行政院國家科學委員會專題研究計畫 期中進度報告
皮質類固醇影響上皮細胞癌生長及化學藥物敏感性機轉之
研究並探討與癌細胞反應模式相關之分子分類(1/3)
計畫類別: 個別型計畫 計畫編號: NSC91-2314-B-002-166- 執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日 執行單位: 國立臺灣大學醫學院內科 計畫主持人: 鄭安理 計畫參與人員: 盧彥伸 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 92 年 6 月 3 日
2
行政院國家科學委員會補助專題研究計畫
■ 期 中
進度報告
皮質類固醇影響上皮細胞癌生長及化學藥物敏感性機
轉之研究並探討與癌細胞反應模式相關之分子分類
計畫類別:■ 個別型計畫 □ 整合型計畫
計畫編號:NSC 91-2314-B-002-166
執行期間: 91 年 8 月 1 日至 92 年 7 月 31 日
計畫主持人:鄭安理
共同主持人:盧彥伸 郭明良 葉坤輝 陳健尉
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整
報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究
計畫、列管計畫及下列情形者外,得立即公開查詢
3
□涉及專利或其他智慧財產權,□一年□二年後可公
開查詢
執行單位:國立台灣大學醫學院 內科
中 華 民 國 92 年 5 月 31 日
4
行政院國家科學委員會專題研究計畫期中進度
報告
皮質類固醇影響上皮細胞癌生長及化學藥物敏感性機
轉之研究並探討與癌細胞反應模式相關之分子分類
Studies on the mechanisms of glucocorticoids on the growth and drug
sensitivity of carcinomas, and exploring relevant molecular classification
一、中文摘要 皮質類固醇除本身對於某些血液腫瘤 具細胞毒性之外,也常與抗癌化學藥物併 用以治療因化學藥物引起之噁心、嘔吐及 過敏反應等副作用。雖然類固醇己被証實 可以影響多種細胞之重要訊息傳遞徑路, 其中有些與癌細胞抗藥性有關。然而我們 對類固醇類藥物對於與一般癌細胞生長以 及化學藥物感受性可能產生之影響仍所知 極少。釐清這個問題對臨床腫瘤治療將會 有重要影響。 我們隨機選擇了十四株癌細胞株有系 統地進行研究以解答這個問題。 Dexamethasone(DEX)被選為皮質類固 醇代表藥物。我們發現: 1. DEX 確實對癌細胞株(十四株之中的 七株)的生長以及化學藥物感受性有影 響。DEX 對癌細胞的影響呈現異質性 而且似乎是彼此互斥的。DEX (0.01~1.0uM)抑制四株細胞的生長 (MCF-7,MCF/MXR1,MCF/TPT300 及 HeLa 細胞),提高了一株細胞對 cisplatin 的化學藥物感受性(SiHa),並 降低兩株細胞對 cisplatin, doxorubicin, 5FU,及 taxol 的化學藥物感受性(H460 及 Hep3B)。 2. 此影響是皮質類固醇受體—依賴性 的。因為 DEX 只有在含有高濃度皮質 類固醇受體(≧2.1x104 /細胞)的七株細 胞才有影響。在其他七株不受 DEX 影 響的細胞中,有五株細胞皮質類固醇受 體濃度範圍僅在 2.0~5.7x103 /細胞之 間。而另二株含有高濃度皮質類固醇受 體但不受 DEX 影響的細胞中(TW01, TW04),我們發現其皮質類固醇受體不 具有功能。 3. DEX 在 SiHa 細胞所造成的化學藥物致 敏感效應與其對 NF-kB 的調控有著高 度相關。透過轉殖含有 dominant negative IkB 的 plasmid 進入 SIHa cells 中以抑制 NFkB 活性,我們發現原 先 DEX 提高 SiHa 細胞對 cisplatin 的 化學藥物感受性的現象消失。 4. 利用即時定量 RT-PCR 測定十位乳癌病 患癌細胞檢體,我們發現在不同病人的 癌細胞中的皮質類固醇受體含量有高 有低。我們推測臨床上有一定比例之癌 細胞含有高濃度皮質類固醇受體,並可 能對 DEX 有感受性。 關鍵詞:皮質類固醇、癌細胞、化學藥物 感受性
Abstract
Objectives: Glucocorticoids (GCs) are commonly co-administered with anti-cancer drugs such as cisplatin to prevent drug-induced allergic reaction, nausea, and vomiting. But little is known regarding the effects of GCs on the growth and chemosensitivity of common carcinomas cells. Methods: Fourteen carcinoma cell lines representing breast (MCF-7, MCF-7/MXR1, MCF-7/TPT300), gastric (AGS, N87,SNU1), lung (H460), cervical (SiHa, HeLa, Caski), liver (Hep3B, Hut7), and nasopharyngeal (NPC-TW01, NPC-TW04) cancer were selected to assess the effects of dexamethasone (DEX) on the cell growth and cisplatin chemosensitivity of common human cancers. Results: DEX had mutually exclusive effects on either growth or cisplatin sensitivity in 7 of the 14 cell lines. DEX inhibited cell growth of 4 (MCF-7, MCF-7/MXR1, MCF-7/TPT300, and HeLa), increased cisplatin cytotoxicity of one (SiHa), and decreased cisplatin cytotoxicity of 2 (H460 and Hep3B) cells lines. Although the effect of DEX on these carcinoma cells was unexpectedly diverse, it remained GC receptor (GCR) dependent. The GCR contents of the 7 cell lines affected by DEX were significantly higher than those of the other 7 cell lines unaffected by DEX (5.2±2.5 × 104
vs 1.3 ± 1.4 × 104, P=0.005).Only two DEX-unresponsive cell lines (NPC-TW01 and NPC-TW04) had GCR contents at the high range as those of the 7 DEX-responsive cell lines. On further examination, the function of the endogenous GCR of these two cell lines was found to be impaired. Further, transfection and expression of a vector encoding GCR to AGS, a GCR low-expressing and GC non-responsive cell line, increased its susceptibility to DEX manifested as an increased resistance toward cisplatin. The cytotoxicity-enhancing effect of GC in SiHa cells correlated well with its effect on abrogating the cisplatin-induced activation of NF-B. Expression of a dominant-negative truncated IB gene in SiHa cells completely abolished the cytotoxicity-enhancing effect of DEX. Conclusions: GCs may affect growth or
chemosensitivity of carcinoma cells containing high concentration of functional GCR. Although the effects are heterogeneous and currently unpredictable , our data underscore the importance of clarifying the impact on tumor control by the co-administed GCs to carcinoma patients receiving chemotherapy. It is mandatory to identify the molecular and cellular markers that help predict the diverse effect of GCs on carcinoma cells.
Keywords: Glucocorticoids, Glucocorticoid receptor, Carcinoma. Cell growth,
Chemosensitivity, Drug resistance.
