About 98% of human DNA polymorphisms are SNPs, with about 3 million in the human genome.
i. SNPs are typed by oligonucleotide hybridization analysis, where an oligo complementary to the common sequence detects polymorphisms by failing to bind at high stringency (Figure 9.2).,
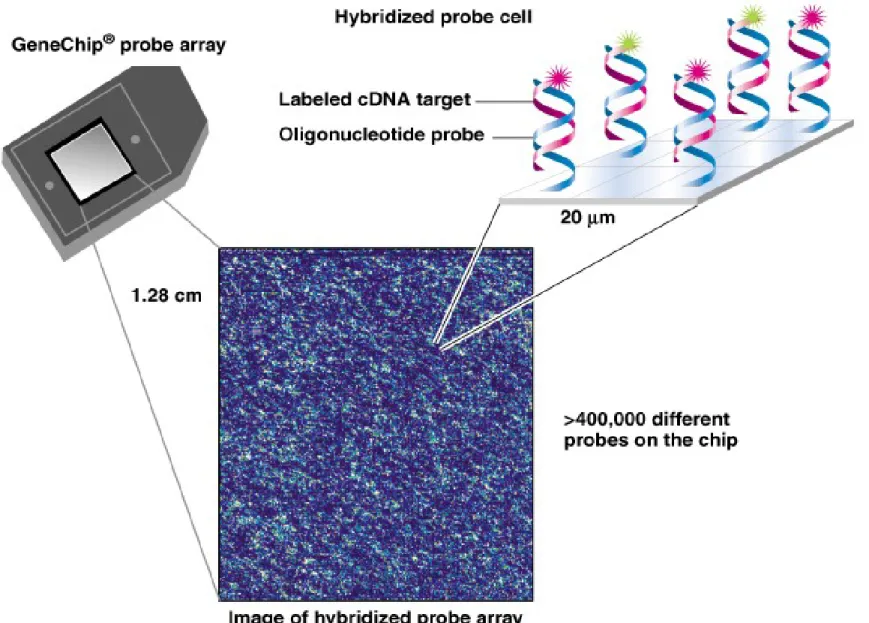
ii. DNA microarrays or oligonucleotide arrays can type hundreds of SNPs in one experiment.
(1) A DNA microarray has DNAs of known sequence fixed at known locations to a solid substrate (usually a silicon chip or glass).
(2) There are two major types of DNA microarray technology:
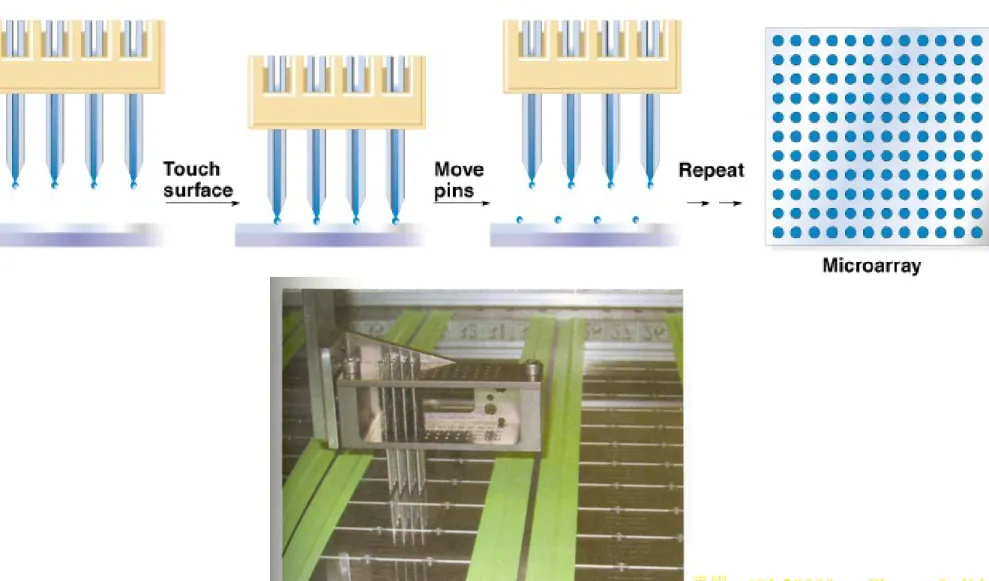
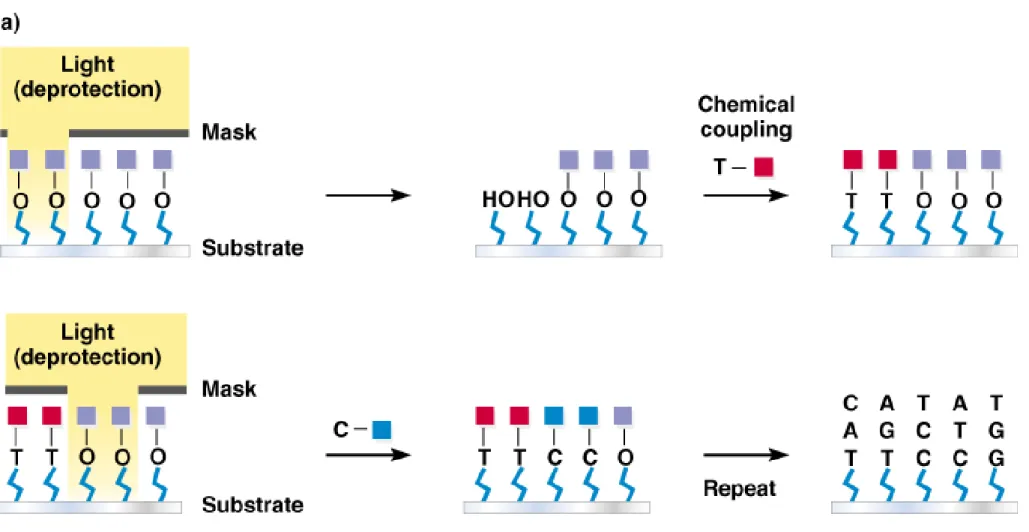
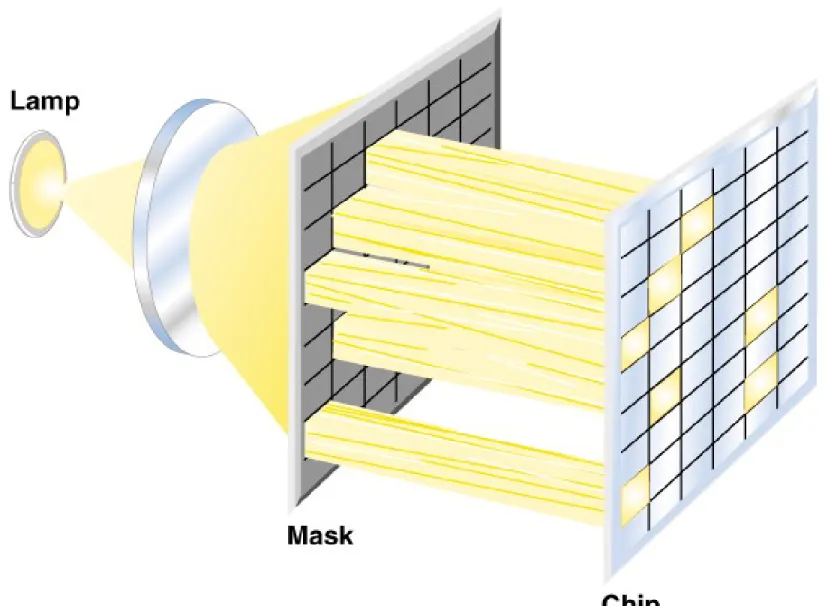
(a) Mechanical spotting of DNAs onto the substrate using specially designed spotting pins or ink jets. (b) Oligonucleotides synthesized at defined positions on the substrate using a light-directed process,
creating an even greater density of DNA sequences in the array (Figure 9.4).
(3) An experiment using a commercially available
GeneChips® probe array (the fixed DNAs are the probes, and the unknown
free DNA that binds is the target) is outlined (Figure 9.5):
(a) In this experiment the chip has an array of oligonucleotide probes, and the target is a population of cDNAs.
(b) Target cDNAs are labeled with a fluorescent tag, and after hybridization the fluorescence pattern is recorded by laser scanning and analyzed. Combinations of fluorescent dyes may be used depending on the goals of the experiment.
(c) In just one experiment, fluorescence patterns can show allele identities for thousands of loci, and indicate whether the individual is homozygous or heterozygous for each.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 10
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 11
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
Fig. 9.3 Preparing a DNA microarray by using robot-driven, mechanical microspotting
台大農藝系 遺傳學 601 20000 Chapter 9 slide 12
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 13
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 14
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 15
A High-density Genetic Map of the Human
Genome
1. Human genetic mapping was revolutionized by discovery of many polymorphic DNA markers, and development of molecular tools to type them. Hundreds may be typed in a given cross, and computer algorithms then determine linkage relationships.
