Anti-
Hepatitis C Virus Dinorditerpenes from the Roots of Flueggea virosa
Chih-Hua Chao, *,†,‡ Ju-Chien Cheng,§ De-Yang Shen,┴ and Tian-Shung Wu*,┴
† School of Pharmacy, China Medical University, Taichung 40402, Taiwan
‡ Chinese Medicine Research and Development Center, China Medical University Hospital, Taichung 40402,
Taiwan
§Department of Medical Laboratory Science and Biotechnology, China Medical University, Taichung 40402,
Taiwan
Department of Chemistry, National Cheng Kung University, Tainan 70101, Taiwan
ABSTRACT
the roots of Flueggea virosa. The absolute configurations of 4–6 were determined by the Mosher’s method and that of 5 confirmed by single-crystal X-ray diffraction analysis. Using the Hepatitis C virus cell culture (HCVcc) infection system, compounds 1, 3, 11, and 12 exhibited significant anti-HCV activity with EC50 values
of 5.6, 5.0, 7.5, and 6.6 μM, respectively. Compounds 11 and 12 were non-toxic toward the tested Huh7.5 cell lines.
Hepatitis C virus (HCV) infection is of international concern and can lead to chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma.1 The current standard HCV therapeutic regimen composed of
interferon-α and ribavirin is associated with significant side effects.2,3 Although the sustained virologic response
is improved by direct-acting antiviral protease inhibitors, drug-resistance is likely to emerge after extended periods of time.4 Traditional Chinese medicines, including Chinese herbs, may serve as a source of potential
new drugs. The roots of Flueggea virosa Roxb. ex Willd. (Euphorbiaceae) have been used in China for the treatment of rheumatism, pruritus, cephalic eczema, leucorrhoea, and bruising.5 The plant is known to be a rich
source of securinine and norsecurinine alkaloids, attracting much attention due to their wide spectrum of biological activities6-10 and novel structures.9,11-13 However, little is known about the nonalkaloid constituents,
except for the presence of bergenin in aerial parts, which exhibited both antiarrhythmic14 and typanocidal15
activities. In this study, screening assays using a J6JFH-based HCVcc infection system revealed that the nonalkaloid extract from the roots of F. virosa exhibited anti-HCV activity with an EC50 value of 17.2 μg/mL.
In the course of investigating potential anti-HCV agents from the nonalkaloid extract of this plant, four known terpenoids (1–4) and eight new dinorditerpenes (5–12) were characterized. Herein we report the isolation, structural elucidation, and anti-HCV activity of the isolated terpenoids.
RESULTS AND DISCUSSION
The known compounds were identified using spectroscopic methods as 3β,12-dihydroxy-13-methylpodocarpa-6,8,11,13-tetraene (1),16 3β,12-dihydroxy-13-methylpodocarpa-8,11,13 -triene (2),17
spruceanol (3),18 and ent-3β,12α-dihydroxypimara-8(14),15-diene (4).19 According to Mosher’s method,20 the R
configuration at C-12 was assigned for 4 (Figure 1).
(3408 cm–1), double bond (1643 cm–1), and aromatic (1608, 1560, 1494 cm–1) functionalities. The NMR data
(Table 1) and the UV absorption bands at 213 and 278 nm are typical of a methylpodocarpane with an aromatic C ring.16 Comparison of the NMR data of 5 with those of a known methylpodocarpane 1 suggested that 5 is a
methylated derivative of 1.16 This was confirmed by the 1H–1H COSY and HMBC correlations (Figure 2).
The relative configuration of 5 was determined by the NOE correlations observed in a NOESY experiment. The NOE correlation of H-3/H-5 suggested that ring A adopted a chair-form conformation and these two protons were both axially orientated. The absence of a correlation between H3-20 and H-5 indicated
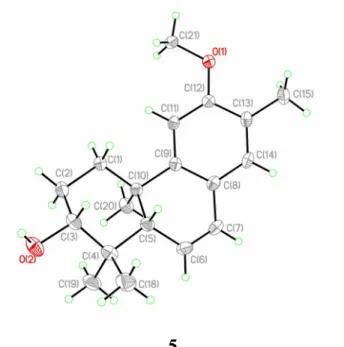
they are oppositely oriented. The 3R absolute configuration was defined by the Mosher’s method (Figure 1). Thus, the absolute configurations of 5 were established as 3R, 5S, 10R. This was confirmed by a single-crystal diffraction analysis with the small Flack parameter, -0.02(17) (Figure 3), and 5 was identified as 3α-hydroxy-12-methoxy-13-methyl-ent-podocarp-6,8,11,13-tetraene.
(+)-HRESIMS analysis of 6 established a molecular formula of C19H26O3. The NMR spectra of 6 resembled
those of 5. Comparison of the 1H NMR data of these two compounds indicated that the C-13 methyl group in 5
was replaced by a hydroxymethyl group in 6. This was confirmed by the HMBC correlations from H2-15 to
C-12, C-13, and C-14. The absence of NOE correlation between H3-20 and H-5, as well as the presence of
correlations between H-3 and H-5, H3-18 and H-3, and H3-18 and H-5, suggested that 6 and 5 shared the same
relative configuration. The 3R absolute configuration was again established by the Mosher ester procedure (Figure 1).20 Thus, compound 6 was identified as 3α-hydroxy-13-hydroxymethyl-12-methoxy
-ent-podocarp-6,8,11,13-tetraene.
13C NMR spectrum of 7, the C-1 and C-5 resonances were shifted upfield by 5.3 and 5.9 ppm, respectively,
compared to those of 6. The γ-effect resulting from the axially oriented C-3 hydroxy functionality is responsible for the aforementioned carbon shift. In addition, the small coupling constants between H-3 and both H2-2
protons (Table 2), as well as NOE correlations of H3-18/H-3, H3-19/H-3, and H3-18/H-5, further corroborated
the β orientation of 3-OH. Thus, 7 was determined as 3β-hydroxy-13-hydroxymethyl-12-methoxy -ent-podocarp-6,8,11,13-tetraene, a C-3 epimer of 6.