二、緣由與目的
Although GCs are effective in inducing apoptosis via yet uncharacterized pathways in many hematological malignancies 1, 2, 3, they are generally not effective in the
treatment of non-hematological solid tumors. However, in such tumors, co-administration of GC with anti-cancer drugs is a common clinical practice to prevent drug-induced allergic reaction or nausea/vomiting. Although GCR is ubiquitous in cancer cells and GCR has been linked to signal
transduction pathways pertinent to their growth, defense, and apoptosis 4, 5, 6, little is known regarding the effects of GC on the growth and chemosensitivity of common human carcinomas. Several studies have shown diverse effects of GC on
chemosensitivity in non-hematological neoplastic cells. Wolff et al. reported that DEX induced drug resistance toward cisplatin in C6 glioma cells 7. Weller et al. also reported a DEX-mediated cytoprotection in glioma cell lines 8, 9. However,
Benckhuijsen et al. reported an enhancement of melphalan cytotoxicity by DEX in
melanoma cells 10. A more comprehensive study is needed to clarify the role of GC on chemosensitivity of non-hematological neoplastic cells.
In the present study, we examined the effects of DEX on the chemosensitivity of 14 carcinoma cell lines. We found that GC exerted a GCR-related differential effect on the growth or chemosensitivity of the majority of carcinoma cells. The results of this study indicate that, while GC may be
co-administered with anti-cancer drugs for other reasons, the possible effect of GC on the chemosensitivity of some selected cancers may be clinically significant and requires further investigation.
三、方法
Cell Culture and Chemicals
SiHa cells (human cervical carcinoma), HeLa, Caski cells (human cervical
carcinoma), H460 cells (human lung carcinoma), Hep3B, Hut 7 cells (human hepatocellular carcinoma), and MCF-7 cells (human breast cancer) were obtained from the American Type Culture Collection (Rockville, MD). The MCF-7/MXR1 and MCF-7/TPT300 cells were derived from MCF-7 by selection for growth in increasing concentrations of mitoxantrone or topotecan, respectively. MCF-7/MXR1 cells were gifts from Dr. Kenneth Cowan (National Cancer Institute, USA). MCF-7/TPT300cells were selected as previously described 11. NPC-TW01 and NPC-TW04 cells
(nasopharyngeal cancer) were obtained as previously described 12,13. They were maintained in Dulbecco's Modified Eagle's Medium supplemented with 2 mM glutamine, 100 U/ml penicillin and 100 g/ml
streptomycin (Sigma Chemical Co., St. Louis, MO), and 10% heat-inactivated fetal bovine serum (Life Technologies, Inc., Gaithersburg, MD). AGS, N87, and SNU1 cells (human gastric cancer) were obtained from the
American Type Culture Collection (Rockville, MD) and maintained in RPMI1640 (Sigma Chemical Co.) supplemented with 2 mM glutamine, 10% fetal bovine serum, 100 U/ml penicillin and 100 g/ml streptomycin. Cisplatin was obtained from
Pharmacia-Upjohn (Kalamazoo, MI). DEX was purchased from Sigma Chemical Co., and 3H DEX (specific activity 35-50
Ci/mmol) was from Blossom
Biotechnologies Inc (Blossom, TX).
Cytotoxicity Assay
The in vitro growth inhibitory effects of
the drugs in all 14 cell lines were determined by the MTT assay as previously described with slight modification 14. Briefly, cells were plated in 96-well plates at 5 x 103 cells/well. After overnight incubation, various concentrations of drugs were added in triplicate samples to each culture. Cells were exposed to drugs continuously. After 3-4 days of culture, when cells in drug-free wells reached 90% confluency, 50 l of 2.5 mg/ml MTT (Sigma Chemical Co.) in PBS was added to each well, followed by
incubation for 4 hours at 37C. The formazan crystals were dissolved in DMSO. The
absorbance was determined with an ELISA reader at 540 nm. Absorbance values were normalized to the values obtained for the vehicle-treated cells to determine the percentage of survival. Each assay was performed in triplicate.
Measurement of GCR Content
The GCR content was measured by a whole-cell binding assay as previously described with minor modification 15. Briefly, cells with 90% confluency were subcultured and allowed to grow overnight, and then trypsinized and suspended in culture medium containing 10% fetal bovine serum (pH 7.2) to a density of 1~10 × 106 cells per ml. Cells were exposed to various
concentrations of 3H DEX from 1 to 100
nM in the presence or absence of 10 M unlabeled DEX, followed by incubation for 1 hr at 37C and harvested by centrifugation at 1,200 x g for 1 min. Cells were then washed three times in 3.0 ml of Hank’s balanced salt solution and finally suspended in 1.6 ml of the same solution. A 0.2 ml aliquot of this suspension was used for the determination of cell number, and 1.0 ml was assayed for radioactivity by a liquid scintillation counter. The presence of at least 200-fold excess of unlabeled DEX effectively competed out all of the binding of 3H DEX to specific GCR.
The difference in disintegrations per minute per cell between those samples incubated with 3H DEX alone and those with 200-fold
excess of unlabeled DEX represented the binding of 3H DEX to specific GCR. Using
the specific activity of 3H DEX, the number
of receptors per cell was calculated, assuming that each receptor binds to one DEX molecule.
Transfection of Reporter Gene and
Expression Vector
The human GCR-expressing plasmid, pS-hGR, and the luciferase reporter plasmid, MMTV reporter plasmid were gift from Prof. Chawnshang Chang (George H.Whipple Laboratory for Cancer Research, University of Rochester, Rochester). The MMTV reporter plasmid contains the 1.4-kb MMTV LTR which encompasses the natural GRE sequences, fused to the sequence coding for luciferase 16. It has been demonstrated previously that GR stimulate the rate of transcription of MMTV LTR . Moreover, GCR bind specifically to DNA sequences mapped within the MMTV LTR (i.e. GRE), which can confer GC responsiveness to a heterologous promoter. MCF-7, TW01, TW02, AGS, and SiHa cells were transfected by Lipofectamine 2000 (Life Technologies, Inc. GIBCO BRL, Gaithersburg, MD) according to the manufacturer’s protocol. The stable clones was selected by 400 g/ml hygromycin for 20 days. Single cell clones were obtained by limiting dilution of the hygromycin-resistant cells. In the MMTV reporter assay, MCF-7, TW01 and TW04 cells were either transiently transfected with MMTV reporter plasmid cells, or
co-transfected with MMTV reporter plasmid and pS-hGR (in a ratio of 5:1). Forty eight hours after transfection, 1 x 105 transfected cells were stimulated with 1 M DEX and incubated for an additional 6 hours. Reporter gene activity was determined with the
Reporter Luciferase Assay System (Packard, Netherlands). In the study of increasing the GCR content in AGS cells, the AGS cells were transfected with the pS-hGR. The stable clones were selected by 400 g/ml
hygromycin for 20 days. Single cell clones were obtained by limiting dilution of the hygromycin-resistant cells.
Western Blot Analysis
Cells were plated in 6 cm dishes at a density of 1 x 106 cells/dish. After incubation with DEX for the indicated time periods, the cells were harvested. Whole cell lysates and nuclear extracts were prepared according to the method of Staal et al 17. Protein concentration was determined by Bradford assay 18. Immunoblotting of GR and β-actin was performed using rabbit
polyclonal antibodies purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Signals were visualized with an enhanced chemiluminecence kit followed by exposure to X-ray films.
Electrophoretic mobility shift assay (EMSA) for NF-B.
