Residue-based Design of Small Molecule
Inhibitor for H1N1, H5N1 and H7N1
Mutants
Weng Ieong Tou
1,
Kun-Lung Chang 2#, Tung-Ti Chang 3,4#,Cheng-Chun Lee 5,6, and Yu-Chian Chen 2,7*
1 School of Medicine, College of Medicine, China Medical University, Taichung,
40402, Taiwan
2 Department of Bioinformatics and Medical Engineering, Asia University, Taichung,
41354, Taiwan
3 School of Post-Baccalaureate Chinese Medicine, College of Chinese Medicine,
China Medical University, Taichung 40402, Taiwan.
4 Department of Chinese Pediatrics, China Medical University Hospital, Taichung
40402, Taiwan.
5 School of Medicine, College of Medicine, China Medical University, Taichung,
40402, Taiwan.
7Research Center for Chinese Medicine and Acupuncture, China Medical University,
Taichung 40402, Taiwan
* Corresponding Authors
Address correspondence to: Calvin Yu-Chian Chen, Ph.D.
School of Medicine, College of Medicine, China Medical University, Taiwan Department of Biomedical Informatics, Asia University
Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
TEL: 886-4-22052121-4 E-mail: [email protected]
ABSTRACT
The largest point mutation, which is seen in the strains H274Y and N294S, makes
influenza viruses resistant to some molecule drugs. Recently, there have been a large
number of experiments finding many frameworks and catalytic residues considered to
prevent the efficacy of anti-flu drugs. In the past, much research focused on the role
of drugs in rigid protein rather than in flexible protein. In this study, we used
molecular dynamics simulation (MD) combined with structure- and the ligand-based
drug design (SBDD and LBDD) method to study the dynamics interaction and protein
dynamics correlation statistics between compounds and the framework, catalytic
residue in N1. Drug candidates are screened by the predicted IC50 of the docking
result made by Support Vector Machine (SVM) and Multiple Linear Regression
(MLR) and Genetic function approximation (GFA), p <0.001. Saussureamine C and
Diiodotyrosine shown by MD have a protein dynamics correlation similar to that of
sialic acid, both can participate in hydrogen bond formation in loop, Framework, and
catalytic residues. In silico finding suggested Saussureamine C can generate inhibition
on H274Y and N294S mutants and Diiodotyrosine can also generate inhibition on
N294S mutants. Therefore, in silicon drug design results, Saussureamine C and
Diiodotyrosine may be potential drugs to produce inhibitory effects on N1 mutants
and the wild-type.
Key words:
1 INTRODUCTION
Influenza viruses belong to a class of viruses called orthomyxoviruses, divided into
A, B, and C types according to their particular antigenicity [1]. Influenza A is the
most powerful influenza virus since it can infect cross species. The spherical surface
of Influenza A shows two glycoproteins: hemagglutinin (HA) and neuraminidase
(NA)[2]. Influenza A viruses infect organs concentrated in the lower respiratory tract.
When the virus infects the host, NA will remove sialic acid and destruct its
glycoconjugates with mucins in the final step of replication, to accelerate the release
and infection of progeny viruses. Subclass NAs (N1-N9) are phylogenetically
classified into two groups: group-1 and group-2. Group-1 includes N1, N4, N5 and
N8 whereas the rest are classified in group-2 [3].
H1N1, H5N1, and H7N1 belong to the influenza viruses type A and have become
the world's largest pandemic threat due to their cross-species transmission
characteristics, high prevalence, and high mortality rates. Mutation and antigenic drift
allow the influenza viruses type A frequently produce new strains and resistant
viruses. In the NA crystal, it is found that residues called catalytic residues-- R118,
D151, R152, R224, E276, R292, R371, and Y406-- can directly contact the substrate
and produce hydrogen bonds. It is found that another framework residues, (E119,
R156, W178, S179, D198, I222, E277, N294, and E425) can stabilize active site of
the structure[4]. The major role of Oseltamivir (Tamiflu) is to compete for the binding
site of sialic acid in the neuraminidase N1 subtype [5]. Therefore, if a mutation occurs
in the binding site, Tamiflu will not easily be accepted by the binding sites, which
then leads to a drug resistance [6-12]. In the past, framework residues of H274 and
N294 mutants were found to make the virus resistant. Mutation points, such as
rotation, the destruction of the bonding of E276 and R224, and destabilization of
hydrophobic pocket, disallowing Oseltamivir to combine with N1; thus drug
resistance is generated [13]. Although zanamivir can be the last line of defense against
the Oseltamivir-resistant viruses, resistance is still inevitable. Furthermore, because of
possible side effects of respiratory dysfunction, zanamivir is not recommended for
chronic respiratory diseases such as asthma or chronic obstructive pulmonary disease.
Therefore, there is a need to design new drugs for N1 mutation points.
