Method for Protein–Ligand Docking
JINN-MOON YANGDepartment of Biological Science and Technology & Institute of Bioinformatics, National Chiao Tung University, Hsinchu, 30050, Taiwan
Received 25 July 2003; Accepted 21 November 2003
Abstract:
We have developed a generic evolutionary method with an empirical scoring function for the protein–ligand docking, which is a problem of paramount importance in structure-based drug design. This approach, referred to as the GEMDOCK (Generic Evolutionary Method for molecular DOCKing), combines both continuous and discrete search mechanisms. We tested our approach on seven protein–ligand complexes, and the docked lowest energy structures have root-mean-square derivations ranging from 0.32 to 0.99 Å with respect to the corresponding crystal ligand structures. In addition, we evaluated GEMDOCK on crossdocking experiments, in which some complexes with an identical protein used for docking all crystallized ligands of these complexes. GEMDOCK yielded 98% docked structures with RMSD below 2.0 Å when the ligands were docked into foreign protein structures. We have reported the validation and analysis of our approach on various search spaces and scoring functions. Experimental results show that our approach is robust, and the empirical scoring function is simple and fast to recognize compounds. We found that if GEMDOCK used the RMSD scoring function, then the prediction accuracy was 100% and the docked structures had RMSD below 0.1 Å for each test system. These results suggest that GEMDOCK is a useful tool, and may systematically improve the forms and parameters of a scoring function, which is one of major bottlenecks for molecular recognition.©2004 Wiley Periodicals, Inc. J Comput Chem 25: 843– 857, 2004
Key words: empirical scoring function; generic evolutionary method; protein–ligand docking; hybrid-solution docking method; structure-based drug design
Introduction
A computer-aided docking process, identifying the lead com-pounds by minimizing the energy of intermolecular interactions, has greatly advanced an understanding of the molecular recogni-tion phenomenon, and has been demonstrated to play an important role for structure-based drug design.1,2 Protein–ligand docking simulations need to yield the binding energy of the bound complex crystal structure. In general, solving a protein–ligand docking problem involves two critical elements:3a good scoring function and an efficient search algorithm for finding a global minimum on the binding energy landscape of a simulation scoring function that is often complex and rugged funnel shapes.4
A good scoring function should be fast and simple for screen-ing large potential solutions and effectively discriminatscreen-ing be-tween correct binding states and nonnative docked conformations. Various scoring functions have been developed for calculating binding free energy, such as empirical-based,5,6 knowledge-based,5,7,8 physic-based,9 –11 solvent-based scoring functions,12 and consensus scoring function.13A search algorithm should con-sist of global and local search strategies for covering the
confor-mation and orientation spaces fast and efficiently, including the deterministic,14,15stochastic,5,16,17and hybrid approach.18
Many automated docking approaches have been developed and can be roughly divided into rigid docking, flexible ligand docking, and protein flexible docking methods. The rigid-docking meth-ods14treated both ligands and proteins as rigid. In flexible ligand docking methods, such as evolutionary algorithms,5,11,17,19 simu-lated annealing,16and fragment-based approach,15the ligand is flexible and the protein is rigid. For reasonably addressing protein flexible problems, where both ligands and proteins are flexible, most of the docking methods often allowed a limited model of protein variations, such as the side-chain flexible or small motions of loops in the binding site.20Among these search algorithms, an evolutionary-based approach is a very promising direction.
Correspondence to: J.-M. Yang; e-mail: [email protected]
Contract/grant sponsor: National Science Council of Taiwan; contract/ grant number: NSC-91-2320-B-009-001.
Contract/grant sponsor: Department of Health of Taiwan; contract/grant number: DOH92-TD-1132.
Here, we developed a generic evolutionary approach with an empirical scoring function, referred to as the Generic Evolutionary Method for molecular DOCKing (GEMDOCK), to address several issues of protein–ligand docking problems. First, we have reported the validation and analysis of GEMDOCK on various search spaces and scoring functions to understand which factors (e.g., search algorithms, scoring functions, or experimental errors) are mainly responsible for the molecular docking errors. Second, we have analyzed the nature and influences of a hybrid-solution
dock-ing method, which evolves simultaneously both rigid and flexible docked conformations, because most of the current methods are either flexible or rigid docking methods. Finally, our approach is likely to help in making a good choice or in improving the scoring function which is one of the major bottlenecks in protein–ligand problems. The GEMDOCK is an extended work of our recently developed evolutionary algorithm which was more robust than three standard evolutionary approaches, including genetic algo-rithms,21evolution strategies,22and evolutionary programming.23 We have now substantially enhanced the original method, and there are four main differences in methodology between the present work and our previous study.24First, GEMDOCK could be a flexible or a hybrid-solution docking method. Second, we devel-oped an empirical scoring function, which was specifically de-signed for fast docking applications. Third, GEMDOCK combines both continuous and discrete search mechanisms to improve the
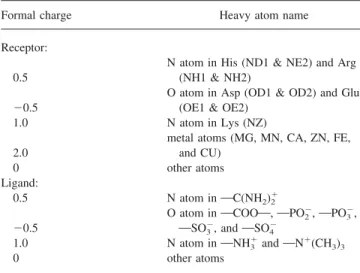
Table 1. Atom Formal Charges of GEMDOCK.
Formal charge Heavy atom name
Receptor: 0.5
N atom in His (ND1 & NE2) and Arg (NH1 & NH2)
⫺0.5
O atom in Asp (OD1 & OD2) and Glu (OE1 & OE2)
1.0 N atom in Lys (NZ)
2.0
metal atoms (MG, MN, CA, ZN, FE, and CU)
0 other atoms
Ligand:
0.5 N atom inOC(NH2)2⫹
⫺0.5
O atom inOCOOO, OPO2⫺,OPO3⫺,
OSO3⫺, andOSO4⫺
1.0 N atom inONH3⫹andON⫹(CH3)3
0 other atoms
Table 2. Atom Types of the GEMDOCK.
Atom type Heavy atom name
Donor primary and secondary amines, sulfur, metal atoms, and atom with positive formal chargea
Acceptor oxygen and nitrogen with no bound hydrogen, and atom with negative formal chargea
Both water and hydroxyl groups
Nonpolar other atoms (such as carbon and phosphorus)
aThe atom formal charge defined in Table 1.
performance via two new genetic operators. Finally, GEMDOCK is an automatic system that is able to prepare all required materials, such as the atom formal charge, the atom type, and the ligand binding site of a protein.
To evaluate the performance and limitations of GEMDOCK, we tested it on seven protein–ligand complexes. The docked low-est energy structures have root-mean-square derivations ranging from 0.32 to 0.99 Å with respect to the corresponding crystal ligand structures. GEMDOCK was compared to five stochastic approaches applying the very similar scoring function.25In addi-tion, GEMDOCK was tested on two crossdocking ensembles of protein structures, 10 complexes of the dihydrofolate reductase,26 and six complexes of the trypsin,15to evaluate GEMDOCK on a problem in which a protein structure is small motion during docking processing. Experimental results indicate that GEM-DOCK is robust and the empirical scoring function is simple and fast to recognize compounds. Furthermore, it may be used to systematically evaluate and thus improve scoring functions.
Method
Here, we present the details of our GEMDOCK for the protein– ligand docking (Fig. 1). GEMDOCK, an automatic docking tool, is able to generate all experimental variables and serve as a flexible or hybrid-solution docking tool. We designed a new rotamer-based mutation operator for reducing the search space of ligand structure conformations, and used a differential evolution operator27 for reducing the disadvantages of Gaussian and Cauchy mutations. First, we specified the coordinates of ligand and protein atoms, the ligand binding area, atom formal charge (Table 1), and atom types (Table 2). Crystal coordinates of the ligand and protein atoms were taken from the Protein Data Bank, and were separated into differ-ent files. The size and location of the ligand binding site was determined by considering the protein atoms located⬍10 Å from each ligand atom when preparing the proteins. GEMDOCK then automatically determined the center of the receptor and the search cube of a binding site according to the maximum and minimum of coordinates of these selected protein atoms.
