The Classical Surface in Modern Theories of Solid Surfaces:
II. Subband Formalism and Dimensionality Theory
Der-Ruenn Su
Department of Physics, National Taiwan University, Taipei, Taiwan 106, R.O.C.
(Received May 25, 2001)
In a previous paper [Chin. J. Phys. 29, 49 (1991)], we found that in front of a classical surface, the dimensionality has some ambiguity. Because we treat the surface or interface as a classical surface for semiconductors, etc., we find some dimensionality discriminations in our theory. We study the one-dimensional energy to incorporate subbands, reduced from the three-dimensional space. Because the surface conditionµ(z), as a one-dimensional-boundary condition is not mathematically compatible with the one-dimensional Schr¨odinger equation in 3D, the plane subband theory thus obtained is not the 2D Bloch theory of electrons. There are different structural formulation influences when reduced from 3D. They imply that we can have, for any one crystal energy, three different appearances and nature: a three-dimensional E3Dand two one-dimensionalEz n¤ andE
(sl)
n . The latter two provide envelope functions, with reductions in dimensionality. This suggests a new picture having various dimensionalities for energy quanta packets. This implies that in the statistical method we have three different Fermi energies: from E3D, EF(3D); from Ezn¤ , EF 1(1D); and fromEn(sl), EF 3(1D), for running waves. This also implies that the physical contents of the crystal momentum include (a) a complex nature to indicate the mean free path, and (b) an extension to other (extra or deviate) dimensionalities. The origins of the mobilities are mentioned and related to the electron scatterings with 2D structure from thek(2D)Z , thek( 2D)Z pz-perturbations. They are related to some energy eigen-equation with energy value E¤
zn which has the peculiarities: (a) it is not exactly one-dimensional for the envelope function; and (b) thek( 2D)Z pz-perturbation for this Ezn¤ for all kinds of perfect crystals with a plane-surface, has a peculiark
(2D)
Z coming from the other dimensions (2D). This reveals a new dimension and is a dimensionality-breakthrough from 2D into 3D. One obvious reason for this dimensionality-breakthrough is the existence of lattice lateral-vibration dynamics on the static surface µ(z). This resolves the problem that subband theories are 2D, e.g., in thexy-plane while the surface equation is expressed in one-variable with one degree of freedom in variable-variations, such asµ(z). Consequently a two-dimensional plane,µ(z), is physically measurable in three-dimensional space, by a certain periodic potential condition in terms of the primitive vectors in an (xy)-plane. Contrary to mathematics, the area integral is immeasurable in the formulation of volume integrals in Riemann integral theory due to the lattice dynamics vibrating into new dimensions.
PACS. 05.30.–d – Quantum statistical mechanics.
PACS. 73.20.At – Surface state, band structure, electron density of states.
PACS. 71.18.+y – Fermi surface: calculations and measurements; effective mass, g factor.
498 ° 2001 THE PHYSICAL SOCIETYc OF THE REPUBLIC OF CHINA
I. Introduction
Investigations of the growth and the adsorption mechanisms of surfaces lead us [1] to understand that we must have a surface or interface of dimensions (geometrical) or
two-degrees of freedom (lattice dynamical) (2D) (2D-xy-plane) to formulate an energy eigen-equation, which is eventually in 1D in computational physics at least, nowadays within computor capabilities. Based on the underlying concepts in Sec. II, eqn. (24a), which is an equation equating a constant to another constant, we have the following argument: First the adsorption process is mainly a 2D-surface penetrated into the third dimension by growth. To this 2D-surface, the subband energy is correlated to the growing third dimension in a realistic process, although the energy equation is finally only 1D. How to understand the physics on account of the dimensionality? We find that we incorporate the 1D in the energy equation as our third dimension (with adsorbate growth). However from (29), we find that there are quantities, as physical constants, i.e. LZ precisely, belonging to the (xy)-plane. Hence we discover the subband energy includes dynamical terms obtained from the 2D-xy-plane. Moreover this (xy)-plane is not precisely the xy-plane in 3D fxyzg- or r-space. It penetrates from the (xy)-directions into z-dependency. This z-dependency forms a new dimension in (29). This point will be clearer in (29). The separability of the above 2D-x; y-plane and the 3D r-space is a kind of dimension theory discussed an p. 30 in Ref. [2] and verification was tried by metal gate-charges in MOSFET in the discussion below (29b). That means, we find, in the theoretical prediction framework of this paper, that this new dimension (z-dependency) is, in fact, not exactly the conventional third z-direction. What we mean by the theoretical framwork is the periodic structure kinematics such as the crystal periodic structures in the Bloch electron theory for energy band calculations. Intuitively the statement above is clear when we have a semi-infinite medium with periodic structure (occupying z ¸ 0) vs. vacuum space (withz < 0). Physically the half-space z > 0 is entirely different from the other half-space z < 0. Mathematically there are no differences for the whole z-axis, since it is only along a 1D axis. In this paper, the above physical distinguishability, forz < 0 and z > 0, makes us to deduce and to induce some dimensional ‘consequences’ such that for z > 0 and z < 0 there are some physical ways to provide two different measures along this one single axis. These measures make it possible for us to reveal some new dimension (like surface charges), mathematically along the z-axis (similar to volume charges). However this new dimension is only for some ‘thickness-in-z-direction’decided by the geometry of the xy-plane (e.g. a quantum size effect of the metal gate in MOSFET, etc.) (eqns. (29), (29a) below).
The subband energy, or the subband ladder energy, or subband bottom energy, is usually in the free particle form, E3D¡ Ezn¤ = ~2(k21+ k22)=2m¤ as in (24a) below. We find in this paper that the remaining 1D the in the 3D-space with an energyE3D¡ Ezn¤ in (24a), is actually determined, in part, by some quantities from other dimensions. This is the main theoretical subject of study in Sec. II, after eqn. (24a).
In recent discoveries and the current treatment of the surface growth process [1], we find that the surface plays the dominate role, physically and mathematically. This is because, by physical methods, the detectable thickness from the surface top into the bulk is limited anyhow, say from a few to less than 100 layers due to multiple scattering losses. More than 100 layers below the surface or interface, we really do not know its chemical composition, and its lattice structure in form and in magnitude, the electron-form-factors, even the matter densities, etc. This point is judged by the coverage increased toµ » 100, and/or moderate high pressure measurements. Up
to the present, this point is determined by the structures near the surface and those ‘atoms’ located well inside the bulk, cf. e.g. [3] by a synchrotron light source, etc. These recent studies are in addition to the traditional LEED, x-ray measurements, etc. and are prepared for the verification of future near-field optic-measurements and femtosecond laser physics results.
We believe that the whole bulk or 3D properties are essentially determined by the ‘near-surface-few-hundred-layers’. Structural distinctions, from single-layer delta-doped quantum wells (as characterized by the layer-localized or surface- or interface-localized phonons [4]) to a surface or interface of a semi-infinite medium (as characterized by the bulk-surface phonons, as given in nearly all current textbooks, such as in the electron-energy-loss-spectroscopy (EELS) [5]), etc. are subjects of minor importance. In fact, the relevant common property is only that they have the subband energy structure on or near the surface or interface relative to the 2D incorporation. The difference is the results from one-layer or from a hundred layers. The realistic dominating factor derived from these considerations is that ‘geometric-flatness’, determinated by the crystallographi-cal Miller indices, impurities, foreign contaminations, defects, missing row, cracks and vacancies, etc., does really lead us to decide to choose some kind of physics ruling the way to prescribe our theories, and leave the strain-stress problems apart. A viewpoint of this kind is explained by an example below:
Current existing debate about the high Miller indices: For stepped surfaces of high Miller’s
indices, e.g. W(750), we have a more than usual Laue diffraction pattern, viz., there are extra diffraction spots due to the lateral steps of W(750) [6] apart from those spots existing due to the ordinary regular net of the upmost layer of terrace platform. Directly from p. 166, Fig. 2.9 in [6], we have two extra spots for W(750) than is common in W(110). Usually we consider that a (110)-surface is a 2D plane. For W(750) described in [6], it is determined by measurements: a terrace width 12.6 § 0.1Å , and a step height 2.19Å . W(750) is typically a 2D surface plus a third penetrated direction. From Fig. 2.9(b), the two extra spots for W(750) are on the same 2D plane which shows the LEED pattern of (11) and (20), etc. for W(110). Therefore we have 2D-reciprocal-momentum-space patterns to exhibit diffraction patterns from both a 2D-crystal surface
and a (2D-terrace +a third penetrated direction)-stepped-surface, typically 3D appearence (stepped)
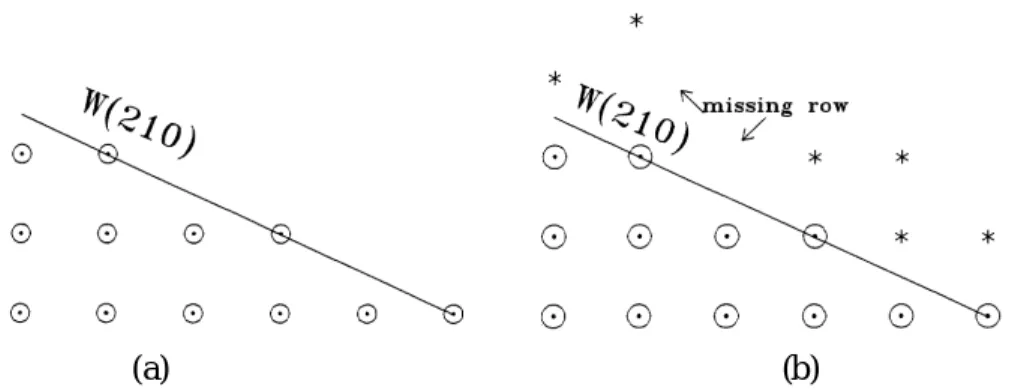
surface. We can not trust that “seeing is believing”. Here we think that both the 3D ‘stepped-surface’ and the 2D-plane-surface make diffraction patterns on the same 2D k-plane(s). There is a dimensionality-transformation or a breakthough in dimensionality from the 2D k-plane into the 3D stepped-surface in reality! Experiments of this kind made us believe that the mathematical surface formed by the outmost steps (750) is our standard “surface” existing in nature for minerals or for growth after deposition processes, etc. We illustrate in Fig. 1(a): a mathematical surface of such a kind, not for (750) but for (110). By ‘mathematical surface’, we mean surfaces existing by Miller indices predictions. They are a kind of “natural” surfaces existing themselves in nature without any artificial processes. They do not include the polished surfaces, capped surfaces and special cut-surfaces [7], etc.