2. High-density genetic mapping has been important in the human genome project. Some aspects of this procedure:
a. A sequence tagged site (STS) is a unique genomic DNA sequence used as a genetic marker. STRs (short tandem repeats) are extensively used for STS mapping, but nonpolymorphic markers are also used.
b. A consortium of laboratories works on the same set of DNA samples (mapping panel), so their data may be combined.
c. A high-density human genetic map completed in 1994 localizes 5,264 STRs to 2,335 chromosomal loci, with an average density of one
台大農藝系 遺傳學 601 20000 Chapter 9 slide 16
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
Fig. 9.6 A high-density genetic map with 5,264 microsatellites localized to 2,335
台大農藝系 遺傳學 601 20000 Chapter 9 slide 17
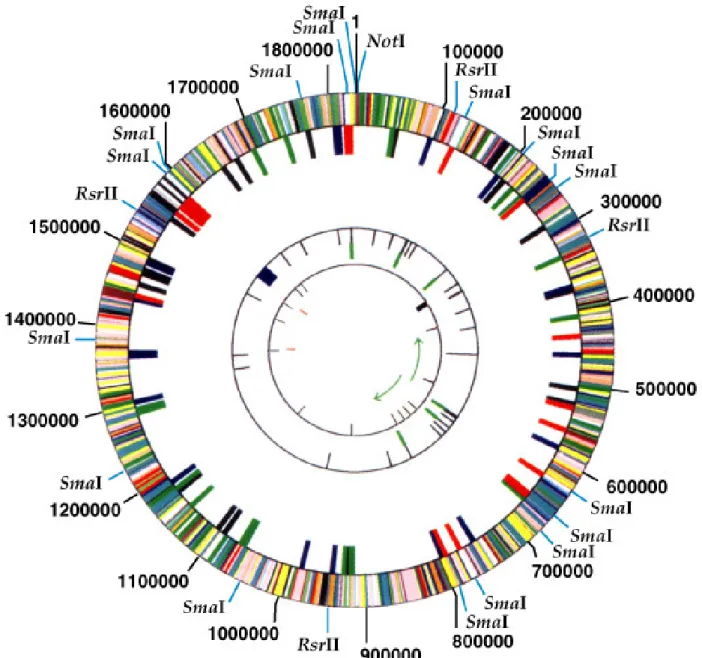
Physical Mapping of a Genome
1. Genetic maps generated for some species (e.g.. E. coli )
are sufficient to begin sequencing, but in humans even
the detailed genetic map described above lacks the
required resolution. Therefore, a physical map derived
directly from genomic DNA rather than analysis of
recombinants has been generated.
2. As in human genetic mapping, there are 24 physical
maps, for the autosomes plus X and Y. Types of physical
maps are presented in order of increasing resolution:
台大農藝系 遺傳學 601 20000 Chapter 9 slide 18
Cytogenetic Maps: Chromosomal Banding
Patterns
1. Microscopic examination of stained chromosomes
reveals a pattern of bands that average about 6
Mb. Regions are designated based on their
chromosomal position relative to the centromere:
a. Regions designated “q” are on the chromosome’s long
arm.
b. Regions designated “p” are on the short arm.
c. Regions are numbered from the centromere outward,
with “q1” and “p1” nearest.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 20
FISH (Fluorescent in situ Hybridization) Maps
•
1. Individual metaphase chromosomes are probed in situ
with specific fluorescently labeled DNA sequences,
identifying homologous sequences in the chromosome.
•
2. Different probes labeled with different fluorescent dyes
may be used in the same experiment. Fluorescence
microscopy provides data for computer imaging analysis
to determine binding site(s) for each probe.
•
3. With a resolution of 2-5 Mb in metaphase
chromosomes, FISH can localize markers to subregions
of chromosomal bands. Less condensed chromosomes
may be. resolved in the 5-700 kb range.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 22
Restriction Maps
1. Restriction enzymes are used that cut rarely, due
either to a large (7-8 bp) recognition sequence or
to scarcity of the recognition sequence in the
DNA under study.