In the past, research has focused on resistance mechanisms and mutant analysis
[14]. However, there is little research about the dynamics process of N1 mutants and
substrates with drug candidates. In the past, structure-based drug design (SBDD)[15]
and ligand-based drug design (LBDD) [16] have been widely used in the design of
different target drugs to be able to quickly and accurately screen drugs. However,
drug binding is a dynamic process so that traditional rigid docking cannot be used to
accurately study flexibility of the drug-receptor interaction. Therefore, with the aid of
molecular dynamics (MD) simulation, the molecular mechanisms of protein and
ligand can be explored in depth so in order to better understand how the drug inhibits
activity of proteins. Additionally, it allows the computer to design drugs better. In this study, SBDD and LBDD are explored for a fast and accurate screening of the
natural compound binding capacity with N1 in the world's largest database of
traditional Chinese medicine TCM database@Taiwan[17]. With the help of molecular
dynamics, the inhibitory mechanisms can be further explored and we can elucidate
how inhibitors suppress the function and role of N1. Besides, molecular dynamics
simulations of point mutations are conducted to find out potential binding sites and
2 RESULT AND DISCUSSION 2.1 Homology Modeling
The protein structures of mutants H274Y and N294S are created by the homology
model. In the homology models of the wild-type, H274Y, and N294S, the mutant
models are more accurate than the wild-type’s. In the wild-type, 5.4% of the residues
are of the allowed region, 1.4% of the outlier region, 2.9% of H274Y and 2.4% of
N294S respectively of the allowed region, and 0.5% of H274Y and N294S of the
outlier region (as shown in Figure 1 and Table 2). The residues’ topology often affects
demolition and stack of protein structure and molecular complementarity. In the
wild-type and mutants, the residues with similar disfavored residue topology include S125,
I225, T225, C292, W296, C331, S400, N449, and W455.
2.2 Structure based drug design
The homology models of the wild-type and mutants were further used to perform
receptor docking (table 1) (Figure 2) where the dock score was used as a primary
standard for evaluating ligand binding affinity. The sum of the scoring function of
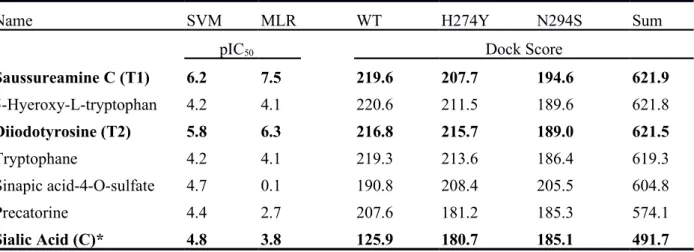
ligand in the individual protein model was used in ranking. Saussureamine C (T1) and
Diiodotyrosine (T2) (Figure 3) were both found to generate a high binding affinity in
both wild-type and mutants, where the sum of dock score of T1 and T2 was 621.887
and 621.524; this was higher than N1 nature ligand- sialic acid (C). The binding
affinity of sialic acid in the wild-type was significantly lower than in the mutants.
There were several ligands such as 5-Hyeroxy-L-tryptophan, Tryptophane, Sinapic
acid-4-O-sulfate, and Precatorine that performed with a high binding affinity but with
a low predicted bioactivity (also listed at Table 1). These ligands were not considered
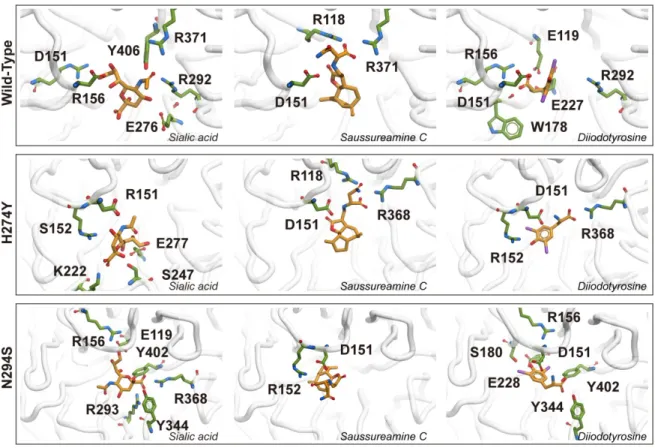
In H274Y, sialic acid generates Hydrogen bonds (H-bond) with I222 and D247,
which are classified in the outlier region. There is a structural difference between
Neuraminidases Group 1 and Group 2 in the ’150 loop’ (i.e. residue 147-152), in
which the ‘150-cavity’ is adjacent to the active site K150. The D151, which is located
on 150 loop, adheres to the active site can generate h-bonds with all ligands in each
mutant except for the sialic acid in the N294S mutant. Compared to R152, D151 can
generate fewer hydrogen bonds. It was found that E276 plays an important role in the
drug resistance. The E276 rotation in H274Y mutants makes Oseltamivir unable to be
combined in a hydrophobic pocket so that the drug resistance is generated. Sialic acid
can produce stable hydrogen bonds in the wild-type. E119 makes participation in the
Hydrogen bond formation of D151. Sialic acid can produce H-bonds only with E119
of N294S. In TCM candidate, with the catalytic residues hydrogen bond formation,
both T1 and T2 can form hydrogen bonds with D151 in H274Y and N294S mutants,
in which sialic acid cannot form H-bond with N294S. On the other hand, T1 can form
stable H-bonds with catalytic residues like R118 in the wild-type and H274Y and
R152 in N294S. T2 is different with both sialic acid and T1, and it can generate
H-bond with framework residue such as E119 in wild-types.