When we formulate our calculation framework, we think the surface of this kind is not only a standard but the only correct one to use everywhere. Furthermore it is the only one in the pattern of this kind by which we can have the Bloch theory of periodic structures presented in the solid state. Based on this conception, from the Bloch theory for 1D, the surface of this kind is the only one able to formulate and to become a 3D-Bloch theory. Actually there are other surfaces such as some classical surfaces, etc. which are applicable or also satisfied by the Bloch theory.
(a) (b)
FIG. 1. Side views of tungsten with the stepped surface W(210) shown. (a) W(210) in a mathematical model. (b) W(210) in a defect model.
We include the classical surfaces of this kind as “cut-surfaces” below.
A contrary viewpoint, started by Mullins, we call Mullins’ defect model [8]: a ‘high Miller indices surface’ is an irregular step surface, as shown in Fig. 1(b) with many additional adsorbates, as surface impurities. It can be a “classical surface” without regular or ‘flat’ Miller indices predictions. There can exist many missing rows or line defects with a deep crack possibly. If the surface, as shown in Fig. 1(b), is the one predicted by Mullins’ model as the outmost surface next to the vacuum space as a natural surface, we really do not know how to explain it by the periodic structure, as used in Bloch theory. Mathematically one of reasons that the subband ladder energy appeared here, instead of the simple parabolic band energy of free electrons, for a (semi-infinite) crystal structure, is the dominate role of the above ‘classical surface’ involved. This classical surface will change our mathematics related to dimensionality in both prediction and calculation. Similar properties were reported in our first paper [9] with a positronium in front of a classical surface and some charge density problems for image potentials.
As a theorist, for the subband manipulation for a metal cap structure, we would think the structure like the one shown in Fig. 1(a) was a proper one. After we introduce the classical
surface for the cap or substrate, we find that the one shown in Fig. 1(b) is also a possible one,
moreover it is more proper to have used Fig. 1(b) than the one given in Fig. 2. The reason, apart from experimental preparation ones, is given as follows: A 2D-surface in the form of a classical plane, such as z = 0 (46) below, is sometimes not a part of the description in the framework of a 3D perfect periodic solid crystal. This description (a) is not unique; and (b) does not include the whole content inside which z = 0 say, does exist. It forms a breakthrough to break the above ‘incomplete description’, in fact the 3D perfect periodic structures, as defined by the kinetic energy in (45); meanwhile the potential energy being (45a), splits the total-periodicity in (49b). The mathematical reason, for the non-existence of a z = 0 plane inside the perfect periodic structure, is that a 2D-plane has “zero measure” inside a 3D volume and aside from the measures of a periodic potential, etc. We might think that a 3D perfect crystal is some ‘volume’ mathematically 3D with the coexistence of many 2D-planes. This intuitive viewpoint has been changed for a crystal either with a periodic structure or with a plane surface below. Putting both the periodic structure and the plane surface together in a consistent theory is the subject of this paper. A surface or interface is 2D, thus of zero measure, similar to a surface integral equal to a
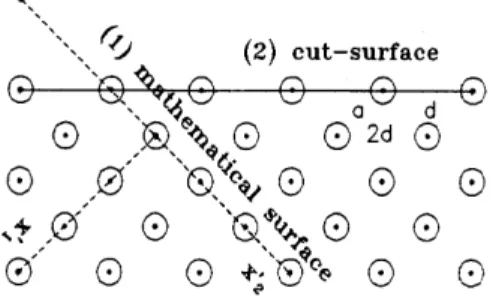
FIG. 2. A two-dimensional illustration of surfaces forV (z) and V (x0
1; x02) in (47c). (1) A “mathematical surface” to express the perfect 3D crystal forV (x01; x02) in (49) for some C+A+,C¡A¡ and proper di inx0i-directions. (2) A physical “cut-surface” forV (z) for different d in the z-direction.
volume integral of zero thickness, and has various uncertain influences on the 3D energy cal-culations. Between the whole-2D-plane and whole-3D-volume, stepped surfaces, as an interface between the material and the vacuum, are subjects for testing influences of this kind. From the viewpoints above, our decompositions into fa,b,Cg primitive translations in (21) below, fit the experimental set-ups in order to fit our “classical surface”.
Primitive and non-primitive cells in crystal structures: The above discussion is conventional.
For a pictorial interpretation, refer to the figures in Kittel’s ISSP [7], Figs. 14, 15 in Ch. 1, conventional non-primitive cells or Bravais or space lattices are given in Fig. 11. All simple cubic, body-centered cubic (bcc), and face-center-cubic (fcc) are given for a lattice constant a. Their corresponding primitive cells are all simple cubic in form and are given in Figs. 12, and 13. Recall that these simple cubics have entirely different oriented faces or surfaces with respect to conventional cell surfaces! They are not oriented in the way of “cut-surfaces”. The periodicity lengths arep3a=2 for bcc and a=p2 for fcc. When we talk about a (classical cut) surface which has a metal cap (which are mostly a surface after it has been polished or laser-evaporated), it is a surface faced on the conventional cell faces, not on the primitive cell faces. While the latter is mostly the Miller indices surface, because we apply to this primitive cell surface the crystal periodic boundary condition in Bloch theory. It must be a ‘mathematical surface’ defined as the right one for Bloch theory. The discussion here shows that Fig. 1(a) is a primitive or Miller indices ‘mathematical surface’ while Fig. 1(b) and Fig. 2 are capped surfaces.
The crystal structure for some currently used surface: Contrary to the above discussion, in
many semiconductor heterostructures, we cut a surface which makes a few degrees (say, 2±) with the conventional surface [10, 11]. The technique substantially extends the scope of our discussion above. For the present author, we should consider that they are classical surfaces. Furthermore they fall into the category of Mullins’ defect model (for surfaces) from an argument of reastically experimental preparations. In experiments, say in [11], we may use Al0:7Ga0:3As. It seems that it is similar to a simple-metallic surface of Mullins’ model. On the theoretical side, we can have quantum transport due to lower gap-energies forX and L points thereby having indirect transitions involving phonon processes. Further there may be uncertainties of energy entering the quantum transport regime, cf. p. 175 in [12]. It is natural to include umklapp processes according to the
reciprocal lattice vector space formulation. However it makes the PL peak intensity increase, say by 10 times as in [11].
Current Fermi-Dirac statistics applied in physics: We introduce the relevant current theoretical
quotations on various topics we need to use in the following for the total value of a dynamical variableA(E), as a function of E. Then the total value of this A over the occupied energy levels labelled byn of E¹ n¹ is
< A(E) >=X ¹ n
A(En¹)P(En¹) (1)
for a (normalized) population (or probability)P (En¹). For many electron systems, we use Fermi-Dirac statistics with the distribution function f(F D)(E) for some energy value E. Hence, with the density of states (DOS) D(E), we have
< A(E) >= R
D(E)dEA(E)f(F D)(E)
RD(E)dEf(F D)(E) : (2)
Current Fermi levels of 3D and 1D in statistics from a periodic structure: (I) For exact 3D
systems, we replace (1) by changingn in the summation to (n¹ x; ny; nz)´ n which appeared in the crystal momentumk = n2¼=L. We then have (1) becoming
X n ¢¢¢) (2¼)L33 Z dk¢¢¢)(2¼)L33 Z E(3D)F 0 4¼(2E3D)d ³p 2E3D ´ ¢¢¢ (3)
for 3DE3D= k2=2, without counting the spins. Here EF(3D)is the usual “3D Fermi energy in 3D systems”. The necessity of (3) is also required for the band energy calculations for crystals when they are of three dimensions, not the one in the Bloch theory of 1D as say in the Kronig-Penney model, for which we have many debates about the changing from 1D to 3D.