2. The map for even a rarely cutting restriction
enzyme is very complex, and so far has been
obtained for only the smallest human
台大農藝系 遺傳學 601 20000 Chapter 9 slide 23
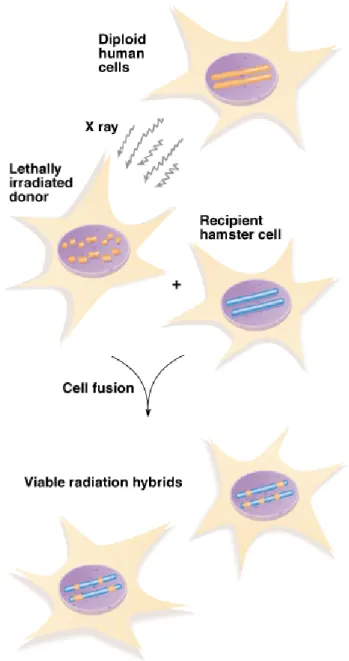
Radiation Hybrid Maps
1. A radiation hybrid (RH) is a rodent cell line carrying a
small genomic DNA molecule from another organism
(e.g., a human). In this technique (Figure 9.8):
a. Exposure to X rays breaks the DNA in human cells. The
fragments become smaller with more X ray exposure, and
fragment length determines the, map resolution.
b. Irradiation kills the human cells, which are then fused with
rodent cells, rescuing chromosomal fragments that are typically a
few Mb in length.
c. Human DNA in the RH is analyzed for gene and/or DNA
markers. The closer two markers are to each other on the
chromosome, the more likely they are to be found together in an
RH.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 24
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 25
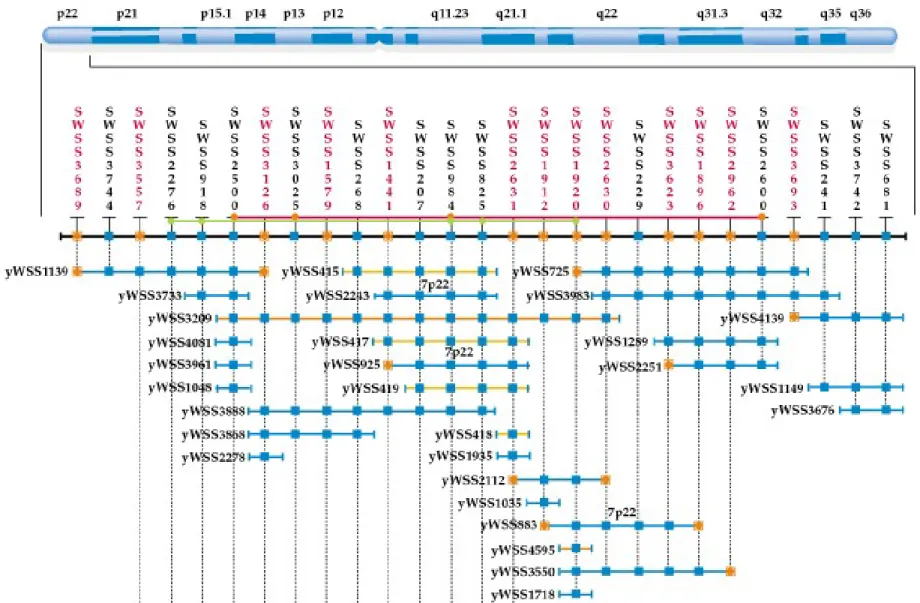
Clone Contig Maps
1. A partial restriction digest produces a set of large, overlapping DNAs, which are cloned into YAC vector cut with a compatible restriction enzyme. Shearing may also be used to make high-molecular-weight DNA that is blunt-end cloned into a YAC.
2. An entire genome or a single chromosome may be represented in a YAC clone library.
3. YAC clones are then assembled into a map either by matching with a FISH-generated chromosome map or by DNA fingerprinting and assembly based on overlaps. Nonpolymorphic STSs are especially useful for YAC contig mapping (Figure 9.9).
4. A complete library should yield a complete contig map that indicates the order in which the cloned fragments occur in the chromosome.
5. Problems arise when some of the YAC inserts contain DNA from more than one chromosomal location. This has complicated efforts at
generating a YAC contig map of human chromosomes.
6. Many labs have switched to BAC (bacterial artificial chromosome) vectors with a capacity of 300 kb and the ability to replicate in E. coli as a resource for their sequencing projects.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 26
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 27
Generating the Sequence of a Genome
1. When a high-resolution map is available, sequencing is possible. (Automated DNA sequencing is discussed in Chapter 7.) Briefly:
a. Dideoxy sequencing is used. DNA is synthesized from a template, and terminates with incorporation of a fluorescently labeled ddNTP.
b. All four reactions (ddA, ddG, ddC and ddT) occur in the same tube. Each ddNTP carries a different fluorescent label.
c. Products are separated electrophoretically, colored bands are detected with lasers and the data are converted to a computer sequence file. d. PCR-based sequencing uses one oligonucleotide primer and
thermostable DNA polymerase. The advantages of this approach are: i. Double-stranded DNA is sequenced directly.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 28
2. One sequencing reaction is limited to about 500 nucleotides, and for accurate sequences both strands must be sequenced several times.
