國立台中教育大學科學應用與推廣學系碩士論文
國立台中教育大學科學應用與推廣學系碩士論文
國立台中教育大學科學應用與推廣學系碩士論文
國立台中教育大學科學應用與推廣學系碩士論文
指導老師
指導老師
指導老師
指導老師:
:
:張嘉麟
:
張嘉麟
張嘉麟
張嘉麟
博士
博士
博士
博士
硫甲醛
硫甲醛
硫甲醛
硫甲醛分子與負離子
分子與負離子
分子與負離子
分子與負離子光電子光譜
光電子光譜
光電子光譜
光電子光譜
的理論研究
的理論研究
的理論研究
的理論研究
Theoretical study of the photoelectron spectra of
thioformaldehyde and thioformaldehyde anion
研究生
研究生
研究生
研究生:
:
:
:江祥印
江祥印
江祥印
江祥印
撰
撰
撰
撰
中 華 民 國 九 十 八 年 一
中 華 民 國 九 十 八 年 一
中 華 民 國 九 十 八 年 一
中 華 民 國 九 十 八 年 一 月
月
月
月
摘要 摘要 摘要 摘要
本 研 究 目 的 在 於 運 用 密 度 泛 函 理 論 的 B3LYP 泛 函 數 , 以 6-311++G(d,p) 、 6-311++G(2d,p) 、 6-311++G(3d,p) 、 6-311++G(3df,p) 、 6-311++G7(3df,pd)和 aug-cc-pVDZ 與 aug-cc-pVTZ 等七個基組,計算硫甲醛 H2CS 分子以及正、負離子的平衡結構和振動頻率,並利用計算所得之基態 和離子態的結構和振動頻率,以本研究室發展的方法,計算法蘭克-康登因 子,進而模擬其光電子光譜,並與實驗光譜相比對。研究結果發現:計算的 分子平衡結構與實驗結果相符,而振動頻率與實驗值的差距小於 5%。另外, 模擬光譜與實驗光譜相符,顯示法蘭克-康登因子的計算,有助於解析或預 測分子光譜。同時,經由理論計算結果發現,H2CS 游離成 H2CS+的平衡結 構變化極小,判斷電子是由非鍵結軌域所游離的;而從 H2CS -脫電子成 H 2CS 其 SC 鍵長縮短了 10 pm,我們認為是電子是由 π*軌域脫離。最後,我們以 CCSD(T)/aug-cc-pVTZ 方法計算分子與離子的單點能,算得分子的游離
能為 9.313 eV、電子親和力為 0.459 eV,與實驗值的 9.338 eV、0.465 eV 相
近,其差異分別為 0.02 eV 和 0.005 eV。
Abstract
The equilibrium geometries and harmonic vibrational frequencies of neutral,
cationic and anionic thioformaldehyde (H2CS, H2CS+ and H2CS-) have been
calculated using the B3LYP method and the basis sets of 6-311+G(d,p),
6-311++G(2d,p), 6-311++G(3d,p), 6-311++G(3df,p), 6-311++G(3df,pd), aug-cc-pVDZ
and aug-cc-pVTZ. Franck-Condon factors have been computed using a method
developed by our research group. The photoelectron spectra of H2CS and H2CS
-have been simulated and compared with the experiment. It was found that the
calculated equilibrium geometries are in agreement with the experiment, and the
error of computed vibrational frequencies is less than 5%. Meanwhile, the
simulated photoelectron spectra are in accord with the experiment, meaning that
the calculation of Franck-Condon factors is useful for interpreting or predicting
molecular spectra. It was also inferred that the ionization of H2CS involves
removing a nonbonding electron, whereas H2CS- a π* electron. Finally, we
calculated the adiabatic ionization energy and electron affinity of
thioformaldehyde by using the CCSD(T) method. The errors of the aug-cc-pVTZ
calculations are small and within 0.02 and 0.005 eV for the adiabatic ionization
energy and electron affinity, respectively.