(II) For the 1D case of standing waves, in the zero temperature formalism for a convention, we change the label n in (1) to a 1D crystal vector (in the standing wave) E¹ ¹n ) E(k) with k = km= m¼=L, m = 1; 2;¢¢¢ for a length L of the 1D periodic structure. (2) becomes
< A(E) >= R
D(E(km))dE(km)A(E(km))f(F D)(E(km)) R
D(E(km))dE(km)f(FD)(E(km))
: (4)
Here the DOSD(E(km)) is calculated by the counting of m instead of ¹n as (in atomic units: ~ = me=jej = aBohr= 1 and, in the zero temperature formalism)
X m ¢¢¢) L¼ Z dkm¢¢¢)L ¼ Z E(1D)F2 0 d³p2E(km) ´ ¢¢¢; (5)
where “¢¢¢” means other functions included in the sum or in the integrand, as in (1) and (2), etc. ForD(E), we need to multiply by another factor 2 for counting the spin-occupations. The upper
limitEF 2(1D) in (5), the “1D Fermi energy in 1D systems” has many applications in solid surface theory [13].
As developed below in this paper for the first time, E3D becomes Ezn¤ in (26) or (24a), which is a 1D energy in a 3D system; (3) is calculated as for a given subband energy(k2
1+k22)=2, X n ¢¢¢)2¼L Z dkn¢¢¢) L 2¼ Z EF 1(1D ) 0 (2Ezn¤ )d³p2E¤ zn ´ ¢¢¢; (6)
for a 1D periodic running wave in a 3D system with crystal momentumkn= n2¼=L for E¤zn= E¤
zn(kn) = kn2=2. The upper limit E (1D)
F1 of the integral in (6) is not the sameE (1D)
F 2 given in (5). However thisEzn¤ -statistics is different from the Kronig-Penney model metioned above.
(III) As a remark,EF 2(1D)in (5) is a different 1D Fermi energy (in a low-dimensional system) in comparison with the “1D Fermi energy (in lower-dimensional systems) in 3D systems” [14], as we shall suggest for the first time in this paper. The difference comes from (30) below for
Ezn¤ = En(sl)+ ~ 2 2mz
kZ(2D) 2: (7)
Here both Ezn¤ and En(sl) are 1D energies. The eigenvalue of Ezn¤ indicates 3D energy quatuam packet while that ofEn(sl)1D energy packet. When we use either E¤znor En(sl), each of them can formulate a kind of statistical mechanics of its own. They can have their own Fermi energy. For instance, the Fermi energy of theEzn¤ -formalism isEF 1(1D), which is a lower-dimensional system including a term 2m~2
zk
(2D) 2
Z , which is in the z-direction mathemaically but comes from other dimensions, viz., withk1, k2 from the x; y-dimensions physically. Therefore for En(sl)-statistics, we can have, similar to (6),
X n ¢¢¢)2¼L Z dkn¢¢¢)2¼L Z EF 3(1D ) 0 (2En(sl))d µ q 2En(sl) ¶ ¢¢¢: (8)
Here we haveEF 3(1D) for a 1D Fermi energy, which is a (pure 1D) low-dimensional system, not a lower-dimensional system in 3D.
Conclusively for 1D Fermi energies: we haveEF 1(1D) for Ezn¤ -statistics, E(1D)F2 for standing wave E(km)-statistics, and EF 3(1D) for E
(sl)
n -statistics. The category-classification here has been applied to the free-electron-model for density-functional theories from Pauli paramagnetism [1, 15]. The main influence in setups is on the evaluation of the workfunctions Ái. Here we have three different workfunctions for 1D-surface-systems, or the familiar lower-dimensional system of a solid surface of a crystal. From the above three kinds of statistics: explicitly
Ái= vacuum level energy ¡ EF i(1D); (9)
where EF i(1D) is the three values of the Fermi energies above. From the experimental side or instrument side, the Fermi level determination is a major subject in heterostructures [14]. The
Fermi levels are determined by the charge neutralization of donor ND or/and acceptor densities NA, i.e., for the electron densityn and hole density p, we determine the Fermi levels by
n + NA= p + ND
forp-GaAs and N-AlxGa1¡ xAs heterostructures. As to the built-in potentialVD, it determines the difference between the Fermi levels of the two sides of an interface of the heterostructure. Clearly this calculation is based on Fermi-Dirac statistics, with the proper chemical potential as the Fermi energy calculated from the total number of particles in the system. As a comment from the above results, we really do not know that the Fermi levels determined here are E(3D)F or E(1D)Fi , since the dimensionalities of the particle numbers above are really relevant. The experimental setups or deposition designs of the electron density [14] are actually based on the formula of electron density n. The Fermi energy EF, as a function of the conduction band bottomEc for a setup-parameter Nc which is usually referred to 300 K, is
EF =Ec+ kBT · ln µ n Nc ¶ + 3.53553£ 10¡ 1µ n Nc ¶ ¡ 4.95009 £ 10¡ 3µ n Nc ¶2 +1.48386£ 10¡ 4 µ n Nc ¶3 ¡ 4.42563 £ 10¡ 6 µ n Nc ¶4¸ ;
in which the Fermi energy we are referring to is really important! But we are not sure about which one of the above Fermi energies we are referring to, 3D or 1D?
To be explicit, our viewpoint or comments on the currently existing framework on semi-conductor, such as (Al,Ga)As heterostructures, is as follows: The flat-band energy band diagrams show only incomplete 1D energy levels for interface physics with interface-barriers, such as the Schottky barriers, etc. in Ref. [14]. For 3D, energy value levels must be those in Ref. [16]. For GaAs with a surface or interface aside, we have energy diagrams with the Fermi-level (here the 3D Fermi energy in the 1D diagram) aligned, such as p. 209 in [14]. This way of treatment is perfectly alright. However current texts argue differently. They have stated some deviated appli-cations followed by some deviated plots, such as a 3D Fermi energy for 1D plots in [14], which seem to lead us to arrive at conclusions in non-equilibrium viewpoints! For heterostructures, near the surface or the interface, we consider that it is at a¡-point for a surface-plane [100] or [111] for GaAs. The system occupiesz > 0, the semi-infinite medium. The energy-band diagram along the whole z-axis is plotted according to the ¡-point energy levels in the band structure. Far enough from the heterojunction plane of heterostructures, we have various flat-bands in various regions along the z-axis. The plot in this way is: (a) different from the usual band-energy diagram for bulk solid state materials in which it is plotted fromX to ¡ to L-points with high symmetries, but reducing its dimensionality, in k-space; and (b) not theoretically acceptable or explainable based on interface physics. However in the so-called flat-band regions, we can calculate the electron distributions in a statistical mechanics way, as on p. 203 in [14]. In this calculation, we claim usually that it is an energy-band application on the ‘solid with surface’ from current solid state physics, which treats solids without surfaces. In the author’s opinion, it is not right since in prac-tice, the semiconductor with surface or interface is only one-dimensional Fermi-Dirac statistics with EF i(1D), as in (8) pure 1D. Not the Fermi energy used in [14] in which EF(3D) is supposed, such as in (3).
Furthermore in the discussion of the band-tail states from the DOS consideration [16], the present author trys to believe that this is a case of one-dimensional statistical mechanics instead of an interpretation in the band picture. On many occasions, this is not related to the energy-band theories at all. In Kane’s theory or the theory of Halperin and Lax say, we have energy-energy-bands (tail) extensions in energy band diagrams, such as the exciton levels say, cf. p. 81 in Ref. [16]. The troublesome thing is the energy distribution extended from 0 (atEc of the conduction band) to some negative energy values in the Fermi-Dirac distribution with a DOSE1=2. In this DOS, we are not able to write a real expression fromp¡ jEj. Here we have put zero energy at Ec whereas negative E means a level slighter lower than Ec, actually in donor states region. This DOS is expressible as a one-dimensional distribution in numerical continuation, with a deviation from the parabolic energy bands, cf. [16]. As given on p. 95 in [16] for nonradiative recombinations, say for CCCH Auger process, etc., the energy-band plot is different from the ones for the ‘solid with surface’, given above in the last paragraph and the ‘flat-band plot’ given throughout the plots in Ref. [14]. In Auger processes of this kind in [16], the energy-band is plotted from the ¡-point to theL-point, such as the one extended from the energy at ¡6 (as the zero-value ofE at Ec) to negative (exciton levels) values. They are for 3D bulk structures, as occurs in current solid state physics not in surface or interface physics. For the Fermi energy, the use of EF(3D) must be a misuse. We need to useEF i(1D) in (9). The subject which needs to have deeper investigations is the same as above, viz., which one ofE(nD)Fi n = 1, or 3 or 2; i = 1?2?3? is proper in practice? The theory that needs to be studied is in what maximum compatible capabilities are we able to interpret and predict the physical properties of semiconductor systems when the energy-band theory and the one-dimensional-statistical theory are put together? Do not mix up the 1D systems and 3D systems!