Compound 8 was assigned a molecular formula of C18H22O2, according to the (–)-HRESIMS and 13C NMR
data (Table 1). The IR absorption bands at 3400 and 1712 cm–1 indicated the presence of hydroxy and carbonyl
groups, respectively. The NMR data of 1 and 8 were similar, with major differences for the signals in ring A. Comparison of the NMR data of these two compounds indicated that the C-3 hydroxy group in 1 was replaced by a carbonyl functionality in 8. This was further demonstrated by the HMBC correlations from both H3-18 and
H3-19 to C-3, C-4, and C-5; H3-20 to C-1, C-5, C-9, and C-10, as well as the 1H–1H COSY correlations between
H2-1 and H2-2. Thus, 8 was identified as 12-hydroxy-13-methyl-ent-podocarp-6,8,11,13- tetraen-3-one.
The (–)-HRESIMS data of 9 suggested a molecular formula of C19H24O2. The IR spectrum revealed the
presence of a carbonyl group (1712 cm–1), which was corroborated by the 13C NMR resonance at δ
C 215.6 (qC)
(Table 1). The NMR data of 9 were similar to those of 8, with the methoxy group at δH 3.84 (3H, s), suggesting
that 9 is the 12-O-methyl derivative of 8. The HMBC correlations from the methoxy protons to the carbon resonance at δC 157.6 (qC, C-12) indicated that this methoxy group was located at C-12. Consequently, 9 was
identified as 12-methoxy-13-methyl-ent-podocarp-6,8,11,13-tetraen-3-one.
H]–). The IR spectrum showed absorption bands of hydroxy (3388 cm–1), carbonyl (1697cm–1), and aromatic
(1506 and 1456 cm–1) groups. The 1H NMR data of 8 and 10 were similar, with differences observed in the
signals of ring B. The 1H NMR spectrum showed the characteristic signals of a tetrasubstituted benzene ring at
δH 6.61 (1H, s), 6.81 (1H, s), and methyl groups at δH 2.19 (3H, s), 1.42 (3H, s), 1.39 (3H, s), and 1.14 (3H, s)
(Table 2). In addition, a hydroxy group was assigned at C-6 due to the proton resonance at δH 4.19 (1H, td, J =
10.0, 5.6 Hz), which correlated with a methine proton at δH 2.11 (1H, d, J = 10.0 Hz) and a benzylic methylene
at δH 3.12 (1H, dd, J = 15.6, 5.6 Hz, H-7a) and 2.78 (1H, dd, J = 15.6, 10.0 Hz, H-7b) in the COSY spectrum.
The relative configuration of 10 was determined based on the analyses of 3J
H,H values and NOE correlations.
The large coupling constants of H-6/H-5 and H-6/H-7b (each 10.0 Hz) suggested a pseudo-axial orientation of H-6. This was substantiated by the NOE correlations between H3-20 and H-6, suggesting the β-orientation of
6-OH. Thus, the structure of 10 was identified as 6β,12-dihydroxy-13-methyl-ent-podocarp-8,11,13-trien-3-one. Compound 11 had a molecular formula of C19H26O3, as determined from (+)-HRESIMS and 13C NMR data
(Table 1), appropriate for seven indices of hydrogen deficiency. The IR spectrum showed absorption bands for hydroxy (3417 cm–1) and aromatic (1620, 1585, 1494, 1454 cm–1) functionalities. In the 1H NMR spectrum, one
downfield methyl singlet [δH 2.22 (3H, s)], two aromatic proton singlets [δH 6.67 (1H, s), 7.01 (1H, s)], one
methoxy [δH 3.86 (3H, s)], two oxymethines [δH 3.38 (1H, dd, J = 11.2, 3.6 Hz) and 4.82 (1H, dd, J = 3.6, 2.0
Hz)], one oxymethylene (δH 4.29 (1H, d, J = 8.8 Hz), 2.83 (1H, d, J = 8.8, 1.6 Hz), and two upfield methyl
groups [δH 0.96 (3H, s) and 1.16 (3H, s)] were observed (Table 3). These data suggested that 11 is a derivative
of 5 with an ether linkage between a methine and a methylene groups. 1H−1H COSY correlations from H 2-1 to
oxymethylene protons (H2-20) showed correlations with C-5, C-9, C-10, and C-7, while C-7 was correlated
with H-14 and H2-20, and C-20 with H2-1 (Figure 2), suggesting an ether linkage between C-7 and C-20. The
NOE correlations between H-20a/H3-19, H-20a/H-2b, H3-19/H-2b, H-3/H-5, and H3-18/H-3 (Figure 4)
suggested that both the 7,20-oxa functionality and the 3-OH group were α–oriented. The upfield-shifted H-20b (δH 2.83), which had a 4J W-coupling (1.6 Hz) with H-5, was due to the shielding effect arising from the
aromatic C ring. Thus, the structure of 11 was identified as
7α,20-epoxy-3α-hydroxy-12-methoxy-13-methyl-ent-podocarp-8,11,13-triene.
The molecular formula of 12 was the same as that of 11, according to the (+)-HRESIMS and 13C NMR data
(Table 1). The IR spectrum indicated the presence of hydroxy (3400 cm–1) and aromatic (1614, 1517, 1504,
1444 cm–1) groups. Comparison of the NMR data of 11 and 12 suggested that they shared the same aromatic C
ring. In the HMBC spectrum of 12 (Figure 2), two oxymethylene protons [δH 4.10 (1H, dd, J = 9.2, 2.8 Hz),
4.00 (1H, d, J = 9.2 Hz)] showed correlations with C-1, C-5, C-9, C-10, and a hemiacetal carbon [δC 99.1(C),
C-3], suggesting the presence of an ether linkage between C-20 and the C-3 hemiacetal carbon. The NOE correlations of H-20b/H-11, H-20a/H-6b, H3-19/H2-6, H3-18/H-5 suggested the β-orientation of H-5 and
α,α-orientation of the 3,20-oxa functionality (Figure 4). The 2.8 Hz coupling between H-20a and H-1b was inferred to be a W-coupling, confirming the relative configuration of 12 (Figure 4). The structure of 12 was, therefore, identified as 3α,20-epoxy-3β-hydroxy-12-methoxy- 13-methyl-ent-podocarp-8,11,13-triene.