2.3 Ligand based drug design
The most representative descriptors were calculated by the Genetic function
approximation (GFA) that generated a confident model with a coefficient of
determination (R2) of 0.9884, RMS residual error of 0.0992. The GFA generated
sNH 2 3 mag AM 3 GFA=−1.2352−13.254∗ES¿2.613∗ES∑ sNH 2+10.494
∗CHI¿C−0.073883∗E¿0.6209∗Kappa¿1.0261∗SC2+0.071953∗Jurs¿
Seven descriptors are calculated and four of them are topology descriptors, which do
not rely on a three-dimensional model, including CHI_V_3_C (Connectivity Indices),
E_ADJ_mag (Edge Adjacency Magnitude), Kappa_3_AM (Kappa Shape Indices) and SC_2 (Subgraph Counts). Jurs_PNSA_3, dipole descriptors, calculate the atomic
charge weighted negative surface area. ES_Sum_sNH2 and ES_Count_sNH2 calculate
the sums of the Electrotopological State (E-state) values and the counts of each nitrogen-hydrogen single bond. The NH-bond consist hydrogen bond donor property
that provide the fundamental element for generating hydrogen bond in N1
hydrophobic pocket. The SVM and MLR models employed the aforementioned
descriptors, which GFA’s ***p < 0.001, to generate training, test, and prediction set.
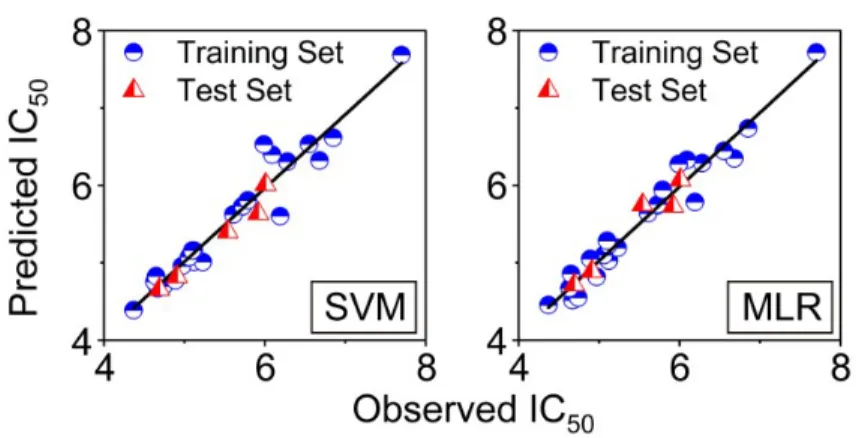
The R2 of SVM and MLR model were 0.9337 and 0.9563, respectively (Figure 4).
The models of SVM and MLR are both accurate enough (R2>0.6) for prediction. The
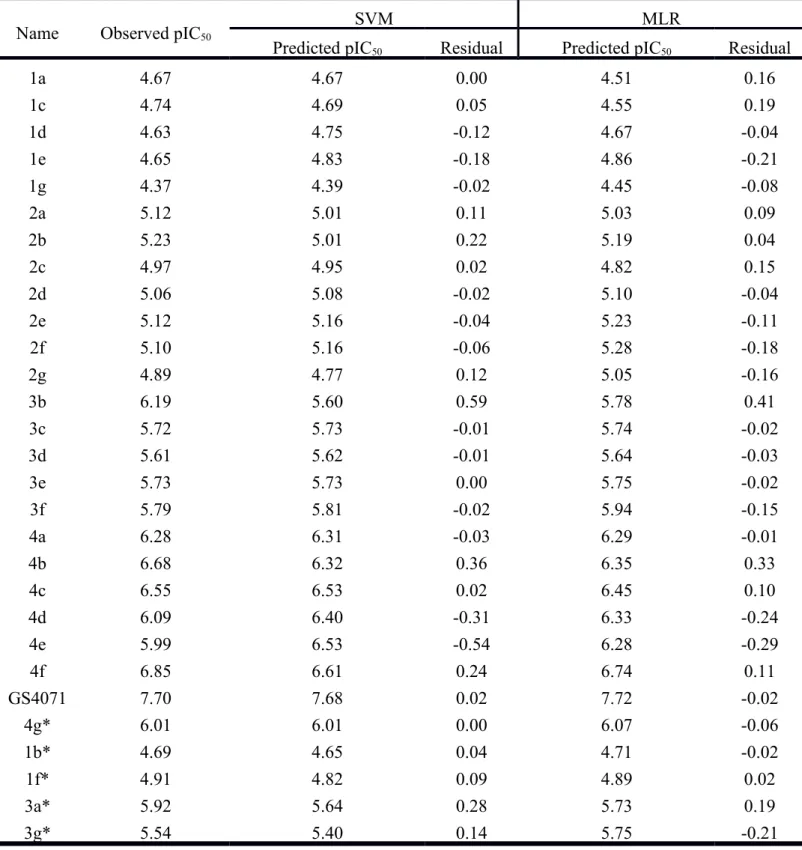
residuals of predicted IC50 of the training set and test set in SVM and MLR were
±0.6 and ±0.5 (Table 3), respectively; these results suggest that MLR shows more
promise in prediction. The predicted IC50 (pIC50) of sialic acid and TCM candidate are
listed in Table 1. Although the sum of dock score of Diiodotyrosine (T2) is lower than
5-Hyeroxy-L-tryptophan, the high pIC50 of Diiodotyrosine show a higher bioactivity
than 5-Hyeroxy-L-tryptophan. Saussureamine C and Diiodotyrosine, candidates
which perform a significant good bioactivity and binding affinity, were further used to
2.4 Molecular dynamics simulation
The analysis results of 12 groups of protein-ligand complex interaction mechanisms
were further explored by molecular dynamics simulation (including:
unbound-wild-type(U-WT), unbound-H274Y(U-H274Y), unbound-N294S (U-N294S) complex;
Sialic acid-wild-typeWT), Sialic acid-H274YH274Y), Sialic acid-N294S
(C-N294S) complex; Saussureamine C-wild-type(T1-WT), Saussureamine
C-H274Y(T1-H274Y), Saussureamine C-N294S (T1-N294S) complex; Diiodotyrosine-wild-type
(T2-WT), Diiodotyrosine-H274Y (T2-H274Y), Diiodotyrosine-N294S (T2-N294S)