Furthermore on the physics side, we need to pay much attention to our theory of semi-conductors. There are investigations to fulfill the interpretations of theory, such as the flat-band concept. Recent attacks of the same author are: (i) From discussion in Refs. [1, 14, 26], it is more natural to interpret the space charge layers near the interfaces by the charge-transfer of semiconductor materials, such as III-V compounds. By an interpretation of this kind, we can have the “flat-band” charge distributions in the flat band regions. This leads us to have Fermi-Dirac statistics for the carrier, hole and electron distributions in place of a pure energy band theory as concluded below. Many of them are discussed in this paper. For a Thomas-Fermi study of this flat-band problem, we have results in x[3.4] and p. 32 in Ref. [2]. (ii) For adsorption theory in front of a surface for charge transfer to an adatom, we have results in [26]. But for the bulk-charge transfer for a semiconductor, which implies experimental results displaying the bulk-space charge near interfaces, we are intensively studying their peculiarities.
Because we have various energies in various dimensions as discussed above, we have some correction to their derived quantities, such as the effective mass definition. In eqn. (4.1) on p. 289 in Ref. [2], we have derived the effective mass for an isotropic material. 1=m¤= (1=3)r2
kE(k) instead of the usual value1=m¤= @2E(k)=@k2. This is clear from the definition of the reciprocal effective mass tensor in (16) below. Generally the correct effective mass for isotropical materials should be derived from the radial part of the Laplacian r2r, viz., 1=m¤ = (1=3)r2
kE(k), not from the wrong expression: borrowed from @2=@r2 which corresponding to the current value 1=m¤= @2E(k)=@k2.
The surface state from the complex k of Kohn: From an analytical function theory for Bloch
electrons of periodic potentials for 3D perfect crystals, in Ref. [17], Kohn introduced the complex crystal momentum and concluded that, for pure imaginary crystal momentumk, the band energy En;k is inside the energy gaps between the bands or the allowed ranges of En;k. The Wannier function is the subject to be studied, particularly for surface states. In Refs. [18, 19], the authors formulated a basis from the “generalized Wannier functions”an(x). In turn, an is expanded in terms of gn0(x¡ n0), the Gaussian distribution functions at various lattice points, x = n0. In these references, this group obtained some surface state above or below the lowest band, just like the acceptor or donor states, inside the band energy gap, which is along one particular direction in crystal momentum space and is essentially 1D in a statistical sense, the same as EF 1(1D). Therefore it is natural to have some imaginary part for the crystal momentum. This quotation is from a background of Gaussian-distribution-bases which had been applied to magnetic field systems extensively in these years. They are always referred to as the localized-states. For dynamical Wannier functions, see eqns. (4.2.6) and (4.3.14) in Ref. [12] for quantum transport in semiconductors.
Resolution of new-dimension hidden coincidently along the z-axis by complex k: From the conclusion (29b) below, 2D behaviours such as subbands, etc. are considered as a kZ(2D) in the z-direction, to have its new dimensionality because it is derived from the x; y-plane quantities, i.e. the constants kx; ky from thex; y-dimensions. The dimensionality is thus conceptually extended to some abstract or implicit dimensions. In the other version, it may be considered as a way to include the complex momenta. For a complex extension of a real physical quantity, such as k above, we can have the real axis together with an imaginary axis. It is thus a possibility to make a breakthrough to break into physical configurations, such as from the bulk crystal (wave function) to be extended to include a surface (envelope function), for bulk material (a 3D volume) and a surface (a 2D envelope). We have a detailed dimension theory in [2]. They are separated in measure theory, such as in the Riemann integral theory in which the 2D-plane is immeasurable in the 3D volume integral for a simplified version of the mathematical origin. For this we can have anx; y; z-axes for the 3D material (such as volume-charges) while there are x; y-coordinates for a 2D surface (such as surface-charges, with zero thickness in thez-direction). Another example is that the complexk may provide a complex E(k) whose imaginary part is related to the lifetime, etc. cf. p. 247 in [2]. All of them exhibit a new extension to various new directions, or degrees of freedom or dimensionality, such as surface charge distributions wrapping a volume charge. Further it incorporates surface behaviour (in surface localized-state conceptions in a delta doped layer) together with 2D peculiarities, one of them for the surface of semi-infinite structures. It is directly related to the mean free path ¸, cf. p. 247 in Ref. [2], by
kI = 1
2¸ in k = kR+ ikI: (10)
It is usual that the plane wave wave function will be changed to
à » eikx= eikRxe¡ kIx (11)
with decreasing length » 1=kI. Their applications to mobilities through either Kohn’s way discussed above, or through (10), as given in Ref. [2], will be given with a physical implication in (29a), or (29b).
Mobility for intrinsic semiconductors: Currently subband theory is extensively applied, e.g.
related to mobility problems, such as microscopic mobility, Hall mobility etc., incorporating two-dimensional behaviour [20, 21] and/or high magnetic fields. Historically, from Seitz’s result of the mobility for intrinsic semiconductors [7], it is related to the collision with phonons and is related to kinetic energy in its fomalism in the initial equations. It is related to the so-called k¢p-perturbation for the intraband calculation. Originally the k ¢p-perturbation was for the uk(r) of the Bloch function bn = ukexp ik¢r in the Schr¨odinger equation. It is a kind of scattering interaction with phonons inside the particular kind of structure. This can be seen from the band electron (i.e., with its motional momentum p) interacting with the phonon (i.e. with the crystal momentum k evaluated from a given structural framework). Or we can see it from
~2 2m Z drjrukj2= ~ 2 2m Z drjrbnj2+ ~ 2k2 2m + 2 ~Re Z drb¤n(r)(k¢p)bn(r) = K(B)+ K(class)+ 2
~ £ Real part of k ¢p-expectation
(12)
in the formula in Ref. [7]. The classical nature of kinetic energy K(class) is given on p. 347 in Ref. [2]. In view of (12) for the periodic uk(r), the kinetic energy appeared twice, once in the quantum form of the Bloch function and another as a classical form. From the concepts of deriving the dimensionality of classical generalized coordinates in classical mechanics, the kinetic energy appears twice (and in different forms) so this must have the degrees of freedom = 6, twice as many as the one particle freedom = 3 in the kinetic part. This is to be understood in the following:
Resolution of a new-dimension by direct addition of the new dimension: This is familiar in
molecular physics [15] for a diatomic molecular system. Below 1000 K, the heat capacity shows 3 translational motional degrees of freedom. Near 2000 K and below 3000 K, the rotational motions enter, so that the total degrees of freedom becomes 5. Higher than 3000 K, the vibrational motions enter so that the total degrees of freedom become 6, 7, or more. When we write the Schr¨odinger equation for this diatomic molecule, the dimensionality changes from 3 to 5 to 6, 7, or more in the above order for various temperatures. We see that, when the temperature increases in molecular physics and thermodynamics, we have to increase the dimensionality for the same molecule. This illustrates that we can add directly the new-dimensions to our system quite naturally and with nearly no limitations.
