國 立 交 通 大 學
生 物 資 訊 及 系 統 生 物 研 究 所
博 士 論 文
預測T細胞後天免疫反應
Prediction of adaptive T-cell immune response
研究生:童俊維
預測 T 細胞後天免疫反應
Prediction of adaptive T-cell immune response
研 究 生:童俊維 Student:Chun-Wei Tung
指導教授:何信瑩 Advisor:Shinn-Ying Ho
國 立 交 通 大 學
生 物 資 訊 及 系 統 生 物 研 究 所
博 士 論 文
A ThesisSubmitted to Institute of Bioinformatics and Systems Biology College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Doctor of Philosophy
in
Bioinformatics and Systems Biology
July 2010
Hsinchu, Taiwan, Republic of China
預測 T 細胞後天免疫反應
學生:童俊維
指導教授:何信瑩 教授
國立交通大學生物資訊及系統生物研究所博士班
摘 要
發展電腦輔助疫苗設計系統能幫助免疫學家能快速有效的辨識候選疫苗, 並且是免疫資訊學的終極目標之一。而精準的預測T 細胞後天免疫反應是發 展電腦輔助疫苗設計系統的關鍵。本研究之核心為發展能適用於探勘致免疫 路徑(immunogenic pathways)中各種反應之重要物化特性的高性能大量參數 最佳化演算法。此重要物理化學特性探勘系統之研發過程包含了三個重要的 步驟:(1)蒐集各種能夠有效解釋生物現象之物理化學特性;(2)結合生物 知識與演算法技巧來建立最佳化問題;(3)發展特定的高性能演算法來解決 最佳化設計問題。重要特徵探勘系統可從大量資訊中探勘重要特徵來解釋各 種免疫反應,並幫助建立T 細胞後天免疫反應預測系統。 T 細胞後天免疫反應包括有細胞毒性與輔助 T 細胞免疫反應。對於預測 T 細胞後天免疫反應,過去的研究多專注於建立主要組織相容性複合物(MHC) 第一及第二型分子的抗原處理與表現路徑之預測模型。然而被主要組織相容 性複合物結合的胜肽抗原並不一定能引起免疫反應。對於更複雜的T 細胞免 疫反應需要有更深入的研究並建立其預測模型。另外,對於抗原表現有重要 影響的蛋白質泛素化(Ubiquitylation),至今仍未有預測模型。高度泛素化的 蛋白因較容易被裂解,因而容易產生可供T 細胞辨認用的抗原。因此準確的 蛋白質泛素化預測將有助於辨識容易引起免疫反應的蛋白質抗原。 本研究專注在研究抗原的內生性物化特性,研發出第一套使用物化特性 來預測與主要組織相容性複合物結合之蛋白質引起的T 細胞免疫反應預測系 統POPI 與泛素化預測系統 UbiPred。並發現過去普遍認同的抗原與主要組織 相容性複合物的結合親和力並不足以準確預測T 細胞免疫反應。針對影響細 胞毒性與輔助T 細胞免疫反應的重要物化特性之分析比較對於了解免疫反應 有極大助益。本研究接著提出基於字串核函數的細胞毒性T 細胞免疫反應預 測模型 POPISK。藉由融入主要組織相容性複合物與胺基酸位置的資訊,POPISK 不僅能加強細胞毒性 T 細胞免疫反應之預測,同時也能準確預測由 單一胺基酸突變引起的免疫反應變化。本研究並利用POPISK 之特性來研究 蛋白抗原上被T 細胞辨認的重要位置。本篇研究結果將能幫助了解免疫系統 並加速新疫苗的發展。
Prediction of adaptive T-cell immune response
Student: Chun-Wei Tung Advisor: Shinn-Ying Ho
Institute of Bioinformatics and Systems Biology
National Chiao Tung University
Abstract
The development of computer-aided vaccine design systems is a goal of immunoinformatics that can largely accelerate the design of vaccines. Accurate prediction of adaptive T-cell immune response is the critical step to develop computer-aided vaccine design systems. The core of this study is to develop high-performance optimization algorithms for solving large-scale parameter optimization problems of bioinformatics to mine informative physicochemical properties from known experimental data for predicting immunogenic pathway. The development of these algorithms involves three major phases: (a) collection of physicochemical properties for encoding peptide sequences; (b) formulation of optimization problems using domain knowledge and computing techniques and, and (c) development of efficient optimization algorithms for solving optimization problems. The developed informative feature mining algorithms can be used to mine informative physicochemical properties for predicting peptide immunogenicity.
There are two major T cells including cytotoxic and helper T cells. For the prediction of adaptive T-cell immune response, previous studies mainly focused on modeling antigen processing and presentation pathways of MHC class I and II. However, the prediction of T-cell response is much harder and less addressed because of the complex nature of T-cell response. Moreover, because over-ubiquitylated protein correlated with its half life, ubiquitylation plays an
important role in providing antigen sources. Accurate prediction of ubiquitylation sites is helpful to identify immunogenic peptides.
This study proposed the first prediction systems POPI and UbiPred for predicting T-cell response and ubiquitylation sites, respectively. The poor performance of a well recognized affinity-based method shows that binding affinity only is not sufficient for predicting T-cell response. The informative physicochemical properties for cytotoxic and helper T cells are identified and analyzed. Subsequently, an improved prediction system POPISK is proposed to predict cytotoxic T-cell response. The POPISK prediction system incorporating MHC allele information is used to identify important positions for T-cell recognition, and can predict immunogenicity changes made by single residue modifications. This study yields insights into the mechanism of immune response and can accelerate the development of vaccines.
Keywords: immunogenic pathway; physicochemical properties; intelligent genetic
Acknowledgement
First of all, I would like to thank my supervisor, Prof. Shinn-Ying Ho, whose guid-ance and support enabled me to pursue this interesting research. He is the principal investigator of Intelligent Computing Lab (ICLAB). Without his profound know-ledge in research experience, it is impossible for me to finish this dissertation. His patience and kindness are greatly appreciated. I learned so much from him in every aspect, especially in the presentation of research ideas and professional ethics.
I am very grateful to my supervisor, Prof. Oliver Kohlbacher, during the time of my research visits to Germany. He showed me different ways to rethink a prob-lem. He always encouraged me and supported me to pursue my research interests. I greatly enjoyed our discussions and learned a lot from his valuable guidance and open mind.
Besides my supervisors, I would like to show my gratitude to my dissertation committees, Prof. Jenn-Kang Hwang, Prof. Jinn-Moon Yang, Prof. Hsien-Da Huang, Prof. Hsueh-Fen Juan and Prof. Hsuan-Cheng Huang for giving me valuable sugges-tions and criticisms.
Furthermore, I would like to thank the National Science Council (NSC) of Taiwan and the German Academic Exchange Service (DAAD) for supporting the Scholarship of Sandwich Program for research visits to Germany (97-2911-I-009-016-2).
Prof. Hui-Ling Huag, Kuan-Wei Chen, Shiou-Bang Chang, Jhong-Chi Guo, Chih-Guo Yeh, Yi-Hsiung Chen, Kai-Ti Hsu, Chia-Ta Tsai, Kuo-Ching Kao, Yu-Hsiang Lin, Chiung-Hui Hung, Yi-Jer Lin, Te-Fen Kao, Yin-Yin Yu, Pin-Chen Lion, Chih-Hung Hsieh, Chia-Yun Chang, and Fu-Chieh Yu for nice collaborations and discussions.
For giving me enthusiastic support in Germany, I am very grateful to Andreas Bertsch, Sebastian Briesemeister, Magdalena Feldhahn, Nina Fischer, Sandra Gesing, Andreas Kaemper, Erhan Kenar, Sven Nahnsen, Lars Nilse, Marc Roettig, Marc Sturm, Nico, Pfeifer, Nora Toussaint, Matthias Ziehm, Jan Schulze, and Claudia Wal-ter. There is so much to thank them for their valuable research and daily life sup-ports.
I owe my deepest gratitude to my Sandwich Program partners Shu-Hsien Li, Chun-Chi Lee, Tung-Hui Hsu, Chih-Chun Hsieh, Ning-Ying Wang, Win-Bin Huang, Tzai-Tang Tsai, Yu-Jen Huang, Hsien-Yi Hsiao, Hsiao-Ching Li, Hui-Yi Shiao, Guang-Yin Chen, Hao-Peng Lin for their daily life support in Germany.