Compounds 1–12 were tested for their inhibitory activity toward HCV (measured as EC50) using the HCVcc
infection system, while the cytotoxicity assay against Huh7.5 cell lines (measured as IC50) was also evaluated
(Table 4). Honokiol, a known inhibitor of HCV infection,21 was used as a positive control with an EC
9.4 μM and an IC50 value of 58.5 μM. Compounds 4–10 showed weak inhibition activity against HCVcc
infection (20 < EC50 < 50 μM), while compound 2 was more potent with an EC50 value of 12.0 μM. Compounds
1 and 3 significantly inhibited HCVcc infection (EC50 = 5.6 and 5.0 μM, respectively) at non-toxic
concentrations (IC50 = 54.9 and 52.4 μM, respectively), while 11 and 12 exhibited significant anti-HCV activity
(EC50 = 7.5 and 6.6 μM, respectively) with non-toxicity (IC50 = 419.2 and 297.1 μM, respectively). Compounds
11 and 12 have higher therapeutic index (TI) values (TI = 55.9 and 45.0, respectively) than honokiol (TI = 6.2).
Thus, these results provide the impetus to investigate further the mechanisms of anti-HCV action and the clinical utility of 13-methyl-ent-podocarpanes from the roots of F. virosa as an adjunct to the current standard treatments for HCV infection.
The 13-methyl-ent-podocarpanes isolated from F. virosa represent a new class of anti-HCV agents, as most natural products that exert anti-HCV activity are found among the flavonoids,22 lignans,22 polyphenols,22
iridoids,23 and pseudoguaianolides.24 The skeleton of 13-methyl-ent-podocarpane is rare in nature, being
described from only two species of plants of the family Euphorbiaceae, that is Flueggea suffruticosa16 and
Jatropha curcas.17 The skeleton can be rationalized by losing a C
2 unit (C-15-C-16) of ent-pimarane (i.e., 4)
during the process of aromatization. In addition, 5 is a major constituent, suggesting its efficacy as a chemotaxonomic marker for F. virosa. The cytotoxicity assay toward the Huh7.5 cell line provides some powerful inferences of structure-activity relationships. Specifically, it revealed that oxidation on the angular methyl group at C-10 dramatically reduced toxicity (e.g., 11 and 12).
EXPERIMENTAL SECTION
melting point apparatus. Optical rotations were determined with a JASCO P1020 digital polarimeter. IR spectra were measured using a Shimadzu IR Prestige-21 FT-IR spectrometer. UV spectra were recorded on a Shimadzu UV-1700 UV/Vis spectrometer. The NMR spectra were recorded on a Bruker AVIII instrument at 400 MHz for
1H (referenced to δ
H 7.26 for CDCl3) and 100 MHz for 13C (referenced to δC 77.0 for CDCl3). ESIMS and
APCIMS were measured with a Finnigan LCQ ion-trap mass spectrometer. HRESIMS and HRAPCIMS were measured with an LTQ Orbitrap XL mass spectrometer. Silica gel 60 (Merck, 230400 mesh) and SiliaBond C18 silica gel (40–63 µm, 60 Å, 17% carbon loading, Silicycle) were used for column chromatography. Precoated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) and precoated silica gel RP-18 plates (Merck,
Kieselgel 60 F254S) were used for TLC analysis.
Plant Material. The roots of F. virosa were collected in September 2011 from Pingtung County, Taiwan and
were identified by Prof. C.-S. Kuoh (Department of Life Sciences, National Cheng Kung University, Tainan, Taiwan). A voucher specimen (specimen no. FV-Chao001) was deposited in the Chinese Medicine Research and Development Center, China Medical University Hospital.
Extraction and Isolation. Air-dried roots of F. virosa (13.0 kg) were cut into slices (ca. 0.5 cm thick),
minced, and extracted exhaustively with MeOH (3 × 20 L). The solvent was concentrated to an aqueous suspension and further partitioned between CHCl3 and H2O. The CHCl3 extract was washed with aqueous
tartaric acid (3 × 3% solution) to remove alkaloids. The resulting nonalkaloid extract (93 g) was fractionated using silica gel column chromatography (CC) with a gradient of n-hexane–EtOAc and EtOAc–MeOH to yield 26 fractions. Fraction 5 (1.7 g), eluted with n-hexane–EtOAc (3:2), was further purified using silica gel CC (gradient, n-hexane–EtOAc, 19:1 to 17:3), resulting in 15 subfractions (5A to 5O). Subfraction 5G was purified
on an RP-18 column (MeOH–H2O, 75 to 90%) to yield 9 (50.3 mg). Fraction 7 was fractionated by silica gel
CC (gradient, n-hexane–EtOAc, 13:1 to 5:1) to afford 18 subfractions (7A to 7R). Compound 5 (513.0 mg) was obtained from subfraction 7K, using a Toyopearl HW-40 C column (2 × 60 cm, MeOH). After further purification by RP-18 CC eluted with MeOH–H2O (gradient, 75 to 79%), 8 (75.1 mg) was obtained from
subfraction 7N. Compounds 2 (7.9 mg), 3 (28.7 mg), 10 (3.8 mg), and 12 (27.0 mg) were obtained from fraction 8 by repeated silica gel CC with n-hexane–EtOAc (gradient, 10:1 to 6:1) and RP-18 gel with MeOH– H2O (gradient, 65 to 70%). Fraction 9 was fractionated on a silica gel column using gradient elution (n-hexane–
EtOAc, 10:1 to 3:1) followed by RP-18 CC eluted with MeOH–H2O (gradient, 60 to 75%) to give compound 1
(82.7 mg.) Fraction 10 was subjected to silica gel CC (gradient, n-hexane–EtOAc, 9:1 to 3:1) to give 14 subfractions (10A to 10N). Subfraction 10H was further fractionated on an RP-18 column using gradient elution with MeOH–H2O (60 to 75%) to afford compound 4 (7.2 mg). Subfraction 10I was purified by RP-18
CC with MeOH–H2O (gradient, 60 to 70%) to afford compounds 6 (7.8 mg) and 7 (9.0 mg). Compound 11 (2.5
mg) was obtained from subfraction 10E using RP-18 CC (MeOH–H2O, gradient, 60 to 72%).