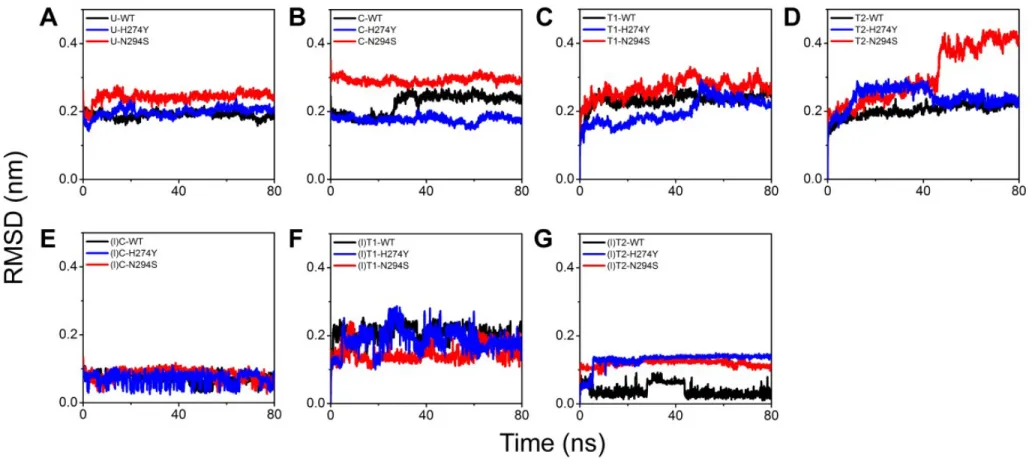
complex). Root-mean-square deviation (RMSD) show that ligand unbound wild-type,
H274Y and N294S are relatively more stable than ligand bound N1 (Figure 5A). After
25ns, the RMSD of Sialic acid bound wild-type N1 rises from 0.2 nm to 0.25 nm.
Like unbound H274Y, sialic acid, bound H274Y does not produce prompting changes
in 80ns, and N294S maintains at 0.3nm in 80ns (as shown in Figure 5B). T1 bound
wild-type rises to 0.25 nm in 5ns. After 45ns, T1-H274Y rises from 0.15nm to 0.3nm
and then declines to 0.2nm till 80ns. T1-N294S maintains between 0.2nm and 0.25nm
(as shown in Figure 5C). H274Y increases from 0.25nm to 0.4nm at 42nsd.
T2-N294S decreases from 0.28nm to 0.2nm at 40nsd (as shown in Figure 5D). T2-N294S is
less stable than H274Y and the wild-type no matter in the ligand bound or the
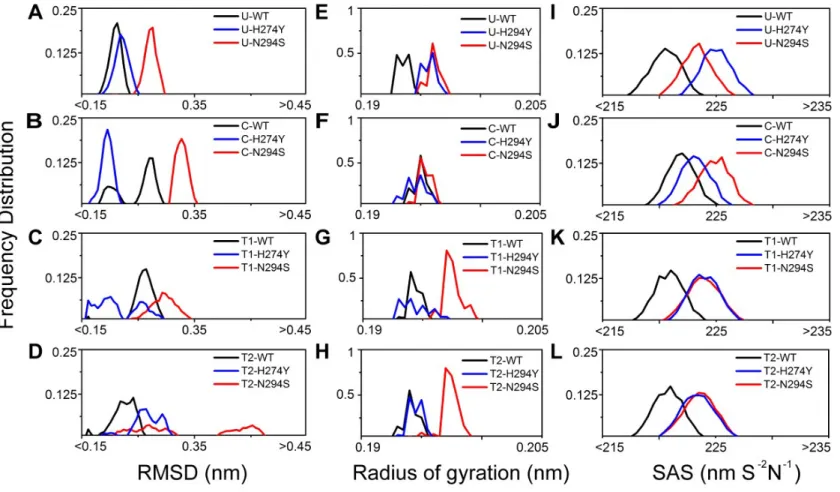
unbound (as shown in Figure 6A-D). The Radius of gyration (Rg) of C-N294S is very
close to C-WT and C-H274Y. Several key residues have been found to be important
in maintaining ligand binding affinity. To maintain a high binding affinity, in the rigid
docking, hydrogen bonds, or pi-pi interaction must be generated and maintained,
andnew hydrogen bonds or pi-pi interactions must also be generate. Although docking
only sialic acid does not react and produce D151 hydrogen bonds, in MD, only
which C-WT can generate hydrogen binds with relatively high frequency (Figure 7).
C-H274Y, T1-WT, T2-WT, and T2-N294S cannot produce ligand-D151 hydrogen
bond. Furthermore, sialic acid can generate stable hydrogen bonds with high
frequency with I149, K150, and D151 in the wild-type and produce H-bonds with
resistance residue E276 (i.e. which is also found in docking). This can explain why
sialic acid can be identified by N1 in nature. Interestingly, sialic acid can generate
H-bonds with low frequency only with D151 in H274Y and N294S. In addition to
through D151, sialic acid maintains the stability in binding in mutants, sialic acid also
produces hydrogen bonds with low frequency with two catalytic residues, E276 and
R292. In the '150 loop', T1 can only participate in the formation of hydrogen bonds
with low frequencies in the wild-type. T1 can participate in more hydrogen bond
formations in mutant species for the 150 loop. T1 participates in formations of
hydrogen bonds with low frequency (< 5%) in D151 and I149; and in N294S, T1 can
generate hydrogen bonds with higher frequency with D151 and R152 than H274
(>5%). T1 can also be involved in a number of non- hydrogen loop 150 formation.