Last, but no least, I am deeply grateful to my parents for their unconditional support and encouragement to pursue my interests.
Table of Contents
摘要 ... i
Abstract ... iii
Acknowledgement ... vi
Table of Contents ...viii
List of Figures ...xi
List of Tables ... xiv
List of Abbreviations ... xvi
Chapter 1 Introduction ... 1
1.1 Motivation ... 1
1.2 Overview of the research ... 4
1.3 Organization ... 7
Chapter 2 Related Work ... 9
2.1 Highly Ubiquitylated Proteins as Antigen Sources ... 9
2.2 Immunogenic Pathway of MHC class I ... 10
2.3 Immunogenic Pathway of MHC class II ... 11
Chapter 3 Informative Physicochemical Property Mining Algorithm ... 13
3.1 Physicochemical properties ... 13
3.2 Support vector machines ... 14
3.3 Orthogonal experimental design ... 15
3.5 Performance evaluations ... 19
Chapter 4 Prediction of ubiquitylation sites ... 21
4.1 Motivation ... 21
4.2 Assessment of features and classifiers ... 22
4.3 Informative physicochemical properties ... 27
4.4 Prediction system UbiPred ... 30
4.5 Knowledge of data mining ... 33
4.6 Screening promising ubiquitylation sites ... 37
4.7 Follow-up works ... 38
4.8 Summary ... 39
Chapter 5 Predicting immunogenicity of MHC binding peptides... 40
5.1 Motivations ... 40
5.2 The proposed prediction systems ... 42
5.3 POPI for predicting immunogenicity of MHC class I binding peptides ... 44
5.4 POPI-MHC2 for predicting immunogenicity of MHC class II binding peptides ... 47
5.5 Analysis of informative physicochemical properties ... 48
5.6 Comparison of physicochemical properties responsible for CTL and HTL responses ... 51
5.7 Peptides capable of inducing both CTL and HTL responses ... 53
5.8 Independent test performance of POPI-MHC2 ... 53
5.9 Follow-up works ... 55
5.10 Summary... 55
Chapter 6 Identification of T-cell receptor recognition sites ... 57
6.1 Motivation ... 57
6.2 Datasets ... 59
6.4 Prediction of peptide immunogenicity ... 61
6.5 Comparison to POPI... 61
6.6 Identification of important positions for immunogenicity ... 63
6.7 Analysis of physicochemical properties... 66
6.8 POPISK ... 67
6.9 Prediction and analysis using POPISK ... 67
6.10 Summary... 69 Chapter 7 Conclusions ... 71 7.1 Summary ... 71 7.2 Future works ... 72 Reference ... 74 Curriculum Vitae ... 86 Publications ... 88
List of Figures
Figure 1.1 Comparison between conventional vaccine development and reverse
vaccinology [7]. ... 2
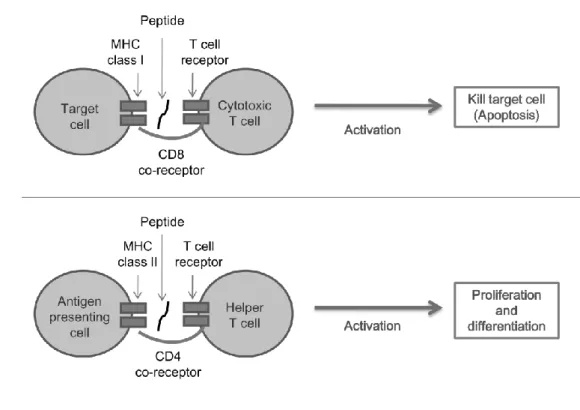
Figure 1.2 A simple illustration of helper and cytotoxic T cell responses. ... 4 Figure 1.3 The immunogenic pathways of MHC class I and II. ... 5 Figure 1.4 The architecture of proposed peptide immunogenicity prediction system
for computer-aided vaccine design. ... 6
Figure 3.1 An illustration example of fitness function evaluation from decoding a
GA-chromosome. ... 18
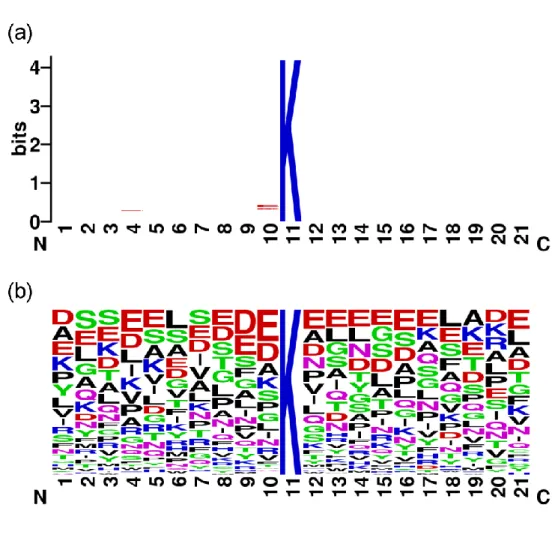
Figure 4.1 The sequence logo of the 151 positive samples with w=21. (a)
information content and (b) frequency plot. ... 22
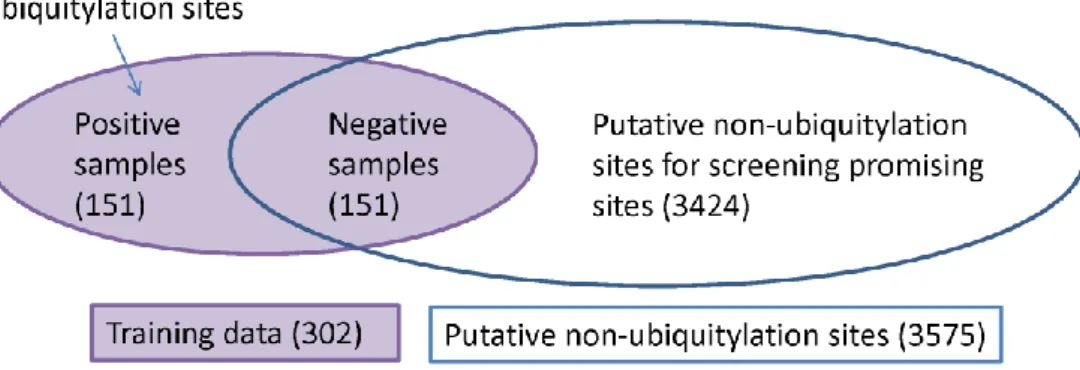
Figure 4.2 The schema for the training and an independent of 3424 putative
non-ubiquitylation sites in dataset of w=21. ... 23
Figure 4.3 Performance comparisons among amino acid identity, evolutionary
information and physicochemical property with various classifiers. ... 24
Figure 4.4 Performance comparisons between the SVM with informative
physicochemical properties (SVM+IPCP) and other compared classifiers. ... 26
Figure 4.5 The best 10-CV accuracies of prediction using SVM with the window
size 21 for various numbers of features (properties) selected by IPMA from 30 independent runs. ... 28
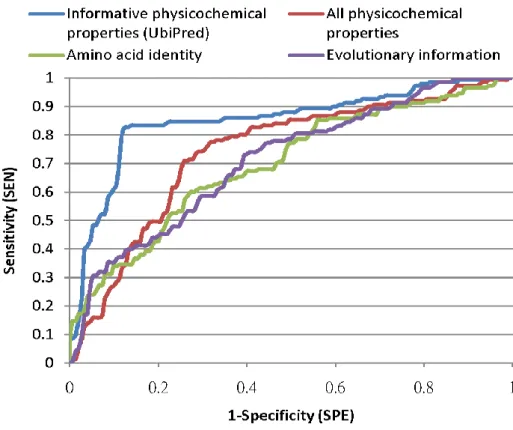
Figure 4.6 The system flow of prediction system UbiPred. ... 29 Figure 4.7 Comparison of receiver operating characteristic curves among
informative physicochemical properties (UbiPred), amino acid identity,
evolutionary information and all physicochemical properties. ... 30
Figure 4.8 The derived decision tree by using C5.0 and the features of informative
physicochemical properties for classification of ubiquitylation sites. ... 34
Figure 4.9 Histogram result of UbiPred using prediction scores from evaluating
3424 putative non-ubiquitylation sites in an independent dataset. The site with a score close to 1 has a high possibility to be an ubiquitylation site. ... 35
Figure 4.10 The sequence logo of the 23 peptides of promising ubiquitylation sites
with w=21. (a) Information content and (b) Frequency plot. ... 36
Figure 5.1 Averaged accuracies (AAs) of 10-CV for IPMA, rank-based methods
(RankD and RankI) and the alignment-based method (ALIGN) for MHC class I binding peptides. ... 43
Figure 5.2 Averaged accuracies (AAs) of 10-CV for IPMA, rank-based methods
(RankD and RankI) and the alignment-based method (ALIGN) for MHC class II binding peptides. ... 47
Figure 5.3 Pie-chart representations of compositions of categorized
physicochemical properties of peptides responsible for CTL and HTL
responses. ... 52
Figure 6.1 Comparison of nested 10-CV performances of POPISK and
POPI-modified and POPI-IPMA. ... 59
Figure 6.2 The decrease in MCC performances evaluated on datasets without using
residues in specific positions. ... 62
Figure 6.3 Two Sample Logo representation of over- (upper half) and
underrepresented (lower half) residues in immunogenic peptides ... 64
Figure 6.4 The over- (upper half) and underrepresented (lower half)
position-specific properties in immunogenic peptides. (A) Hydrophobicity. (B) Normalized van der Waals volume. The symbols S, M and L indicate residues
with small, medium and large hydrophobicity/volume, respectively. ... 65
Figure 6.5 Structures of PDB IDs 1ao7 and 1qrn. Structures of PDB IDs 1ao7 and
1qrn share high structural similarity presenting complexes of
List of Tables
Table 3.1 An illustration example of orthogonal array L8(27) and factor analysis. ... 16
Table 4.1 Summary of used parameters and LOOCV performances of the methods
using informative physicochemical properties (UbiPred), amino acid identity, evolutionary information, and all physicochemical properties. ... 25