3β,12-Dihydroxy-13-methylpodocarpa-6,8,11,13-tetraene (1): pale yellow oil; [α]24
D +52 (c 1.23, acetone);
lit. [α]20
D +86 (c 0.91, acetone).16
3β,12-Dihydroxy-13-methylpodocarpa-8,11,13-triene (2): colorless oil; [α]24
D –57 (c 0.59, MeOH); lit. [α]25D
–12 (c 0.5, MeOH).17
Spruceanol (3): pale yellow oil; [α]24
D –14 (c 1.19, CHCl3); lit. [α]25D –3 (c 0.52, CHCl3).18
ent-3β,12α-Dihydroxypimara-8(14),15-diene (4): amorphous solid; [α]24
D –75 (c 0.72, CHCl3); [α]D was not
provided in lit.19
[α]24
D +54 (c 1.32, CHCl3); UV (MeOH) λmax (log ε) 213 (4.21), 278 (3.83) nm; IR (KBr)
v
max 3408, 2966, 2933,2868, 1643, 1608, 1560, 1496, 1463, 1363, 1305, 1259, 1074, 1056, 995 cm1; 13C NMR and 1H NMR data, see
Tables 1 and 2; (+)-APCIMS m/z 287 [M+H]+; (+)-HRAPCIMS m/z 287.2017 [M+H]+ (calcd for C
19H27O2,
287.2006).
3α-Hydroxy-13-hydroxymethyl-12-methoxy-ent-podocarp-6,8,11,13-tetraene (6): pale yellow oil; [α]24
D +74
(c 0.78, CHCl3); UV (MeOH) λmax (log ε) 222 (4.33), 278 (4.04) nm; IR (KBr)
v
max 3381, 2997, 2964, 2933,2870, 1608, 1556, 1494, 1454, 1371, 1259, 1211, 1072, 1055, 1039, 1029, 991 cm1; 13C NMR and 1H NMR
data, see Tables 1 and 2; (+)-ESIMS m/z 325 [M+Na]+; (+)-HRESIMS m/z 325.1785 [M+Na]+ (calcd for
C19H26O3Na, 325.1774).
3β-Hydroxy-13-hydroxymethyl-12-methoxy -ent-podocarp-6,8,11,13-tetraene (7): pale yellow oil; [α]24
D +34
(c 0.43, CHCl3); UV (MeOH) λmax (log ε) 222 (4.29), 276 (3.90) nm; IR (KBr)
v
max 3406, 2999, 2935, 2870,1608, 1562, 1494, 1454, 1386, 1371, 1259, 1205, 1085, 1047, 1026, 983 cm1; 13C NMR and 1H NMR data, see
Tables 1 and 2; (+)-ESIMS m/z 325 [M+Na]+; (+)-HRESIMS m/z 325.1782 [M+Na]+ (calcd for C
19H26O3Na,
325.1774).
12-Hydroxy-13-methyl-ent-podocarp-6,8,11,13-tetraen-3-one (8): pale yellow oil; [α]24
D +21 (c 1.31,
CHCl3); UV (MeOH) λmax (log ε) 207 (4.17), 280 (3.78) nm; IR (KBr)
v
max 3400, 2970, 2935, 1712, 1645, 1608,1583, 1454, 1386, 1273, 1274, 1205, 1147, 1022 cm1; 13C NMR and 1H NMR data, see Tables 1 and 2;
(–)-ESIMS m/z 269 [M – H]–; (–)-HRESIMS m/z 269.1546 [M – H]– (calcd for C
18H21O2, 269.1536).
12-Methoxy-13-methyl-ent-podocarp-6,8,11,13-tetraen-3-one (9): pale yellow oil; [α]24
D +14 (c 1.31,
and 2; (–)-ESIMS m/z 283 [M – H]–; (–)-HRESIMS m/z 283.1682 [M – H]– (calcd for C
19H23O2, 283.1698).
6β,12-Dihydroxy-13-methyl-ent-podocarp-8,11,13-trien-3-one (10): colorless oil; [α]24
D –136 (c 0.38,
CHCl3); UV (MeOH) λmax (log ε) 222 (3.87), 283 (3.48) nm; IR (KBr)
v
max 3388, 2966, 2933, 1697, 1506, 1456,1242, 1215, 1149, 1076, 1020, 1008, 999 cm1; 13C NMR and 1H NMR data, see Tables 1 and 2; (–)-ESIMS m/z
287 [M – H]–; (–)-HRESIMS m/z 287.1641 [M – H]– (calcd for C
18H23O3, 287.1642).
7α,20-Epoxy-3α-hydroxy-12-methoxy-13-methyl-ent-podocarp-8,11,13-triene (11): colorless oil; [α]24
D +54
(c 0.25, CHCl3); UV (MeOH) λmax (log ε) 277 (3.45), 224 (3.84) nm; IR (KBr)
v
max 3417, 2951, 2933, 2868,1620, 1585, 1494, 1454, 1365, 1286, 1238, 1205, 1184, 1155, 1124, 1076, 1051, 1029 cm1; 13C NMR and 1H
NMR data, see Tables 1 and 3; (+)-ESIMS m/z 303 [M + H]+; (+)-HRESIMS m/z 303.1961 [M + H]+ (calcd for
C19H27O3, 303.1955).
3α,20-Epoxy-3β-hydroxy-12-methoxy-13-methyl-ent-podocarp-8,11,13-triene (12): amorphous solid; [α]24
D –
146 (c 1.48, CHCl3); UV (MeOH) λmax (log ε) 281 (3.45), 221 (3.92) nm; IR (KBr)
v
max 3400, 2929, 2866, 1614,1517, 1504, 1444, 1321,1257, 1215, 1168, 1136, 1101, 1047, 1026 cm1; 13C NMR and 1H NMR data, see
Tables 1 and 3; (+)-ESIMS m/z 303 [M + H]+; (+)-HRESIMS m/z 303.1957 [M + H]+ (calcd for C
19H27O3,
303.1955).