3. Progress on the human genome and other projects has been accelerated by improved technologies for sequencing and analysis.
4. Human genome sequencing by the mapping approach used BACs, but a BAC insert is far too large to sequence in one reaction. Instead, the
inserts were each sequenced using a shotgun approach:
a. Each insert is cut from the vector, sheared into fragments that will be partially overlapping and cloned into a plasmid vector.
b. Each subclone is sequenced, and overlaps are used by a computer to assemble the data into one contiguous sequence representing the BAC insert.
c. Using the chromosomal map for BAC clones, the BAC insert sequences are put in order to yield the complete chromosome sequence.
5. In theory, sequencing contigs for a total length of 6.5-8 times the genome will span more than 99.8% of the genomic sequence.
6. In practice, the HGP (human genome project) did its sequencing 7-
times over, and has obtained 97% of the genome, although assembly of the sequences is still incomplete.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 29
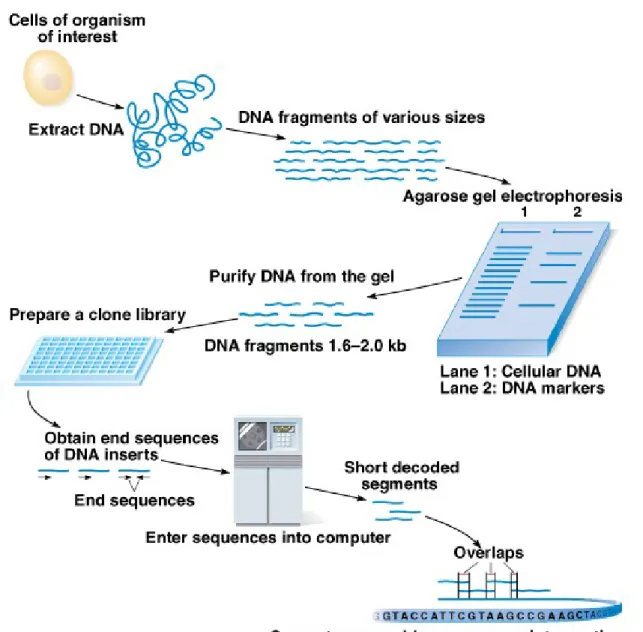
Genome Sequencing Using a Direct Shotgun
Approach
Animation: Direct Shotgun Sequencing of Genomes
1. The shotgun approach obtains a genomic sequence by
breaking the genome into overlapping fragments for
cloning and sequencing. A computer is then used to
assemble the genomic sequence.
2. Advances that have made this approach practical for large
genomes include:
a. Better computer algorithms for assembling sequences.
b. Automation in the actual sequencing.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 30
3. A pioneer of this approach is J. Craig Venter, whose Celera Genomics has also sequenced (5-fold) the human genome to 97%, with complete assembly of the fragments except for gaps caused by the missing 3%. 4. Direct shotgun sequencing involves (Figure 9.10):
a. Mechanical shearing and cloning of small (about 2 kb) genomic DNA fragments.
b. Sequencing about 500 bp on each end of the insert DNA. Sequences in the center of the cloned DNA are obtained from an overlapping clone rather than directly.
c. Computer analysis gives the sequence of most of the genome, with gaps caused by sequences missing from the library.
d. A second library is made with larger (about 10 kb) random fragments, allowing resolution of repeated sequences.
5. The shotgun approach is a successful option for genomic sequencing that does not require genetic mapping. With both approaches, however, finishing is required to correct errors and fill in gaps.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 31
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
Fig. 9.10 The direct shotgun approach to obtaining the genomic DNA sequence of an
台大農藝系 遺傳學 601 20000 Chapter 9 slide 32
Overview of Genomes Sequenced
1. The human mitochondrial genome was the first
sequenced, in 1981. Many viral genomes have
been sequenced, as have a number of genomes
from living organisms, the focus of this section.