For the extension of atomic or molecular vibrations on a surface, we see that the vibration frequencies on a surface do not depart from the free status very much. For instance, a CO-molecule on Cu(111) shows a C–O vibration with frequency 2074 cm¡ 1 (coverage< 0.33) [22]. This can happen in a wall-collision of CO on a Cu-wall, stuck or adsorbed, and detached. From a standard table, the vibrational frequency of gaseous-phase C–O is 2169.8233 cm¡ 1, not very different from that of the surface value. Because, as a picture, C is between O and the Cu surface, there are two harmonic potential energies acting on C, viz., one is the C–O bondVbondand another is the whole CO inside a harmonic potential VCO due to the surface. When written inside the system total Hamiltonian, the C is affected by two harmonic potentials, added together as Vbond+ VCO=Cu. However in a status of motion, we obtained the frequency of the C–O vibration which occurs for the so-called localized vibrations, cf. p. 346 in [22], not the localized phonon band of surface
[4], in a mode which is nearly not influenced byVCO=Cu. Now we write the coordinates as that between C–O, Vbond= 1 2kr 2 bond; (12a)
and between CO and the Cu-surface we write VCO=Cu =
1 2KR
2; (12b)
where R is the center of mass coordinates of CO with respect to the surface. For brevity, we write it only in 1D along the z-axis without considering the bond-bending etc. with C at z from the Cu-surface, so that R is perpendicular to the Cu-surface and
R = z + zCR; zCR=
mC(R¡ z) + mO(zO¡ R) mC + mO
; (12c)
where z is the coordinate of C and zCR the distance between C and the center of mass of CO; zO is the coordinate of O. Therefore
VCO=Cu = 1
2K(z + zCR)
2: (12d)
One of the vibration frequencies of C is nearly pk(mC+ mO)=mCmO » 2074 cm¡ 1 » 2169 (standard) although the total potential affects on C is
Vbond+ VCO=Cu= 1 2 £ k(zO¡ z)2+ K(z + zCR)2 ¤
; same mathematical z (12e) as in the 1D expression. That means, in this (normal) mode of motion, C–O vibrates withVbond independently fromVCO=Cuno matter that it is the samez in the two potentials in (12e). If the potential energy in (12e) is summable for all z as a 1D-variable, we must have
Vbond+ VCO=Cu= K1 2 (z¡ z1) 2+ C; with K1= k + K 1 (¹ + 1)2; ¹ ´ mC=mO; z1= zOk(¹ + 1)2¡ [(¹ ¡ 1)R + zO]K (¹ + 1)2k + K ; 2C =¡ [zOk(¹ + 1)¡ f(¹ ¡ 1)R + zOgK=(¹ + 1)] 2 (¹ + 1)2k + K +kz2 O+ K (¹ + 1)2[(¹¡ 1)R + zO] 2; (12f)
with an entirely different but complicated combined vibrational frequency from K1, etc. This forces us to consider that rbondis in a separated dimension from the CO–Cu-surface, viz., the z + zCR coordinate. While for the other potentialVCO=Cu in (12e), because of the complications
of zCR which itself depends on z in (12c), by simplification, we may consider this degree of freedom or the new dimensionality by way of a mean-free-path or mobility, etc. such as the one given in (29b). In the author’s treatment, it provides a new dimension. For the kinetic energy term, similar to (12), we have a kinetic energy for the relative coordinates of C–O and another kinetic energy for the center of mass of CO/Cu.
The separation is particularly clear when the C-O makes a detachment from the surface to become a gas-phase particle. Since the mathematical surface is infinite in any direction, so to speak and measure, the ratiozCR=surface dimension is always zero. It is a scale-free-measure, used in many different ways and academic branches. We must separate rbond from zCR as a new dimension since these quantities are not scale-free, particularly when a surface dimension, included as scale-free dimension, or surface quantities are involved. Furthermore, for zCR À 10Å , C-O is a gas-phase or free particle, and it must be determined by new dimensions or a new system of coordinates for this isolated system. Cf. Ref. [26].
Mathematics of linear independency: Dimensionality is determined mathematically by the
judg-ment of the existence of the so-called “linear independency”. It is the mathematics of linear vector space theory: the linear independency of vectors in mathematics is restricted by the so-called “Gram determinant condition”. Rigorously in mathematics it must be an if-and-only-if or suffi-cient and neccessary condition for linear independency. However the Gram determinant condition is only a condition, not a definition or a theorem. Linear dependency is determined by two ‘only if’conditions, cf. Ch. 4 in Ref. [15]. We can say that linear independency is still an open problem in mathematics. Forn-linearly-independent vectors, the non-vanishing of the Gram determinant is definitely not equivalent to the existence ofn-mutually-perpendicular vectors, which constitute an orthogonal basis in the linear vector space. The mathematical linear independency is questioned by physical periodicity-split rule, (rule 2) below, cf. below (47a).
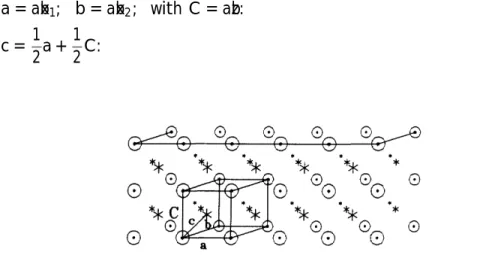
Many points referred in this paper can be compared with the review article of Ando, Fowler and Stern [23] for 2D electron systems. We quote some derivations from the text of the same author [15] which give a consistent results in deductions. The usual geometry of systems here is as follows: in parallel to the interface, we have the primitive translational vectors: a and b. For the third direction, it is usual to have a C-axis as shown in Fig. 3 instead of c which is an edge of the unit-cell for the standard fcc structure in Fig. 15 in Ch. 1 of Kittel’s ISSP [7]. It is generally the case that
a¢c 6= 0 (13)
and
b¢c 6= 0; (14)
so that c is not used as the third direction to indicate the layer separations. However the original condition from the periodic system is to have the periodic potential
Vperiodic(r) = Vperiodic(r + n1a + n2b + n3c); for sets of integers ni: (15) From the usual theory, essentially applied to the subband theory in semiconductor physics, we
have the kinetic energy expressed in a general form as K(1)=¡ ~ 2 2 X i;j uij @2 @xi@xj (16)
for the system reciprocal effective-mass tensor[uij] for consideration near the particular conduction-band minimum or valley. In (16), we propose to consider, in a general case, that the lattice system hasa? b ? c in the unit cell. However the physical configuration becomes nearly impossible to treat mathematically from (45)–(46) below, when there is a plane-surface introduced for a crystal, such as a metal cap or gate on a semiconductor ‘crystal’. Further it is well-known that many times we cut the crystals at some small angles inclined to the routine (111)-plane, see e.g., [10, 11]. It is noted that this kind of set-up of crystals in papers of this kind is in fact a general-ization of the high Miller’s indices discussed above, say for W(750), but sometimes mixed with the impurities such as was mentioned in Mullins’ defect model, cf. Fig. 1. On many occasions, we discuss strains of the surface and mismatch of the interface. We must try to understand solid physics applications to these systems: their relationship between a ‘cut-surface’ and the routine periodic configuration for periodic potentials. This is done in Sec. III below. We shall abandon the usual crystal classifications above, and in some fabrication processes of deposition, say from chemical solutions, etc. This argument, we shall illustrate, by a kind of reduced-fcc lattice as in our consideration in Sec. II below, is for the general cases. Further we change the unit cell to be a realistic one to fit the surface or interface plane, so as to treat Fig. 1(b) as being some structure as in Fig. 3 with a metal cap or gate on the top. Incidentally for high-Miller-indices-or stepped-surfaces without any contaminations high-Miller-indices-or irregular depositions, we have other kinds of special treatments, such as in the stepped surface atoms movements, etc.
But for this reduced-fcc lattice and for the purpose to have a surface parallel tox1x2-plane, as shown in Fig. 3, we choose first a unit cell fa,b,cg to fulfill the requirement of (15)
a = abx1; b = abx2; with C = abz: c = 1 2a + 1 2C: (17)
FIG. 3. Reduced-face-centered-cubic structure with substrate-plane on the top. Solid state primitive trans-lation vectorsfa,b,cg are shown together with surface physics translational vectors fa,b,Cg.
We consider the variables xi (i = 1,2) in the directions of a and b. But z is in its usual direction of C not in the c-direction. This is to emphasize the active potential exhibited in the form of
Vact = V (z) (18)
evaluated according to the variation in the layer-change direction (i.e. z-direction). This potential, according to the subband theory does have the periodicity
V (z) = V (z + d); (19)
for layer separationd. Now we have to reformulate the kinetic energy to replace (16) by K =¡ ~2 2 X i;j wi;j @2 @xi@xj (20)
for system reciprocal effective-mass tensor fwi;jg for a coordinate system
x1 = x; x2= y; x3= z; i:e: fa; b; Cg-system; (21)
where thexi are perpendicular to each other.
In Sec. II, we formulate the subband theory from the kinetic energy tensor with a particular relation between tensor elements. In Sec. III, the compatibility of the surface equation, as the boundary condition µ(z) or by the validity of space-coordinate value z >0, with the potential energy V (z), is introduced. We find that for kinetic energy tensor elements: w1;z 6= 0 and w2;z 6= 0, this compatibility does not exist. The one-dimensional potential energy V (z) is equal to a two-dimensionalV (ax01+ bx02) in (49) although z = ax01(t) + bx02(t) for static z and lattice dynamicalx0i(t). Further investigation is left for future work.