Table 4.2 The MEDs for 31 mined physicochemical property. ... 31 Table 4.3 The LOOCV performances of the SVM with 31 informative
physicochemical properties on datasets of various sequence identity thresholds ... 32
Table 4.4 Five concise if-then rules with confidence larger than 0.5 obtained by
using C5.0 and 31 informative physicochemical properties ... 33
Table 4.5 List of 23 promising ubiquitylation sites identified from an independent
dataset of 3424 putative non-ubiquitylation sites ... 37
Table 5.1 The dataset PEPMHCI and PEPMHCII of peptides associated with
human MHC class I and II molecules ... 42
Table 5.2 Performance comparisons of ALIGN, PSI-BLAST and POPI using
LOOCV on the whole dataset PEPMHCI ... 44
Table 5.3 Performance comparisons between AFFIPRE and POPI ... 44 Table 5.4 Performance comparisons of ALIGN, PSI-BLAST and POPI-MHC2. .. 46 Table 5.5 Performance comparison between AFFIPRE and POPI-MHC2. ... 46 Table 5.6 Individual effects of identified properties for CTL responses in terms of
main effect difference (MED) ... 49
Table 5.7 Individual effects of identified properties for HTL responses in terms of
main effect difference (MED) ... 50
Table 4.8 Predicted levels of peptides to induce both CTL and HTL responses ... 54 Table 6.1 Physicochemical properties with feature usage larger than 50% ... 66
List of Abbreviations
AUC Area under Receiver Operating Characteristic Curve APC Antigen Presenting Cell
ER Endoplasmic Reticulum GA Genetic Algorithm
DNA Deoxyribonucleotide Acid CTL Cytotoxic T Lymphocytes HTL Helper T Lymphocytes HLA Human Leukocyte Antigen
IBCGA Inheritable Bi-objective Genetic Algorithm
IPMA Informative Physicochemical Property Mining Algorithm MHC Major Histocompatibility Complex
MED Main Effect Difference SVM Support Vector Machine
Chapter 1
Introduction
This study aims to develop computer-aided vaccine design systems to help the design of vaccines. For peptide-based vaccine design, the most critical step is to select im-munogenic peptides capable of inducing immune responses. Therefore, accurate prediction of adaptive T-cell immune response can greatly accelerate vaccine designs. For this propose, efficient optimization algorithms were developed and applied to mine informative features for the predictions of adaptive T-cell immune response. This dissertation presents a comprehensive study of the developed prediction sys-tems for identifying peptide vaccine candidates.
1.1 Motivation
The most significant advance of medicine is the utilization of vaccines against dis-eases. Vaccines can help to prevent infections and prolong peoples‟ life. There are mainly five types of vaccines: live attenuated vaccines, killed vaccines, purified sub-unit vaccines, recombinant subsub-unit vaccines and gene-based vaccines [1-4].The live attenuated vaccines consist of the pathogens with reduced toxicity to prevent the risk of infections. However, it is hard to develop live attenuated vaccines with high safety. The killed vaccines consist of killed or deactivated pathogens have higher safety, compared to live attenuated vaccines.
The killed vaccines suffered from incapability of replication result in low im-munogenicity. The deficiency of these two types of vaccines is caused by using whole pathogens as vaccines that will dilute the immunogenicity of vaccines. Thus, the subunit vaccines using identified protective antigens are safer and more efficient to focus the immune response on specific target. For preventing or curing cancers,
Figure 1.1 Comparison between conventional vaccine development and reverse
the traditional vaccines described above are less effective. By applying deoxyribonuc-leotide acid (DNA) as vaccines, it can provide effective protections against cancers with high immunogenicity of cytotoxic T lymphocyte (CTL) [1].
For developing subunit vaccines and DNA vaccines, traditional experiment methods to identify protective antigens cost a lot and is time consuming with often five to fifteen years duration. The growing needs of identifying protective antigen to develop vaccines result in the emergence of reverse vaccinology (shown in Figure1.1). Vaccine design using bioinformatics methods can largely reduce the cost of time and money [2-4].
Peptide immunogenicity, its ability to induce immune responses, determines the effectiveness of vaccines. T-cell activation, one kind of immune responses, plays important roles in developing adaptive immunity. An immunogenic peptide should be processed and presented to a cell surface by antigen processing and presentation pathway, and then induce T-cell responses. Major histocompatibility complex (MHC) molecules are responsible for both recognition of antigens and presentation of anti-gens to T cells. MHC class I molecules can present processed endogenous peptides of antigen to cytotoxic T lymphocytes (CTL), while processed exogenous peptides of antigen are presented to helper T lymphocytes (HTL) by MHC class II molecules.
Immune responses will be triggered when CTL or HTL recognize immunogen-ic antigens. CTL response is mainly characterized by killing target cells (e.g. tumor cell and infected cell), presenting immunogenic antigens by the activated CTL. In contrast, the activated HTL will induce resting HTLs to proliferate and differentiate into memory cells or effector cells, and provide specific help for CTL, B lymphocytes and phagocytic cells, as known as HTL response. A simple illustration of helper and cytotoxic T cell responses is shown in Figure 1.2.
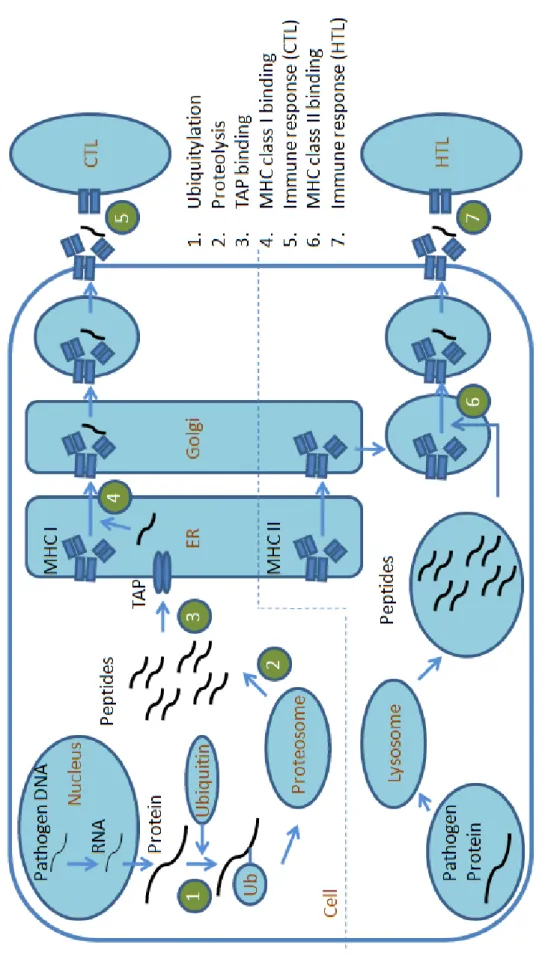
For computer-based vaccine design, previous studies pocus on modeling anti-gen processing and presentation pathways of MHC class I and II. The works for modeling the pathway of MHC class I (shown in reaction 2-4 of Figure1.3) include predictions of antigen proteasomal cleavage sites, binding affinities of peptides and the transporter associated with antigen processing (TAP) and binding affinities of peptides and MHC class I molecules. The major work for modeling the pathway of MHC class II (shown in reaction 6 of Figure1.3) is the prediction of binding affini-ties between peptides and MHC class II molecules.