After GEMDOCK prepares the ligand and protein, GEM-DOCK randomly generates a starting population with N solutions by initializing the orientation and conformation of the ligand relating to the center of the receptor according to the search cube. Each solution is represented as a set of three n-dimensional vectors (xi
, i , i
), where n is the number of adjustable variables of a docking system and i⫽ 1, . . . , N. The vector x represents the adjustable variables to be optimized in which x1, x2, and x3are the three-dimensional location of the ligand; x4, x5, and x6 are the rotational angles; and from x7to xnare the twisting angles of the rotatable bonds inside the ligand. and are the step-size vectors of decreasing-based Gaussian mutation and self-adaptive Cauchy mutation, respectively. In other words, each solution x is associ-ated with some parameters for step-size control. The initial values of x1, x2, and x3are randomly chosen from the search box, and the others ( x4to xn) are randomly chosen from 0 to 2 in radians. The initial step sizes is 0.8 and is 0.2. The ligand conformations (i.e., twisting angles of the rotatable bonds inside a ligand) are randomly generated if GEMDOCK is a flexible docking method. If GEMDOCK works as a hybrid-solution docking method, the ini-tial ligand conformations of rigid solutions (0.2N) are set to the conformation of the ligand crystal structure in PDB and the others (flexible solutions with 0.8N) are randomly generated.
GEMDOCK enters the main evolutionary loop which consists of three main stages in every iteration: decreasing-based Gaussian mutation, differential equation, and self-adaptive Cauchy mutation after GEMDOCK initializes the solutions. Each stage is realized by generating a new quasi-population (with N solutions) as the parent of the next stage. As shown in Figure 1, these stages apply a general procedure “FC_adaptive,” with only different working population and the mutation operator.
The FC_adaptive procedure (Fig. 1) employs two parameters, namely, the working population (P, with N solutions) and muta-tion operator (M), to generate a new quasi-populamuta-tion. The main work of FC_adaptive is to produce offspring and then conduct the family competition. Each individual in the population sequentially becomes the “family father.” With a probability pc, this family father and another solution that is randomly chosen from the rest of the parent population are used as parents for a recombination operation. Then the new offspring or the family father (if the recombination is not conducted) is operated on by the rotamer mutation and then by the working mutation, i.e., decreasing-based Gaussian mutation (Mdg), differential equation (MDE), or self-adaptive Cauchy mutation (Mc). For each family father, such a procedure is repeated L times called the family competition length.
Table 3. Parameters of the GEMDOCK.
Parameter Value of parameters
Initial step sizes
⫽ 0.8, v ⫽ ⫽ 0.2 (in radius)
Family competition length L⫽ 2
Population size N⫽ 400
Recombination rate pc⫽ 0.3
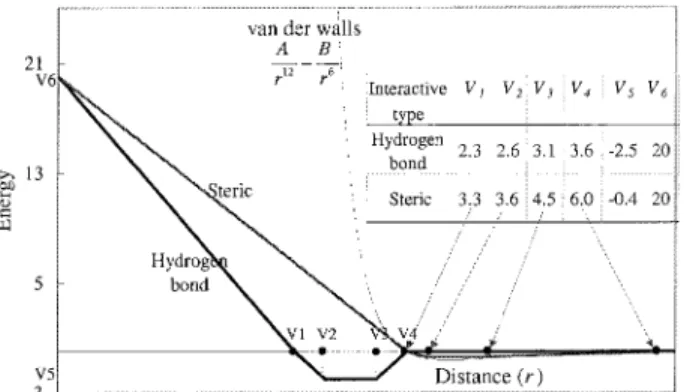
No. of the maximum generation 60 Figure 2. The linear energy function of the pair-wise atoms for steric
and hydrogen bonds in GEMDOCK (bold line) with a standard Len-nard–Jones potential (light line).
Among these L offspring and the family father, only the one with the lowest scoring function value survives. Because we create L children from one “family father” and perform a selection, this is a family competition strategy. This method avoids the population prematureness but also keeps the spirit of local searches. Finally, the FC_adaptive procedure generates N solutions, because it forces each solution of the working population to have one final off-spring.
When GEMDOCK is a rigid docking, these values of x7to xn, conformations of rotatable bonds inside a ligand, are fixed and set to the ligand conformations of the crystal bound complex. GEM-DOCK is a flexible docking tool if it evolves the conformation variables ( x7, . . . , xn) of each solution in a population.
GEM-DOCK is a hybrid-solution approach if the conformation variables of part of solutions (e.g.,N solutions) are set to the values of the crystal bound complex. In this article, is 0.2 when GEMDOCK is a hybrid-solution method.
In the following, genetic operators are briefly described. We use a⫽ ( xa
,a ,a
) to represent the “family father” and b⫽ ( xb , b, b) as another parent. The offspring of each operation is represented as c⫽ ( xc ,c ,c ). The symbol xj s is used to denote the jth adjustable optimization variable of a solution s, @j 僆 {1, . . . , n}.
Recombination Operators
A recombination operator selected the “family father (a)” and another solution (b) randomly selected from the working popula-tion. GEMDOCK implemented both modified discrete recombina-tion and intermediate recombinarecombina-tion.22 The former generates a child as follows: xj c⫽
再
xj a with probability 0.8 xj bwith probability 0.2. (1)The generated child inherits genes from the “family father” with a higher probability 0.8. Intermediate recombination works as:
wj c ⫽ wj a ⫹共wj b ⫺ wj a 兲/ 2, (2)
where w is or based on the mutation operator applied in the FC_adaptive procedure. The intermediate recombination only op-erated on step-size vectors, and the modified discrete recombina-tion was used for adjustable vectors ( x).
Mutation Operators
After the recombination, a mutation operator, the main operator of GEMDOCK, is applied to mutate adjustable variables ( x).
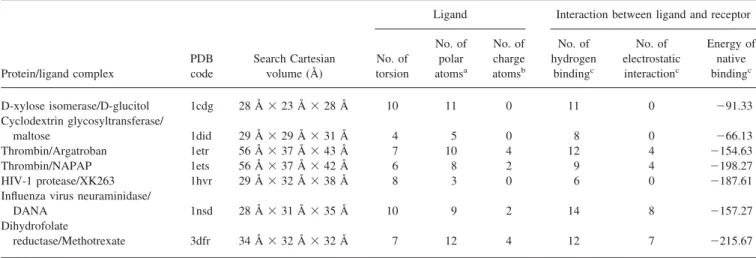
Gauss-Table 4. Test Systems Used in Docking Experiments.
Protein/ligand complex
PDB code
Search Cartesian volume (Å)
Ligand Interaction between ligand and receptor
No. of torsion No. of polar atomsa No. of charge atomsb No. of hydrogen bindingc No. of electrostatic interactionc Energy of native bindingc D-xylose isomerase/D-glucitol 1cdg 28 Å⫻ 23 Å ⫻ 28 Å 10 11 0 11 0 ⫺91.33 Cyclodextrin glycosyltransferase/ maltose 1did 29 Å⫻ 29 Å ⫻ 31 Å 4 5 0 8 0 ⫺66.13 Thrombin/Argatroban 1etr 56 Å⫻ 37 Å ⫻ 43 Å 7 10 4 12 4 ⫺154.63 Thrombin/NAPAP 1ets 56 Å⫻ 37 Å ⫻ 42 Å 6 8 2 9 4 ⫺198.27 HIV-1 protease/XK263 1hvr 29 Å⫻ 32 Å ⫻ 38 Å 8 3 0 6 0 ⫺187.61
Influenza virus neuraminidase/
DANA 1nsd 28 Å⫻ 31 Å ⫻ 35 Å 10 9 2 14 8 ⫺157.27
Dihydrofolate
reductase/Methotrexate 3dfr 34 Å⫻ 32 Å ⫻ 32 Å 7 12 4 12 7 ⫺215.67
aandbare defined in Table 1 and Table 2, respectively.
cStatics are derived from the native crystal conformations of test systems according to our scoring function [eq. (12)].