The catalytic residue is such as R118, R371(>90% in wild-type), Y406; the
framework residue is such as E119 and R156. T2 concentrates hydrogen bonds in
D151 and R152 in the 150 loop and generates in the wild-type > 40% and > 50%
hydrogen bonds respectively in N294S and R152, and in H274Y >15% hydrogen
bonds with D151. D151 of the wild-type and mutants produces large amounts of
hydrogen bonds with T2 in docking. But in the MD process, only H274Y hydrogen
bonds are kept. T2 E119 hydrogen bonds generated in the wild-type docking are
maintained in MD and in H274Y and N294S, new E119 hydrogen bonds are also
generated. In WT, R118 new high-frequency hydrogen bonds are generated (>40%).
or catalyst residues than T1 and T2 can only produce> 40% and > 60% hydrogen
bonds with R156 in the wild-type and N294S.
In the wild-type N1 protein, U-WT, T1-WT (R2 = 0.7136), and T2-WT (R2 =
0.6159) have similar mechanisms of action (Figure 8); C-WT and U-WT have
different mechanisms of action (R2 = 0.4293); and T1 and T2 also retain similar
mechanisms of action (R2 = 0.7004). In the N1 protein of H274Y mutants, besides
from the similarity of T1-H274Y and T2-H274Y drops (R2 <0.5), other ligand-bound
complexes retain high similarity (R2> 0.5). C-H274Y generates mechanisms similar to
unbound H274Y, T1, and T2 in H274Y mutation compounds. The T2 of H274Y
mutants maintains high similarity with C-WT and T1-WT in the wild-type (R2 =
0.7085 and R2 = 0.6076). Also, T1 of H274Y produces high similarity to T2 of the
WT independently (R2 = 0.6798). In the N294S mutation species, T1 and T2 also
decrease like H274Y and retain similarity between C and T1 (R2 = 0.585), showing
that in different mutants, T1 can produce an effect similar to C but in the wild-type,
3 CONCLUSION
In this study, the N1 wild-type and the homology model of mutants are used in
combination with LBDD by SBDD. Sialic acid is as the control for screening the
TCM candidate. In the core of docking and MD for 150 loops, 150 cavities, 8
catalytic residues, and 11 framework residues, the effect of suppressing of TCM in N1
is studied. In the changes of protein structure, combined with wild-type results, it is
speculated that although Saussureamine C (T1) and Diiodotyrosine (T2) have a high
degree of similarity to unbound N1 in the wild-type, indicating that Saussureamine C
and Diiodotyrosine cannot produce an inhibitory effect on N1 wild-type,
Saussureamine C and oseltamivir can produce hydrogen bonds with N347s like
Saussureamine C and oseltamivir. R371 is the key residue that the oseltamivir
wild-type produces usual bidentate interaction (which only Saussureamine C and
oseltamivir can perform such interaction) and Saussureamine C can produce hydrogen
bonds with >80% frequency with R371. Since Saussureamine C may be able to
produce the wild-type N1 suppression effect similarly to oseltamivir, in the
perspective of structural dynamics connection, Saussureamine C is capable of mutant
inhibition to H274Y and N294S, and Diiodotyrosine is capable of mutant inhibition to
N294S mutants. Diiodotyrosine can produce stable hydrogen bonds with the
wild-type and R156 of N294S. In the past, it was considered more important that the
formation of stable hydrogen bonds with R156 than with the 150 loop, which
produces better inhibitory effects for mutants and wild-types than the past inhibitor
such as Oseltamivir. In the 150 loop, Diiodotyrosine can also generate stable
hydrogen bonds for the wild-type and mutants. Therefore, Saussureamine C and
Diiodotyrosine may be effective in generating potential drugs with inhibitory effect
4 METHODS
2.1 Structure-based drug design
The homology models of wild-type N1 (H1N1), H274Y N1 (H5N1), and N294S N1
(H5N1) as well as their mutant models were respectively established with the protein
desired in the experiment from the protein data bank download influenza A virus
neuraminidase (PDB: 2HTY [18]; 3CL0 and 3CL2 [19]). Multiple alignment scoring
matrices employed BLOSUM as primary algorithms with 10 gap open penalty.
RAMPAGE was used to verify validity of the predicted N1 model [20]. Sialic acid
were used to control and establish binding sites. The world's largest Chinese medicine
database, TCM Database@taiwan [17, 21], was used to provide about 60,000 TCM
compounds and N1s to conduct small molecule docking. In the setting of docking,
first, CHARMm was used to add hydrogen atoms of sixty thousand TCM compounds.
Then Lipinski’s Rule of Five was conducted to select the drug-like compounds. Then LigandFit algorithm was used for protein-ligand receptor matching [22]. In
LigandFit, Monte Carlo simulation was used to produce a number of different ligand
docking poses.Smart minimization was started by employing force-field CHARMm.
Minimized docking poses were then clustered with 1.0 RMS Threshold for Diversity.