Crystallographic Data and X-ray Structure Analysis of 5. A colorless crystal (0.540.520.46 mm3) of 5
was grown by slow evaporation in MeOH solution. Diffraction intensity data were acquired with a CCD area detector with graphite-monochromated Cu Kα radiation (λ = 1.54178 Å). Crystal data for 5: C20H30O3, M =
318.44, monoclinic, a = 8.1278(2) Å, b = 6.9825(2) Å, c = 16.1268(4) Å, α = 90.00°, β = 92.741(2)°, γ = 90.00°, V = 914.19(4) Å3, T = 293(2) K, space group P21, Z = 2, μ(CuKα) = 0.597 mm-1, 6740 reflections
wR(F2) values were 0.1030 (I > 2σ(I)). The final R
1 values were 0.0371 (all data). The final wR(F2) values were
0.1036 (all data). The goodness of fit on F2 was 1.086. Flack parameter = –0.02(17). Crystallographic data for 5
have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 933580). Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB21EZ, UK [fax: +44(0)-1223-336033 or e-mail: [email protected]].
Preparation of (S)-and (R)-MTPA Esters of 4-6. (R)-MTPA chloride (25 μL) was added to a solution of
the secondary alcohol (0.5 mg each) in pyridine (0.4 mL), and the mixture stirred at room temperature for 3 h. A 1.0 mL aliquot of H2O was added to quench the reaction, and the mixture was extracted with EtOAc (3 × 1.0
mL). The combined EtOAc layers were dried over anhydrous MgSO4 and evaporated to give a residue, which
was purified using silica gel CC (n-hexane−EtOAc, 5:1) to yield the (S)-MTPA esters, 4a, 5a, and 6a. The (R)-MTPA esters, 4b, 5b, and 6b were prepared using the same procedure with (S)-(R)-MTPA chloride, each prepared from 0.5 mg of its corresponding alcohol. Selected 1H NMR (CDCl
3, 400 MHz) of 4a: δ 7.400−7.550 (10H, m,
Ph), 5.723 (1H, dd, J = 17.0, 10.5 Hz, H-16), 5.111 (1H, m, H-12), 5.048 (1H, d, J = 10.5 Hz, H-17a), 5.040 (1H, s, H-14), 5.034 (1H, d, J = 17.0 Hz, H-17b), 4.735 (1H, dd, J = 11.6, 3.8 Hz, H-3), 3.508 (3H, s, OCH3),
3.571 (3H, s, OCH3), 1.815 (1H, m, H-2a), 1.788 (1H, m, H-11a), 1.692 (1H, m, H-2b), 1.678 (1H, m, H-11b),
0.928 (3H, s, H3-15), 0.844 (3H, s, CH3, H3-18 or H3-19 or H3-20), 0.820 (3H, s, CH3, H3-18 or H3-19 or H3-20),
0.776 (3H, s, CH3, H3-18 or H3-19 or H3-20). Selected 1H NMR (CDCl3, 400 MHz) of 4b: δ 7.400−7.600 (10H,
m, Ph), 5.732 (1H, dd, J = 17.0, 10.5 Hz, H-16), 5.070 (1H, d, J = 10.5 Hz, H-17a), 5.044 (1H, d, J = 17.0 Hz, H-17b), 5.033 (1H, s, H-14), 5.020 (1H, m, H-12), 4.689 (1H, dd, J = 12.0, 4.5 Hz, H-3), 3.541 (3H, s, OCH3),
3.528 (3H, s, OCH3), 1.754 (1H, m, H-11a), 1.727 (1H, m, H-2a), 1.547 (1H, m, H-2b), 1.327 (1H, m, H-11b),
1.010 (3H, s, H3-15), 0.911 (3H, s, CH3, H3-18 or H3-19 or H3-20), 0.820 (3H, s, CH3, H3-18 or H3-19 or H3-20),
0.717 (3H, s, CH3, H3-18 or H3-19 or H3-20). 1H NMR (CDCl3, 400 MHz) of 5a: δ 7.579−7.418 (5H, m, Ph),
6.869 (1H, s, H-14), 6.638 (1H, s, H-11), 6.507 (1H, dd, J = 9.6, 3.0 Hz, H-7), 5.795 (1H, dd, J = 9.6, 2.6 Hz, H-6), 4.836 (1H, dd, J = 11.1, 4.2 Hz, H-3), 3.837 (3H, s, OCH3), 3.600 (3H, s, OCH3), 2.248 (1H, m, H-1a),
2.203 (1H, dd, J = 3.0, 2.6 Hz, H-5), 2.169 (3H, s, H3-15), 2.104 (1H, m, H-2a), 1.956 (1H, m, H-2b), 1.911 (1H, m, H-1b), 1.059 (3H, s, CH3, H3-18 or H3-19 or H3-20) 1.033 (3H, s, CH3, H3-18 or H3-19 or H3-20), 0.926 (3H, s, CH3, H3-18 or H3-19 or H3-20). 1H NMR (CDCl3, 400 MHz) of 5b: δ 7.553−7.423 (5H, m, Ph), 6.868 (1H, s, H-14), 6.635 (1H, s, H-11), 6.511 (1H, dd, J = 9.6, 3.0 Hz, H-7), 5.808 (1H, dd, J = 9.6, 2.5 Hz, H-6), 4.809 (1H, dd, J = 11.1, 4.4 Hz, H-3), 3.833 (3H, s, OCH3), 3.550 (3H, s, OCH3), 2.208 (1H, dd, J = 3.0, 2.5 Hz, H-5), 2.178 (1H, m, H-1a), 2.168 (3H, s, H3-15), 2.035 (1H, m, H-2a), 1.868 (1H, m, H-1b), 1.838 (1H, m, H-2b), 1.045 (3H, s, CH3, H3-18 or H3-19 or H3-20) 1.034 (3H, s, CH3, H3-18 or H3-19 or H3-20), 1.014 (3H, s, CH3, H3-18 or H3-19 or H3-20). 1H NMR (CDCl3, 400 MHz) of 6a: δ 7.353−7.575 (10H, m, Ph), 6.952 (1H, s, H-14), 6.663 (1H, s, H-11), 6.461 (1H, dd, J = 9.6, 3.0 Hz, H-7), 5.818 (1H, dd, J = 9.6, 2.4 Hz, H-6), 5.355 (1H, d, J = 12.3 Hz, H-15a), 5.302 (1H, d, J = 12.3 Hz, H-15b), 4.829 (1H, dd, J = 11.1, 4.2 Hz, H-3), 3.796 (3H, s, OCH3), 3.592 (3H, s, OCH3), 2.238 (1H, m, H-1a), 2.185 (1H, dd, J = 3.0, 2.4 Hz, H-5), 2.101 (1H, m, H-2a),
1.946 (1H, m, H-2b), 1.914 (1H, m, H-1b), 1.055 (3H, s, CH3, H3-18 or H3-19 or H3-20) 1.032 (3H, s, CH3, H3-18
or H3-19 or H3-20), 0.925 (3H, s, CH3, H3-18 or H3-19 or H3-20). 1H NMR (CDCl3, 400 MHz) of 6b: δ
7.353−7.550 (10H, m, Ph), 6.961 (1H, s, H-14), 6.658 (1H, s, H-11), 6.471 (1H, dd, J = 9.7, 3.2 Hz, H-7), 5.833 (1H, dd, J = 9.7, 2.4 Hz, H-6), 5.343 (1H, d, J = 12.2 Hz, H-15a), 5.304 (1H, d, J = 12.2 Hz, H-15b), 4.801 (1H,
dd, J = 11.0, 4.5 Hz, H-3), 3.786 (3H, s, OCH3), 3.546 (3H, s, OCH3), 2.193 (1H, dd, J = 3.0, 2.4 Hz, H-5),
2.192 (1H, m, H-1a), 2.047 (1H, m, H-2a), 1.854 (1H, m, H-1b), 1.844 (1H, m, H-2b), 1.039 (3H, s, CH3, H3-18
or H3-19 or H3-20) 1.030 (3H, s, CH3, H3-18 or H3-19 or H3-20), 1.013 (3H, s, CH3, H3-18 or H3-19 or H3-20).