The above studies assume that peptide binding affinity to MHC molecules cor-relates with its immunogenicity. The prediction problems of CTL and HTL immune responses are rarely studied (reaction 5 and 7 of Figure1.3). However, recent studies
show that peptide binding affinity to MHC molecules is required but not strongly correlated to the strength of immunogenicity. Also, the prediction of protein tylation sites is crucial for the prediction of peptide immunogenicity because ubiqui-tylation plays key roles in antigen supply (reaction 1 of Figure1.3). The investigation of these problems is necessary for accurate prediction of adaptive T-cell immune response and development of computer-aided vaccine design systems.
1.2 Overview of the research
This dissertation presents a comprehensive prediction system for computer-aided vaccine design whose architecture is shown in Figure 1.4. The proposed system is based on several state-of-the-art methods for predicting antigen processing and presentation pathways and newly developed prediction systems for T-cell responses and protein ubiquitylation. To develop prediction systems for T-cell responses and protein ubiquitylation, An informative physicochemical property mining algorithm is proposed to mine informative physicochemical properties for predicting reactions 1, 5 and 7 in this system that are described as follows.
F igu re 1 .3 T he i mm unog eni c pa th w ays of MHC c la ss I a nd I I.
1) (reaction 5 in Figure 1.3) The prediction of immunogenicity of MHC
class I binding peptides (CTL response) is important to understand immune systems and accelerate vaccine design. Previous studies show that moderate binding affinity of peptides to MHC molecules is required but is not the de-terministic factor. Because of the complex effects of intrinsic factors like physicochemical properties and extrinsic factors of MHC repertoire, it is hard to predict immunogenicity. For solving this problem, an informative physicochemical property mining algorithm was proposed to simultaneously mine informative physicochemical properties from existing experimental da-ta and design a support vector machine classifier. By mining a subset of 23 informative physicochemical properties from 531 physicochemical proper-ties, a prediction system of POPI was constructed. POPI performs better than alignment-based methods and traditional affinity-based methods. For
Figure 1.4 The architecture of proposed peptide immunogenicity prediction
HLA-A2-restricted peptides, an improved prediction system POPISK is proposed for predicting immunogenicity using string kernels. The prediction and analysis ability of POPISK gives insights into the underlying mechanism of T-cell recognition. Important positions for T-cell responses are identified and analyzed. POPISK can accurately predict immunogenicity changes made by single residue modifications.
2) (reaction 7 in Figure 1.3) For the prediction of immunogenicity of MHC
class II binding peptides (HTL response), the developed informative physi-cochemical property mining algorithm was applied to mine informative phy-sicochemical properties from experimental data. A prediction system PO-PI-MHC2 for predicting immunogenicity of MHC class II binding peptides was implemented by using 21 informative physicochemical properties. The same as POPI, POPI-MHC2 performs much better than alignment-based methods and traditional affinity-based methods.
3) (reaction 1 in Figure 1.3) Three kinds of features were assessed for their
performances of ubiquitylation site prediction. For classifier selection, three classifiers including k-nearest neighbor classifier, NaïveBayes and support vector machines (SVM) were assessed. Results show that SVM using physi-cochemical properties performs best. Moreover, the informative physico-chemical property mining algorithm was applied to mine 31 informative physicochemical properties from all 531 physicochemical properties. A pre-diction system UbiPred constructed by using 31 informative physicochemi-cal properties shows large improvement in prediction performance.
This dissertation presents the first prediction systems for CTL and HTL res-ponses and protein ubiquitylation. The obtained informative physicochemical prop-erties yield insights into the immune systems and are helpful to develop prediction systems for vaccine designs.
1.3 Organization
In summary, this dissertation focuses on predicting adaptive T-cell immune responses for computer-aided vaccine design. For solving optimization problems of mining informative physicochemical properties for the peptide immunogenicity, efficient evolutionary algorithms were proposed to develop efficient vaccine design system. The rest of this dissertation is organized as follows. Chapter 2 addresses the related works of this dissertation. The proposed algorithm for mining informative
physico-chemical properties is presented in Chapter 3. Chapter 4 presents the prediction sys-tem for predicting ubiquitylation sites. Chapter 5 describes the proposed prediction systems for predicting immunogenicity of MHC class I and II binding peptides. The improved prediction of peptide immunogenicity using string kernels is presented in Chapter 6. Finally, conclusions are given in Chapter 7.
Chapter 2
Related Work
This chapter presents related works for predicting adaptive T-cell immune response including prediction of ubiquitylation sites, immunogenicity prediction of MHC class I binding peptides, and immunogenicity prediction of MHC class II binding pep-tides.
2.1 Highly Ubiquitylated Proteins as Antigen
Sources
Ubiquitin-proteasome system is an important mechanism for protein degradation that the ubiquitylated proteins will be degraded by proteasome. The ubiquitin acts as a specific tag for marking proteins for degradation. The proteasome is a major me-chanism for cells to regulate the concentration of particular proteins and degrade misfolded proteins. The degradation process produces short peptides of about 7~8 amino acids. The resulting short peptides can be further degraded into amino acids that can be used in protein synthesis [2, 3].
The proteasome plays an important role in the function of the adaptive im-mune system. The peptide antigens presented on the surface of antigen-presenting cells are produced by proteasomal degradation of pathogen proteins and displayed by MHC class I molecules [4]. A previous study investigated the role of ubiqui-tin-dependent proteolytic pathway in MHC class I-restricted antigen presentation and concluded that ubiquitin-conjugation (also called ubiquitylation) plays an important role in the presentation of a cytosolic antigen with MHC [5]. Another study found that an amino-terminal modification of a viral protein will promote ubiqui-tin-dependent degradation and lead to the enhancement of presentation with MHC
class I [6].
Some recent studies have similar results that ubiquitin-conjugation will enhance the efficacy of polynucleotide viral vaccines [7] and vaccines against tuberculosis [8]. Another study claimed that the low frequency of memory cytotoxic T lymphocyte and inefficient antiviral protection of DNA immunization with minigenes can be rectified by ubiquitylation [9]. Therefore, accurate prediction of ubiquitylation sites can provide better understandings of ubiquitylation mechanism. The selection of highly ubiquitylated peptides can improve the effectiveness of vaccines. In Chapter 4, three kinds of features and three classifiers were assessed for their prediction per-formances. Subsequently, informative physicochemical property mining algorithm is applied to select informative physicochemical properties and improve the prediction performance. Finally, a prediction system UbiPred was constructed to predict ubi-quitylation sites.
2.2 Immunogenic Pathway of MHC class I
Developing a computer-aided system to design peptide vaccines is one goal of im-munoinformatics. The major work of previous studies for peptide vaccine designs is to identify cytotoxic T lymphocyte (CTL) epitopes and investigate their correspond-ing immunogenicity. The CTL cells play a critical role in protective immunity by re-cognizing and eliminating self-altered cells, which recognize short peptides derived from intracellular degradation of foreign proteins in combination with major histo-compatibility complex (MHC) class I molecules. The immunogenicity of MHC class I binding peptides is their ability to induce CTL responses. Accurate predictions of the CTL epitopes and their corresponding immunogenicity are critical in developing a computer-aided system for vaccine designs.
Direct approach to predicting the CTL epitopes has been studied initially but its accuracy is fairly low [10]. Instead, indirect approach to predicting the MHC-binding peptides is useful because peptides must be processed prior to inducing cellular im-munogenicity. The recent studies of bioinformatics utilized the information about antigen processing pathway to predict the CTL epitopes. At first, the peptides are cleaved by proteasomal cleavage. Several studies elucidating the specificity of pro-teasome have been presented. To predict proteasomal cleavage sites, NetChop used a neural network method [11] and Pcleavage is based on a support vector machine (SVM) learning model [12].