A large number of the complexes after Docking obtained were respectively scored
and selected according to dock scores, the bioactivity predicted by the similarity and
ligand-based control. Scoring functions such as dock score and PMF and the related
N1-ligand complex were further chosen to produce scaffold for molecular dynamics
simulation. Dock Score = - (ligand/receptor interaction energy + ligand internal
energy). Finally, the Top 2 candidate was selected and its docking pose and inhibition
were studied with the control, respectively for H274Y, N294S, and wt-H1N1.
2.2 Ligand-based drug design
Bioactivity Prediction by Support Vector Machine (SVM) and Multiple Linear
Regression (MLR). 28 existing N1 inhibitor structures were used to conduct the
experiment and pIC50 was obtained to calculate individual physical property [26].
After calculation, 552 descriptors were obtained. Then the descriptors with higher
correlation were selected through genetic function approximation (GFA) and sorted
on square correlation coefficient (R2) [27]. Seven descriptors were obtained after the
calculation of the model with the highest R2 and merged with pIC50, and then the
training set and the test set were randomly assigned. The Support Vector Machine
(SVM) and Multiple Linear Regression (MLR) models were used to analyze
descriptors[23]. The nonlinear functions and predictions are determined by:
The SVM method with non-linear functions and the
MLR method with linear functions were used to build
models to predict the biochemical activity of TCM compounds. In this study,
LibSVM[28] and MATLAB-r2010a were used to respectively build SVM and MLR
models. The predicted bioactivity obtained by SVM and MLR prediction was
compared with the observed bioactivity to calculate square correlation coefficient
(R2), with R2> 0.5 classified as having credibility. Finally, the data were combined
with structure based screening to select the candidate.
f (x )=
∑
i=1 l (αi−αi¿)k( xi, x )+b s .t . 0≤αi¿ ≤C ,0≤αi≤C max . −1 2i,j=1∑
l (αi−αi¿)(αj−α¿j)k ( xi, xj) −ε∑
i=0 l (αi−αi¿)+∑
j=0 l yi(αj−α¿j)+ ¿{¿}¿{} ¿s . t .∑
i=0 l (αi−αi¿)=0 and αi−αi¿∈[0, C ]¿ ¿2.3 Molecular Dynamics Simulation
Full atom molecular dynamics simulations were performed on Gromacs platform
[29]. Cubic box was generated for immersing buffer solution, coupled ligand-N1
complex and uncoupled N1 (solvated with TIP3P water model). Ligands were
prepared by via SwissParam, force field CHARMm and parameters were added
through pdb2gmx protocol[30]. The distance was set to 1.2nm between the complex
and the edge of cubic box and 0.145M NaCl ion were added to neutralize the complex
system. Max(|Fn|)<ε was defined for determining minimization which was calculated
by steepest descent algorithm(i.e. time steps was set to 5000). Restrained dynamics
production generated by energy-minimization after 5000 time steps. Dynamics
production utilized methods such as Particle-Mesh Ewald (PME), Berendsen weak
thermal coupling methods, and NVT equilibration. Calculation of electrostatic
interaction employed PME. The total simulation time was set to 80ns under 2fs PME
option where the cut off was set to 1.0nm. Simulation trajectories analysis was
conducted by secondary structure database (DSSP) to analyze protein secondary
structure change. Plugin program g_gyrate, g_sas[31], g_rms, g_rmsf was used to
measure the radius of gyration, solvent accessible surface area, root mean square
ACKNOWLEDGEMENTS
The research was supported by grants from Asia University (ASIA102-CMU-1,
ASIA102-CMU-2, ASIA102-CMU-3) and China Medical University Hospital
(DMR-104-001, DMR-104-084, DMR-104-118). This study is also supported in part by
Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of
Excellence (MOHW104-TDU-B-212-113002) and CMU under the Aim for Top
REFERENCES
[1] M.B. Ritchey, P. Palese, J.L. Schulman, Mapping of Influenza-Virus Genome .3. Identification of Genes Coding for Nucleoprotein, Membrane-Protein, and Nonstructural Protein, J Virol, 20 (1976) 307-313.
[2] J.A. Griffin, S. Basak, R.W. Compans, Effects of Hexose Starvation and the Role of Sialic-Acid in Influenza-Virus Release, Virology, 125 (1983) 324-334.
[3] J.D. Thompson, D.G. Higgins, T.J. Gibson, Improved Sensitivity of Profile Searches through the Use of Sequence Weights and Gap Excision, Comput Appl Biosci, 10 (1994) 19-29.
[4] P.M. Colman, J.N. Varghese, W.G. Laver, Structure of the Catalytic and Antigenic Sites in Influenza-Virus Neuraminidase, Nature, 303 (1983) 41-44.
[5] M. Enserink, Oseltamivir becomes plentiful - But, still not cheap, Science, 312 (2006) 382-383.
[6] J.D. Bloom, L.I. Gong, D. Baltimore, Permissive Secondary Mutations Enable the Evolution of Influenza Oseltamivir Resistance, Science, 328 (2010) 1272-1275. [7] A.J. Hay, F.G. Hayden, Oseltamivir resistance during treatment of H7N9 infection, Lancet, 381 (2013) 2230-2232.
[8] A.H. Gaur, B. Bagga, S. Barman, R. Hayden, A. Lamptey, J.M. Hoffman, D. Bhojwani, P.M. Flynn, E. Tuomanen, R. Webby, Intravenous Zanamivir for Oseltamivir-Resistant 2009 H1N1 Influenza, New Engl J Med, 362 (2010) 88-89. [9] R.K. Gupta, J.S. Nguyen-Van-Tam, Oseltamivir resistance in influenza A (H5N1) infection, New Engl J Med, 354 (2006) 1423-1424.