Cell Culture and Cell Viability Assay.25 The Huh7.5 cells derived from human hepatoma Huh7 cells were
maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS) and 1% non-essential amino acid (NEAA). For the cell viability assay, serial concentrations of the indicated compounds were added into the 96-well culture plates which were pre-seeded with Huh7.5 cells at a cell density of 1 × 104cells/well for 24 h. After 72 h, the viable cells were determined by the CellTiter 96
Aqueous One Solution Cell Proliferation Assay Kit as described by the manufacturer (Promega).
Infectious HCV Particles Production and Infection Inhibition Assay. The production of infectious
JC1-Luc2A HCV reporter viral particles was performed as described previously.26 Briefly, in vitro-transcribed viral
genomic RNA was transfected into Huh7.5 cells by electroporation. The virus-containing supernatant collected at days 2 and 4 was applied to low-speed centrifugation, passed through a filter (pore size 0.45 μm), and concentrated by ultracentrifugation. For the infection inhibition assay, the HCV reporter virus (with 0.1 MOI) was added into the 96-well culture plates which were pre-seeded with Huh7.5 cells at a cell density of 1 × 104cells/well for 24 h. After 4h incubation, the virus-containing supernatant was removed, washed (× 3) and
then replaced with the fresh medium containing the compounds. After 72 h, the cell lysates were collected and the luciferase activity and cell viability were determined. The luciferase activity assay was performed using the Bright-Glo™ Luciferase Assay System as described by the manufacturer (Promega). The relative firefly
luciferase versus cell viability activity is reported as the mean plus or minus the standard deviation of three independent reactions. The honokiol used as a positive control was isolated from the roots of Magnolia
officinalis.27
ASSOCIATED CONTENT Supporting Information
1H and 13C NMR spectra for 512, 1H NMR spectra of Mosher esters of 46, and the CIF file of 5 are available
free of charge via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION Corresponding Author
* Tel: +886-4-22053366, ext. 5157. Fax: +886-4-22078083. E-mail: chaochihhua @ hotmail.com (C.-H. C.) *Tel: +886-6-2747538. Fax: +886-6-2740552. E-mail: [email protected]. (T.-S. W.)
ACKNOWLEDGEMENTS
The authors are grateful for financial support from the National Science Council of Taiwan (NSC101-2113-M-039-003) awarded to C.-H.C. This study was also supported in part by the China Medical University Hospital (DMR-101-106) and China Medical University (CMU99-S-35).
.
REFERENCES
(1) Saito, I.; Miyamura, T.; Ohbayashi, A.; Harada, H.; Katayama, T.; Kikuchi, S.; Watanabe, Y.; Koi, S.; Onji, M.; Ohta, Y.; Choo, Q.L.; Houghton, M.; Kuo, G. Proc. Natl. Sci. Acad. USA 1990, 87,
6547–6549.
(2) Mangia, A.; Santoro, R.; Minerva, N.; Ricci, G.L.; Carretta, V.; Persico, M.; Vinelli, F.; Scotto, G.; Bacca, D.; Annese, M.; Romano, M.; Zechini, F.; Sogari, F.; Spirito, F.; Andriulli, A. N. Engl. J.
Med. 2005, 352, 2609–2617.
(3) Lai, M. Y. Intervirology 2006, 49, 91–95.
(4) Welsch C.; Zeuzen S. Curr. Opin. Virol. 2012, 2, 651–655.
(5) Li, B. T. Chinese Flora (Zhongguo Zhiwu Zhi); Science Press: Beijing, 1994; Vol. 44 (1), pp 68-74. (6) Chen, M. J.; Hou, L. L. J. Integr. Plant Biol. 1985, 27, 625– 629.
(7) Gan, L. S.; Yue, J. M. Nat. Prod. Commun. 2006, 1, 819– 823.
(8) Dehmlow, E. V.; Guntenhoner, M.; Van, R. T. Phytochemistry 1999, 52, 1715– 1716.
(9) Gan, L. S.; Fan, C. Q.; Yang, S. P.; Wu, Y.; Lin, L. P.; Ding, J.; Yue, J. M. Org. Lett. 2006, 8, 2285– 2288.
(10) Iketubosin, G. O.; Mathieson, D. W. J. Pharm. Pharmacol. 1963, 15, 810– 815.
(11) Zhao, B. X.; Wang, Y.; Zhang, D. M.; Jiang, R. W.; Wang, G. C.; Shi, J. M.; Huang, X. J.; Chen, W. M.; Che, C. T.; Ye, W. C. Org. Lett. 2011, 13, 3888– 3891.