Figure 3. The ligands used for docking in this article with rotatable bonds are indicated. The lower-case four-letter and upper-case three-letter symbols are the PDB code and ligand code in PDB, respectively.
ian and Cauchy Mutations are continuous search operators and the rotamer mutation is a discrete operator.
Gaussian and Cauchy Mutations
Gaussian and Cauchy Mutations are accomplished by first mutat-ing the step size (w) and then mutatmutat-ing the adjustable variable x:
w⬘j⫽ w⬘jA, (3)
x⬘j⫽ xj⫹ w⬘jD, (4) where wj and xj are the ith component of w and x, respectively, and wjis the respective step size of the xjwhere w is or . If the mutation is a self-adaptive mutation, A is evaluated as exp[⬘N(0, 1) ⫹ Nj(0, 1)] where N(0, 1) is the standard normal distribution, Nj(0, 1) is a new value with distribution N(0, 1) that must be regenerated for each index j. When the mutation is a decreasing-based mutation A is defined as a fixed decreasing rate ␥ ⫽ 0.95. D is evaluated as N(0, 1) or C(1) if the mutation is, respectively, Gaussian mutation or Cauchy mutation. Our decreasing-based Gaussian mutation uses the step-size vector with a fixed decreasing rate ␥ ⫽ 0.95 and works as
c⫽␥a , (5) xj c⫽ x j a⫹c Nj共0, 1兲. (6)
The self-adaptive Cauchy mutation is defined as j c ⫽j a exp关⬘N共0, 1兲 ⫹ Nj共0, 1兲兴, (7) xj c ⫽ xj a ⫹j c Cj共t兲. (8)
We set and ⬘ to (公2n)⫺1 and (公2公n)⫺1, respectively, according to the suggestion of evolution strategies.22A random variable is said to have the Cauchy distribution [C(t)] if it has the density function:
f共 y; t兲 ⫽ t/
t2⫹ y2, ⫺⬁ ⬍ y ⬍ ⬁, where t is set to 1.
Differential Evolution
An offspring of differential evolution is generated as
xj c⫽
再
uj m , if rand关0, 1兲 ⱕ CR xj a, otherwise (9) and uj m ⫽ xj a ⫹ F共 xj b ⫺ xj c 兲, (10)where a is the “family father”; b and c are two solutions randomly selected from the working population subjected to a⫽ b ⫽ c. In this work, F and CR are set to 0.5 and 0.9, respectively.
Rotamer-Mutation
This operator is only used for x7to xnto find the conformations of the rotatable bonds inside the ligand. For each ligand, this operator mutates all of the rotatable angles according to the rotamer distri-bution and works as:
xj⫽ rkiwith probability pki, (11)
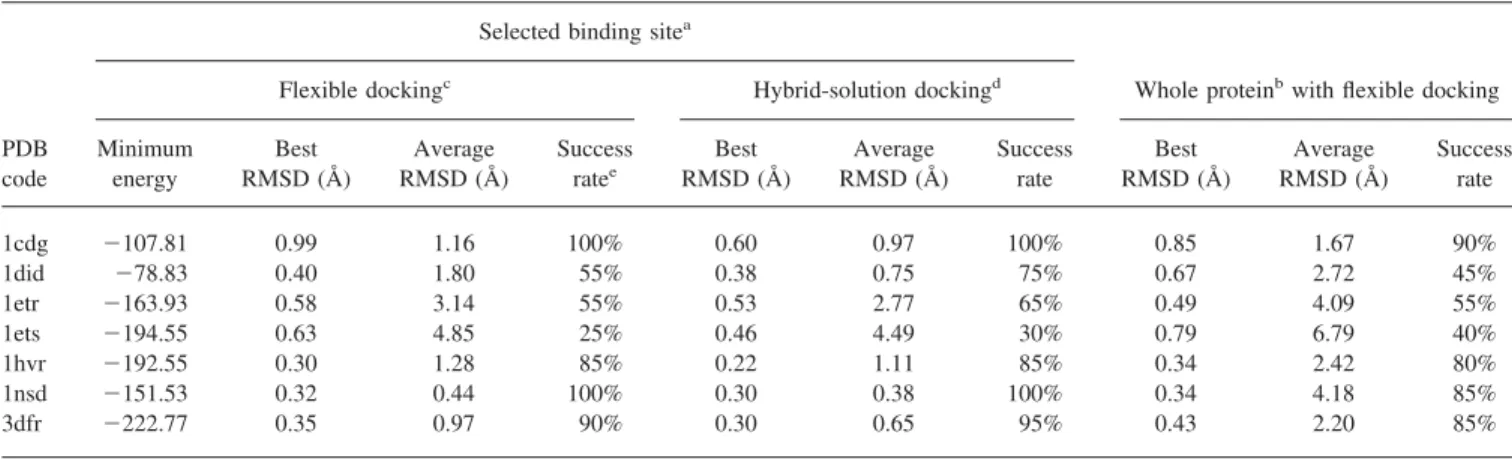
Table 5. GEMDOCK Results on the Test Cases Presented in Table 4.
PDB code
Selected binding sitea
Whole proteinbwith flexible docking
Flexible dockingc Hybrid-solution dockingd
Minimum energy Best RMSD (Å) Average RMSD (Å) Success ratee Best RMSD (Å) Average RMSD (Å) Success rate Best RMSD (Å) Average RMSD (Å) Success rate 1cdg ⫺107.81 0.99 1.16 100% 0.60 0.97 100% 0.85 1.67 90% 1did ⫺78.83 0.40 1.80 55% 0.38 0.75 75% 0.67 2.72 45% 1etr ⫺163.93 0.58 3.14 55% 0.53 2.77 65% 0.49 4.09 55% 1ets ⫺194.55 0.63 4.85 25% 0.46 4.49 30% 0.79 6.79 40% 1hvr ⫺192.55 0.30 1.28 85% 0.22 1.11 85% 0.34 2.42 80% 1nsd ⫺151.53 0.32 0.44 100% 0.30 0.38 100% 0.34 4.18 85% 3dfr ⫺222.77 0.35 0.97 90% 0.30 0.65 95% 0.43 2.20 85%
All results are derived from 20 independent docking runs, and the docked lowest energy conformation is considered to calculate the best RMSD and average RMSD.
aandbthe selected binding site and the whole protein are considered as the search binding areas, respectively. cGEMDOCK evolves a population with N flexible solutions.
dGEMDOCK evolves a population with 0.2N rigid solutions and 0.8N flexible solutions.
eThe percentage of the docking runs that find a docked lowest energy structure within 2.0 Å RMSD with respect to the
where rkiand pkiare the angle value and the probability, respec-tively, of ith rotamer of kth bond type including sp3Osp3 and
sp3Osp2bonds. The values of r
kiand pkiare based on the energy distributions of these two bond types.
Scoring Function
In this work, we used an empirical scoring function given as
Etot⫽ Einter⫹ Eintra⫹ ECO, (12) where Einterand Eintraare the intermolecular and intramolecular energy, respectively, ECOpenalizes a solution if the relative con-tract order between ligand and receptor is less than a predefined value.