- 32PdCTP end-labeled double-strand
oligo-deoxyribonucleotides
(5’-GGATTGGGACTT TCCCCTCC-3’ and 3’-CCTAACCCTGAAAGGGGAGG-5’ ) were used as the binding substrates for NF-B. The preparation of nuclear extracts for EMSA was performed according to the method of Andrews and Faller (12). Nuclear extracts of SiHa cells (10 g per assay) were incubated with 10,000 cpm of probe (0.1 to 0.5 ng) and 1 g poly(dI-dC) for 30 min at room temperature with a final reaction mixture of 15 l containing 20 mM HEPES, (pH7.5), 100 mM KCl, 0.2 mM EDTA, 20% glycerol, 1mM dithiothreitol, and 1 g/l BSA. Samples were analyzed in a 5% polyacrylamide gel with 0.25 x TBE as running buffer, and run at room temperature at 150 V for 2~ 2.5 h. The nuclear extract from TNF--treated SiHa cells was used as positive control. Antibody to p65 (Rel A) was added to the reaction mixture before the addition of labeled probe for supershift analysis. After electrophoresis, gels were dried and autoradiographed for 12 hours at -70C.
Transfection of reporter plasmid and measurement of luciferase and reporter gene activity.
The luciferase reporter plasmid, pM-Luc, contains the 1.4-kb MMTV LTR which
encompasses the natural GRE sequences (13). The other luciferase reporter plasmid,
pRB-Luc, contains five NF-B sites followed by a TATA box. These plasmids both contain the hygromycin resistance gene from SV2hygro. SiHa cells were transfected by Lipofectamine 2000 (Life Technologies, Inc. GIBCO BRL, Gaithersburg, MD) according to the manufacturer’s protocol. The stable clone was selected by 400 g/ml hygromycin for 20 days. Single cell clones were obtained by limiting dilution of the hygromycin-resistant cells. The
SiHa/B-reporter cell line was selected on the basis of TNF- induced luciferase activity and constitutive -galactosidase activity. For each time point, 1x105
SiHa/B-reporter cells were stimulated with 10 ng/ml TNF or cisplatin (20 and 200 M) and incubated for an additional 6 hours. Reporter gene activity was determined with the Reporter Luciferase Assay System (Packard, Netherlands).
Transfection of dominant-negative IB. The dominant-negative truncated IB (dnIB) cDNA was constructed by deletion of amino acids residues 1 to 70, which contain the phosphorylation sites (serine residues 32 and 36) of IB kinases (IKKs) and ubiquitin binding sites (lysine residues 21 and 22). This cDNA was inserted into the vector pRCMV (purchased from Invitrogen) followed by the CMV promoter. The empty vector was used for the generation of control cells. SiHa cells were transfected by
lipofectamine 2000 (Life Technologies, Inc. GIBCO BRL, Gaithersburg, MD)
according to the manufacturer’s protocol. The stably transfected SiHa cells were pooled by hygromycin selection for 20 days after transfection. The experiments
examining the effect of DEX on the growth of these cells were performed within 30 days of each transfection.
Quantification of GCR mRNA Expression Using RTQ RT-PCR
An RNA extraction kit (Rneasy Minit kit; Qiagen, Valencia, CA) was used to extract total RNA from the frozen resected tumor tissue.
RTQ RT-PCR, a newly developed kinetic quantitative RT-PCR method, is considered to be one of the most sensitive and accurate methods for the quantification of nucleic acid (DNA and RNA) in tissue samples. This
method (the TaqMan reaction) is based on the 5' nuclease activity of Taq polymerase, which
cleaves a specific dual-labeled fluorogenic hybridization probe during the extension phase of the PCR. As long as this sequence-specific probe is intact, emission by a reporter dye at its 5' end is quenched by a second fluorescent dye at the 3' end. During the extension phase of the PCR, Taq polymerase hydrolyses the probe and
releases the reporter dye, resulting in an increase in peak fluorescence emission that is directly proportional to the number of amplified copies and is detected and quantified by a detector in real time. A higher starting copy number of the nucleic acid results in an earlier increase in fluorescence. The threshold cycle (CT) is defined as the fractional cycle number at
which the fluorescence generated by cleavage of the probe exceeds a fixed threshold above baseline. For a chosen threshold, a smaller starting copy number results in a higher CT
value. In this study, we used RTQ RT-PCR for the relative quantification of GCR mRNA in tumor specimens, with TATA box-binding protein (TBP, a component of the DNA-binding protein complex TFIID) mRNA as an internal control.
Primers, Probes, and Reference Internal Control mRNA
Primers and probes were chosen using the computer program Primer Express
(Perkin-Elmer Applied Biosystems, Forster City, CA). Primers and probes were synthesized by and purchased from Perkin-Elmer Applied Biosystems. On the basis of the cDNA sequence (gene bank accession no. m32977),
the sequences of the primers and probe used for RTQ RT-PCR of GCR mRNA were as follows: (1) forward primer, 5'-TGG CAG CGG TTT TAT CAA CTG-3' (in exon 8); (2) reverse primer, 5'-ATG TTT GGA AGC AAT AGT TAA GGA GAT TT-3' (in exon 9); and (3) probe, 5'-CAA AAC TCT TGG ATT CTA TGC ATG AAG TGG TTG-3' (spanning the exon 8-exon 9 junction to avoid quantification of the PCR product of contaminating GCR genomic DNA). TBP in the tumor sample was quantified in the same way as the endogenous RNA control, using forward and reverse primers and a probe designed for TBP mRNA analysis, the forward primer sequence being 5'-CAC GAA CCA CGG CAC TGA TT-3', the reverse primer sequence being 5'-TTT TCT TGC TGC CAG TCT GGA C-3', and the probe sequence being 5'-TGT GCA CAG GAG CCA AGA GTG AAG A-3'.
Standard Curve Sample Preparation
The standard curve samples used for RTQ RT-PCR were prepared by serial dilution of a specific RNA sample to cover the range of 5 to 500 ng. The serially diluted samples were aliquoted and stored at -80°C until use.
RT-PCR Procedure
The amplification mixture (50 μL) contained 50 ng of sample RNA, × 5 TaqMan EZ buffer (10 μL), 25 mmol/L manganese acetate (6 μL), 300 μmol/L dATP, dCTP, and dGTP, 600 µmol/L dUTP, 5 units of rTth DNA polymerase, 0.5 units of AmpErase uracil N-glycosylase (UNG), 200 nmol/L GCR (or TBP) forward and reverse primers, and 100 nmol/L GCR (or TBP) probe (all from Perkin-Elmer Applied Biosystem). The rTth DNA polymerase had both RTase and Taq polymerase activity. The thermal cycling parameters were an initial step of 2 minutes at 50°C, 30 minutes at 60°C for reverse
transcription, 5 minutes at 95°C for deactivation, and then 40 cycles at 94°C for 20 seconds and 62°C for 1 minute for the melting and combined
annealing and extension phases of the PCR reaction. Each assay included duplicate standard curve samples, a no-template control, and triplicate total RNA samples. All samples with a coefficient of variation (CV) higher than 10% were retested.
Detection of Fluorescence Emission and Quantification of VEGF mRNA and IL-8 mRNA
Fluorescence emission from the reporter dye (FAM-6-carbosy-fluorescein, peak fluorescence emission at 518 nm) was detected online in real time using an ABI prism 7,700 sequence detection system (Perkin-Elmer Applied Biosystem). The amount of GCR mRNA in the cell lines or tumor sample, standardized to the TBP mRNA, was expressed as follows: -[DELTA]CT = -[CT GCR - CT TBP]. The ratio of the
amount of GCR mRNA/amount of TBP mRNA was then calculated as 2-[DELTA]C
T × K (K indicates
constant).