第一章 第一章 第一章 第一章 緒論緒論 ...緒論緒論... 1111 1.1 1.1 1.1 1.1 研究動機與重要性研究動機與重要性研究動機與重要性 ...研究動機與重要性... 1... 111 1.2 1.2 1.2 1.2 硫甲醛文獻探討硫甲醛文獻探討硫甲醛文獻探討...硫甲醛文獻探討... 3333 1.3 1.3 1.3 1.3 法蘭克法蘭克法蘭克-法蘭克--康登因子-康登因子 ...康登因子康登因子 ... 8888 1.4 1.4 1.4 1.4 量子計算發展沿革量子計算發展沿革量子計算發展沿革 ...量子計算發展沿革... 12... 1212 12 1.5 1.5 1.5 1.5 論文架構論文架構論文架構...論文架構... 14... 1414 14 第二章 第二章 第二章 第二章 理論計算方法理論計算方法 ...理論計算方法理論計算方法... 15151515 2.1 2.1 2.1
2.1 密度泛函理論密度泛函理論密度泛函理論 (density functional theory, DFT)密度泛函理論 (density functional theory, DFT) (density functional theory, DFT) (density functional theory, DFT) ... 15... 1515 15
2.1.1 Kohn-Sham 方程式 ... 19 2.1.2 泛函數(Functional)... 22 2.1.3 基底函數組(basis set)... 23 2.1.4 耦合簇理論 (Coupled-cluster theory)... 27 2.2 2.2 2.2 2.2 中性分子中性分子中性分子、中性分子、、正離子、正離子正離子正離子、、負離子的平衡結構計算、、負離子的平衡結構計算負離子的平衡結構計算負離子的平衡結構計算 ... 29... 2929 29 2.3 2.3 2.3 2.3 法蘭克法蘭克-法蘭克法蘭克---康登因子康登因子康登因子康登因子... 313131 31 2.4 2.4 2.4 2.4 馬克斯威爾馬克斯威爾馬克斯威爾─馬克斯威爾───波茲曼分布函數波茲曼分布函數 ...波茲曼分布函數波茲曼分布函數... 333333 33 2.5 2.5 2.5 2.5 光譜模擬光譜模擬光譜模擬...光譜模擬... 35... 3535 35 第三章 第三章 第三章 第三章 研究結果與討論研究結果與討論 ...研究結果與討論研究結果與討論... 37373737 3.1 3.1 3.1 3.1 硫甲醛分子硫甲醛分子硫甲醛分子、硫甲醛分子、、、正離子正離子正離子正離子、、負離子平衡結構的計算結果、、負離子平衡結構的計算結果負離子平衡結構的計算結果負離子平衡結構的計算結果 ... 373737 37 3.1.1 平衡結構計算值與實驗值差距比較結果 ... 37 3.1.2 分子與離子結構的改變 ... 42 3.2 3.2 3.2 3.2 硫甲醛分子硫甲醛分子硫甲醛分子、硫甲醛分子、、、正離子與負離子的振動模式與頻率正離子與負離子的振動模式與頻率正離子與負離子的振動模式與頻率正離子與負離子的振動模式與頻率 ... 454545 45 3.2.1 硫甲醛分子的振動模式與頻率 ... 45
目次
目次
目次
目次