The intermolecular energy is defined as
Einter⫽
冘
i⫽1 lig冘
j⫽1 pro冋
F共rij Bij兲 ⫹ 332.0qiqj 4rij册
, (13)where rijis the distance between the atoms i and j, qiand qjare the formal charges and 332.0 is a factor that converts the
electro-static energy into kilocalories per mol. The lig and pro denote the numbers of the heavy atoms in the ligand and receptor, respec-tively. The formal charge of atom type of receptor and ligand are defined in Table 1. F(rij
Bij) is a simple atomic pair-wise potential function (Fig. 2) modified from previous works5,28and given as
F共rij Bij兲 ⫽
冦
V6⫺ V6rij Bij V1 , if rij Bijⱕ V1 V5共rij Bij⫺ V1兲 V2⫺ V1 , if V1⬍ rij Bijⱕ V2 V5, if V2⬍ rij Bijⱕ V3 V5⫺ V5共rij Bij⫺ V3兲 V4⫺ V3 , if V3⬍ rij Bijⱕ V4 0, if rij Bij⬎ V4 . (14) rijBijis the distance between the atoms i and j with bond type B ij which is the interaction bonding type forming by the pair-wise heavy atoms of a ligand and a protein. Bij is either hydrogen binding or steric state. The values of parameters, V1, . . . , V6, are given in Figure 2. In this atomic pair-wise model, the interactive types are only hydrogen binding and steric potential which have the same function form but with different parameters, V1, . . . , V6.
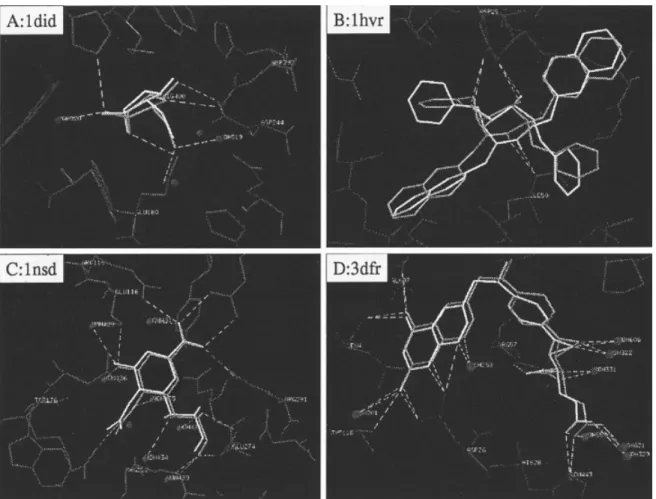
Figure 4. GEMDOCK results of four protein–ligand complexes. The RMSD values of these four complexes are less than 1.0 Å, and most of the docked ligand groups (white) are identical with the crystal ligand structures (gray). The white dotted lines are hydrogen bonds.
The energy value of hydrogen binding should be larger than the one of steric potential. In this model, the atom is divided into four different atom types (Table 2): donor, acceptor, both, and nonplar.
The hydrogen binding can be formed by the following atom-pair types: donor–acceptor (or acceptor– donor), donor– both (or both– donor), acceptor– both (or both–acceptor), and both– both. Other atom-pair combinations are to form the steric state.
The intramolecular energy of a ligand is
Eintra⫽
冘
i⫽1 lig冘
j⫽i⫹2 lig 关F共rij Bij兲兴 ⫹冘
k⫽1 dihed A关1 ⫺ cos共mk⫺0兲兴, (15) where F(rijBij) is defined as eq. (14) except the value is set to 1000 when rij
Bij⬍ 2.0 Å for penalizing the unreasonable ligand confor-mations and dihed is the number of rotatable bonds. We followed the work of Gehlhaar et al.5to set the values of A, m, and0. For the sp3Osp3bond A, m, and0are set to 3.0, 3, and; and A ⫽ 1.5, m ⫽ 6, and0⫽ 0 for the sp3Osp2bond.
The relative contract order is defined as RCO⫽ f/T, where f is the frequency of the atom-pair distance less than 8 Å, and T is the total number of interactions between the ligand and receptor. The penalty ECOis based on RCOand is given
ECO⫽
再
1000共K ⫺ RCO兲, if RCOⱕ K
0, if RCO⬎ K. (16)
In this article, the K is set to 0.025 and 0.075 when the whole protein and the selected binding site as the search binding areas, respectively.
Results and Discussions
Parameters of GEMDOCK
Table 3 indicates the setting of GEMDOCK parameters, including initial step sizes, family competition length (L ⫽ 2), population size (N⫽ 400), and recombination probability ( pc⫽ 0.3) in this work. The GEMDOCK optimization stops when either the con-vergence is below certain threshold value or the iterations exceed a maximal preset value which was set to 60. Therefore, GEM-DOCK generated 2400 solutions in one generation and terminated after it exhausted 144,000 solutions in the worse case. These parameters were decided after experiments conducted to recognize complexes of test docking systems with various values.
Test Complexes and Docking Protocols
We chose seven protein–ligand complexes shown in Figure 3 and Table 4 to illustrate the effectiveness of our approach and to allow comparison with other docking approaches,6,25 which used the similar empirical scoring functions. These ligands have between 4 and 10 rotatable bonds, between 3 and 12 polar atoms, and be-tween 0 and 4 formal charge atoms. The native binding energy of a crystal complex is calculated by using our scoring function [eq. (12)], which consists of three major kinds of protein–ligand inter-actions (i.e., the hydrophobic interinter-actions, electrostatic interac-tions, and hydrogen bindings). The number of the hydrogen bonds ranges between 6 and 14 and the number of the electrostatic interactions ranges between 0 and 8. These statics are derived from
Figure 5. Results of docking the ligand argatroban (MQI) into throm-bin (1etr) with (a) the while protein and (b) the selected throm-binding site as the search binding areas. The docked ligand conformations are yellow, and the crystal ligand structures are red. (a) Shows three main clusters of the docked ligand conformations, 55% results are near the native binding state (yellow), 30% results are near the pocket (green), and 15% results are in the other positions.
the crystal binding conformations and the distant thresholds of a hydrogen bond and an electrostatic interaction are set to 3.2 and 4.5 Å in this article, respectively. GEMDOCK was also tested on two crossdocking ensembles of protein structures, 10 complexes of the dihydrofolate reductase, and six complexes of the trypsin (serine proteinase) complex, to evaluate it on unbound docking problems and on the problem in which a protein structure is small motion during docking processing.
Crystal coordinates of ligand and protein atoms were taken from the Protein Data Bank, and were separated into different files. The protein atoms are selected if they are located less than 10 Å apart from each ligand atom. We followed the work25to retain the metal atoms and water molecules. GEMDOCK automatically de-cided the cube of a binding site based on the maximum and minimum of atom coordinates of a selected binding site. Among these seven test systems, the minimum cube is 28⫻ 23 ⫻ 28 Å (1cdg) and the maximum cube is 56 ⫻ 37 ⫻ 44 Å (1etr). Our program also automatically assigned the formal charge (Table 1) and the atom type (Table 2) of each atom in the ligand and protein. The bond type (sp3Osp3, sp3Osp2, or others) of a rotatable bond inside a ligand is also assigned. The energies of the native crystal conformations of test systems are indicated for referring based on our scoring function [eq. (12)].
Accuracy of Docking Prediction
The overall accuracy of GEMDOCK in predicting the docked conformations of seven test cases is shown in Table 5. We used two performance criteria to evaluate the accuracy and robustness of a docking method. The first is the root-mean-square deviation (RMSD) error in ligand heavy atoms between the docked confor-mation and the crystal ligand structure. The second criterion is the success rate, which is the percentage of the trials that find a solution within 2.0 Å RMSD. The RMSD is commonly used and is given
再
冘
i⫽1M关共Xi⫺ xi兲2⫹ 共Yi⫺ yi兲2⫹ 共Zi⫺ zi兲2兴/M
冎
1/ 2, (17)
where M is the heavy atom number of a ligand; (Xi, Yi, Zi) and ( xi, yi, zi) are the coordinates of the ith atom of X-ray crystal and docked structures, respectively. All results were derived from 20 independent docking runs and the docked lowest-energy structure was considered for each test case. In this work GEMDOCK runs on Pentium 1.4 GHz personal computer with single processor. On average GEMDOCK took 410 s, the maximum time was 652 s for the complex, 1ets, and the shortest time was 130 s for 1did. When we analyzed the characteristics of GEMDOCK and compared GEMDOCK with other methods, GEMDOCK executed 100 and 500 docking runs and the docked lowest energy structure was considered for each test system, respectively.