[10] A.C. Hurt, K. Hardie, N.J. Wilson, Y.M. Deng, M. Osbourn, N. Gehrig, A. Kelso, Community Transmission of Oseltamivir-Resistant A(H1N1)pdm09 Influenza, New Engl J Med, 365 (2011) 2541-2542.
[11] V.J. Lee, J. Yap, A.R. Cook, M.I. Chen, J.K. Tay, B.H. Tan, J.P. Loh, S.W. Chew, W.H. Koh, R. Lin, L. Cui, C.W.H. Lee, W. Sung, C.W. Wong, M.L. Hibberd, W.L. Kang, B. Seet, P.A. Tambyah, Oseltamivir Ring Prophylaxis for Containment of 2009 H1N1 Influenza Outbreaks., New Engl J Med, 362 (2010) 2166-2174.
[12] A. Moscona, Global Transmission of Oseltamivir-Resistant Influenza, New Engl J Med, 360 (2009) 953-956.
[13] A. Moscona, Oseltamivir resistance - Disabling our influenza defenses, New Engl J Med, 353 (2005) 2633-2636.
[14] L. Le, E.H. Lee, D.J. Hardy, T.N. Truong, K. Schulten, Molecular Dynamics Simulations Suggest that Electrostatic Funnel Directs Binding of Tamiflu to Influenza N1 Neuraminidases, Plos Computational Biology, 6 (2010).
[15] J. Robertus, Structure-Based Drug Design 10 Years On, Nat Struct Biol, 1 (1994) 352-354.
[16] P.M. Dean, D.G. Lloyd, N.P. Todorov, De novo drug design: Integration of structure-based and ligand-based methods, Curr Opin Drug Disc, 7 (2004) 347-353. [17] C.Y.C. Chen, TCM Database@Taiwan: The World's Largest Traditional Chinese Medicine Database for Drug Screening In Silico, PLoS One, 6 (2011).
[18] R.J. Russell, L.F. Haire, D.J. Stevens, P.J. Collins, Y.P. Lin, G.M. Blackburn, A.J. Hay, S.J. Gamblin, J.J. Skehel, The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design, Nature, 443 (2006) 45-49. [19] P.J. Collins, L.F. Haire, Y.P. Lin, J.F. Liu, R.J. Russell, P.A. Walker, J.J. Skehel, S.R. Martin, A.J. Hay, S.J. Gamblin, Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants, Nature, 453 (2008) 1258-U1261.
[20] S.C. Lovell, I.W. Davis, W.B. Adrendall, P.I.W. de Bakker, J.M. Word, M.G. Prisant, J.S. Richardson, D.C. Richardson, Structure validation by C alpha geometry: phi,psi and C beta deviation, Proteins-Structure Function and Genetics, 50 (2003) 437-450.
[21] T.Y. Tsai, K.W. Chang, C.Y.C. Chen, iScreen: world's first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan, J Comput Aid Mol Des, 25 (2011) 525-531.
[22] C.M. Venkatachalam, X. Jiang, T. Oldfield, M. Waldman, LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites, J Mol Graph Model, 21 (2003) 289-307.
[23] W.I. Tou, C.Y.C. Chen, In Silico Investigation of Potential Src Kinase Ligands from Traditional Chinese Medicine, PLoS One, 7 (2012).
[24] W.I. Tou, C.Y.C. Chen, Traditional Chinese medicine as dual guardians against hypertension and cancer?, J Biomol Struct Dyn, 30 (2012) 299-317.
[25] W.I. Tou, S.S. Chang, C.C. Lee, C.Y.C. Chen, Drug Design for Neuropathic Pain Regulation from Traditional Chinese Medicine, Sci Rep-Uk, 3 (2013).
[26] Y. Liu, F.B. Jing, Y.Y. Xu, Y.C. Xie, F.Y. Shi, H. Fang, M.Y. Li, W.F. Xu, Design, synthesis and biological activity of thiazolidine-4-carboxylic acid derivatives as novel influenza neuraminidase inhibitors, Bioorgan Med Chem, 19 (2011) 2342-2348.
[27] D. Rogers, A.J. Hopfinger, Application of Genetic Function Approximation to Quantitative Structure-Activity-Relationships and Quantitative Structure-Property Relationships, J Chem Inf Comp Sci, 34 (1994) 854-866.
[28] C.C. Chang, C.J. Lin, LIBSVM: A Library for Support Vector Machines, Acm T Intel Syst Tec, 2 (2011).
[29] S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M.R. Shirts, J.C. Smith, P.M. Kasson, D. van der Spoel, B. Hess, E. Lindahl, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics, 29 (2013) 845-854.
[30] V. Zoete, M.A. Cuendet, A. Grosdidier, O. Michielin, SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules, Journal of Computational Chemistry, 32 (2011) 2359-2368.
[31] F. Eisenhaber, P. Lijnzaad, P. Argos, C. Sander, M. Scharf, The Double Cubic Lattice Method - Efficient Approaches to Numerical-Integration of Surface-Area and Volume and to Dot Surface Contouring of Molecular Assemblies, Journal of Computational Chemistry, 16 (1995) 273-284.