The -[DELTA]CT was analyzed as both a
continuous and dichotomous variable. The median value was used as the cutoff to distinguish between low and high levels of GCR mRNA expression.
Statistical Analysis
Independent t test was used to assess the correlation of GCR contents with the effect of DEX.
四、結果
DEX Affects Either Growth or
Chemosensitivity in 7 of the 14 carcinoma Cell Lines
DEX (0.01-1.0M) inhibited cell growth in MCF-7, MCF-7/MXR1,
MCF-7/TPT300, and HeLa cells. However, DEX alone, up to 20 M, was not toxic to the other 10 cell lines, including AGS, N87, SNU1, SiHa, Caski, Hep3B, Hut 7, TW01,
TW04, and H460. The latter 10 cell lines were further tested for the effect of GC on the chemosensitivity of carcinoma cells
toward cisplatin, doxorubicin, 5FU, and taxol. Pretreatment of SiHa cells with 1 M DEX for 3 hours decreased the IC50 of cisplatin
from 18.6±1.9 M to 9.7±2.0 M. This cytotoxicity-enhancing effect could be observed even when the concentration of DEX was as low as 1 nM (data not shown). In contrast, DEX slightly decreased
chemosensitivity toward cisplatin,
doxorubicin, 5-FU, and taxol in H460 and Hep3B cells. DEX had no effect on the
chemosensitivity of AGS, N87, SNU1, Hut-7, Caski, NPC-TW01, and NPC-TW04 cells.
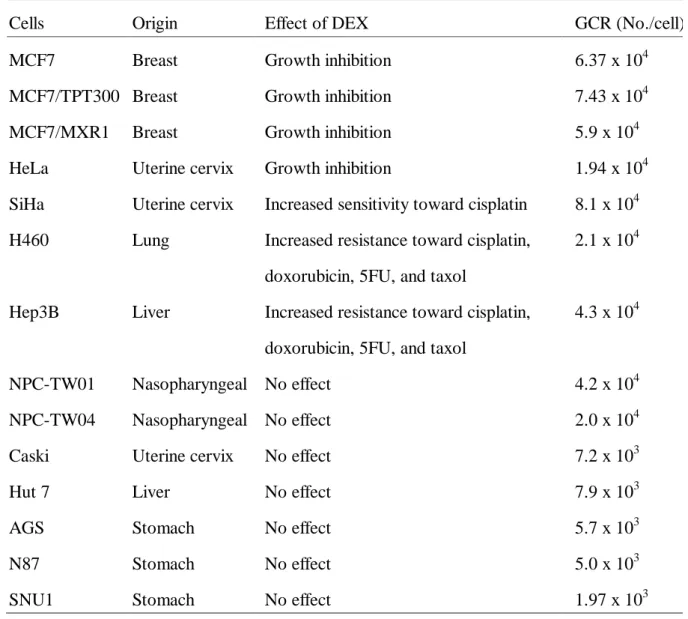
GC Effect Correlates well with GCR Content of the Cells
The GCR contents of these 14 cell lines are listed in Table 1. The GCR contents of the 7 cell lines affected by DEX were significantly higher than those of the other 7 cell lines unaffected by DEX (5.2±2.5 ×104 vs. 1.3±1.4 ×104, P=0.005) suggesting GCR is one of the pivotal mediators of the effect of DEX on carcinoma cell. The GCR content of human lymphocytes, the internal control for these experiments, was parallely tested and was within the reported range (2,500 ~ 5,400 sites/cell) 19.
GC-unresponsive GCR-rich Carcinoma Cells Have Dysfunctional GCR
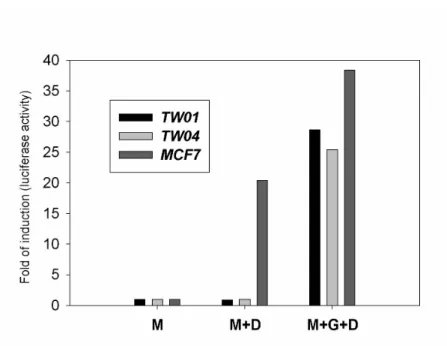
As shown in Table 1, we noticed that DEX had no effect on NPC-TW01 and NPC-TW04, two cell lines with GCR content as high as that of the 7 GC-responsive cell lines. The function of the GCR in these two cell lines was further examined. As shown in Fig. 1, MCF-7 cells contained endogenous DEX-responsive GCR while NPC-TW01 and NPC-TW04 cells did not. Further, when NPC-TW01 and NPC-TW04 cells were co-transfected with MMTV reporter plasmid and pS-GCR, which contains functional human GCR gene, the response to DEX was restored (Fig 1). These data strongly
suggested that the function of endogenous GCR of NPC-TW01 and NPC-TW04 cells
was probably impaired.
Expression of GCR in GCR-poor Cells Increases its Responsiveness to DEX
To further examine whether the GCR content is pivotal in mediating the
susceptibility to DEX in carcinoma cell, we transfected pS-hGR to AGS cells, a GCR low-expressing cell line. The GCR content in empty vector-transfected AGS cells, and pooled stably pS-hGR-transfected AGS cells were 5.2 x 103/cell and 1.42 x 104/cell, respectively. Treatment of DEX alone has no effect on cell growth in these cells. However, as shown in Fig. 2, pS-hGR transfected AGS cells that expressing high GCR content became susceptible to the effect of DEX with increasing drug resistance toward cisplatin. The GCR content in single cell cloned
pS-hGR transfected AGS cells clone 1, 2, and 3 were 1.54 x 104/cell, 1.32 x 104/cell, and 5.8 x 103/cell, respectively. The cells that express high GCR content (clone 1 and clone 2) were susceptible to the effect of DEX with increasing resistance toward cisplatin, but the cells which express low GCR content (clone 3) remained non-susceptible to DEX treatment
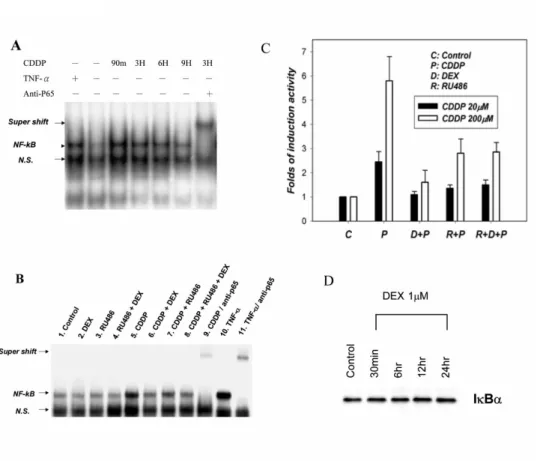
DEX Suppressed Cisplatin-induced NF-B Activation in SiHa Cells
To explore the mechanism underlying the chemosensitizing effect of DEX in SiHa cells, EMSA assay of NF-B DNA binding activity and reporter luciferase assay of NF-B transcription activity were done. As shown in Fig. 3A, NF-B DNA binding activity transiently increased after exposure to 20 M (IC50) cisplatin. This NF-B DNA
binding activity was blocked by
pre-incubating the cells with 1 M DEX (Fig. 3B). While RU486 alone had some intrinsic effect on the suppression of NF-B, it could only partially reverse the effect of DEX on the suppression of NF-B DNA binding activity (Fig. 3B). The transactivating activity of NF-B on its cis elements was further verified in SiHa cells stably
transfected by a reporter construct containing five NF-B binding sites. Treatment with
cisplatin (20~200 M) resulted in the induction of luciferase activity, which could be repressed by pretreatment with DEX. Again, RU486 could partially reverse the effect of DEX on the repression of NF-B activity, while RU486 alone had some intrinsic effect on the repression of NF-B activity (Fig. 3C). The effect of DEX on the IB expression in SiHa cells was also examined. Western blot analysis of
whole-cell protein showed that DEX did not upregulate the expression of IB in SiHa cells (Fig. 3D). By contrast, in other cell lines, NF-B was either not activated by TNF- or cisplatin (H460), or cisplatin-activated NF-B was not suppressible by DEX
(Hep3B, AGS, SNU1, and N87). These data suggest that protein-protein interaction between activated GCR and NF-B plays a central role in the chemosensitizing effect of DEX in SiHa cells.