3.2.2 硫甲醛正離子的振動模式與頻率 ... 49 3.2.3 硫甲醛負離子的振動模式與頻率 ... 54 3.3 3.3 3.3 3.3 游離能與電子親和力游離能與電子親和力游離能與電子親和力 ...游離能與電子親和力... 606060 60 3.4 3.4 3.4 3.4 法蘭克法蘭克法蘭克-法蘭克--康登因子-康登因子 ...康登因子康登因子... 63... 6363 63 3.4.1 分子→正離子的法蘭克─康登因子 ... 63 3.4.2 H2CS -負離子→中性分子的法蘭克─康登因子 ... 66 3.5 3.5 3.5 3.5 馬克斯威爾馬克斯威爾馬克斯威爾─馬克斯威爾───波茲曼分布函數波茲曼分布函數 ...波茲曼分布函數波茲曼分布函數... 707070 70 3.6 3.6 3.6 3.6 模擬光電子光譜和實驗光譜的比對模擬光電子光譜和實驗光譜的比對模擬光電子光譜和實驗光譜的比對 ...模擬光電子光譜和實驗光譜的比對... 727272 72 3.6.1 硫甲醛的光電子光譜與實驗光譜的比對 ... 72 3.6.2 硫甲醛負離子的光電子光譜與實驗光譜的比對 ... 75 第四章 第四章 第四章 第四章 結論結論 ...結論結論... 83838383 參考文獻 參考文獻 參考文獻 參考文獻...85...85...85...85 附 附 附 附錄錄錄...89錄...89...89...89
表一 表一 表一
表一、、、、DUNNINGDUNNINGDUNNINGDUNNING 的基底函數包含的極化函數類型的基底函數包含的極化函數類型的基底函數包含的極化函數類型...的基底函數包含的極化函數類型... 26262626 表二 表二 表二 表二、、、、HHHH2222CS 不同計算基組之幾何優選與實驗值之比較CSCSCS不同計算基組之幾何優選與實驗值之比較不同計算基組之幾何優選與實驗值之比較不同計算基組之幾何優選與實驗值之比較 ... 39393939 表三 表三 表三 表三、、、H、HHH2222CSCSCSCS++++不同計算基組之幾何優選不同計算基組之幾何優選不同計算基組之幾何優選不同計算基組之幾何優選 ... 40404040 表四 表四 表四 表四、、、H、HHH2222CSCSCSCS----不同計算基組之幾何優選與實驗值不同計算基組之幾何優選與實驗值...不同計算基組之幾何優選與實驗值不同計算基組之幾何優選與實驗值... 41414141 表五 表五 表五 表五、、、H、HHH2222CSCS 的正規振動模式CSCS的正規振動模式的正規振動模式的正規振動模式... 46464646 表六 表六 表六 表六、、、、HHHH2222CS 在不同計算基組的振動頻率與實驗值的比較CSCSCS在不同計算基組的振動頻率與實驗值的比較在不同計算基組的振動頻率與實驗值的比較在不同計算基組的振動頻率與實驗值的比較 ... 48484848 表七 表七 表七 表七、、、、HHHH2222CSCSCSCS++++的正規振動模式的正規振動模式的正規振動模式的正規振動模式... 49494949 表八 表八 表八 表八、、、H、HHH2222CSCSCSCS++++在不同計算基組的振動頻率與實驗值的比較在不同計算基組的振動頻率與實驗值的比較在不同計算基組的振動頻率與實驗值的比較在不同計算基組的振動頻率與實驗值的比較 ... 51515151 表九 表九 表九 表九、、、H、HHH2222CS 與CSCSCS與與與 HHHH2222CSCSCSCS++++的振動頻率比較 ...的振動頻率比較的振動頻率比較的振動頻率比較 ... 52...525252 表十 表十 表十 表十、、、、HHHH2222CSCSCSCS----的正規振動模式的正規振動模式的正規振動模式的正規振動模式... 54545454 表十一 表十一 表十一 表十一、、H、、HHH2222CSCSCSCS----不同計算基組的振動頻率與實驗值的比較不同計算基組的振動頻率與實驗值的比較不同計算基組的振動頻率與實驗值的比較不同計算基組的振動頻率與實驗值的比較 ... 56565656 表十二 表十二 表十二