After cleavage, peptide fragments are transported into endoplasmic reticulum by TAP which is the transporter associated with antigen processing. Some studies of
investigating the TAP transport efficiency were presented such as the affinity predic-tion of TAP binding peptides using the cascade SVM [13] and the predicpredic-tion of TAP transport efficiency of epitope precursors using a simple scoring matrix [14]. Finally, the peptide fragments that bound to MHC class I molecules are subsequently trans-located to the cell surface, where these complexes may active CTL. Some methods have been developed to predict MHC class I binding affinity, such as the SVM-based SVMHC [15] and Gibbs sampling method [16]. Moreover, the hybrid approaches integrated the above-mentioned methods like the prediction of proteasomal cleavage, TAP transport efficiency and MHC binding to advance the prediction performance [17, 18].
The problem of predicting immunogenicity of MHC class I binding peptides is crucial to further identify highly immunogenic peptides. The selection of highly im-munogenic peptides can save many experimental efforts and accelerate the develop-ing progress. In Chapter 5, a prediction system POPI was developed to predict im-munogenicity of MHC class I binding peptides. POPI performs better than align-ment-based and affinity-based methods.
In Chapter 6, an improved prediction system POPISK was constructed to pre-dict T-cell responses induced by HLA-A2-restricted peptides. POPISK using string kernels is useful for predict peptide immunogenicity and immunogenicity changes made by single residue modifications that is especially useful for optimizing pep-tide-based vaccines.
2.3 Immunogenic Pathway of MHC class II
The immunogenic pathway of MHC class II includes four steps. First, antigens are engulfed by endocytosis forming endosome. Second, endosome fuses with lysosome and is cleaved by peptidase in lysosome. Third, the peptide fragments bound to MHC class II will be translocated to cell surface. Finally, immune responses (also called immunogenicity) will be triggered when helper T lymphocyte (HTL) recognize non-self antigens presented by antigen presenting cell (APC). The activated HTL will induce the resting HTLs to proliferate and differentiate into memory cells or effector cells and provide specific help to CTL, B lymphocytes and phagocytic cells [19, 20].
Previous studies for predicting immunogenic pathway of MHC class II focus on the prediction of MHC class II-restricted peptides (qualitative methods) and the binding affinity of peptide-MHC complex (quantitative methods). Many methods are proposed to predict MHC class II binding peptides. The evolutionary algorithms in-cluding ant colony algorithms [21], evolutionary algorithms combined with artificial
neural networks [22] and multi-objective evolutionary algorithms [23] are developed for optimizing a matrix for predicting binding affinity. Other methods including the neural network based methods [22, 24, 25], Bayesian neural networks [26], fuzzy neural networks [27], the hidden Markov model [28], Gibbs samplers [16], support vector machines [29-31] and alignment-based method SMM-align that is a stabiliza-tion matrix alignment method for predicting MHC class II binding affinity [32].
However, the problem of predicting immunogenicity of MHC class II binding peptides is also important to understand immunogenicity and design effective vac-cines. In Chapter 5, a prediction system POPI-MHC2 based on informative physi-cochemical properties was developed to predict immunogenicity of MHC class II binding peptides. The informative physicochemical properties are mined by using the informative physicochemical property mining algorithm (described in Chapter 3). This study shows similar results to POPI that the traditional affinity-based method and alignment-based methods are less effective than the proposed method PO-PI-MHC2.
Chapter 3
Informative Physicochemical
Property Mining Algorithm
For mining informative physicochemical properties from experimental data, a genetic algorithm based method was proposed to simultaneously determine optimal subset of physicochemical properties and design a support vector machine classifier.
3.1 Physicochemical properties
Physicochemical properties of amino acids were extensively and successfully used in sequence-based prediction methods [33-37]. There are 544 physicochemical proper-ties of amino acids extracted from amino acid index database version 9.0 (AAindex), which is a collection of published amino acid indices representing different physico-chemical and biological properties of amino acids [38, 39]. Each physicophysico-chemical property consists of a set of 20 numerical values for amino acids. The property hav-ing the value „NA‟ in a value set of amino acid index was discarded. Finally, 531 properties were used for the following mining method.
To encode an input vector from peptide sequences for machine learning clas-sifiers, a two-step method is utilized. The first step determines a vector Dt of 531 index values for each amino acid of peptides. A peptide of length l has 531
l-dimensional vectors that can be defined as:
1 2
Dt d dt , t ,...,dtl ,t1,...,531,
Informative Physicochemical
Property Mining Algorithm
where t denotes the t-th physicochemical properties. In the second step, a vector V of 531 mean values is obtained by averaging these l attributes in each vector, defined as follows:
1 2 V v v, ,...,vt , 1 1 l t ti i v d l
, (3-1) where tv is the averaged value of elements in Dt,.
3.2 Support vector machines
Support vector machines (SVMs) are powerful tools in the field of machine learning. SVMs cope well with the over-fitting problem arising from a small training dataset by finding a linear separation hyperplane that maximizes the distance between two classes to create a classifier. SVMs can efficiently deal with classification, prediction, and regression problems. Given training vectors xi Rn and their class values yi
{-1, 1}, i = 1, …, N, an SVM solves the following problem:
min T 1 1 2 N i i C
w w , subject to T ( ) 1 i i i y w x b , (3-2) 0 i ,where w is a normal vector perpendicular to the hyperplane and i are slack va-riables allowing for some misclassifications. The cost parameter C > 0 controls the trade-off between the margin and the training error. Larger values of C will lead to a higher error penalty.
In order to make linear separation of samples easier, SVM uses one of various kernel functions to transform the samples into a high-dimensional search space. In this work, the commonly-used radial basis function is applied to nonlinearly trans-form the feature space, defined as follows:
2
( ,i j) exp( i j ), 0
The kernel parameter γ determines how the samples are transformed into a high-dimensional search space. These two parameters C and γ must be tuned to get the best prediction performance.
For multi-class classification problems, „one-against-one‟ strategy is applied to transform the multi-class problem into several binary classification problems. Given
h classes, there are h(h−1)/2 classifiers constructed and each one trains the samples
from two classes. A voting strategy is applied to give a final prediction for test sam-ples. In this study, the used SVM is obtained from LIBSVM package version 2.81 [40].
3.3 Orthogonal experimental design
Statistic design of experiments is a process of planning experiments. Orthogonal experimental design with orthogonal array and factor analysis is an efficient method to analyze the effect of several factors simultaneously [41, 42]. The factors are the parameters, which affect response variables, and a discriminative value of a factor is regarded as a level of the factor. A “complete factorial” experiment would make measurements at each of all possible level combinations. However, the number of level combinations is often so large that this is impractical, and a subset of level combinations must be judiciously selected to be used, resulting in a “fractional fac-torial” experiment. Orthogonal experimental design utilizes properties of fractional factorial experiments to efficiently determine the best combination of factor levels to use in design problems.
Orthogonal array is a fractional factorial array, which assures a balanced com-parison of levels of any factor. Orthogonal array can reduce the number of level combinations for factor analysis. Each row of an orthogonal array represents the le-vels of factors in each combination, and each column represents a specific factor that can be changed from each combination. The term “main effect” of one factor de-signates the effect on response variables that one can trace to a design parameter, which does not bother the estimation of the main effect of another factor. After proper tabulation of experimental results, the summarized data are analyzed using factor analysis to determine the relative level effects of factors.