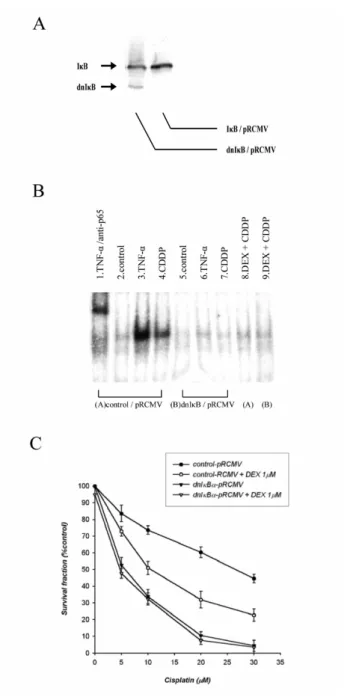
Inhibition of NF-B Activation Blocks the Cytotoxicity-enhancing Effect of DEX in SiHa Cells
To further examine the role of NF-B in the cytotoxicity-enhancing effect of DEX, we generated a re-combinant plasmid containing dominant negative IB (dnIB) gene. This dnIB protein does not contain the residues necessary for signal-induced phosphorylation and proteasome-mediated degradation of IB, thereby preventing dissociation and translocation of NF-B to the nucleus. The expression of the dnIB in pooled stably transfected SiHa cells was verified by Western blot analysis. As shown in Fig. 4A, the control-pRCMV-transfected SiHa cells contained only the endogenous wild-type IB protein, while the dnIB-pRCMV-transfected SiHa cells contained an additional band representing the truncated exogenous IB protein. Results of EMSA showed that NF-B binding activity
was markedly suppressed in the
dnIB-pRCMV-transfected cells after either TNF- or cisplatin treatment (Fig. 4B). In addition, as shown in Fig. 4C, the cytotoxicity-enhancing effect of DEX in
dnIB-pRCMV-transfected SiHa cells was abolished. The dnIB-pRCMV-transfected SiHa cells had also become more sensitive to
cisplatin as compared to the
control-pRCMV-transfected SiHa cells(Fig. 4C). These data confirmed that NF-B plays a central role in the chemosensitizing effect of DEX in SiHa cells.
Expression of GCR mRNA in Carcinoma Cell Lines and Human Breast Cancer Tumor Samples
Real-time RT-PCR of TBP mRNA showed the integrity of the tumoral RNA to be good. GCR mRNA expression in breast cancer tissue (standardized to TBP mRNA) was expressed as -[DELTA] CT = -(CTGCR –CT TBP). Fig 5A show that the level of GCR
mRNA expression correlate well with the GCR number measured by 3H DEX ligand
biding assay. Fig 5B showed that in 10 breast cancer patients tumor samples, the GCR mRNA expression is variable, and some of them has high level of GCR mRNA
expression as compare with the carcinoma cell lines.
四、討論
This study has demonstrated that GC affects either growth or chemosensitivity in a substantial portion of carcinoma cells. Since GCs are commonly co-administered with anticancer drugs such as taxanes and platinums, how GC alter the effect of chemotherapy may have to be taken into consideration in clinical practice. As shown in this study, it may not be difficult to identify those carcinoma patients of whom tumor response is going to be affected by GC, since only cells with high GCR content are affected. However, how GC will actually affect the growth of tumors of these cancer patients remains uncertain since the effects of GC, as disclosed in this study, are extremely diverse and without useful cellular or molecular predictors.
content of the cells and the magnitude of physiologic response to GC has been reported in hematologic malignancies20. The sensitivity of many lymphoid cell lines to GC-induced growth arrest and apoptosis is directly proportional to intracellular receptor content 21-23. Several studies also
identified a correlation between reduced GCR expression and a poor treatment
response as well as poor prognosis in patients with acute lymphocytic leukemia, suggesting that reduced GR expression could lead to clinical glucocorticoid resistance 19, 24-26. Our study demonstrated that the
susceptibility to the effect of DEX on cell growth or chemosensitivity in carcinoma cells is also correlate well with the level of GCR content. However, the GCR contents of the GC-responsive carcinoma cells are
almost 10 times higher than that of lymphoid cells 24-26, suggesting that the cellular contexts or the signal transduction pathways for the interaction of GC and GCR are probably different between these two groups of cells.
In this study, the only two cell lines, NPC-TW01 and NPC-TW04, which have relatively high GCR content but not
susceptible to the growth regulatory effect of DEX, were found to have non-functional endogenous GCR. Previous studies on both human and mouse cell lines have shown that somatic mutation in the GCR gene is the principal mechanism for in vitro acquisition of GC resistance 27-33. Whether
NPC-TW01 and NPC-TW04 contain GCR gene mutations needs to be further
elucidated.
Activated GCR may activate or
suppress gene expression through interaction with respective positive or negative
cis-acting regulatory elements in the
promoters regions 34, 35. Activated GR can also regulate the expression of GC
responsive genes indirectively through protein-protein interactions with other
transcription factors such as NF-B and AP-1 36–38. Inactivation of NF-B or AP-1 has
been shown to alter the vulnerability of cancer cells to several cytotoxic agents 39,
40. Activation of NF-B has been
implicated in mediating drug resistance of cancer cells. NF-Bcould be activated by a variety of stresses, including oxidative stress and DNA damage (41-46). Activated NF-B may prevent the triggering of apoptosis, and thus result in drug resistance against
DNA-damaging agents (47-50). The molecular mechanism of NF-B-mediated protection of cells remains unclear, but may involve the up-regulation of caspase
inhibitors (51). In this study, we have provided evidence that NF-B plays an important role in mediating the drug resistance of SiHa cells. Suppression of NF-B activity by dnIB not only abolished the chemosensitizing effect of DEX, but also increased the chemosensitivity of SiHa cells to cisplatin (Fig. 5B and 5C). DEX had no effect on cells without
discernable changes of NF-B. However, it remains to be clarified why certain
carcinoma cell lines do not have an NF-B response to cisplatin or have an NF-B response which cannot be attenuated by DEX.