表十二、、H、、HHH2222CSCSCSCS----在兩種方法在兩種方法在兩種方法在兩種方法 B3LYPB3LYP、B3LYPB3LYP、、、MP2MP2MP2 下的振動頻率與實驗值的比MP2下的振動頻率與實驗值的比較下的振動頻率與實驗值的比下的振動頻率與實驗值的比較較 ..較... 58585858
表十三 表十三 表十三 表十三、、、H、HHH2222CS 與CSCSCS與與與 HHHH2222CSCSCSCS----的振動頻率比較 ...的振動頻率比較的振動頻率比較的振動頻率比較... 59595959 表十四 表十四 表十四 表十四、、、、計算硫甲醇分子與離子計算硫甲醇分子與離子計算硫甲醇分子與離子計算硫甲醇分子與離子 CCSD(T)CCSD(T)能量表CCSD(T)CCSD(T)能量表能量表能量表... 60606060 表十五 表十五 表十五 表十五、、、、硫甲醛分子絕熱游離能硫甲醛分子絕熱游離能硫甲醛分子絕熱游離能硫甲醛分子絕熱游離能 ... 62626262 表十六 表十六 表十六 表十六、、、、硫甲醛分子電子親和力硫甲醛分子電子親和力硫甲醛分子電子親和力硫甲醛分子電子親和力 ... 62626262 表十七 表十七 表十七
表十七、、、、以基組以基組以基組以基組 augaugaugaug----cccccc-cc---pVTZpVTZpVTZ 所計算的pVTZ所計算的所計算的所計算的 Duschinsky matrixDuschinsky matrixDuschinsky matrix...63Duschinsky matrix...63...63...63
表目次
表目次
表目次
表十八 表十八 表十八 表十八、、、、HHHH2222CS 分子電離成離子基態的法蘭克康登因子CSCSCS分子電離成離子基態的法蘭克康登因子分子電離成離子基態的法蘭克康登因子 ...分子電離成離子基態的法蘭克康登因子 ... 65656565 表十九 表十九 表十九
表十九、、、、以基組以基組以基組以基組 augaugaugaug----cccccc-cc-pVTZ--pVTZpVTZ 所計算的pVTZ所計算的所計算的所計算的 Duschinsky matrixDuschinsky matrix...Duschinsky matrixDuschinsky matrix...66...6666 66 表二十 表二十 表二十 表二十、、、、HHHH2222CSCSCSCS----從基態ν從基態從基態從基態ννν=0=0=0=0 躍遷的躍遷的 FCF 躍遷的躍遷的FCF FCF FCF ... 67676767 表二十一 表二十一 表二十一 表二十一、、、、HHHH2222CSCSCSCS----從從ν從從ννν3333====1111 躍遷至分子基態的躍遷至分子基態的 FCF 躍遷至分子基態的躍遷至分子基態的FCF FCF FCF ... 68686868 表二十二 表二十二 表二十二 表二十二、、、、HHHH2222CSCSCSCS----從從ν從從ννν4444====1111 躍遷至分子基態的躍遷至分子基態的 FCF躍遷至分子基態的躍遷至分子基態的FCFFCFFCF... 69696969 表二十三 表二十三 表二十三 表二十三、、、、HHHH2222CSCSCSCS----從從ν從從ννν4444====2222 躍遷至分子基態的躍遷至分子基態的 FCF躍遷至分子基態的躍遷至分子基態的FCFFCFFCF... 69696969