2. Some features of genomic sequences are noted
when the sequence is published. Published
genomic sequences are usually only mostly
complete, and work continues to fill in gaps and
resolve ambiguities.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 33
Bacterial Genomes
1. Haemophilus influenzae, the first cellular organism to have its genome sequenced, was selected for its typical bacterial genome size and its GC content close to humans.
a. No genetic or physical map existed, so a shotgun approach was used. b. The H. influenzae genome is 1.83 Mb, with 38% GC content.
c. Annotation of the sequence involved computer analysis to find significant sequences, including:
i. Open reading frames, regions with no stop codon in a particular reading frame. Arbitrarily, ORFs over 100 codons are considered likely to encode proteins (Figure 15.2).
ii. Repeated sequences. iii. Operons.
iv. Transposable elements. v. rRNA and tRNA genes.
d. Nearly half of the predicted genes have no “role assignment” meaning that no function is yet verified for them.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 34
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 35
2. Mycoplasma genitalium was selected because
mycoplasmas have the smallest known genomes
of any living cells, and they often are significant
pathogens.
a. A shotgun approach was used.
b. The genome is 0.58 Mb with a GC content of 32%.
c. Only 470 genes occur in this organism, comprising
台大農藝系 遺傳學 601 20000 Chapter 9 slide 36
3. Escherichia coli was selected because it is an important
model system for molecular biology, genetics and
biotechnology, as well as a common bacterium in animal
intestines and the environment.
a. A shotgun approach was used.
b. The genome is 4.64 Mb with a GC content of 50.8%.
c. Analysis of the genome sequence shows that:
i. 88% of the genome is ORFs.
ii. 0.8% encodes rRNAs (7 operons) and tRNAs (86 genes).
iii. 0.7% is repeated sequences.
iv. About 11% is regulatory and other sequences.
d. Of 4,288 ORFs, 38% are of unknown function.
e. The sequence correlates with the extensive genetic mapping
already in existence for this well-studied organism.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 37
4. Examples of other bacterial genomes sequenced,
and associated disease or traits:
a. Treponema pallidum (syphilis).
b. Rickettsia prowazekii (typhus).
c. Deinococcus radiodurans (survives heat, cold, poison
and radiation).
台大農藝系 遺傳學 601 20000 Chapter 9 slide 38
Archaeon Genomes
1. Methanococcus jannaschii is an anaerobic, hyperthermophilic methanogen that reduces CO2 to methane.
a. A shotgun approach was used.
b. The genome has 31% GC, and three parts:
i. A large circular chromosome of about 1.66 Mb, with 1,682 ORFs. ii. A circular extrachromosomal element (ECE) of about 58 kb, with
44 ORFs.
iii. A smaller circular ECE of about 17 kb, with 12 ORFs. c. Only 38% of the 1,738 ORFs have assigned functions.
2. Analysis of the sequence confirms Archaea’s unique taxonomic position, showing that:
a. Most M. jannaschii genes involved in energy production, metabolism and cell division are similar to those of eubacteria.
b. Most of the genes involved in DNA replication, transcription and translation are similar to those of eukaryotes.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 39
Eukaryotic Genomes
1. The yeast, Saccharomyces cerevisiae, is a model eukaryote for many types of research. It was the first eukaryotic genome to be completely sequenced.
a. The mapping approach was used.
b. The 16-chromosome genome is 12 Mb, with individual chromosomes ranging from 230 kb to 1.5 Mb. An estimated 969 kb of repeated
sequences are missing from the published sequence. c. Analysis reveals:
i. 6,183 ORFs, 233 with introns. ii. 120–150 rRNA genes.
iii. 37 snRNA genes.
iv. 262 tRNA genes, 80 with introns.
d. ORFs comprise about 70% of the total genome, and about 1⁄3 have no known function.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 40
2. Caenorhabditis elegans, a nematode, has been important
in both genetic and molecular study of embryogenesis,
morphogenesis, development, nerve development and
function, aging and behavior.
a. The nearly-complete genome sequence spans 97 Mb distributed
between six chromosomes (five autosomes and an X
chromosome).
b. Analysis shows:
i.19,099 ORFs, with an average of five introns; 27% of the
sequenced genome consists of exons.
ii. 659 tRNA genes.
iii. One tandem array of rRNA genes.
iv. One tandem array of 5S rRNA genes.
v. A number of non-coding RNA genes in introns of
protein-coding genes.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 41
3. Drosophila melanogaster, the fruit fly, has been
important in both classical genetics and the molecular
genetics of development.
a. Sequencing used the direct shotgun approach, supported by
clone-based sequencing and a BAC-derived physical map.
b. The genome is estimated at 180 Mb. About 1⁄3 (60 Mb) is
heterochromatin located near centromeres. This heterochromatin
is so far unclonable, blocking completion of genomic
sequencing.