II. Theory
We model a kind of structure for an easier and clearer formulation. It is not a model, as required in the theory and in experimental setups, which is perfectly general. This structural model is defined as a “reduced-fcc”. It is a face-centered-cubic but without the face-lattice-sites at all the parallelxy or ab-planes. It is shown in Fig. 3. For the interface or surface, it is shown on the top of Fig. 3. Then we have ordinary rectangular coordinatesx = x1; y = x2; z, and (20) then provides the Schr¨odinger equation in the form
[K + V (z)]ª1(x1; x2; z) = · ¡ ~22 X i;j=1;2;3 wi;j @2 @xi@xj + V (z) ¸ ª1(x1; x2; z) = E3Dª1(x1; x2; z); (22)
to fit our model of a reduced-fcc in Fig. 3 with a square net on the ab-surface so that w1;1= w2;2= m1¤; wi;j= wj;i»= 0; for i 6= j = 1;2 and
otherwisew1z= wz16= 0; w2z= wz26= 0; wzz = 1 mz
:
Sometimes we argue that we have small angle cut inclined from the ab-plane, etc. [10, 11] as described in the Introduction. Thus (22) becomes
[K + V (z)]ª1(x1; x2; z) = · ¡ ~ 2 2m¤ µ @2 @x2 1 + @ 2 @x2 2 ¶ ¡ ~ 2 2 ¢2 X i=1;2 wi;z @ 2 @xi@z ¡ ~2 2mz @2 @z2+ V (z) ¸ ª1(x1; x2; z) = E3Dª1(x1; x2; z): (24)
When we use a statistical model, we useE3Das the one to be used in (3). We need to have a 3D Fermi energyEF(3D) fromE3D. Sometimes the intuitive wave function is put as
ª1(x1; x2; z) = ³(z)eik1x1+ik2x2 (25)
by the ‘separable of variables’ method, conventionally called ‘separable’ instead of ‘separation’ for some reasons. A treatment of this kind is particularly suitable for all kinds of two dimensional electron gas systems (2DEG) [23]. We obtain an equation for³(z),
· ~2 2m¤(k21+ k22)¡ i~2(wx1;zk1+ wx2;zk2) @ @z ¡ ~2 2mz @2 @z2 + V (z) ¸ ³(z) = E3D³(z) (25a)
for the function³(z), which thus obeys a one-dimensional differential equation in a sense. The Fermi energy required in (25a) is still the 3DEF(3D), the same one as in (3). This one-dimensional Schr¨odinger equation must be reconciled with its complete one-dimensional behaviour by the following processes: First
· ¡ µ ~2m2 z @2 @z2 + i~ 2(w 1;zk1+ w2;zk2) @ @z ¶ + V (z) ¸ ³(z) = µ E3D¡ ~ 2 2m¤(k 2 1+ k22) ¶ ³(z) ´ E¤ zn³(z); (26)
after quantization to have 1D energy eigenvaluesE¤
zn. For a statistical model we referred to (6) for the DOS. We need to defineE(1D)F1 from this energy. Sometimes we refer to~2(k21+ k22)=(2m¤) or even the whole term in the second line of (26) as the subband energy. It is possible to have the matrix coefficientswi;z = wi;z(x; y) appearing somehow in (26) after reduction from (24). For (50)–(51) below, this possibility does exist. It must be this case for the cut-surface at a few degrees with respect to the conventional crystal basal plane, as given in Refs. [10, 11] introduced in the Introduction above. If we consider a way to indicate some pseudo-crystal-momentum-in-z-direction
kZ(2D)´ [w1;zk1+ w2;zk2]mz » ¹w 2¼
from (23), we have used this quantity for the second term in the first line in (26). This shows thatEzn¤ measures the k1; k2-coupling from other dimensions, viz., the x and y dimensions for a z-dependent equation (26). Thus it is not simply 1D. The role of k(2D)Z is to breakthrough from the subbands in 2D into the third dimension. Similar tok¢p in current solid state physics, this k(2D)Z denotes some “structural effect” indicated by crystal momentum k. This makes us completely confused whether the subband theory is 1D (in the final eigen-equation), or 2D (for its own energy value), or 3D (after the breakthrough in (26) with all 3D quantities included). This shows a failure-point in dimensionalities physically for the ‘separable of variables’ methods which are pure mathematics in x; y; z, while the system is different for physics, say k1; k2 in kZ(2D) in (27). A physical conterpart of this point, as given in pp. 234-235 in [2], is for the Fermi energy in transports. In semiconductors, the driving-force of movements is derived from Fermi energy. Dynamically, the transport-movement is directional and thus 1D. This induces that the used Fermi energy should be 1D. For (24) to apply to the complete picture of the ‘separable of variables’, we must have for (25)
E3D+ 1 ³(z) · ~2 2mz @2 @z2 + i~2 mzk (2D) Z @ @z ¡ V (z) ¸ ³(z) = 1 eik1x1+ik2x2 ~2 2m¤(k 2 1+ k22)eik1x1+ik2x2 = E3D¡ Ezn¤ : (24a)
We have separated the coordinate variable z positionally in contrast to x1; x2 in a certain way: in the first two lines of (24a), z appeared only on the first line while x1; x2 appeared only on the second line. Further, because of the equality, we have that they are independent of anyxi; z, variation from point to point, viz., constants. Thus we have a constant E3D¡ Ezn¤ after the final equality which is exactly the subband energy. In physics, the physical properties indicated by constants, or constant dynamical variables, such as k(2D)Z includingk1; k2, are still included on the first line of (24a). That is why we have (26) which is not simply one-dimensional because we fail in separation of the dimensions xi i = 1; 2, in contrast to z. The connection between them is through physical reasons. It is obvious here that this way of treatment has generated some inclusion of interpretation in the new dimension.
For the quantum momentum in thez-direction pz=¡ i~@=@z, we have (26) become " ¡ Ã ~2 2mz @2 @z2 ¡ ~k(2D) Z pz mz ! + V (z) # ³(z) = Ezn¤ ³(z): (28)
The difference between (26) and (28) forEzn¤ , is that we consider in (28) the~k(2D)Z pz=mz-term to have the form of ak¢p-perturbation in conventional solid state physics. This becomes general for perfect crystals of all kinds, when it is formulated from 3D reduced to 1D by (25). For instance, it is well-known that a k¢p-perturbation changes the effective mass at the conduction band edge incorporated with the valence band states for 3D wave functions, (not the envelope ³(z)), coupling two bands, as much current literature indicates. Here, there may be two subbands [21].
The peculiarity here is that kZ(2D) may not be purely one-dimensional or may not be the routine crystal momentum: kz = 2¼nz=Lz in the solution ofuk(r) in the usual Bloch function (not the kZ(2D) above). It may couple properties from the z-direction into the xy-directions. In addition to thiskzpz, we moreover have the simplekZ(2D)pz-interaction in this subband theory. It is this term that makes the mobility of intrinsic semiconductors in Ref. [7] appear. However the difference of k(2D)Z in (27) from thekz in 1D Bloch electron theory shown in eqn. (12), makes the equation here predict some peculiar mobilities. The effective mass of this case is supposed to be detectable by the cyclotron resonance frequency, and other experimental evidence.
Since our model is for a ‘reduced-fcc’, in parallel to the current theory [23], the wave function³(z) in their consideration, is changed to an envelope function ´(z) subjected to some simplified structural conditions by
³(z) = ´(z) exph¡ ikZ(2D) ¢z i
» ´(z)e¡ imzw(2¼ =L)(n¹ 1+n2)z; (29) for n1+ n2´ nz = integers. Here the effective thickness in thez-direction is equal to
LZ= L mzw¹;
(29a) forL along the x; y-directions! This is regardless of whether the structure is a delta-doped or semi-infinite material. This reveals rather a new dimension than the standard third direction. In the author’s intuition, the situation here is like an enveloping ‘surface chargeq±(z¡ z0)’, represented by the wave function exp¡ i2¼nzz=LZ, rather than a factor in new dimension, wrapping over a volume which is characterized by ´, described above in dimension theory ideas. Because the wave functions here indicate the behaviour of electrons, therefore the pictorial new idea here is exactly that described below (9) in the Introduction for 2D-surface charges in 3D-space. The whole entity is described by³. Therefore ´(z) should be the “enveloped wave function”. Whereas LZ is something like the value ofq±(z¡ z0) at z = z0. This was emphasized in the Introduction regarding the dimensionality conception in (12) – (12e). It is not a property along thez-axis alone physically.
The probability distribution from (29) is j³(z)j2=j´(z)j2£ ¯¯
¯exph¡ ik(2D)Z ¢zi¯¯¯ 2
: (29b)
The second factor with LZ actually may be controlled from sizes in the (x; y)-directions and indicates the pseudo-thickness in the z-direction by Bloch theory for finite crystal influencing the (pseudo-surface) charge density contributed from the wave function probability, i.e. the j exp[¡ ikZ(2D) ¢z]j2-part in its own (new) dimension. However from the electric potential cal-culation in electrodynamics, this new dimensional ‘surface charge’ does still contribute a potential electrically similar to the exact surface charges. The application is also applicable to semicon-ductor devices, such as the MOSFET. We replace the semiconsemicon-ductor oxide layer by polarizable materials, we are able to form a ‘new dimension’ charge layer described above with a certain prescribed thickness, to produce a (signal) “gate-charges” from some metal gate to replace the (signal) electric field in this field-effect transistor. Theoretically, the above interpretation of the wave function together with the 2D gate-charges, if the experiments show the existences correctly,
makes a judgment in favor of having a new dimension theory. A further development can be to investigate the ‘homogeneity-dimensions’ given on pp. 30, 49 in Ref. [2] for quantum physics.