As shown in Table 5, GEMDOCK yielded the best RMSD values ranging between 0.30 Å (1hvr) and 0.99 Å (1cgd) and the average RMSD values ranging between 0.44 Å (1nsd) and 4.85 Å (1ets) when GEMDOCK worked as a flexible docking and the selected binding site was considered as the binding search area. The success rates range between 100% (1cdg) and 35% (1ets). Figure 4 shows four docked solutions (i.e., 1did, 1hvr, 1nsd, and 3dfr) in which GEMDOCK predicted correct positions for most of the ligand groups. The docked and crystal ligand conformations are white and gray, respectively, and the white dotted lines indicate hydrogen bonds. The RMSD values of these four docked confor-mations are less than 1.0 Å. According to these docked conforma-tions, we observed that GEMDOCK often yielded more number of hydrogen bonds than native states to minimize the docking energy based on our energy function [eq. (12)]. The energy of the docked conformation (Table 5) obtained by GEMDOCK was often lower than the energy of the crystal conformation (Table 4). Although GEMDOCK worked as a hybrid-solution docking method, evolv-ing 260 flexible ligand solutions and 40 rigid ligand solutions, it consistently yielded lower RMSD values and higher success rates than the ones of the flexible docking method for all test cases.
Although the whole protein was considered as the search bind-ing area, GEMDOCK used the same parameter values, shown in Table 3, except that the population size was 700 to improve the success rates and reduce the average RMSD values. Table 5 indicates that GEMDOCK yields slightly different performance on
Table 6. Comparisons GEMDOCK with Different Energy Functions on Test Cases.
PDB code
Einter[eq. (13)] without electrostatic
energy Etot[eq. (12)] without penalty ECO RMSD [eq. (17)] scoring function
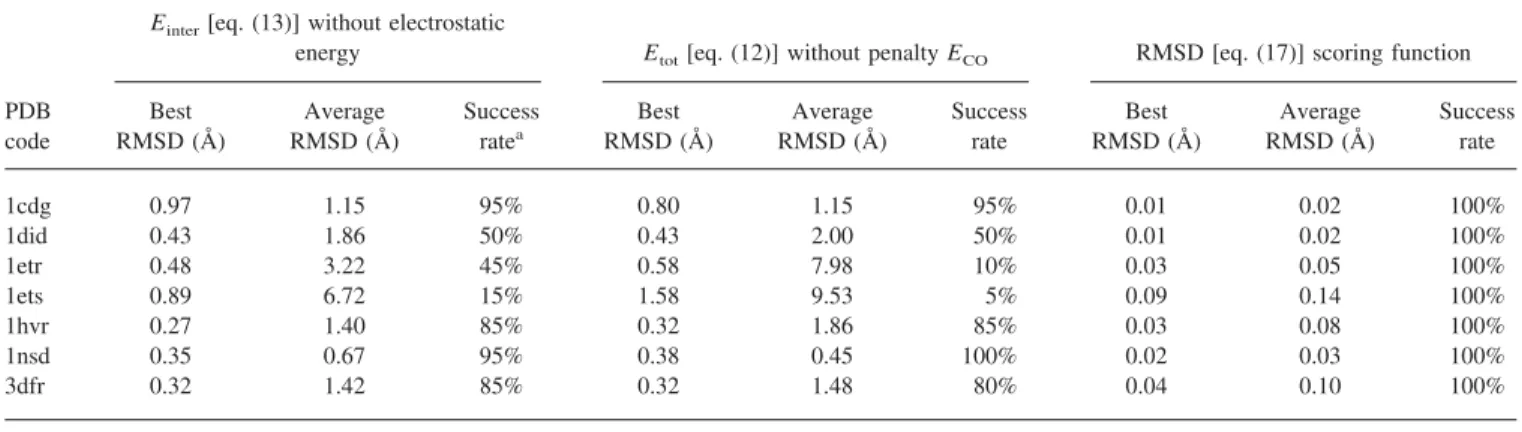
Best RMSD (Å) Average RMSD (Å) Success ratea Best RMSD (Å) Average RMSD (Å) Success rate Best RMSD (Å) Average RMSD (Å) Success rate 1cdg 0.97 1.15 95% 0.80 1.15 95% 0.01 0.02 100% 1did 0.43 1.86 50% 0.43 2.00 50% 0.01 0.02 100% 1etr 0.48 3.22 45% 0.58 7.98 10% 0.03 0.05 100% 1ets 0.89 6.72 15% 1.58 9.53 5% 0.09 0.14 100% 1hvr 0.27 1.40 85% 0.32 1.86 85% 0.03 0.08 100% 1nsd 0.35 0.67 95% 0.38 0.45 100% 0.02 0.03 100% 3dfr 0.32 1.42 85% 0.32 1.48 80% 0.04 0.10 100%
All results are derived from 20 independent docking runs and the docked lowest energy conformation is considered for each test case.
aThe percentage of the trials that find a docked lowest energy structure within 2.0 Å RMSD with respect to the crystal
these two kinds of the search binding sites, the selected binding site, and the whole protein. Figure 5a and b shows the results of docking the argatroban (MQI) into the thrombin (1etr) with the whole protein and selected binding site as the search binding areas, respectively. The docked ligand conformations are yellow and the crystal ligand structures are red. The docked lowest energy con-formation is identical with the crystal structure for most of ligand
groups. Figure 5a shows that the docked ligand conformations can be divided into three main clusters: the first (55% solutions) is near the native binding state (yellow), the second (30% solutions) is near the pocket (green), the final (15% solutions) is in the other locations.