c. Remaining 2⁄3 (120 Mb) contains more than 99% of
Drosophila’s 13,600 genes. Comparison with genomic sequences
from other species indicates:
i. Drosophila (fruit fly) has about twice the number of genes
found in Saccharomyces cerevisiae (yeast).
ii. Of 289 genes known to be involved in human disease,
Drosophila has homologs for 177.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 42
4. Homo sapiens. DNA from a variety of anonymous donors has been sequenced. The “human genome sequence” does not exactly match the genome of any human being.
a. A “working draft” of the human genome was announced in June 2000 jointly by: i. Francis Collins for the HGP (Human Genome Sequencing Project Consortium),
an effort involving 16 institutions located in 5 countries. ii. J. Craig Venter of Celera Genomics.
b. By June 2000, the sequencing effort had generated 7-fold coverage of the genome, with about 50% of the genome sequence considered to be near-finished, and 24% completely finished.
c. The sequencing approaches:
i. The HGP consortium focused on sequencing the gene-rich euchromatin regions, ignoring the generally unclonable heterochromatin, using existing genetic and physical maps.
ii. Celera Genomics used shotgun sequencing followed by a very large computer calculation looking for overlaps in the random DNA fragments (enough to represent 4.6-fold coverage of the human genome). Shotgun assembly results were verified by comparison with BAC clone sequences available in public databases.
d. The next step in the human genome project is annotating the sequence, analyzing its genes and other features.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 43
Functional Genomics
1. Functional genomics analyzes all genes in genomes to determine their functions and their gene control and expression.
2. Classically, genetic analysis has started with a phenotype and gone in search of a gene. New approaches are needed to work in the opposite direction, from gene to phenotype.
3. Current functional genomics relies on molecular biology lab research and sophisticated computer analysis by bioinformatics researchers.
4. This fusion of biology with math and computer science is used for many things. Examples:
a. Finding genes within a genomic sequence.
b. Aligning sequences in databases to determine matching. c. Predicting structure and function of gene products.
d. Describing interactions between genes and gene products in the cell, between cells and between organisms.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 44
Identifying Genes in DNA Sequences
1. Annotation begins the process of assigning
functions to genes, especially protein-coding
genes, using computer algorithms to search both
strands for ORFs. Introns complicate analysis of
eukaryotic genes.
2. ORFs exist in all sizes, and not all encode
proteins. To focus on sequences most likely to
encode proteins, a minimum ORF size is
arbitrarily set and shorter sequences are not
analyzed.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 45
Homology Searches to Assign Gene Function
1. Computers are used to find homology between sequences in a database (e.g., a BLAST search). Similarity reflects evolutionary relationships and often shared functions.
2. Either DNA or amino acid sequences can be searched, but amino acids yield more
specific information, since there are 20 possible matches, rather than just four. Often no convincing match is found, due in part to the limitations of current databases.
3. Sometimes matches are found only at the domain level, when a region in the new protein matches protein domains in the database. This provides clues to the new protein’s
function and the evolution of its gene.
4. As databases grow, so does our knowledge of gene functions. The current distribution of knowledge about the genes of yeast is (Figure 9.14):
a. About 30% of the genes have known functions. b. Of the remaining 70% of ORFs:
i. 30% encode a protein that either has homology to protein(s) of known function, or has domains related to functionally characterized domains.
ii. 10% are FUN (function unknown) genes. They have homologs in databases, but function(s) of the homologs are unknown. Groups of homologous genes of unknown function are orphan families.
iii. 30% of ORFs have no homologs in the databases. These include 6–7% that may not actually encode proteins. The remainder may represent genes known only in yeast, the single orphans.
5. Every genome sequenced contains “function unknown” genes, but as databases are expanded the problem should decrease.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 46
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 47
Assigning Gene Function Experimentally
1. One approach to determining gene function is to delete the gene, and observe the phenotype when that gene’s function is knocked out. PCR may be used to produce a gene knockout (Figure 9.15):
a. Using known genome sequences, PCR primers are designed to construct an artificial linear DNA deletion module. It consists of:
i. The gene sequence upstream and through the start codon.
ii. A kanR (kanamycin) marker gene conferring resistance to a chemical, G418.
iii. The gene sequence downstream of and including the stop codon.
b. The amplified linear DNA is transformed into yeast, and G418-resistant colonies selected. These are generated when the new DNA replaces the gene of interest in the genome by homologous recombination.
c. They now express kanR instead of the gene under study, producing a loss-of-function
mutation.