(12) Zhao, B. X.; Wang, Y.; Zhang, D. M.; Huang, X. J.; Bai, L. L.; Yan, Y.; Chen, J. M.; Lu, T. B.; Wang, Y. T.; Zhang, Q. W.; Ye, W. C. Org. Lett. 2012, 14, 3096– 3099.
(13) Zhang, H.; Zhang, C. R.; Zhu, K. K.; Gao, A. H.; Luo, C.; Li, J.; Yue, J. M. Org. Lett. 2013, 15, 120– 123.
(14) Pu, H. L.; Huang, X.; Zhao, J. H.; Hong, A. Planta Med. 2002, 68, 372–374.
(15) Nyasse, B.; Nono, J.; Sonke, B.; Denier, C.; Fontaine C. Pharmazie, 2004, 59, 492–494.
(16) Yuan, W.; Lu, Z.; Liu, Y.; Meng, C.; Cheng, K. D. Zhu, P. Chem. Pharm. Bull. 2005, 53, 1610–1612. (17) Ravindranath, N.; Reddy, M.R.; Ramesh, C.; Ramu, R.; Prabhakar, A.; Jagadeesh, B.; Das, B. Chem.
Pharm. Bull. 2004, 52, 608–611.
(18) Gunasekera, S.P.; Cordell, G.A.; Farnsworth, N.R. J. Nat. Prod. 1979, 42, 658–662. (19) Sakai, T.; Nakagawa, Y. Phytochemistry 1988, 27, 3769–3779.
(20) Randazzo, A.; Bifulco, G.; Giannini, C.; Bucci, M.; Debitus, C.; Cirino, G.; Gomez-Paloma, L. J.
Am. Chem. Soc. 2001, 123, 10870–10876.
(21) Lan, K. H.; Wang, Y. W.; Lee, W. P.; Lan, K. L.; Tseng, S. H.; Hung, L. R.; Yen, S. H.; Lin, H. C.; Lee, S. D. Liver Int. 2012, 32, 989–997.
(22) Calland, N.; Dubuisson, J.; Rouillé, Y.; Séron, K. Viruses 2012, 4, 2197–2217.
(23) Zhang, H.; Rothwangl, K.; Mesecar, A.D.; Sabahi, A.; Rong, L.; Fong, H. H. S. J. Nat. Prod. 2009,
72, 2158–2162.
(25) Cheng, J. C.; Yeh, Y. J.; Tseng, C. P.; Hsu, S. D.; Chang, Y. L.; Sakamoto, N.; Huang, H. D. Cell.
Mol. Life Sci. 2012, 69, 2621–2633.
(26) Huang, J. T.; Tseng, C. P.; Liao, M. H.; Lu, S. C.; Yeh, W. Z.; Sakamoto, N.; Chen, C. M.; Cheng, J. C. J. Virol. 2013, 87, 4994–5004.
(27) Shih, H. C.; Hwang, T. L.; Chen, H. C., Kuo, P. C.; Lee, E. J.: Lee, K. H.; Wu, T. S. PLoS One 2013,
Table 1. 13C NMR Spectroscopic Data (100 MHz, CDCl 3) of Compounds 512 # 5, δC 6, δC 7, δC. 8 δC 9, δC 10, δC 11, δC 12, δC 1 34.3, CH2 34.3, CH2 29.0, CH2 35.1, CH2 35.2, CH2 37.6, CH2 26.8, CH2 34.9, CH2 2 27.7, CH2 27.7, CH2 25.5, CH2 34.6, CH2 34.5, CH2 33.5, CH2 27.2, CH2 29.4, CH2 3 78.7, CH 78.6, CH 74.9, CH 216.3, C 215.6, C 218.4, C 78.9, CH 99.1, C 4 38.3, C 38.3, C 37.2, C 47.2, C 47.2, C 47.6, C 39.4, C 40.3, C 5 50.5, CH 50.3, CH 44.4, CH 51.5, CH 51.6, CH 55.3, CH 42.8, CH 48.4, CH 6 126.5, CH 127.0, CH 127.4, CH 125.2, CH 125.3, CH 67.4, CH 29.9, CH2 20.6, CH2 7 127.7, CH 127.6, CH 127.6, CH 128.4, CH 128.5, CH 40.7, CH2 69.9, CH 30.0, CH2 8 125.5, C 126.3, C 126.0, C 125.4, C 125.1, C 124.2, C 131.1, C 129.2, C 9 146.5, C 148.6, C 149.1, C 145.3, C 144.8, C 144.9, C 144.6, C 136.6, C 10 37.8, C 38.0, C 37.9, C 37.3, C 37.6, C 39.3, C 37.6, C 36.2, C 11 104.3, CH 104.6, CH 104.6, CH 109.3, CH 104.3, CH 111.7, CH 101.8, CH 108.0, CH 12 157.3, C 157.1, C 157.0, C 153.9, C 157.6, C 152.7, C 157.4, C 156.0, C 13 123.7, C 125.9, C 125.9, C 121.3, C 124.3, C 122.2, C 124.0, C 125.1, C 14 128.9, CH 127.2, CH 127.1, CH 129.4, CH 129.2, CH 130.8, CH 125.5, CH 131.1, CH 15 15.6, CH3 61.8, CH2 61.9, CH2 15.2, CH3 15.7, CH2 15.3, CH3 15.9, CH3 15.7, CH3 18 27.8, CH3 27.8, CH3 27.1, CH3 24.8, CH3 24.9, CH3 31.1, CH3 28.1, CH3 26.9, CH3 19 16.5, CH3 16.5, CH3 23.1, CH3 22.6, CH3 22.7, CH3 20.0, CH3 14.3, CH3 18.0, CH3 20 20.2, CH3 20.1, CH3 19.8, CH3 19.9, CH3 19.8, CH3 26.2, CH3 67.5, CH2 72.4, CH2 OMe 55.4, CH3 55.4, CH3 55.4, CH3 55.4, CH3 55.6, CH3 55.4, CH3