Our preliminary data have indicated that suppression of NF-B is one of the major mechanism of interacting cisplatin sensitivity in SiHa cell. However, it remains difficult to explain the diverse effect of GC on GCR-rich carcinoma cells 52. Although high-GCR content is necessary for a response of the cells to GC, the diversified and even contradictory effects of GC on these cells cannot be simply explained by the amount of GCR. Our findings suggest that an upstream switch point at the level of GC-GCR
interaction may work to segregate the direction of downstream pathways. Recent studies on the action of androgen receptor (AR) may provide a possible example for the diverse effects of GC on carcinoma cells. Co-regulators of AR have played crucial roles in determining the ultimate activity of AR, and the presence or absence of certain co-regulators may even change the activity of anti-androgens to become androgens 53, 54. Several novel co-regulators of GC have also been found to play important roles in the signaling pathway of the GCR 55. The
possibilities that the specific presence of certain co-regulators of GCR in different carcinoma cells may dictate the ultimate effect of GC need to be clarified.
After a single oral dose of 7.5 mg of DEX, the serum concentration of DEX was found to be around 0.12 M lasting for 1~3 hours 56. However, the serum concentration of DEX may reach 2 M after a single
intravenous infusion of 80~100 mg of DEX 57. Since the administration of relatively high-dose DEX, at the range of 10-50 mg/day, or its equivalents, is widely used for the
prevention of cisplatin-induced
nausea/vomiting and taxanes-induced allergic reactions, the possible effect of GC on the chemosensitivity of some cancer patients needs to be seriously considered. In summary, the results of this study suggest that GC exerts a GCR-dependent effect on the growth or chemosensitivity in a substantial portion of carcinoma cells. The clinical relevance and the cellular mechanisms that dictate the disparate effects of GC need to be further clarified.
五、計畫成果自評
In summary, the results of this study suggest that GC exerts a GCR-dependent effect on the growth or chemosensitivity in a substantial portion of carcinoma cells. The clinical relevance and the cellular
mechanisms that dictate the disparate effects of GC need to be further clarified. The current results have be submitted for publication in the peer-reviewed journal.
六、參考文獻
1. Haskell, C. M. Antineoplastic agents. In: Haskell, C. M., Break, J. S.(eds.), Cancer Treatment, forth edition, pp. 105-106. Philadelphia: W. B. Saunders, 1995. 2. Wood, A. C., Waters, C. M., Garner, A.,
and Hickman, J. A. Changes in c-myc expression and the kinetics of
dexamethasone-induced programmed cell death and apoptosis in human lymphoid leukemia cells. Br. J. Cancer, 69: 663-669, 1994.
3. Schwartzman, R. A. and Cidlowski, J. A. Mechanism of tissue-specific induction
of internucleosomal deoxyribonucleic acid cleavage activity and apoptosis by glucocorticoids. Endocrinology, 133: 591-599, 1993.
4. Wright, A. P., Zilliacus, J., McEwan, I. J., Dahlman-Wright, K., Almlof, T., and Carlstedt-Duke, J. Structure and function of the glucocorticoid receptor. J. Steroid Biochem. Mol. Biol., 47: 11-19, 1993. 5. Schwartzman, R. A. and Cidlowski, J. A.
Glucocorticoid-induced apoptosis of lymphoid cells. Int. Arch. of Allergy Immunol., 105: 347-354, 1994. 6. McEwan, I. J., Wright, A. P., and
Gustafsson, J. A. Mechanism of gene expression by the glucocorticoid receptor: role of protein-protein interactions.
Bioessays, 19: 153-160, 1997.
7. Wolff, J. E., Denecke, J., and Jurgens, H. Dexamethasone induces partial resistance to cisplatinum in C6 glioma cells.
AntiCancer Res., 16: 805-809, 1996. 8. Weller, M., Schmidt, C., Roth, W., and
Dichgans, J. Chemotherapy of human malignant glioma: prevention of efficacy by dexamethasone? Neurology, 48: 1704-1709, 1997.
9. Naumann, U., Durka , S., and Weller , M. Dexamethasone-mediated protection from drug cytotoxicity: association with p21 WAF1/CIP1 protein accumulation? Oncogene, 17: 1567-1575, 1998. 10. Benckhuijsen, C., Osman , A. M., Hillebrand, M. J., and Smets, L. A. "Glucocorticoid effect on melphalan cytotoxicity, cell-cycle position, cell size, and [3H]uridine incorporation in one of three human melanoma cell lines". Cancer Res., 47: 4814-4820, 1987. 11. Yang, C. H., Schneider, E., Kuo, M. L.,
Volk, E. L., Rocch, E., and Chen, Y. C. BCRP/MXR/ABCP expression in topotecan-resistant human breast carcinoma cells. Biochem. Pharmacol., 60: 831-837, 2000.
12. Lin, C.T., Wong, C.I., Chan, W.Y., Tzung, K.W., Ho, J. K. C., Hsu, M.M. & Chuang, S.M. (1990). Establishment and
characterization of two nasopharyngeal carcinoma cell lines. Lab. Investig. 62, 713-724.
H.-M., Wu, H.-C., Hsu, M.-M., Chuang, S.-M. & Wang, C.-C.(1993).
Characterization of seven newly established nasopharyngeal carcinoma cell lines. Lab. Investig. 68, 716-727. 14.Carmichael, J., DeGraff, W. G., Gazdar, A.
F., Minna, J. D., and Mitchell, J. B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res., 47: 936-942, 1987. 15. Harmon, J. M. and Thompson, E. B.
Isolation and characterization of dexamethasone-resistant mutants from human lymphoid cell line CEM-CT. Mol. Cell Biol., 1: 512-521, 1981.16
16 Scheidereit, C., Geisse, S., Westphal, H. M., and Beato, M. The glucocorticoid receptor binds to defined nucleotide sequences near the promoter of mouse mammary tumour virus. Nature 304: 749-752, 1983.
17. Staal, F. J., Roederer, M., Herzenberg, L. A., and Herzenberg, L. A. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA, 87: 9943-9947, 1990. 18. Bradford, M. M. A rapid and sensitive
method for the quantitation of microgram quantities of protein utilizing the
principle of protein-dye binding. Anal. Biochem., 72: 248-254, 1976. 19. Lippman, M. E., Yarbro, G. K., and
Leventhal, B. G. Clinical implications of glucocorticoid receptors in human leukemia. Cancer Res., 38: 4251-4256, 1978.
20. Vanderbilt, J. N., Miesfeld, R., Maler, B. A., and Yamamoto, K. R. Intracellular receptor concentration limits
glucocorticoid-dependent enhancer activity. Mol. Endocrinol., 1: 68-74, 1987.
21. Gehring, U., Mugele, K., and Ulrich, J. Cellular receptor levels and
glucocorticoid responsiveness of lymphoma cells. Mol. Cell Endocrinol.,
36: 107-113, 1984.
22. Bourgeois, S. and Newby, R. F. Diploid and haploid states of the glucocorticoid receptor gene of mouse lymphoid cell
lines. Cell, 11: 423-430, 1977. 23. Chapman, M. S., Askew, D. J.,
Kuscuoglu, U., and Miesfelt, R. L.
Transcritional control of steroid-regulated apoptosis in murine thymoma cells. Mol. Endocrinol., 10: 967-978, 1996.
24.Bloomfield, C. D., Smith, K. A., Peterson, B. A., and Munck, A. Glucocorticoid receptors in adult acute lymphoblastic leukemia. Cancer res., 41: 4857-4860, 1981.