2. Work is underway to systematically analyze by knockout mutation each gene in the genome of yeast and other organisms.
a. Each knockout must be screened for possible phenotype change in every area of cell function, making these studies a substantial undertaking.
b. Knockout mutations analyzed in yeast to date indicate about 1⁄3 of the genes are essential, 1⁄3 are nonessential but affect phenotype, and 1⁄3 show no significant change in phenotype
台大農藝系 遺傳學 601 20000 Chapter 9 slide 48
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 49
Describing Patterns of Gene Expression
1. Genomic sequencing makes it possible to
determine all genes that are expressed in a cell by
analyzing the total RNA transcripts of the cell, its
transcriptome. The transcriptome is an indicator
of cell phenotype and function. Similarly, the
complete set of proteins in a cell is its proteome.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 50
The Transcriptome
1. The transcriptome changes as the cell responds to stimulus and moves through its cell cycle, and so is a tool for understanding cellular function.
2. Probe arrays are used to study gene expression. Yeast sporulation is one example:
a. Yeast sporulation produces four haploid spores, and involves four stages, each associated with its own transcripts (Figure 9.16).
i. DNA replication and recombination. ii. Meiosis.
iii. Meiosis II.
iv. Spore maturation.
b. Samples of mRNA taken at intervals during sporulation were converted to cDNAs and analyzed on microarrays of PCR-amplifled ORF sequences. The. results were correlated with cellular events.
c. Control cDNA was made from pre-induction mRNAs, and labeled green. The cDNAs from post-induction mRNAs were labeled red. Microarrays were probed with a mix of both, and results were interpreted as follows:
i. Red spots indicate a gene induced during sporulation. ii. Green spots indicate a gene repressed during sporulation.
iii. Yellow spots mark genes whose expression is unchanged during sporulation.
d. Results show more than 1,000 genes with altered expression during sporulation, about 1/2 repressed and the other 1/2 not repressed. Patterns of expression over time become apparent in this type of experiment.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 51
Peter J. Russell, iGenetics: Copyright © Pearson Education, Inc., publishing as Benjamin Cummings.
Fig. 9.16a, b Global gene expression analysis of yeast sporulation using a DNA
台大農藝系 遺傳學 601 20000 Chapter 9 slide 52
3. DNA microarrays are now widely used, although still
expensive. Examples of studies . that currently use this
technology:
a. Changes in Drosophila gene expression during morphogenesis.
b. Human cancers and their characteristic patterns of gene
expression (transcriptional fingerprints) that reveal distinctions
between different types of cancer.
c. Screening for genetic diseases, especially those resulting from
one of many alleles. A patient’s blood, for example, can be
screened for hundreds of possiblc mutations in the BRCA1 and
BRCA2 genes associated with breast cancer.
d. Optimizing drug therapies for patients using pharmacogenomics,
analyzing changes in transcription when the drug is present as a
means of developing drugs that target specific mutations.
台大農藝系 遺傳學 601 20000 Chapter 9 slide 53
The Proteome
1. Proteomics is cataloging and analysis of the proteome, or complete set of expressed proteins in a cell at a given time. Proteomics focuses on which proteins are made and in what quantities, and their interactions with other proteins.
2. Goals of proteomics are to:
a. Identify every protein in the proteome, using 2-D PAGE mapping, isolating each protein and analyzing it by mass spectrometry.
b. Develop a database with the sequence of each protein.
c. Analyze protein levels in different cell types and stages of development.
3. Protein identification and sequencing is very complex. Celera Genomics is involved in identification, sequencing and computer analysis of the data.
4. Proteomics stands to make a major contribution to understanding of human diseases and development of biopharmaceutically based
台大農藝系 遺傳學 601 20000 Chapter 9 slide 54
Comparative Genomics
iActivity: Personalized Prescriptions for Cancer
Patients
1. Comparative genomics provides a way to study
functions of human genes by working with
non-human homologs. Genes and their arrangement
also provide valuable clues to evolutionary
台大農藝系 遺傳學 601 20000 Chapter 9 slide 55
Ethics and the Human Genome Project
1. The ability to identify human genes raises complex ethical issues
involving the right to information about one’s own genome, access to genomic information by employers, insurance companies and
government agencies, and concerns about the ability to diagnose but not treat genetic disorders.
2. Federal agencies funding the HGP devote 3–5% of their budgets to study of ethical, legal and social issues (ELSI), producing the world’s largest bioethics program. Areas currently emphasized by the ELSI program:
a. Privacy of genetic information.
b. Appropriate use of genetic information in the clinical setting. c. Fair use of genetic information.