圖一 圖一 圖一 圖一 硫甲醛硫甲醛 (H硫甲醛硫甲醛 (H (H (H2222CS) 分子示意圖CS) CS) CS) 分子示意圖分子示意圖分子示意圖 ... 7777 圖二 圖二 圖二 圖二 硫甲醛分子與正離子示意圖硫甲醛分子與正離子示意圖 ...硫甲醛分子與正離子示意圖硫甲醛分子與正離子示意圖 ... 42424242 圖三 圖三 圖三 圖三 硫甲醛分子與負離子結構示意圖硫甲醛分子與負離子結構示意圖 ...硫甲醛分子與負離子結構示意圖硫甲醛分子與負離子結構示意圖 ... 43...434343 圖四 圖四 圖四 圖四 HHHH2222CSCSCSCS----的游離成的游離成的游離成的游離成 HHHH2222CSCSCSCS 的溫度的溫度 ...的溫度的溫度... 70...707070 圖五 圖五 圖五 圖五 模擬硫甲醛光電子光譜與實驗光譜比較圖模擬硫甲醛光電子光譜與實驗光譜比較圖...模擬硫甲醛光電子光譜與實驗光譜比較圖模擬硫甲醛光電子光譜與實驗光譜比較圖... 72...727272 圖七 圖七 圖七 圖七 HHHH2222CSCS 在不同基組下的模擬光電子光譜CSCS在不同基組下的模擬光電子光譜在不同基組下的模擬光電子光譜在不同基組下的模擬光電子光譜... 74747474 圖八 圖八 圖八 圖八 HHHH2222CSCSCSCS----從基態從基態從基態從基態νννν==0==000 躍遷的模擬光電子光譜躍遷的模擬光電子光譜躍遷的模擬光電子光譜躍遷的模擬光電子光譜 ... 76...767676 圖九 圖九 圖九 圖九 HHHH2222CSCSCSCS----從基態從基態從基態從基態νννν3333==1==1 躍遷的模擬光電子光譜11躍遷的模擬光電子光譜躍遷的模擬光電子光譜躍遷的模擬光電子光譜... 77...777777 圖十 圖十 圖十 圖十 HHHH2222CCCCSSSS----從基態從基態從基態從基態νννν4444==1==1 躍遷的模擬光電子光譜11躍遷的模擬光電子光譜躍遷的模擬光電子光譜躍遷的模擬光電子光譜... 78...787878 圖十一 圖十一 圖十一 圖十一 HHHH2222CSCSCSCS----從基態從基態從基態從基態νννν4444==2==2 躍遷的模擬光電子光譜22躍遷的模擬光電子光譜躍遷的模擬光電子光譜躍遷的模擬光電子光譜... 79...797979 圖十二 圖十二 圖十二 圖十二 HHHH2222CSCSCSCS----從不同模式躍遷的模擬光譜圖從不同模式躍遷的模擬光譜圖從不同模式躍遷的模擬光譜圖從不同模式躍遷的模擬光譜圖,,,,與最後計算波與最後計算波與最後計算波與最後計算波茲曼分布茲曼分布茲曼分布茲曼分布 的合成光譜圖 的合成光譜圖 的合成光譜圖 的合成光譜圖。。。。... 80808080 圖十三 圖十三 圖十三 圖十三 計算計算計算 H計算HHH2222CSCSCSCS----波茲曼分布的合成光譜圖與實驗室光譜進行比波茲曼分布的合成光譜圖與實驗室光譜進行比波茲曼分布的合成光譜圖與實驗室光譜進行比波茲曼分布的合成光譜圖與實驗室光譜進行比對對對對。。。。 ... 82828282
圖目次
圖目次
圖目次
圖目次
第一章
第一章
第一章
第一章
緒論
緒論
緒論
緒論
本論文旨在運用密度泛函理論,對硫甲醛分子、正離子與負離子進行量 子計算的理論研究,並以本研究室發展的方法計算法蘭克-康登因子 (Franck-Condon factor, FCF),預測其光電子光譜。本章共分成五節,第一 節介紹本研究的動機與重要性,第二節與第三節分別進行硫甲醛分子與法蘭 克-康登因子的文獻探討,第四節介紹量子計算發展的歷史沿革,第五節說 明本論文的架構。
1.1 研 究 動 機 與 重 要 性
研 究 動 機 與 重 要 性