25. Pui, C. H., Dahl, G. V., Rivera, G., Murphy, S. B., and Costlow, M. E. The relationship of blast cell glucocorticoid receptor levels to response to single-agent steroid trial and remission response in children with acute lymphoblastic
leukemia. Leuk. Res., 8: 579-585, 1984. 26. Pui, C. H., and Costlow, M. E. Sequential
studies of lymphoblast glucocorticoid receptor levels at diagnosis and relapse in childhood leukemia: an update. Leuk. Res. 10: 227-229, 1986.
27. Wells, R.J., Mascaro, K., Young, P. C., and Cleary, R. E. Glucocorticoid
receptors in the lymphoblasts of patients with glucocorticoid-resistant childhood acute lymphoblastic leukemia. Am. J. Pediatr. Hematol. Oncol., 3: 259-264, 1981.
28. Powers, J. H., Hillmann, A. G., Tang, D. C., and Harmon, J. M. Cloning and expression of mutant glucocorticoid receptors from glucocorticoid-sensitive and glucocorticoid-resistant human leukemic cells. Cancer Res., 53: 4059–4065, 1993.
29. Danielsen, M., Northrop, J. P., and Ringold, G. M. The mouse glucocorticoid receptor: mapping of functional domains by cloning, sequencing and expression of wild-type and mutant receptor proteins. EMBO J., 5: 2513–2522, 1986.
30. Ashraf, J., and Thompson, E. B. Identification of the activation-labile gene: a single point mutation in the human glucocorticoid receptor presents as two distinct receptor phenotypes. Mol. Endocrinol., 7: 631–642, 1993.
31. Lee, S., Duncan, A., Chou, H., Chen, D., Kohli, K., Huang, C. F., and Stallcup, M. R. A somatic cell genetic method for
identification of untargeted mutations in the glucocorticoid receptor that cause hormone binding deficiencies. Mol. Endocrinol., 9: 826–837, 1995. 32. Strasser-Wozak, E. M. C.,
Hattmannstorfer, R., Hala, M., Hartmann, B. L., Fiegl, M., Geley, S., and Kofler, R. Splice site mutation in the glucocorticoid receptor gene causes resistance to
glucocorticoid-induced apoptosis in a human acute leukemic cell line. Cancer Res., 55: 348–353, 1995.
33. Hala, M., Hartmann, B. L., Bock, G., Geley, S., and Kofler, R.
Glucocorticoidreceptor- gene defects and resistance to glucocorticoid-induced apoptosis in human leukemic cell lines. Int. J. Cancer, 68: 663–668, 1996.
34. Karkera, J. D., Taymans, S. E., Turner, G., Yoshikawa, T., Detera-Wadleigh, S. D., and Wadleigh, R. G. Deletion of a consensus oestrogen response element half-site in the glucocorticoid receptor of human multiple myeloma. Br. J.
Haematol., 99: 372–374, 1997. 35. Beato, M., Chavez, S., and Truss, M.
Transcriptional regulation by steroid hormones. Steroids, 61: 240–251, 1996. 36. Reichardt, H. M., and Schutz, G.
Glucocorticoid signalling–multiple
variations of a common theme. Mol. Cell. Endocrinol., 146: 1–6, 1998.
37. McEwan, I. J., Wright, A. P. H., and Gustafsson, J-Å. Mechanism of gene expression by the glucocorticoid receptor: role of protein-protein interactions.
Bioessays, 19: 153–160, 1997.
38. Dumont, A., Hehner, S. P., Schmitz, M. L., Gustafsson, J-Å., Liden, J., Okret, S., van der Saag, P. T., Wissink, S., van der Burg, B., Herrlich, P., Haegeman, G., De Bosscher, K., and Fiers, W. Cross-talk between steroids and NF-k: what language? Trends Biochem. Sci., 23: 233–235, 1998.
39. Gottlicher, M., Heck, S., and Herrlich, P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J. Mol. Med., 76: 480–489, 1998.
40. Van Antwerp, D. J., Martin, S. J., Kafri, T., Green, D. R., and Verma, I. M. Suppression of TNF-alpha-induced
apoptosis by NF-kappa B. Science, 274: 787-789, 1996.
41. Sen, C. K. and Packer, L. (1996)
Antioxidant and redox regulation of gene transcription. FASEB J. 10, 709-720. 42. Friedberg, E. C., Walker, G. C., and Siede,
W. (1995) DNA repair and mutagenesis. pp. 14-19. ASM Press: Washington D.C. 43. Bertoncini, C. R. and Meneghini, R.
(1995) DNA strand breaks produced by oxidative stress in mammalian cells exhibit 3'-phosphoglycolate termini. Nucleic Acids Res. 23, 2995-3002. 44. Piret, B. and Piette, J. (1996)
Topoisomerase poisons activate the transcription factor NF-kappaB in ACH-2 and CEM cells. Nucleic Acids Res. 24, 4242-4248.
45. Legrand-Poels, S., Bours, V., Piret, B., Pflaum, M., Epe, B., Rentier, B., and Piette, J. (1995) Transcription factor NF-kappa B is activated by
photosensitization generating oxidative DNA damages. J. Biol. Chem. 270, 6925-6934.
46. Quinto, I., Ruocco, M. R., Baldassarre, F., Mallardo, M., Dragonetti, E., and Scala, G. (1993) The human immunodeficiency virus type 1 long terminal repeat is activated by monofunctional and bifunctional DNA alkylating agents in human lymphocytes. J. Biol. Chem. 268, 26719-26724.
47. Van Antwerp, D. J., Martin, S. J., Kafri, T., Green, D. R., and Verma, I. M. (1996) Suppression of TNF-alpha-induced
apoptosis by NF-kappa B. Science 274, 787-789.
48. Beg, A. A. and Baltimore, D. (1996) An essential role for NF-kappaB in
preventing TNF-alpha-induced cell death. Science 274, 782-784.
49. Baringo M. (1996) A life-death balance within the cell. Science 274, 724.
50. Wang, C. Y., Mayo, M. W., and Baldwin, A. S. (1996) TNF-alpha and cancer therapy-induced apoptosis: potentiation by inhibition of NF-B. Science 274, 784-787.
51. Chu, Z. L., McKinsey, T. A., Liu, L., Gentry, J. J., Malim, M. H., and Ballard, D. W. (1997) Suppression of tumor
necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc. Natl. Acad. Sci. USA 94, 10057-10062.
52. Lu, Y. S., Yeh, P. Y., Chuang, S. E., Yeh, K. H., Yang, C. H., Kuo, M. L., and Cheng, A. L. Glucocorticoids Affect Growth and Chemosensitivity of Carcinoma Cells via Diverse Glucocorticoid Receptor-related Pathways. Proc. American Association for Cancer Res. 42: 2951, 2001.
53. Wang, C. Y., Mayo, M. W., and Baldwin, A. S. TNF-alpha and cancer
therapy-induced apoptosis: potentiation by inhibition of NF-B. Science, 274: 784-787, 1996.
54. Kang, H. Y., Yeh, S., Fujimoto, N., and Chang, C. " Cloning and characterization of human prostate coactivator ARA54, a novel protein that associates with the androgen receptor". J. Biol. Chem., 274: 8570-8576, 1999.