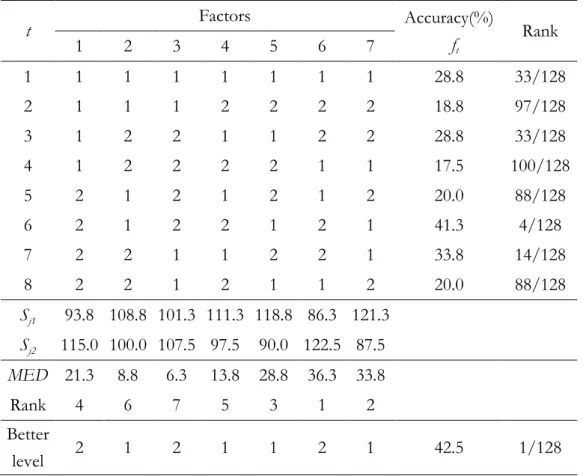
Factor analysis can evaluate the effects of individual factors on the evaluation function, rank the most effective factors, and determine the best level for each factor such that the evaluation function is optimized. Table 3.1 shows an illustrative exam-ple of orthogonal experimental design using a two-level orthogonal array LM(2M-1) with M rows and M-1 columns. In this example of M=8, there are 7 factors where
each corresponds to a physicochemical property and its two levels correspond to ex-clusion and inex-clusion of the feature in the proposed feature selection. Let ft denote a function value (prediction accuracy of 10-CV in this study) of the combination t. Define the main effect of factor j with level k as Sjk where j = 1, …, M-1 and k = 1, 2:
Sjk=
ft∙ Ft , t = 1, …, M, (3-4) where Ft = 1 if the level of factor j of combination t is k; otherwise, Ft = 0. Since the objective function is to be maximized, the level 1 of factor j makes a better contribu-tion to the funccontribu-tion than level 2 of factor j does when Sj1>Sj2. The main effect re-veals the individual effect of a factor. After the better one of two levels of each fac-tor is determined, a good combination consisting of all facfac-tors with the better levels can be easily reasoned [43].The Rank in Table 3.1 shows the rank of the combination t in all 128 (=27)
Table 3.1 An illustration example of orthogonal array L8(27) and factor analysis.
t Factors Accuracy(%) ft Rank 1 2 3 4 5 6 7 1 1 1 1 1 1 1 1 28.8 33/128 2 1 1 1 2 2 2 2 18.8 97/128 3 1 2 2 1 1 2 2 28.8 33/128 4 1 2 2 2 2 1 1 17.5 100/128 5 2 1 2 1 2 1 2 20.0 88/128 6 2 1 2 2 1 2 1 41.3 4/128 7 2 2 1 1 2 2 1 33.8 14/128 8 2 2 1 2 1 1 2 20.0 88/128 Sj1 93.8 108.8 101.3 111.3 118.8 86.3 121.3 Sj2 115.0 100.0 107.5 97.5 90.0 122.5 87.5 MED 21.3 8.8 6.3 13.8 28.8 36.3 33.8 Rank 4 6 7 5 3 1 2 Better level 2 1 2 1 1 2 1 42.5 1/128
possible combinations. In this example, the reasoned combination gets the best ac-curacy with Rank 1. Notably, the reasoned combination is not guaranteed to be the best one in general cases. The most effective factor j has the largest main effect dif-ference MED=|Sj1 –Sj2|. The 6th factor having the largest main effect difference 36.3 is the most effective factor.
3.4 Inheritable bi-objective genetic algorithm
Selecting a minimal number of informative features while maximizing prediction ac-curacy is a bi-objective 0/1 combinatorial optimization problem. An efficient inhe-ritable bi-objective genetic algorithm (IBCGA, [43]) is utilized to solve this optimiza-tion problem. IBCGA consists of an intelligent genetic algorithm [44] with an inhe-ritable mechanism. The intelligent genetic algorithm uses a divide-and-conquer strategy and an orthogonal array crossover to efficiently solve large-scale parameter optimization problems. In this study, the intelligent genetic algorithm can efficiently explore and exploit the search space of C(n, r). IBCGA can efficiently search the space of C(n, r1) by inheriting a good solution in the space of C(n, r) [43].
There-fore, IBCGA can economically obtain a complete set of high-quality solutions in a single run where r is specified in an interesting range such as [5, 45].
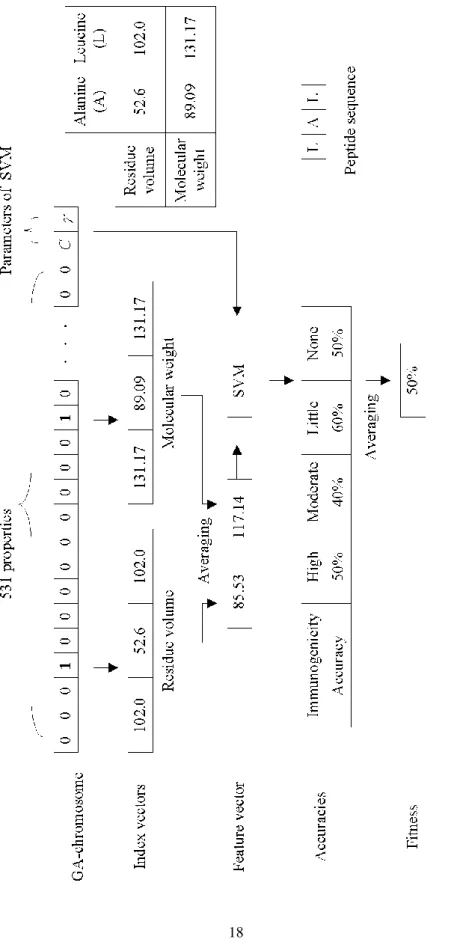
The proposed chromosome encoding scheme of IBCGA consists of both bi-nary genes for feature selection and parametric genes for tuning SVM parameters, where the gene and chromosome are commonly-used terms of genetic algorithm (GA), named GA-gene and GA-chromosome for discrimination in this paper. The GA-chromosome consists of n=531 binary GA-genes bi for selecting informative properties and two 4-bit GA-genes for tuning the parameters C and γ of SVM. If
bi=0, the i-th property is excluded from the SVM classifier; otherwise, the i-th prop-erty is included. This encoding method maps the 16 values of and C into {2-7, 2-6…,
28}. Figure 3.1 shows the encoding scheme of GA-chromosome and process of
constructing feature vectors for fitness function evaluation using a concise example. The feature vector for training the SVM classifier is obtained from decoding a GA-chromosome using the following steps. Consider a given peptide sequence, e.g., LAL. At first, the index vectors for all selected physicochemical properties (Residue volume and Molecular weight in this example) are constructed from AAindex for each amino acid. Feature vector of a peptide consists of the selected features whose values are obtained by averaging the values in their corresponding index vectors. Fi-nally, all values of the feature vectors are normalized into [-1, 1] for applying SVM.
F igu re 3.1 A n ill us tr ation e xa mple of f itnes s f uncti on ev alua tion from dec odi ng a GA -c hr omos ome .
avoid from the prediction bias for some classes, the averaged accuracies (AA) of all classes, defined in (3-10), is adopted as the fitness function. The performance of se-lected properties associated with the parameter values of SVM is measured by 10-CV. Therefore, the fitness value of a GA-chromosome is obtained by computing the mean accuracy of 10 runs.
IBCGA with the fitness function f(X) can simultaneously obtain a set of solu-tions, Xr, where r=rstart, rstart+1, …, rend in a single run. The algorithm of IBCGA
with the given values rstart and rend is described as follows:
Step 1) (Initiation) Randomly generate an initial population of Npop individuals. All the n binary GA-genes have r 1‟s and n-r 0‟s where r = rstart.
Step 2) (Evaluation) Evaluate the fitness values of all individuals using f(X). Step 3) (Selection) Use the traditional tournament selection that selects the
winner from two randomly selected individuals to form a mating pool.
Step 4) (Crossover) Selectpc·Npop parents from the mating pool to perform orthogonal array crossover on the selected pairs of parents where pc is the crossover probability.
Step 5) (Mutation) Apply the swap mutation operator to the randomly se-lected pm·Npop individuals in the new population where pm is the muta-tion probability. To prevent the best fitness value from deteriorating, mutation is not applied to the best individual.
Step 6) (Termination test) If the stopping condition for obtaining the solu-tion Xr is satisfied, output the best individual as Xr. Otherwise, go to
Step 2).
Step 7) (Inheritance) If r < rend, randomly change one bit in the binary GA-genes for each individual from 0 to 1; increase the number r by one, and go to Step 2). Otherwise, stop the algorithm.
3.5 Performance evaluations
Four measurements are applied to evaluate developed prediction systems including accuracy (ACC) and Matthew‟s correlation coefficient (MCC) for each class, and overall accuracy (OA) and averaged accuracy (AA) for all classes, defined as follows:
100% i i i i TP ACC TP FN , (3-5)
i TP TN FP FN TP FN TP FP TN FP TN FN i i i i MCC i i i i i i i i , (3-6) i TP OA N
, (3-7) i ACC AA h
, (3-8)where i is the number of classes and TPi, TNi, FPi and FNi are the numbers of true positives, true negatives, false positives and false negatives, respectively. N is the total number of sequences and h is the number of classes.