Table 2. 1H NMR Spectroscopic Data (400 MHz, CDCl 3) of Compounds 510 # 5, δH (J in Hz) 6, δH (J in Hz) 7, δH (J in Hz) 8, δH (J in Hz) 9, δH (J in Hz) 10, δH (J in Hz) 1 2.20, m 2.19, m 2.13, m 2.39, ddd(13.2, 6.0, 2.8) 2.49, m 2.25, m 1.80, m 1.78, m 1.92, m 2.04, ddd(13.6, 13.2, 4.8) 2.11, ddd (13.6, 13.2, 4.8) 2.12, m 2 1.90, m 1.87, m 2.13, m 2.86, ddd (15.2, 13.6, 6.0) 2.89, ddd (15.6, 13.6, 6.4) 2.77, ddd (15.6, 2.8, 2.8) 1.78, m 1.79, m 1.85, dt (10.8, 3.2) 2.50, ddd (15.2, 4.8, 2.8) 2.52, m 2.49, ddd (15.6, 9.2, 4.8) 3 3.38, dd (11.2, 4.4) 3.32, m 3.56, br s 5 2.10, (3.2, 2.8) 2.08, dd (3.2, 2.8) 2.54, dd (3.2, 2.8) 2.52, dd (3.2, 2.8) 2.54, t (2.8) 2.11, d (10.0) 6 5.86, dd (9.6, 2.8) 5.89, dd (9.6, 2.8) 5.86, dd (9.6, 2.8) 5.78, dd (9.6, 2.8) 5.79, dd (9.6, 2.8) 4.19, td (10.0, 5.6) 7 6.50, dd (9.6, 3.2) 6.52, dd (9.6, 3.2) 6.53, dd (9.6, 3.2) 6.55, dd (9.6, 3.2) 6.56, dd (9.6, 2.8) 3.12, dd (15.6, 5.6) 2.78, dd (15.6, 10.0) 11 6.66, s 6.71, s 6.75, s 6.63, s 6.67,s 6.61, s 14 6.86, s 6.99, s 6.98, s 6.87, s 6.89, s 6.81, s 15 2.17, s 4.63, s 4.66, d (12.4) 2.21, s 2.18, s 2.19, s 4.61, d (12.4) 18 1.09, s 1.08, s 1.06, s 1.15, s 1.16, s 1.39, s 19 1.025, s 1.02, s 1.07, s 1.27, s 1.28, s 1.42, s 20 1.034, s 1.03, s 1.04, s 1.22, s 1.24,s 1.14,s OM 3.83, s 3.87, s 3.87, s 3.84,s
Table 3. 1H NMR Spectroscopic Data (400 MHz, CDCl3) of Compounds 11 and 12 # 11, δH (J in Hz) 12, δH (J in Hz) 1 a 2.15, m 2.44, td (12.0, 5.6) b 1.83, m 1.67, tdd (12.0, 3.2, 2.8) 2 a 1.86, m 2.28, td (12.0, 5.6) b 1.75, m 1.93, td (12.0, 3.2) 3 3.38, dd (11.2, 3.6) 5 1.27,ddd (11.6, 6.0, 1.6) 1.64, m 6 a 2.13, m 1.83, m b 1.58, ddd (13.2, 11.6, 2.0) 1.65, m 7 4.82, dd (3.6, 2.0) 2.63, m 2.74, m 11 6.67, s 6.68, s 14 7.01, s 6.85, s 15 2.22, s 2.15, s 18 0.96, s 1.12, s 19 1.16, s 1.05, s 20 4.29, d (8.8) 4.00, d (9.2) 2.83, dd (8.8, 1.6) 4.10, dd (9.2, 2.8) OMe 3.86, s 3.79, s
Table 4. Anti-HCV activities of 1–12
compd EC50 (μM)a IC50 (μM)b therapeutic index (TI)c
1 5.6 ± 0.4 54.9 ± 2.2 9.8 2 12.0 ± 0.8 54.3 ± 6.1 4.5 3 5.0 ± 0.9 52.4 ± 8.5 10.5 4 52.7 ± 6.4 166.9 ± 3.3 3.2 5 24.9 ± 4.5 70.0 ±14.1 2.8 6 27.4 ± 1.9 125.4 ± 2.5 4.6 7 30.5 ± 2.3 150.9 ± 7.8 4.9 8 25.5 ± 1.7 86.5 ± 9.1 3.4 9 47.3 ± 1.5 81.8 ± 9.4 1.7 10 52.8 ± 6.6 291.6 ± 3.7 5.5 11 7.5 ± 0.9 419.2 ± 9.5 55.9 12 6.6 ± 0.5 297.1 ± 2.0 45.0 honokiol 9.4± 0.9 58.5 ± 4.9 6.2 a EC
50: concentration that inhibits HCVcc infection by 50%. b IC50:
concentration that inhibits cell growth by 50%. c TI = IC
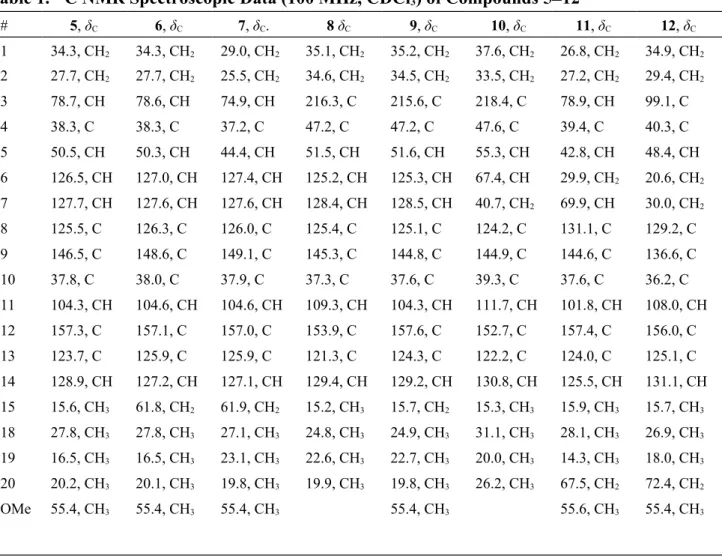
Figure 1. 1H NMR chemical shift differences of MTPA esters of 4–6.
5
Figure 3. X-ray ORTEP drawing of compound 5.