55. Kino, T., Gragerov, A., Kopp, J. B., Stauber, R. H., Pavlakis, G. N., and Chrousos, G. P. The HIV-1
virion-associated protein vpr is a
coactivator of the human glucocorticoid receptor. J. Exp. Med., 189: 51-62, 1999. 56. McKenna, N. J., Lanz, R. B., and
O'malley, B. W. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Reviews, 20: 321-344, 1999.
57. Weijtens, O., Schoemaker, R. C., Cohen, A. F., Romijn, F. P., Lentjes, E. G., and Van Rooij, J. Dexamethasone
concentration in vitreous and serum after oral administration. Am. J. Ophthalmol., 125: 673-679, 1998.
58. Brady, M. E., Sartiano, G. P., Rosenblum, S. L., Zaglama, N. E., and Bauguess, C. T. The pharmacokinetics of single high doses of dexamethasone. Eur. J. Clin. Pharmacol., 32: 593-596, 1987.
Table 1 Correlation of glucocorticoid receptor content with the effect of dexamethasone in carcinoma cells
Cells Origin Effect of DEX GCR (No./cell)
MCF7 Breast Growth inhibition 6.37 x 104
MCF7/TPT300 Breast Growth inhibition 7.43 x 104
MCF7/MXR1 Breast Growth inhibition 5.9 x 104
HeLa Uterine cervix Growth inhibition 1.94 x 104
SiHa Uterine cervix Increased sensitivity toward cisplatin 8.1 x 104
H460 Lung Increased resistance toward cisplatin,
doxorubicin, 5FU, and taxol
2.1 x 104
Hep3B Liver Increased resistance toward cisplatin,
doxorubicin, 5FU, and taxol
4.3 x 104
NPC-TW01 Nasopharyngeal No effect 4.2 x 104
NPC-TW04 Nasopharyngeal No effect 2.0 x 104
Caski Uterine cervix No effect 7.2 x 103
Hut 7 Liver No effect 7.9 x 103
AGS Stomach No effect 5.7 x 103
N87 Stomach No effect 5.0 x 103
18
Fig. 1. Functional assay of the GCR in NPC-TW01 and NPC-TW04 cells.
NPC-TW01, NPC-TW04, and MCF-7 cells were transiently transfected with MMTV reporter plasmid (lane M and lane M+D) or co-transfected with MMTV reporter plasmid and pS-hGR (lane M+G). The cells were then treated with 1 M DEX for 6 hours (lane M+D and M+G+D). Then the luciferase activity was assayed and
represented in terms of folds of the induction activity of the control (lane M). All values represent means ± standard deviation of 3 experiments.
Fig. 2. Increased drug resistance to cisplatin in pS-hGR-transfected AGS. AGS
cells were transfected with pS-hGR and MTT assay was performed. a Western blot analysis for GR and β-actin in whole cell lysate of transfected AGS cells. Lane1: AGS/GCR-pool, lane 2: AGS/empty vector, lane 3: AGS/GCR-c1, lane 4:
AGS/GCR-c2, lane 5: AGS/GCR-c3. b, c, and d Pretreatment with DEX 1 M for 3 hrs diminished cisplatin cytotoxicity in AGS/GCR-pool, AGS/GCR-c1, and
AGS/GCR-c2 cells. e and f Pretreatment with DEX 1 M for 3 hrs had no effect on the cisplatin cytotoxicity in AGS/empty vector cells and AGS/GCR-c3.
AGS/GCR-pool: AGS cells transfected with pS-hGR, pooled cells. AGS/empty vector: AGS cells transfected with empty vector. AGS/GCR-c1 to c3: AGS cells transfected with pS-hGR, single cell cloned, clone 1 to clone 3. All values represent mean ± standard deviation of 6 separate wells.
19 GCR β-actin A G S / G C R -p o o l A G S / e m p t y v e c t o r A G S / G C R -c 1 A G S / G C R -c 2 A G S / G C R -c 3
Fig 2
20
Fig. 3. Effect of DEX and RU486 on cisplatin-induced NF-B activity. A, Nuclear
extract was prepared and EMSA was performed as described in the Materials and Methods. Exposure of SiHa cells to cisplatin 20 M for 3 hours resulted in activation of NF-B activity. The TNF- lane represents positive control. Supershift by anti-p65 antibody verifies the correct band of NF-B. B, SiHa cells were pretreated with or without 1 M DEX and with or without 1 M RU486 for 24 hours. Cells were then exposed to 20 M cisplatin for 3 hours. DEX and RU486 had no effect on NF-B
(lane 2,3,and 4). Cisplatin activated NF-B (lane 5), but DEX pretreatment completely abolished cisplatin-induced NF-B activation (lane 6). RU486 partially abrogated the effect of DEX, but RU486 also had some intrinsic effect in reversing cisplatin-induced NF-B activation. C, SiHa cells were stably transfected with luciferase reporter plasmid containing five NF-B sites as described in Materials and Methods. The SiHa/B-reporter cells were pretreated with or without 1 M DEX and with or without 1M RU486 for 24 hours. Cells were then exposed to 20 or 200 M cisplatin for 3 hours. The luciferase activity was assayed and represented in terms of folds of the induction activity of the control. All values in Fig. 3C represent means ± standard deviation of 3 experiments. D, Effect of DEX on IB expression. SiHa cells were exposed to 1 M DEX for different durations before harvesting. Western blot analysis of the whole cell lysates was performed. The protein amount of IB was not changed after DEX treatment. Abbreviations: CDDP, cisplatin; TNF-, tissue necrosis factor-, N.S., non-specific binding.
21
Fig. 4. Effect of DEX on cisplatin-induced NF-B activity in dominant negative IB transfected SiHa
cells. A, Western blot analysis for IB in whole cell lysate of control-pRCMV-transfected SiHa cells and dnIB-pRCMV-transfected SiHa cells. An additional band in the lane of
dnIB-pRCMV-transfected SiHa cells represents of the exogenous truncated IB protein. B,
Nuclear extract was prepared and EMSA was performed as described in Materials and Methods. Exposure of control-pRCMV-transfected cells to cisplatin 20M 3 hours or TNF- 30 minutes resulted in activation of NF-B (lane 3, 4), which was suppressible by DEX 1 M pretreatment (lane 8). Super-shift by anti-p65 antibody verifies the correct band of NF-B (lane 1). The NF-B activity was not increased in dnIB-pRCMV-transfected SiHa cells exposed to TNF- and cisplatin (lane 6, 7). C, Effect of DEX on the chemosensitivity in dominant negative IB transfected SiHa cells. Cell numbers
22
were measured by MTT assay and plotted as a percentage of the control (cells not exposed to the drugs). The control-pRCMV-transfected SiHa cells pretreated with DEX for 24 hours were still more sensitive to cisplatin. However, the cytotoxicity-enhancing effect of DEX in dnIB-pRCMV-transfected SiHa cells was abolished.
1111111
Correlation of GR content and gr mRNA expression
SAOS2 YSL N87 AGS Caski HeLa TW01SNU449TW04 Hep3BMCF-7TPT300 SiHa
Ar b ita ry u n it 0 20 40 60 80 100 120 GR mRNA / TBP mRNA x 102 GR number / cell x 103
Breast Cancer GR mRNA from 10 patients
1 2 11 13 17 24 28 30 33 34 A rb it a ry u n it 0 20 40 60 80 100 120 140 GR mRNA/TBP mRNA x 102