Motivation for introducing a complexk: It is noted that we have the one-dimensional Bloch-type wave function form for ³(z) with k(2D)Z in (27) as our crystal momentum. This is the
second peculiar crystal momentum which may breakthrough into other dimensionalities, apart
from the first one where the complex nature in (11) indicates the mean free path. Since (28) has ak¢p-perturbation form, the mobility is related to
Z dz ¯ ¯ ¯¯dzd ´(z) ¯ ¯ ¯¯2:
Here V (z) is a periodic potential energy in the “z-direction” characterized in (19), with the separation between layersd, in the z-direction, not the c-direction as is usually provided by the Bloch theory.
Here we modify the ordinary energy band theory for the purpose of terms applicable to apply to systems with surfaces or interfaces having a 2D nature, as in a (2+1)-dimensional system. The final Schr¨odinger equation is still one-dimensional in the form of (28) with eigenfunctions´n(z) and eigenvaluesEn(sl) · ¡ ~ 2 2mz d2 dz2 + V (z) ¸ ´n(z)´ Hz(z; pz)´n(z) = ½ E¤zn¡ 2m~2 zk (2D) 2 Z ¾ ´n(z) ´ En(sl)´n(z): (30)
For a statistical model, we introduceEF 3(1D) in (8) for En(sl)-statistics. From a study of (30) we have two points to discuss:
(I) for semi-infinite materials, V (z) = V (z)µ(z); and for delta doped materials, V (z)» V (r)±(z¡ z0), the conditions of the Bloch electron theory of periodic systems in the z-direction are not satisfied. From (30), if the solution´nis localized, i.e., in (29),³(z)» tight-binding or a linear combination in Gaussian distributions, or LCAO as usual in solid state physics, we have no Bloch crystal momentum (BCM). From the appearance of (29), the pseudo-BCM part in³(z) is controlled by thex; y-direction behaviour in kZ(2D). It is a new kind of behaviour, contrary to the Bloch theory for periodic structures in 1D in solid state physics. For instance, we have an ´(z) to replace theuk(r) in the Bloch function with uk(r) = uk(r + R) for a lattice point separation
R. There will be no actual lattice point along the z-direction with layer-separation d, such as in Fig. 2, for the samex; y-coordinates, no lattice point at a distance z = d from the lattice point at z = 0. There is no k¢p-term for a k totally determined by the z-direction property in (30). This is different from the pure 1D Bloch theory where we have Bloch function results for´(z) since we have delta-doped layer or semi-infinite medium, although the periodicityV (z) = V (z + d) still holds. Here we introduce a complexk. However the physical picture of ´n is the same one as given in the transformation of Stern and Howard [24]. Because of the real physical configuration
given above, we need to reconsider the mobility or there are no mobilities for ´n(z) rigorously, with different dimensionalities instead. If we try to put in the mean free path, we need to choose
kZ = kZR+ ikZI= complex;
like the one described in (11) above. Here we have incorporated a surface or interface or 2D plane, but we do not have to be very clear about the physics behaviour. The physical picture is still the one given in (11). This implies (29)
³(z) = ´(z)e¡ i(kZR+ikZ I)z= ´(z)e¡ jzj=2¸e¡ ikZRz (29b) for the mean free path¸ in the z-direction no matter what the structural form of the material and whether ´ is localized or not. Here we have reconciled the sense of Kohn’s Wannier function for a complexk discussion as mentioned below eqn. (9) in the Introduction. The last equation expresses a kind of effective thickness in the z-direction, with a similar restriction as derived from (29) disregarding the actual physical thickness in thez-direction. Here we have the second interpretation for ¸, emphasized in a statistical sense.
(II) From (28), (24a) and (30), we can see that E(sl)n = E¤zn¡ ~
2 2mzk
(2D) 2
Z = subband ladder energy; (30a)
which is the realistic 1D energy of 1D energy quantum packet. The coupling of k1; k2 ink(2D)Z in another 1D energy Ezn¤ is subtracted Here Ezn¤ is of 3D energy quantum packet. This also concludes our discussion ofEF i(nD),n = 1, 2, 3 in (9) in the Introduction. There we have extended the fact forEF i(nD), etc. to be initiated (a) by a complexk in the Introduction; (b) to some special mobilities; or (c) by introducing new dimension(s) directly.
From (26) and (30), the 3D energy packet E3D(n; k1; k2) = Ezn¤ + ~ 2 2m¤[k12+ k22] = En(sl)+ ~ 2 2 ·µ 1 m¤ + w1;z2 mz ¶ k2 1 +2(w1;zw2;zmz)k1k2+ µ 1 m¤ + w 2 2;zmz ¶ k22 ¸ : (31)
It is noted that Ezn¤ is the “z-direction” energy with the mobility calculated from the three-dimensional Bloch energy E3D band theory. It includes kZ(2D) itself. When we apply it to semiconductors to consider the intrinsic semiconductors at 0 K, we have the electrons filled up to the top of the energies En(sl), i.e. EF 3(1D). This is the way we calcualte the hole density in a heterostructure [15]. This EF 3(1D) is exactly the top energy Ev of the valence band from the definition of intrinsic semiconductor when the conduction and valence bands are considered along one particular direction in k-space, or in the perpendicular direction to a surface compatibly. When we assign the Fermi level EF(3D) from the three-dimensional theory of solid state physics, from
E3D, the components of the corresponding Fermi levels in thez-direction must be assigned from E¤zn from (26), (i.e. E(1D)F1 6= E (1D) F 3 above). However EF(3D)= E(1D)F3 + ~2 2m¤(k21+ k22) + ~ 2 2mz kZ(2D) 2= ~2k21 2m¤ + ~ 2k2 2 2m¤ + Ezn¤ (max); Ezn¤ (max)´ E(1D)F3 + ~ 2 2mz kZ(2D) 2= Ev+ ~ 2 2mz k(2D) 2Z : (32)
This is why we have Fermi levels with values in the energy gap between the conduction and valence band topEv = EF 3(1D) from (32), not down toE
(1D) F 1 of E
(sl) n .
Following Stern et al., since Ezn¤ andEn(sl) may be negative, in order to compare which one ofEzn¤ and En(sl) has a lower value of energy according to the energy level assignment on a surface, we investigate and develop the usual calculated energy band diagrams:
(A) It is most common to assign the zeroes of the energy levels at the Fermi energy which
is unique. The exceptions of this uniqueness are for the so-called “non-thermal equilibrium” in semiconductors [14] and pp. 73 in [16], such as (Ga,Al)As quantum wells. There we can have the so-called ‘quasi-Fermi levels’. We can even have two or more quasi-Fermi levels.
(B) The second way is to assign the zero level at the free-particle-zeroes to determine the
space charge layer by the same author [1]. This is particularly useful for the Fermi-Dirac statistics cases. The introduction of negative values of energies in the statistics of this kind has resolved into ‘for energy domain¡ 1 to +1’ for finite temperatures, e.g. in (Ga,Al)AS. This is evident for the paramagnetic cases. However we move on to discuss systems of “free-particles” [25]. Many cases give the bottom of the parabolic band of energy as zero. This includes the ‘conduction band zeroes’ at some Fermi energy =¹, for Fermi-Dirac statistics for electrons,
fF¡ D(E) = 1 1 + e¯ (E¡ ¹ );
where unavoidably¹ = EF(3D) in many current textbooks; or the ‘valence band zeroes’ for holes statistics in semiconductors,
fh(E) = 1¡ fF¡ D(E);
cf. Ch. 4, pp. 195–207 in [14]. Here the zeroes are all three-dimensional Fermi energies for quantum well layers! However these zeroes are in the energy band gap with no electrons occu-pied when the interfaces are concerned, say at the¡-point. Further this assignment is not good for electron density calculations in Thomas-Fermi model. Incidentally, if there are materials or heterostructures in which the Fermi energy is not in the gap for the statistics, e.g., we may apply theEF 3(1D)as our Fermi energy for the Fermi-Dirac statistics, then we can have much more carrier density for such a kind of “1D statistical heterostructures”, say holes, that we are looking for. The electron, or other carrier densities will be drastically changed.
(C) We assign the zero at the potential-energy-zero. For instance, in (30), the energy-zero
of E(sl)n coincides with the zero of V (z). Actually it is the kinetic energy zero in (B) above. However there are dimensionality problems [15]: e.g. in (22), E3D-zero is atV (z)-zero for 3D.
But E¤zn in (26), andEn(sl) in (30), respectively, have zeroes different from that ofV (z) for 1D when any of k1; k26= 0 in the other two dimensions, as shown in (31) for E3D, E¤zn, and E
(sl) n , if the kinetic part is not zero. Further, if the kinetic part is not zero, according to Ref. [26] for energy-levelling-matching, we may have energy-non-conservation problems. This is shown explicitly in (34) below. Another trouble is that for the coulombic potential» 1=r with zero at r =1. For solid calculations there is no such zero-at-infinity points.