Table
Table 1 Candidates were selected based on the DockScore, which are higher than Sialic Acid
amount wide type and two mutants of N1. Sum of DockScore of candidates show that the score are higher than control. The predicted value from SVM and MLR models denote Saussureamine C and Diiodotyrosine with high activity values are higher than Sialic Acid.
Name SVM MLR WT H274Y N294S Sum
pIC50 Dock Score
Saussureamine C (T1) 6.2 7.5 219.6 207.7 194.6 621.9 5-Hyeroxy-L-tryptophan 4.2 4.1 220.6 211.5 189.6 621.8 Diiodotyrosine (T2) 5.8 6.3 216.8 215.7 189.0 621.5 Tryptophane 4.2 4.1 219.3 213.6 186.4 619.3 Sinapic acid-4-O-sulfate 4.7 0.1 190.8 208.4 205.5 604.8 Precatorine 4.4 2.7 207.6 181.2 185.3 574.1 Sialic Acid (C)* 4.8 3.8 125.9 180.7 185.1 491.7 *Control
Table 2. Homology model validation and protein-ligand interaction detail. Hydrogen
bond and pi-pi interaction of each complex is listed below.
Index Residue WT H274Y N294S
Eva C T1 T2 Eva C T1 T2 Eva C T1 T2
118 ARG HH HH 119 GLU H HH 120 PRO a 125 SER o o a 145 SER a 151 ASP HH HHH H HH HHH HHH a HH HH 152 ARG H HH HH 156 ARG HH HHP H P 178 TRP H 180 SER HH 200 GLY a 201 GLY a 202 ALA a 222 ILE o HHH 223 ILE o o 225 THR a 226 THR a a 227 GLU HH 228 GLU H 235 ASN a 247 ASP o H 248 GLY a 249 GLN a 270 ALA o 271 PRO a 276 GLU H 277 GLU HHHH 291 CYS a 292 CYS HH P a a 293 ARG HH 295 TRP a 296 TRP a a
310 LEU a 330 ASP a 331 LYS a a 343 SER a 344 ASN a H H 345 GLY a 347 ASN a 368 ARG HHH HHH HHH 371 ARG H HHH 384 ASP a 387 ASP a 388 ASN a 400 SER a a 402 TYR H H 404 SER a 406 TYR H 448 VAL o 449 ASNa o a 451 SER a 453 THR a 455 TRP a a 456 TRP a
Eva: Evaluation of residues.
a:449ASN(H274Y)/VAL(wt)
H: hydrogen bond. P: pi-pi interaction.
Table 3. Bioactivity predicted by SVM and MLR.
Name Observed pIC50
SVM MLR
Predicted pIC50 Residual Predicted pIC50 Residual
1a 4.67 4.67 0.00 4.51 0.16 1c 4.74 4.69 0.05 4.55 0.19 1d 4.63 4.75 -0.12 4.67 -0.04 1e 4.65 4.83 -0.18 4.86 -0.21 1g 4.37 4.39 -0.02 4.45 -0.08 2a 5.12 5.01 0.11 5.03 0.09 2b 5.23 5.01 0.22 5.19 0.04 2c 4.97 4.95 0.02 4.82 0.15 2d 5.06 5.08 -0.02 5.10 -0.04 2e 5.12 5.16 -0.04 5.23 -0.11 2f 5.10 5.16 -0.06 5.28 -0.18 2g 4.89 4.77 0.12 5.05 -0.16 3b 6.19 5.60 0.59 5.78 0.41 3c 5.72 5.73 -0.01 5.74 -0.02 3d 5.61 5.62 -0.01 5.64 -0.03 3e 5.73 5.73 0.00 5.75 -0.02 3f 5.79 5.81 -0.02 5.94 -0.15 4a 6.28 6.31 -0.03 6.29 -0.01 4b 6.68 6.32 0.36 6.35 0.33 4c 6.55 6.53 0.02 6.45 0.10 4d 6.09 6.40 -0.31 6.33 -0.24 4e 5.99 6.53 -0.54 6.28 -0.29 4f 6.85 6.61 0.24 6.74 0.11 GS4071 7.70 7.68 0.02 7.72 -0.02 4g* 6.01 6.01 0.00 6.07 -0.06 1b* 4.69 4.65 0.04 4.71 -0.02 1f* 4.91 4.82 0.09 4.89 0.02 3a* 5.92 5.64 0.28 5.73 0.19 3g* 5.54 5.40 0.14 5.75 -0.21 *: test set.
Figure
` 1. Homology model validation of (A) wild-type, (B) H274Y and (C) N294S N1
Figure 2. Docking pose of wild-type, H274Y and N294S N1. Figures only show the
residues which perform interaction with ligand. Ligands are colored in orange
(a) (b) (c)
SaussureamineC (T1) Diiodotyrosine (T2) Sialic Acid
Figure 4. The relationship between observed and predicted activities of SVM and
Figure 6. Frequency Distribution of root mean square derivation (RMSD), Radius of gyration (Rg) and Solvent accessible surface area (SAS)
analysis of N1 protein.
Figure 7. Frequency of Hydrogen bond formation between ligand and N1 protein. Seven catalytic residues (R118, D151, R152, E276, R292,
R371, and Y406) are shown. Other framework residue (E119, R156, W178, I222, E227, E277, N294, and E425) are shown. Catalytic residues
Figure 8. Protein RMSF correlation analysis, correlation between each complex are calculated and demonstrated in red (R2<0.5), white
(R2=0.5) and blue (R2>0.5) colors.