In the following subsections, we validated and analyzed the characteristics of GEMDOCK, including energy functions, the
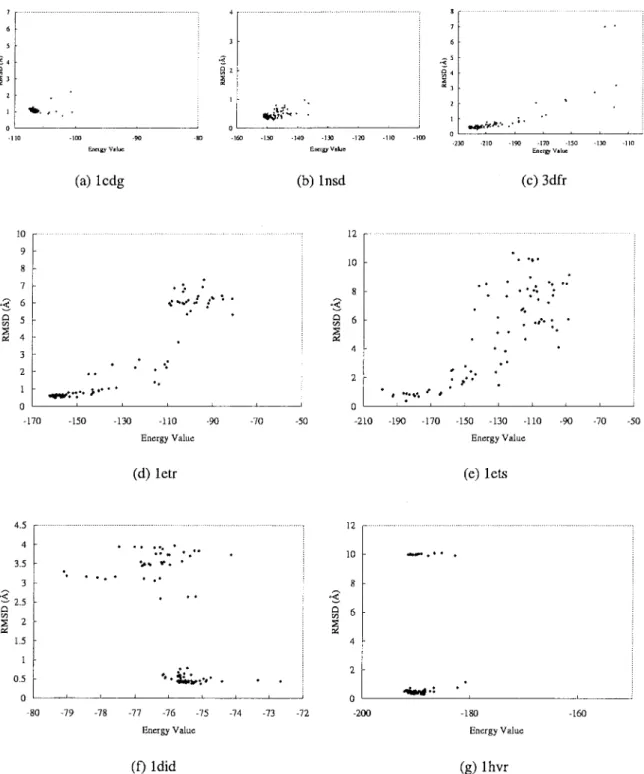
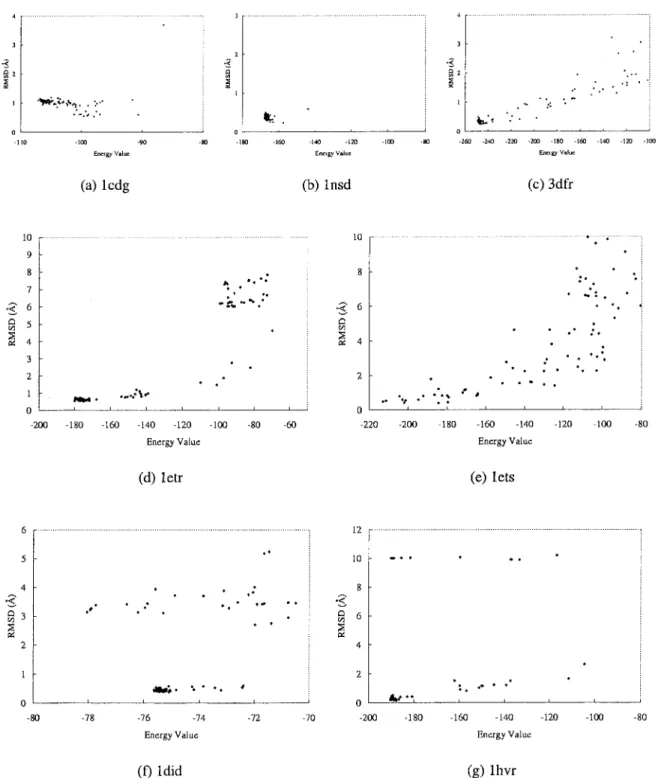
Figure 6. GEMDOCK results are divided into three categories (see text) for the flexible docking in seven complexes with 100 docking runs: (a) 1cdg, (b) 1nsd, and (c) 4dfr are the first class; (d) 1etr and (e) 1ets are the second class; (f) 1did and (g) 1hvr are the final class.
search spaces, and docking materials, and should therefore help to understand the error-free prediction of docked conformations.
Evaluation of the Energy Function Used
One of main objectives of this study was to evaluate whether our empirical scoring function was robust for molecular docking. To
simplify the task, we tested GEMDOCK with various uses and parameter values of our scoring function [eq. (12)] on test com-plexes. The overall accuracy is shown in Tables 5 and 6. GEM-DOCK generally improved the docked quality by considering the electrostatic energy if the protein–ligand interaction has the elec-trostatic energy, such as the 1etr, 1ets, 1nsd, and 3dfr. For complex
Figure 7. GEMDOCK results are divided into three categories (see text) for the hybrid-solution docking in seven complexes with 100 docking runs: (a) 1cdg, (b) 1nsd, and (c) 4dfr are the first class; (d) 1etr and (e) 1ets are the second class; (f) 1did and (g) 1hvr are the final class.
1nsd, eight electrostatic interactions were formed between the atoms O of CO2⫺(ligand) and Arg115 N, Arg291 N, and Arg373 N(receptor). For the complex 1etr, three electrostatic interactions were formed between the atoms N of (NH2)2⫹(ligand) and Asp189 O␦(receptor) and one electrostatic interaction was formed between the atoms O of CO2⫺(ligand) and His57 N(receptor). The ECOis useful for large search Cartesian volume, such as 1etr and 1ets. According to these experimental results, the element, F(rij
Bij), of the Einter[eq. (13)] was the main element of our scoring function. In contrast, the Eintra, ECO, and electrostatic energy were minor elements that influenced some specific docking cases.
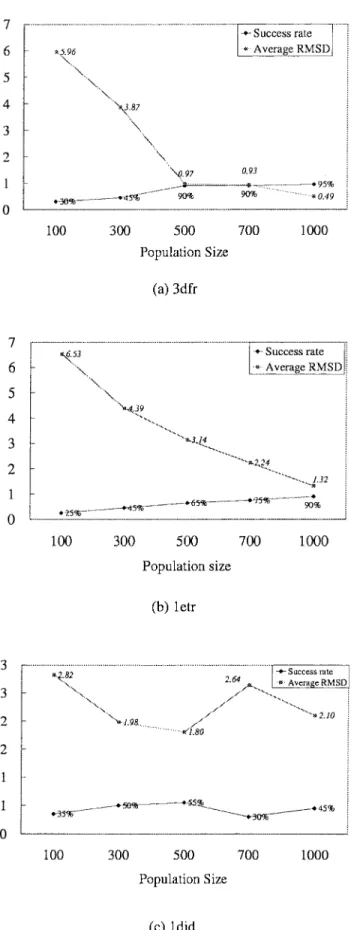
Figures 6 and 7 show the relationships between binding ener-gies and RMSD values of the docked lowest energy structures for flexible and hybrid-solution docking methods with 100 indepen-dent docking runs for each complex. For these two docking meth-ods, GEMDOCK has similar search behaviors and results that are roughly classified into three kinds of typical categories. For the first category, GEMDOCK yielded high success rates (⬎90%) and our scoring function [eq. (12)] is able to discriminate between native and nonnative conformations for complexes 1cdg (Figs. 6a and 7a), 1nsd (Figs. 6b and 7b), and 3dfr (Figs. 6c and 7c). For the second category, GEMDOCK yielded medium success rates (⬍60%) and may trap into local optimal, such as 1etr and 1ets. Figures 6d, 7d, 6e, and 7e show that our scoring function is also able to discriminate between correct binding states and nonnative conformations. In general, GEMDOCK is able to improve the success rate and docked accuracy by enlarging the population size for this category (Fig. 8b). For the final category, including 1did (Figs. 6f and 7f) and 1hvr (Figs. 6g and 7g), our scoring function may be unable to discriminate between correct binding states and incorrect conformations, for example, the lowest energy structures cannot promised to produce good docked conformations. In sum-mary, GEMDOCK is able to achieve good predictions for the complexes of categories 1 and 2 by increasing the population size (Figs. 8a and b) or lengthening the family competition length. In contrast to the protein–ligand complexes of the third category, a modified scoring function is required to improve solution quality. However, with uncertainty in the scoring function, the robust-ness of GEMDOCK was difficult to assess. To address this ques-tion, we made use of the high adaptability of GEMDOCK and simply replaced the empirical scoring function with a RMSD scoring function (i.e., one that would produce zero RMSD in heavy atom positions). As shown in Table 6, using the RMSD scoring function [eq. (17)], GEMDOCK could achieve the best RMSD and the average RMSD of docked structures were below 0.09 and 0.14 Å for each test complex, respectively. It is also worthy of note that GEMDOCK converges much faster with the RMSD scoring func-tion (⬍4 s for a docking run). These results may suggest that the flexible of GEMDOCK should allow us to begin to systematically
Figure 8. The relationships between the solution quality (the success rate and average RMSD value) and the population size. GEMDOCK is able to improve the solution quality of docking (a) MTX into 3dfr and (b) thrombin into 1etr when the population size increases. In contrast, the solution quality of docking (c) cyclodextrin glycosyltransferase into 1did is unrelated to the population size.
improve the forms and parameters of energy function for molec-ular recognition.
Evaluation of Search Spaces Used
Table 5 shows the accuracy of GEMDOCK with various search spaces and environments, including ligand size (i.e., number of heavy atoms), ligand flexibility (i.e., the number of rotatable bonds), sizes of the binding areas (i.e., selected binding site and whole protein), hetero atoms (i.e., water molecules and metal ions), and ligand polarity (i.e., numbers of hydrogen binding and electrostatic interactions between ligands and proteins). When GEMDOCK is a hybrid-solution docking method (Fig. 6), it yielded slight better docked conformations than the ones of the flexible docking method (Fig. 7). These results show that the GEMDOCK performance was somewhat influenced by ligand size and ligand flexibility. For the large search cube (i.e., 1etr and 1ets) or the whole protein as the search binding area, GEMDOCK often yielded a low success rate and a large average RMSD value (Table 5). Fortunately, Figure 8 shows that GEMDOCK is able to im-prove the docked accuracy by enlarging the population size if the scoring function can discriminate between native and nonnative conformations.