(D) Other kinds of rather arbitrary assignments: e.g., in heterostructures we may have the
zeroes at the middle or at some point inside the band-gap between the conduction band and the valence band.
Here is a criticism for judging the property of these choices. In (35) and (36) below, we have to decide which one of E3D, E¤zn, and E(sl)n has the largest magnitude when they have the same zero reference level. These energies are determined by (26) and (30). On the contrary, from (31), if we choose the origin of E3D atEF(3D), and of E
(sl) n atEF 3(1D) E3D¡ EF(3D)= E (sl) n ¡ E(3D)F + ~ 2 2mz kZ(2D) 2+ ~ 2 2m(k 2 1+ k22) 6= En(sl)¡ E(1D)F3 + ~ 2 2mzk (2D) 2 Z + ~ 2 2m(k 2 1+ k22): (33) Here E3D¡ EF(3D)6= En(sl)¡ E (1D) F 3 ; (34)
because of the different reference levels on both sides. An energy-non-conservation result is given in Ref. [26]. An application of the change of energy absolute values of this kind also appeared in Ref. [26]. This is also correct for both positive or negative Ezn¤ . However for the cases: (a) Ezn¤ < 0 or (b) both of E3D < 0 and Ezn¤ < 0, we may have the opposite inequality as in (36) in comparison with (35). These are entirely dependent on the proper zero of the Schr¨odinger equations. Similar comparison can be made forEn(sl). When we treat the subband problem similar to the parabolic band cases, i.e. all energies are positive in (31), we can have
E3D> Ezn¤ >En(sl): (35)
On the other hand, when we consider (24), (26), and (30) in practical calculation cases: i.e. from the tight-binding or LCAO cases. All the energies are negative in (31), as
jE3Dj < jEzn¤ j < jEn(sl)j: (36)
A structural consideration with surface or interfaces is given in next section.
III. Why subband theory is not the 2D Bloch theory of electrons
From purely mathematical methods, we wonder if we can diagonalize (20) to the so-called “principal values”, i.e. for the kinetic energy in our set-up for the surface-cut described between
(18) and (22), i.e. in addition to (23) and (27) for our reduced-fcc in Fig. 3, w1z= wz1= w2z = wz2 = ¹w; K´ ¡ ~22(@)T(K)(@); (@)´ 0 B B @ @ @x1 @ @x2 @ @z 1 C C A ; (@)T ´ transpose of (@); (K)´ 0 B B @ 1 m¤ 0 w¹ 0 m1¤ w¹ ¹ w w¹ m1z 1 C C A ; K´ ¡ ~ 2 2 (@) TSS¡ 1(K)SS¡ 1(@); S¡ 1(K)S = 0 @ a01 a02 00 0 0 a3 1 A ´ A = diagonal; (37)
by standard mathematical methods. The similarity transformation matrixS is to be found by the secular equation of(K), viz.,
j(K) ¡ a ¢1j = 0; S ´ (S1S2 S3): (38)
We find the three eigenvalues ofa, a1= a+; a2 = a¡; a3= 1 m¤; a§ = 1 2 · 1 mz + 1 m¤ ¸ § 12 s· 1 mz ¡ 1 m¤ ¸2 + 8 ¹w2; (39)
and their eigenvectors
S1= C+ 0 @ 11 A+ 1 A ; S2= C¡ 0 @ 11 A¡ 1 A ; S3= 0 B B @ 1 p 2 ¡ p1 2 0 1 C C A ; A§ = 2 ¹w µ 1 mz ¡ a§ ¶¡ 1 ; C§ = q 1 2 + A2 § : (40)
We can have orthonormality of the vectorsS1,S2, S3, since(K) is symmetric, ST i Si = 1; i = 1; 2; 3; or C§2(2 + A2§) = 1; ST i Sj6=i = 0; or 2 + A+A¡ = 0; etc: (41)
From the mathematics of symmetric matrices, we have that
S¡ 1= ST = 0 @ S T 1 ST 2 S3T 1 A = 0 @ C+ C+ C+A+ C¡ C¡ C¡A¡ 1 p 2 ¡ 1 p 2 0 1 A : (42)
In (37), the matrixK includes
S¡ 1(@) = 0 B B @ @ @x0 1 @ @x02 @ @x0 3 1 C C A = 0 B B @ C+@x@1 + C+@x@2 + C+A+@z@ C¡ @x@1 + C¡ @x@2 + C¡A¡ @z@ 1 p 2 @ @x1 ¡ 1 p 2 @ @x2 1 C C A etc: (43)
If we want (K) to be diagonalized, we must have the transformation 0 @ x 0 1 x02 x03 1 A = S¡ 1 0 @ xx12 z 1 A = 0 @ C+x1+ C+x2+ C+A+z C¡x1+ C¡x2+ C¡A¡ z 1 p 2x1¡ 1 p 2x2 1 A : (44)
This transformation makes the kinetic energy in (37), in ‘principal values’, to become
K = ¡ ~22 3 X i=1 ai @2 @x02 i : (45)
It is our conception that the form of the kinetic energy in (35) is the ideal one for our ‘3D perfect
crystal’ in which we can have the periodic condition on the potential energy
¹
V (x01; x02; x03) = ¹V (x01+ d1; x02+ d2; x03+ d3) (45a) for periodicitiesdi. But it also makes the equation of the surface or interface plane,
z = 0; (46) to become z = 0 @S 0 @ x 0 1 x02 x03 1 A 1 A z = C+A+x01+ C¡A¡x02= 0; (47)
which is no longer the plane
For (46), z = 0 is one equation in 3D-space together with one compatible periodicity condition (19); it represents a 2D-surface. Furthermore for most physics cases, we have considered that the first equalityz = 0 of (47) is static, without any time-dependence. Whereas the second equality
C+A+x01(t) + C¡A¡ x02(t) = 0; (47a)
should be “stationary”, i.e. it is time-dependent just like the x; p in the Hamiltonian H(x; p). Either Newton’s equation or Schr¨odinger equation shows thatx = x(t)» x0+ v0t + at2=2 say, or < x > = time-varying in Heisenberg picture, etc. Therefore the x0i(t) here are dynamical variables, this is particularly clear from Fig. 3,x0i(t) are the coordinate (sites) of lattice points! It is a necessary consideration that x0i(t) is needed to consider the degrees of freedom in motions, particularly in kinetics or lattice dynamics.
As usual, one equation in a 3D space represents a 2-dimensional surface. However from the physics and the mathematics for dynamical variables in (47a), this 2D surface is not necessarily 2D. For example, (46)z = 0, a constant always, so that we can have x; y varying arbitrary on the z = 0 plane. We have left two degrees of freedom as free variables. After transforming to (47a), for x01, x02, we can have two choices: (a) 2D, because there is only one equation, always like static, i.e. (46) in 3D space; or
(b) two variables or degrees of freedom ‘used’ so that we can have only one degree of freedom “totally free”. Nevertheless, for the two degrees of freedom used in (b) here, we must judge whether it is:
(b-1) x01; x02 determined only by one linear independency in total. However there is no direct relationship to determine the dimensionality from the linear independency. There may be other non-linear independencies in some other theories (such as our periodicity-split rule in (Rule 2) below), which may lead to making another decision as to the dimensionalities of the spaces.
(b-2) x0
1; x02 used to determine two linearly independent degrees of freedom. From (49) – (49c), if our judgement is to choose the ‘peridicity-split’rule as a decision for linear independency, so thatx01; x02are two linearly independent variables in two directions. This leads us to have two degrees of freedom for the determination of one equation (47a). Totally we have thatx01; x02 are two independent degrees of freedom plus a two-dimensional planez=0 in (46). By an intuitive judgement, we can have four dimensions in total. At the least, we can judge the correctness of a discrete primitive translational space for the four degrees of freedom.
The linear independency is described in the Introduction as being determined by the Gram determinant and is not a necessary condition; we can have non-linear independencies! In fact in Fig. 3, we have x01 and x02, two independent directions provided for a periodicity condition. This configuration is different from current mathematical thoughts: 3D-space is ‘spanned’ by three perpendicular axes, or a 2D-plane plus az-direction perpendicular to the plane. The investigations cease usually at the point that the x01 and x02 directions are not (both) perpendicular to the 2D surface (cut-surface). In a discrete lattice primitive translation space, do they form a 4D space? They are related to the homogeneity-dimensions somehow.
As to the dimensionality investigation, in a Bravais lattice, we can pick the line of the periodic condition along the coordinate axis, explicitly, if there is a condition
V (x0I) = V (x0I+ aI); for various I
then we can assignx0I as an axis. From the existence of axes in this way, by the definition above, we can have one set of basis-vectors corresponding to the above assigned axes. This is what we