研 究 動 機 與 重 要 性
研 究 動 機 與 重 要 性
硫甲醛 (H2C=S, Thioformaldehyde) 是硫醛類中最簡單的分子,在常 溫常壓下是極不穩定的氣態分子,在 0.1mmHg 的化學平衡狀態下的半衰期 約為 6 分鐘[1],因此分子常以三聚體 CH2(SCH2)2S 的方式存在[2]。在天文 學上硫化物是外太空主要的成分之ㄧ,在 1973 年[3]發現 H2CS 是星際物質 之ㄧ,而硫甲醛分子在 Moses 等人研究硫化物在木星表層上的光化學反應 中[4],是一個極為重要的中間產物,因此探討 H2CS 分子在天文與大氣化學 所扮演的角色,有其重要性。同時,了解其分子結構也將有助於鑑別太空中 S=C 化合物的的種類和光化學反應[5-7]。在日常生活應用中,為了使絲織物 增加重量而使用化學處理方法,稱之為「增重處理」,會使用尿素甲醛或硫 甲醛初縮體當作反應試劑[8]。 光譜學上,碳硫鍵 (C=S) 的化學性質在光化學領域中一直受到科學界的重視,其光譜的研究從微波到紫外光的範圍,都已經有了實驗數據,但 是對於電子激發態的光譜標定仍然有著一定的難度。2005 年 Chiang 和 Lin 進行 H2CS 在高雷德堡態 5.6–9.5eV 的光譜研究,因為產生 CS2與 CH4的光 譜干擾,限制了光譜標定的準確性[9],而在光電子光譜的研究上也遇到相 同的困難,1972 年 Kroto 和 Suffolk[10]由二甲基二硫化物 (CH3SSCH3) 的 熱裂解,觀察到 H2CS 的光電子光譜與 CH3SH 的第一個游離態重疊,因此 無法明確標定 H2CS 的光譜。1976 年 Solouki 等人雖然藉由熱裂解 H3CSCl, 成功研究出 H2CS 的光電子光譜[11],但是並未深入解析其光譜的結構。因 此在本研究中,我們想要利用本研究室推導出的法蘭克-康登因子運算公 式,計算其法蘭克-康登因子,並模擬 H2CS 和 H2CS - 的光電子光譜,再與 實 驗 光 譜 進 行 比 對,以 期 能 對 H2CS 和 H2CS - 的光電子光譜做深入的研 究。 法蘭克-康登因子代表分子的電子振動躍遷機率,其計算方式則是運用 分子在躍遷前後的平衡結構差異,與不同的振動模式的角頻率,算出分子在 兩個電子態的振動波函數重疊積分的平方。2005 年 Chang (張嘉麟教授) 提 出另一種公式來計算法蘭克-康登因子[12],與其他方法相較,這個方法更 為簡單易懂,因此本研究採用 Chang 所推導的法蘭克-康登因子的公式, 對硫甲醛進行光電子光譜的理論研究。
1.2 硫 甲 醛
硫 甲 醛
硫 甲 醛 文 獻 探 討
硫 甲 醛
文 獻 探 討
文 獻 探 討
文 獻 探 討
硫甲醛是硫醛家族中基本而且具代表性的分子,尤其在大氣學與天文學 領域中,硫甲醛的分子結構與化學反應性都有助於分析外太空硫化物的種類 與相關的光化學反應。1973 年 Sinclair 等人從人馬星座 B2 星雲 (Sagittarius B2) 觀察到 H2CS 在轉動態 211←212的微波躍遷光譜[13],證實 H2CS 為星際 物質之ㄧ。在 1994 年 SL9 號彗星撞擊木星後,木星平流層硫化物的光化學 反應過程,發現 H2CS 在一連串的光化學反應過程中,扮演著極為重要的角 色。1997 年 Willacy 和 Millar[14]指出,在研究輕質量恆星晚期形成的環星 包 (circumstellar envelopes) 中,H2CS 等硫化物和環星包的形成有密切的關 係。