Chapter 4
Prediction of ubiquitylation
sites
Ubiquitylation plays an important role in regulating protein functions. Recently, expe-rimental methods were developed toward effective identification of ubiquitylation sites. To efficiently explore more undiscovered ubiquitylation sites, this stud aims to develop an accurate sequence-based prediction method to identify promising ubiqui-tylation sites.
4.1 Motivation
Ubiquitylation (also called ubiquitination) is an important mechanism of post-translational modification that ubiquitin will be linked to specific lysine residues of target proteins by forming isopeptide bonds. Three enzymes including activating enzyme (E1), conjugating enzyme (E2), and ubiquitin ligase (E3) are involved in the ubiquitylation process. Another enzyme E4 can help to stabilize and extend polyubi-quitin chain [45, 46]. The first discovered function of ubiquitylation is to target pro-teins for subsequent degradation by the ATP-dependent ubiquitin-proteasome sys-tem. Subsequently, many regulatory functions of ubiquitylation were discovered in-cluding the regulation of DNA repair and transcription, control of signal transduc-tion, and implication of endocytosis and sorting [45, 46].
Because of the important regulatory roles of ubiquitylation, numerous methods were developed to purify ubiquitylated proteins [47]. Also, the growing number of studies of large-scale identification of ubiquitylated proteins and analysis of
ubiqui-Predicting of ubiquitylation
sites
tin-related proteome reflect the importance of identifying ubiquitylation proteins and sites [48-53]. The three steps affinity purification, proteolytic digestion, and analysis using mass spectrometry were applied in most of these studies [54]. These works cost a lot of experimental efforts. Therefore, developing a prediction system using informative features from protein sequences can not only save experimental efforts but also provide insights into the mechanism of ubiquitylation.
4.2 Assessment of features and classifiers
This study focuses on the sequence-based prediction of ubiquitylation sites. There-fore, three kinds of useful features which can be extracted from protein sequences
Figure 4.1 The sequence logo of the 151 positive samples with w=21. (a)
and are widely used in bioinformatics studies are evaluated for prediction of ubiqui-tylation sites: conventional amino acid identity [55], evolutionary information [56, 57], and physicochemical property [58, 59]. For predicting functions of a residue in a protein, it is well recognized that nearby residues will influence the property and structure of a central residue. For machine learning based prediction methods, the environmental information will be useful to enhance prediction accuracy that is ex-tensively used in previous studies [55-57]. The feature representations for applying to the mentioned classifiers are described below.
The conventional feature representation, amino acid identity, uses 20 binary bits to represent an amino acid [55]. For example, the amino acid A is represented by „00000000000000000001‟ and R is represented by „00000000000000000010‟. To deal with the problem of windows spanning out of N-terminal or C-terminal, one addi-tional bit is appended to indicate this situation. A vector of size (20+1)w bits is used for representing a sample where w is the window size.
Evolutionary information has been successfully applied in many studies [56, 57]. To prepare evolutionary information for each protein sequence, the corresponding position-specific scoring matrix (PSSM) is obtained by applying PSI-BLAST [60] against non-redundant SWISS-PROT database using 3 iteration and default values of parameters. For each residue, there are 20 values indicating the probabilities of oc-currences for 20 amino acids at the position. One additional bit is applied to deal with the terminal spanning windows as used for amino acid identity. A vector of size
Figure 4.2 The schema for the training and an independent of 3424 putative
(20+1)w is used for representing a sample.
Using informative features as well as an appropriate classifier is essential to de-sign an accurate prediction system. Three machine learning classifiers including
k-nearest neighbor, NaïveBayes and support vector machine (SVM) are evaluated for
predicting ubiquitylation sites. Two extensively used classifiers including IBk for
k-nearest neighbor classifier and NaïveBayes classifier that are included in the
ma-chine learning tool of WEKA [61] are applied to evaluate prediction performances of features. To optimize the performance of IBk classifier, five numbers of nearest neighbors k=1, 3, …, 9 used to classify samples are evaluated for selecting the best number of k. For NaïveBayes, in addition to normal distribution, a distribution ob-tained from kernel estimation is used to model numeric attributes.
To find the best kind of feature for SVM-based prediction of ubiquitylation sites, the control parameters C and of SVM and associated window size w{11, 13, …, 29} for each kind of features should be tuned to obtain best performance for
Figure 4.3 Performance comparisons among amino acid identity, evolutionary
comparison. The grid search method is applied to tune parameters C and that total 16*16=256 grids are evaluated. The prediction accuracy of 10-CV is used to deter-mine the best parameter values for the three kinds of features for SVM.
To evaluate the proposed methods, a positive dataset UBIDATA consisting of 157 ubiquitylation sites from 105 proteins was established by extracting annotated proteins from the UbiProt database [62]. By mapping the ubiquitylation sites to the corresponding 105 protein sequences retrieved from the UniProt Knowledgebase (Swiss-Prot and TrEMBL), the 3676 lysine residues with no annotation of ubiquityla-tion sites were regarded as putative non-ubiquitylaubiquityla-tion sites. A sliding window me-thod is applied to the central residue to be predicted for gleaning environment in-formation. A positive sample is denoted as a sequence of size w with a central resi-due lysine which is an ubiquitylation site. If the central resiresi-due lysine is not an ubi-quitylation site, the sequence is regarded as a negative sample. Only one of the sam-ples with the same sequences and annotation of ubiquitylation sites was used. All the inconsistent samples which have the same sequences but not the same annotation were discarded. The 10 positive datasets were constructed using various values of w from UBIDATA, which have 149 samples of w=11, 150 samples of w=13 and 15, and 151 samples of w=17, 19, …, 29. Due to the discard of duplicate and inconsis-tent samples, different values of w would result in different sample numbers of data-sets.
For training an SVM classifier, both positive and negative samples are necessary.
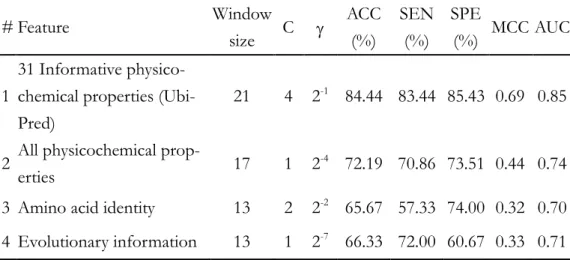
Table 4.1 Summary of used parameters and LOOCV performances of the
me-thods using informative physicochemical properties (UbiPred), amino acid identi-ty, evolutionary information, and all physicochemical properties.
# Feature Window size C ACC (%) SEN (%) SPE (%) MCC AUC 1 31 Informative physico-chemical properties (Ubi-Pred)
21 4 2-1 84.44 83.44 85.43 0.69 0.85
2 All physicochemical
prop-erties 17 1 2
-4 72.19 70.86 73.51 0.44 0.74
3 Amino acid identity 13 2 2-2 65.67 57.33 74.00 0.32 0.70
The dataset of post-translational modification including phosphorylation and ubi-quitylation sites is unbalanced that the number of positive samples is much smaller than that of negative samples. The negative samples for training the SVM classifier were selected randomly from the 3676 putative non-ubiquitylation sites. In this study,
the number of negative samples is the same with that of positive samples in the da-taset. For example, there are 151 negative samples in the dataset of w=21. The rest (e.g., 3424 samples with no annotation of ubiquitylation sites for w=21) are formed as an independent dataset to be scored for identifying promising ubiquitylation sites (see Fig. 8). Notably, since the value of C for tuning the error penalty (see the next section) is determined subsequently according to the performance measurement of SVM, it is not obligatory to select a matched number of negative peptides for train-ing the SVM classifier. The used datasets of various windows sizes can be publicly downloaded from the web server of UbiPred.
Figure 4.1 shows the sequence logo of the 151 positive samples with w=21 generated by the WebLogo tool [63]. The sequence logo with low information con-tent reveals disadvantages of the SVM using the two position-based features, amino
Figure 4.4 Performance comparisons between the SVM with informative
acid identity and evolutionary information, compared with the non-position based features, physicochemical properties using averaged measurement of amino acids in a sequence.
We established ten datasets with window sizes 11, 13, …, 29 from UbiProt, a database of ubiquitylated proteins [62], to evaluate the three kinds of features for applying classifiers. The dataset of window size 21 is shown in Figure 4.2. According to the prediction accuracies using 10-fold cross-validation (10-CV), the physico-chemical property is the best feature to SVM with best performance among all clas-sifiers and all kinds of features shown in Figure 4.3.