When hetero atoms in the binding site were retained, GEM-DOCK generally improved the predicated accuracy for complexes 1did, 1nsd, 1etr, and 3dfr. For the complex 1cdg, Figures 9a and 6a show that the success rates are 31 and 99% for removing and retaining water molecules, respectively. For the complex 1nsd, Figures 9b and 6b show that the success rates are 86 and 100% for removing and retaining water molecules, respectively. The reasons can be attributed to the additional steric, hydrogen binding, and electrostatic interactions between ligands and hetero atoms, which are “both (water molecules)” or “donor (metal ions)” (Table 2). As shown in Figure 4, a water molecule is often able to form hydrogen bonds with ligand atoms and become the search space constraint to reduce the possible docked orientations. For example, the ligand DIG forms two hydrogen bonds with the 519th and 520th water molecules in the complex 1did (Fig. 4A); the ligand DAN forms
five hydrogen bonds with the 429th, 431th, 434th, 435th, and 437th water molecules in the complex 1nsd (Fig. 4C). This obser-vation was also supported by the work of Westhead,25 which showed that the removal of the waters could lead to considerable errors.
Comparison with Other Approaches
Table 7 shows the results of comparing GEMDOCK with five different search methods, which were tested on the very similar scoring function [eq. (12)].25In general, it is neither straightfor-ward nor completely fair to compare the results of different mo-lecular docking methods because different accuracy measures, energy functions, and test complexes. Most of docking methods, with the exception of the studies of Jones et al.17and Kramer et al.,15have performed on a rather small set of complexes. Despite this, here we compared GEMDOCK with several molecular dock-ing methods includdock-ing simulated annealdock-ing (SA), evolutionary programming (EP), Tabu search (TS), genetic algorithm (GA), and random search (RS). These methods were studied by Westhead et
Figure 9. GEMDOCK results for removing the structure water molecules in the complexes (1cdg and 1nsd) with 100 independent runs.
Table 7. Comparison GEMDOCK with Some Heuristic Approaches Based on Success Rate.a
PDB code GEMDOCK SAb EPb TSb GAb RSb 1etr 55% 30% 21% 39% 13% 3% 1ets 21% 3% 9% 8% 11% 2% 1hvr 86% 65% 54% 58% 59% 2% 1nsd 98% 40% 64% 88% 57% 6% 3dfr 92% 90% 76% 93% 76% 9%
aThe percentage of 500 docking runs that find a docked lowest energy
structure within 1.5 Å RMSD with respect to the crystal ligand structure.
bThese results were summarized from ref. 25. SA is simulated annealing,
EP is evolutionary programming, TS is Tabu search, GA is genetic algo-rithm, and RS is random search.
al.25with the similar empirical scoring function and the same test complexes. They calculated the success rate, the percentage of the trials which find a solution within 1.5 Å RMSD, based on 500 independent runs. We followed their criteria to obtain the GEM-DOCK results and the results of comparative approaches were directly summarized from the previous study.25
As shown in Table 7, our approach was more robust than these comparative approaches on this test set. The random search is the worst and GEMDOCK is the best among these approaches on this test set. GEMDOCK seems good for the complexes 1etr and 1ets and the success rate is approaching to 90% if we enlarged the population size to 1000 (Fig. 8). At the same time, the GEMDOCK approach, as discussed above, can be used to analyze elements of molecular docking approaches, such as search schemes, docking
materials, and energy functions. It should help in moving toward error-free prediction of docked conformations and in systemati-cally improving the forms and parameters of a scoring function, which is one of major bottlenecks for molecular recognition.
Crossdocking Results
We used two ensembles of protein structures, i.e., 10 complexes of the dihydrofolate reductase and six complexes of the trypsin (serine proteinase) complex,15 to evaluate GEMDOCK on the unbound docking problem and the problem in which a protein structure is small motion during docking processing. These protein structures differ only on a small variation of side chains and loops on the active site. Figure 10 shows the cross-RMSD matrices (e.g., protein heavy atoms of the binding site) of all paired PDB entries that indicate the protein flexibility in the binding site. The largest RMSD is 0.54 Å and the smallest RMSD is 0.1 Å. Figure 11a and b shows these 10 inhibitors of the dihydrofolate reductase ensem-ble and six inhibitors of the trypsin ensemensem-ble, respectively. The symbol, four lower-case letters with three upper-case letters, was used to denote a ligand. For example the ligand “4dfr.MX,” “4dfr” denotes the PDB code and “MTX” is the ligand name in the Protein Data Bank. Figure 12a and b shows the binding modes of the dihydrofolate reductase and the trypsin complex, respectively. For the dihydrofolate reductase ensemble, the sizes and conforma-tions of all of ligands are similar. Three ligands, 1jol.FFO (5-formyl-6-hydrofolic acid colored dark), 1dyh.DZF (5-deazafolate colored gray), and 4dfr.MTX (methotrexate colored white), are shown in Figure 12a. On the other hand, the binding mode of the trypsin can be divided into two categories, one is the ligand 1tni.PBN (4-phenylbutylamine colored gray) and the other consists of the other five ligands (white).
Figure 10. Cross-RMSD matrices of all paired PDB entries for (a) 10 dihydrofolate reductase complexes, and (b) six trypsin complexes.
Figure 11. Ligands bound to (a) 10 dihydrofolate reductase com-plexes, and (b) six trypsin complexes tested in this work. Coordinates for each complex were obtained from the Protein Data Bank, using the accession codes given here. A four lower-case letter (e.g., 1dhj and 1tng) with a three upper-case letter (e.g., MTX and AMC) denotes a ligand.
The dihydrofolate reductase26is a small enzyme that plays an essential role, in the building of DNA and other processes. There are over 60 crystal structures with good resolution (ⱕ2.8 Å) in the Protein Data Bank. This reductase with diverse inhibitors, which are similar in size and structure flexible (Fig. 11a), is an important medicinal target, and shows the type of flexibility that can pose significant problems in docking simulation. It has been commonly used to evaluate and compare the performance of various docking methods. The trypsin, a kind of serine proteinase, specifically cleaves the peptide bond on the carboxyterminal side of positively charged residues, namely lysine and arginine. The trypsin binding pocket is only small motion and the structures of several trypsin-inhibitor complexes have been solved in the PDB.
Figure 13 shows the results for the crossdocking experiments in which all ligands of a protein ensemble were docked into each protein of this ensemble. For example, we obtained 100 cross-docked results when each of 10 ligands was cross-docked into each of 10 complexes of the dihydrofolate reductase. For the trypsin ensem-ble, we obtained 36 crossdocked solutions. When preparing the proteins for crossdocking experiments, the size and location of the ligand binding site was determined by considering the protein atoms that are located less than 12 Å apart from each ligand atom. We removed all water molecules from the binding sites.