次年,Taylor 等人[15]在天文學上對於 H2CS 的研究有了更近一步的發 現,在研究一顆輕質量恆星形成的早期組成成分中,CS 分子含量高於 NH3 家族,而包括 H2CS 在內的硫醛家族,更是恆星早期形成的重要分子。 綜觀上述文獻,星體的化學組成與化學結構是天文學領域重要的一門研 究,而星際物質間的序列化學反應與星體形成之間有著密不可分的關係,對 於星際物質的光化學反應一直是天文學家的研究重點,其中包括外太空主要 的星際物質「硫碳化合物」,而硫甲醛更是硫化物在光化學反應過程中一個 重要的中間產物,因此研究硫甲醛的光譜與分析其分子結構,有助於鑑別星 際間的硫化物的種類與化學反應性。對於 H2CS 的光譜研究,則是由 Johnson 等人[1]於 1971 年首先觀察到 H2CS 的微波光譜,了解其化學結構與轉動常數 A、B、C。並且確立分子結 構屬於 C2v平面分子,之後對於其光譜的研究便如火如荼的展開,翌年 Beers 等人[16]指出 H2CS 分子在平衡狀態的系統有六分鐘的半衰期,也觀察了 H2CS 在基態的振動微波光譜,並測量出 H2CS 四個同位素 (D212C32S、 H213C32S、H212C34S、H212C32S) 的轉動光譜與轉動常數,而獲得下列基態分 子平衡結構的數據:rS-C=1.6108 Å, rC-H=1.0925 Å,∠HCH=116.87°,並且 藉由 Stark effect 的技術將 J=101←000的能帶分離,獲得分子偶極矩 μ=1.6474
Debye。1983 年 Clouthier 和 Ramsay 統整 H2CS 和 H2CO 以及其同位素的光
譜學[17],並將實驗所得的基態與不同激發態的分子結構與振動模式,與計 算結果做比對。 在實驗室中,探討 S=C 混合物的分解動力學與光化學反應,也多以 H2CS 為典型範例,1972 年 Kroto 和 Suffolk 經由 CH3SSCH3的熱裂解觀察到 硫甲醛的光電子光譜[18],但由於熱裂解產物中的 CH3SH 的光譜與 H2CS 的 電子光譜重疊,在當時只能以概略的方式標定硫甲醛的光電子光譜。 2004 年 Chiang 和 Lin 發現將三聚甲硫醛加熱至攝氏 800 度時,會完全 裂解成 H2CS、C2H4和 CS2等三種主要產物[9],因為在不同的溫度下會裂解 成不同的分子,因此分子在激發態的光譜不易標定。近年來由於硫甲醛對光 化學的重要性,因此許多硫甲醛的光譜研究也受到學界的重視,目前硫甲醛
的光譜研究在微波[1,16,20]、遠紅外光[21-24]、可見光[25-36]、與紫外光 [37-40]等波長範圍皆有豐碩的研究結果。 在振動頻率的研究上,早期因為無法排除光譜互相重疊的影響,只能求 得部分的分子振動頻率,在 1971 年 Johns 和 Olson[21]藉由觀察 H2CS 紅外 線光譜,得到 C-H 的振動頻率ν1=2971.03、ν5=3024.61 cm -1。1980 年 Bedwell 和 Duxbury[41]藉由雷射 Stark 高解析光譜的技術,簡化轉動量子態的躍遷 譜線,減少了譜線重疊的干擾,成功觀察到其他的振動頻率:ν3=1059.2037、 ν4=990.1866、ν6=991.0149 cm-1,並將 Johns 所提出的 2ν6修正為 2ν2,但仍 未確定ν2,直到隔年 Clouthier 等人[42]經由共振螢光分析技術 (resonance fluorescence) 才確定ν2=1457.3(10) cm -1。1987 年由 Moran 與 Ellison 兩人[43] 研 究 出 硫 甲 醛 的 負 離 子 的 光 電 子 光 譜, 其 在 ν3 與 ν4 振 動 頻 率 分 別 為 860 與 450 cm-1。 