In order to provide insight into the underlying mechanism of ubiquitylation and improve the prediction accuracy, IPMA is applied to mine physicochemical proper-ties and tune SVM parameters while maximizing the 10-CV accuracy, a set of 31 in-formative physicochemical properties is obtained. A prediction system UbiPred for identifying ubiquitylation sites is implemented by utilizing the 31 informative physi-cochemical properties. UbiPred performs well with a prediction accuracy of 84.44% using leave-one-out cross-validation (LOOCV), compared with the SVM-based me-thods using amino acid identity (65.67%), evolutionary information (66.33%) and all physicochemical properties (72.19%). The performances and area under the ROC curve (AUC) are shown in Table 4.1
4.3 Informative physicochemical properties
Most of the 531 physicochemical properties may be irrelevant features or even inter-fere with prediction of the SVM classifier. Therefore, it is important to mine infor-mative physicochemical properties for advancing the prediction accuracy. IPMA de-termines a feature set of r informative physicochemical properties and the values of SVM parameters (C and ) for a given window size w. Because of the non-deterministic nature of IPMA, the obtained solutions would be different for each run. To obtain the features with robust performance, 30 independent runs of IPMA were performed for each window size w.The highest, mean, and lowest prediction accuracies of IPMA using 10-CV are shown in Figure 4.4. For comparison, the decision tree method C5.0 [64] with the ability of feature selection based on information gain was also evaluated. The accura-cies of C5.0 and SVM with the properties selected by C5.0 for various window sizes are also given in Figure 4.4. For all window sizes, the accuracies of SVM using in-formative physicochemical properties mined by IPMA are better than those of C5.0, SVM using all 531 physicochemical properties, and SVM using the C5.0-selected
properties. Considering the mean accuracies of SVM with informative physicochem-ical properties in Figure 4.4, the best window size is w=21.
Figure 4.5 shows the best 10-CV accuracies of using IPMA with w=21 for var-ious numbers of features from 30 independent runs. The accuracy of w=21 can be improved from 69.87% to 85.43% by using m=31 out of n=531 physicochemical properties, where the values of SVM parameters are C=4 and =0.5. The 31 infor-mative physicochemical properties constitute a good feature set obtained by consi-dering the inter-correlation among properties.
The quantified effectiveness of individual physicochemical properties on pre-diction is useful to characterize the ubiquitylation mechanism by physicochemical properties. Orthogonal experimental design with factor analysis [41] [42] can be used to estimate the individual effects of physicochemical properties according to the
val-Figure 4.5 The best 10-CV accuracies of prediction using SVM with the window
size 21 for various numbers of features (properties) selected by IPMA from 30 independent runs.
ue of main effect difference (MED) [59] [58]. The property with the largest value of MED is the most effective in predicting ubiquitylation sites.
According to MED, the 31 informative properties are ranked and their descrip-tions are shown in Table 4.2. The most effective property with MED=31.79 is NADH010102 denoting “hydropathy scale based on self-information values in the two-state model of 9% accessibility”. The least effective properties with MED=1.32 are NAKH900101 and QIAN880129 denoting “amino acid composition of total protein” and “weights for coil at the window position of -4”, respectively. The ranked informative physicochemical properties provide valuable information to bi-ologists for further experimental verification.
4.4 Prediction system UbiPred
To implement a prediction system UbiPred for identifying ubiquitylation sites, the 31 informative physicochemical properties with w=21, C=4, and =0.5 were used. The system flow of UbiPred is shown in Figure 4.6. The required input for UbiPred is peptide sequence. UbiPred will automatically encoding the windows with central ly-sine residue using 31 informative physicochemical properties. Subsequently, the lyly-sine residues will be annotated with SVM predicted result and shown in web page.
The prediction accuracy 84.44% of UbiPred shows good performance, com-pared with those of SVM with physicochemical property (72.19%), amino acid iden-tity (65.67%) and evolutionary information (66.33%). The SEN, SPE and MCC of UbiPred are 83.44%, 85.43% and 0.69, respectively. To compare the robustness of
Figure 4.7 Comparison of receiver operating characteristic curves among
infor-mative physicochemical properties (UbiPred), amino acid identity, evolutionary information and all physicochemical properties.
Table 4.2 The MEDs for 31 mined physicochemical property.
AAindex identity
Description MED
NADH010102 Hydropathy scale based on self-information values in the two-state model of 9% accessibility
31.79 BROC820102 Retention coefficient in HFBA 29.80 MEIH800102 Average reduced distance for side chain 28.48 LEVM780101 Normalized frequency of alpha-helix, with weights 25.17 GUYH850104 Apparent partition energies calculated from Janin index 23.84 CORJ870101 NNEIG index 23.18 RACS770102 Average reduced distance for side chain 22.52 GEOR030108 Linker propensity from helical (annotated by DSSP)
da-taset
22.52 HARY940101 Mean volumes of residues buried in protein interiors 21.85 GRAR740102 Polarity 19.87 GUYH850105 Apparent partition energies calculated from Chothia
index
19.87 MEIH800103 Average side chain orientation angle 17.88 KRIW790102 Fraction of site occupied by water 17.88 LEVM780106 Normalized frequency of reverse turn, unweighted 14.57 BULH740102 Apparent partial specific volume 13.25 FAUJ880101 Graph shape index 11.92 PUNT030102 Knowledge-based membrane-propensity scale from
3D_Helix in MPtopo databases
10.60 HUTJ700103 Entropy of formation 9.93 EISD840101 Consensus normalized hydrophobicity scale 8.61 CEDJ970105 Composition of amino acids in nuclear proteins
(per-cent)
7.28 ZIMJ680102 Bulkiness 7.28 CEDJ970103 Composition of amino acids in membrane proteins
(percent)
5.96 CHOC760103 Proportion of residues 95% buried 5.30 CEDJ970102 Composition of amino acids in anchored proteins
(per-cent)
5.30 ROSM880102 Side chain hydropathy, corrected for solvation 4.64 BROC820101 Retention coefficient in TFA 4.64 FAUJ830101 Hydrophobic parameter pi 1.99 NAKH920101 AA composition of CYT of single-spanning proteins 1.99 ZHOH040102 The relative stability scale extracted from mutation
ex-periments
1.99 NAKH900101 AA composition of total proteins 1.32 QIAN880129 Weights for coil at the window position of -4 1.32
UbiPred with other methods, the nonparametric method of ROC curve is applied by using the decision value of SVM as a tuning parameter. The area under the ROC curve (AUC) is calculated, as shown in Figure 4.7. UbiPred with AUC=0.85 performs well, compared with the SVM-based methods using all physicochemical properties (0.74), amino acid identity (0.70) and evolutionary information (0.71).
The quantified effectiveness of individual physicochemical properties on pre-diction is useful to better characterize the ubiquitylation mechanism by physico-chemical properties. According to MED, the 31 informative properties are ranked and their descriptions are shown in Table 4.2. The ranked informative physicochem-ical properties provide valuable information to biologists when further performing experimental verification.
The problem of sequence redundancy may result in overestimation of predic-tion performance. To address this issue, six thresholds of sequence identity (90%, 80%, …, 40%) were applied to construct six additional datasets from the dataset of
w=21 by using CD-HIT [65]. The numbers of positive and negative samples of
da-tasets with various sequence identity thresholds are shown in Table 4.3. By using the strictest threshold 40%, there are only 36 redundant samples and the resulting dataset consists of 145 negative samples and 121 positive samples. By applying LOOCV to evaluate prediction accuracies on these datasets, good performance (>79%) was ob-tained by using SVM with the mined 31 informative physicochemical properties and
Table 4.3 The LOOCV performances of the SVM with 31 informative
physico-chemical properties on datasets of various sequence identity thresholds. Sequence identity
threshold Accuracy(%)
Number of positive samples
Number of negative sam-ples 100% 84.44 151 151 90% 82.71 145 150 80% 81.72 141 149 70% 80.63 136 148 60% 81.23 131 146 50% 80.80 130 146 40% 79.70 121 145
![Figure 1.1 Comparison between conventional vaccine development and reverse vaccinology [7]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8758709.207707/20.892.212.742.146.1026/figure-comparison-conventional-vaccine-development-reverse-vaccinology.webp)