Figure 13a shows the crossdocking results of the dihydrofolate reductase ensemble. All of the diagonal results, docking the ligand
Figure 13. Crossdocking results of all-pair experiments for (a) 10 dihydrofolate reductase complexes and (b) six trypsin complexes. The color-coded table shows the gray-scaling of RMSD values for each ligand (row) docked into each protein (column) of a protein ensemble. Figure 12. Binding modes of (a) dihydrofolate reductase and (b)
trypsin complexes. Three ligands, 1jol.FFO (dark), 1dyh.DZF (gray) and, 4dfr.MTX (white), of dihydrofolate reductase have a similar binding mode. Six ligands of trypsin have similar conformations (white) except the ligand 1tni.PBN (dark). The dotted lines are hydro-gen bonds and white balls are the water molecules.
back into its respective complex, are less than 1.0 Å, and most of off-diagonal results, crossdocking examples, GEMDOCK also yielded good results that the RMSD values are less than 1.5 Å. The largest RMSD value is 1.37 Å when the ligand 2drc.MTX was docked into the complex 1dyi, and the average is 0.77 Å. As shown in Figure 13a and Figures 3 and 4 in ref. 29, GEMDOCK is significantly better than FlexX15and FlexE29on the dihydrofolate reductase ensemble. The largest RMSD values of docked confor-mations predicted by FlexE and FlexX were 5.37 and 7.55 Å, respectively. FlexX and FlexE yielded 44 and 83.3% docked conformations with RMSD less than 2.0 Å, respectively. For six ligands, including 1dhj.MTX 1dra.MTX 1drb.MTX 2drc.MTX 3drc.MTX 4dfr.MTX, both FlexE and FlexX worked well for predicting the docked conformations. On the other hand, FlexX was unable to obtain good enough solutions for other ligands, such as 1dyh.DZF 1dyi.FOL 1dyj.DDF 1jol.FFO. FlexE also yielded little poor predictions for these four ligands, especially, FlexE obtained wrong docked conformations for the ligand 1jol.FFO. By contrast, GEMDOCK achieved 100% correct docked conforma-tions, i.e., the RMSD values less than 1.5 Å, for all 100 cross-docking experiments.
For all 36 docked conformations of the trypsin ensemble, GEMDOCK also obtained good and stable results except when the ligand 1tni.PBN was docked into the complexes 1tnk and 1tnl. The largest RMSD value is 2.72 Å, and the average is 0.82 Å (Fig. 13b). Our approach was more stable than FlexX15on this trypsin ensemble. As shown in Figure 12b, the binding mode of the ligand 1tni.PBN was significantly distinct from other ligands. Although this ligand was docked into six trypsin complexes, GEMDOCK obtained lower successful rates (ⱕ20%) than the rates (ⱖ60%) of other crossdocking experiments in the trypsin ensemble. If water molecules were retained in the binding site, GEMDOCK was able to find corrected conformations, which the RMSD value is less than 0.6 Å and the successful percentage is more than 60%, for docking the ligand 1tni.PBN into other systems. These results show that GEMDOCK may be able to address the problem in which the protein structure makes a slight variation during docking processing.
Conclusions
We have developed a robust evolutionary approach with an em-pirical fitness function for the flexible protein–ligand docking. GEMDOCK seamlessly blends local search and global search to work cooperatively by the integration of a number of genetic operators, each having unique search mechanism. We have vali-dated GEMDOCK on seven test cases and on two crossdocking experimental sets by using various search spaces and scoring functions. Experimental results have demonstrated the robustness and adaptability of GEMDOCK for exploring the conformational space of a molecular docking problem and efficiently finding the solution under the constraint of the fitness function used. GEM-DOCK could indeed yield 100% docking accuracy if the RMSD scoring function was used.
Despite the GEMDOCK’s apparent success, there are a number of problems with the methodology in general. First, our approach is somewhat time-consuming; second, the binding site of the
protein is essentially rigid; and finally, some protein–ligand inter-actions were not considered in our fitness function. In the future, we will investigate three directions to reduce above disadvantages: (1) developing a rapid energy evaluation with grid-based potentials for drug screening; (2) considering the side-chain flexibility in the protein active site; (3) incorporating important function– group interactions between ligands and proteins;17the solvent effect,12 and the hydrogen bond strength for calculating a hydrogen bond-ing energy6into our empirical scoring function.
References
1. Kuntz, I. D. Science 1992, 257, 1078.
2. Wlodawer, A.; Vondrasek, J. Annu Rev Biophys Biomol Struc 1998, 27, 249.
3. Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Proteins 2002, 47, 409. 4. Miller, D. W.; Dill, K. A. Protein Sci 1997, 6, 2166.
5. Gehlhaar, D. K.; Verkhivker, G. M.; Rejto, P.; Sherman, C. J.; Fogel, D. B.; Fogel, L. J.; Freer, S. T. Chem Biol 1995, 2, 317.
6. Verkhivker, G. M.; Bouzida, D.; Gehlhaar, D. K.; Rejto, P. A.; Arthurs, S.; Colson, A. B.; Freer, S. T.; Larson, V.; Luty, B. A.; Marrone, T.; Rose, P. W. J Comput Aided Mol Design 2000, 14, 531. 7. Gohlke, H.; Hendlich, M.; Klebe, G. J Mol Biol 2000, 295, 337. 8. Verdonk, M. L.; Cole, J. C.; Watson, P.; Gillet, V.; Willett, P. J Mol
Biol 2001, 307, 841.
9. Weiner, S. J.; Kollman, P. A.; Case, D. A.; Singh, U. C.; Ghio, C.; Alagona, G.; Profeta, S., Jr.; Weiner, P. J Am Chem Soc 1984, 106, 765. 10. Brooks, B. R.; Bruccoleri, R. E.; Olafson, B. D.; States, D. J.;
Swami-nathan, S.; Karplus, M. J Comput Chem 1983, 4, 187.
11. Taylor, J. S.; Burnett, R. M. Proteins Struct Funct Gene 2000, 41, 173. 12. Shoichet, B. K.; Leach, A. R.; Kuntz, I. D. Proteins Struct Funct Gene
1999, 34, 4.
13. Paul, N.; Rognan, D. Proteins Struct Funct Gene 2002, 47, 521. 14. Kuntz, I. D.; Blaney, J. M.; Oatley, S. J.; Langridge, R.; Ferrin, T. E.
J Mol Biol 1982, 161, 269.
15. Kramer, B.; Rarey, M.; Lengauer, T. Proteins Struct Funct Gene 1999, 37, 228.
16. Sherman, C. J.; Ogden, R. C.; Freer, S. T. J Med Chem 1995, 38, 466. 17. Jones, G.; Willett, P.; Glen, R. C.; Leach, A. R.; Taylor, R. J Mol Biol
1997, 267, 727.
18. Wang, J.; Kollman, P. A.; Kuntz, I. D. Proteins Struct Funct Gene 1999, 36, 1.
19. Morris, G. M.; Goodsell, D. S.; Halliday, R. S.; Huey, R.; Hart, W. E.; Belew, R. K.; Olson, A. J. J Comput Chem 1998, 19, 1639. 20. O¨ sterberg, F.; Morris, G. M.; Sanner, M. F.; Olson, A. J.; Goodsell,
D. S. Proteins Struct Funct Gene 2002, 46, 34.
21. Goldberg, D. E. Genetic Algorithms in Search, Optimization and Machine Learning; Addison-Wesley Publishing Company, Inc.: Read-ing, MA, 1989.
22. Ba¨ck, T. Evolutionary Algorithms in Theory and Practice; Oxford University Press: New York, 1996.
23. Fogel, D. B. Evolutionary Computation: Toward a New Philosophy of Machine Intelligent; IEEE Press: New York, 1995.
24. Yang, J.-M.; Kao, C.-Y. J Comput Chem 2000, 21, 988.
25. Westhead, D. R.; Clark, D. E.; Murray, C. W. J Comput Aided Mol Design 1997, 11, 209.
26. Feeney, J. Angew Chem Int Ed 2000, 39, 290.
27. Storn, R.; Price, K. V. J Global Optimizat 1997, 11, 341.
28. Knegtel, R. M. A.; Antoon, J.; Rullmann, C.; Boelens, R.; Kaptein, R. J Mol Biol 1994, 235, 318.
29. Claussen, H.; Buning, C.; Rarey, M.; Lengauer, T. J Mol Biol 2001, 308, 377.