在分子的鍵長與鍵角的研究上,從基本理論觀點而言,碳原子與硫原子 形成雙鍵,C 以 sp2混成軌域與兩個氫原子鍵結構成,所以 H 2CS 是一個平 面型分子,∠HCH應為 120°,屬於 C2v點群,分子示意圖如圖一;1998 年 Kuchitsu [44]整理實驗室數據∠HCH 為 116.52°,∠SCH 為 121.74°;鍵長方 面,C=S 的鍵長為 161.1 pm,C-H 鍵長為 108.7 pm。 本研究希望藉由理論計算的方式,模擬出 H2CS→H2CS++e-與 H2CS -→ H2CS+e -的光電子光譜,再和實驗光譜進行比對,並且運用上述的實驗數據
來驗證理論計算結果是否有偏差,以求得更準確的理論計算方式,冀望本研
究在光譜模擬領域,能有所貢獻,對於往後的硫甲醛分子相關研究,也能有
1.3 法 蘭 克
法 蘭 克
法 蘭 克
法 蘭 克 -
-
-
- 康 登 因 子
康 登 因 子
康 登 因 子
康 登 因 子
法蘭克─康登因子代表分子在兩電子態間,振動躍遷的機率,FC 原理 發 展 最 早 是 由 Franck[45] 和 Condon[46 、 47] 所 提 出 來 的 , 在 1930 年 Hutchisson[48]則提出 FCF 的計算方法,可藉由電子躍遷後,分子結構所產 生的位移∆d與扭曲來計算推得。約 20 年後 Manneback[49]使用數學上的遞 迴公式來計算 FCF 的積分,在 1959 年 Wagner[50]則推導出一種計算 FCF 的積分公式,後來由 Ansbacher [51]進一步提出一個明確的法蘭克-康登因子 計算公式,而被廣泛的採用 在早期,FCF 的計算由於牽涉艱深的理論與波函數的計算,對於 FCF 原理的推廣與應用有限。在 2005年Chang[12]提 出 更 為 簡 單 易 懂 的 公 式 來 計 算 FCF, 該 方 法 做 了 兩點假設 (1) 基態和激發態的位能面皆為諧振子。 (2) 忽略不同振動模式之間混合 (mode-mixinging) 的影響,亦即忽略杜 辛斯基效應 (Duschinsky effect)。 因為分子在平衡結構附近的振動,其真實的位能面和假設的諧振子位能面是 相當趨近的,所以 Chang 藉此假設簡化 FCF 的計算過程。另外,Chang 在 計算過程中,使 用 了 數 學 上 已 知 的 Hermite 多項式公式:(
)
∑
( )
( )(
)
= − = + n k k n k n k n y x H y x H 0 2 (1-1) 將 FC 積分展開成高斯積分的線性組合,使得計算上更 為 簡 單 易 懂 。到了 2008 年 Chang[53]提出新的計算方法,承襲了先前簡單易懂的計算 方式,並且考慮不同振動模式之間的混合,也就是杜辛斯基效應,使得計算 的結果更為精準。所謂杜辛斯基效應指的是,多原子分子的諧振子在兩個電 子態之間產生了位移(平衡結構不同)、扭曲(振動頻率不同)、旋轉(正規 座標不平行)的現象,也因此在計算振動波函數重疊積分的過程中,不同模 式間的座標會產生交互作用,使得計算多模式混合的 FCF 變的非常困難。 目前能處理杜辛斯基效應的多維度諧振子 FCF 的計算方法,已有許多文獻 的發表,我們將其中比較重要的文獻整理如下。1951 年 Sharp 和 Rosenstock 以 Hutchisson 計算一維諧振子的 FCF 方法為基礎,利用生成函數 (generating function) 的方法推導出了一個數學展開式,其展開式的係數可用於計算多
重模式混合的 FCF[54]。Kikuchi 等人以 Sharp 和 Rosenstock 的方法為基礎,
藉由一個數學技巧,導出了一個較為簡單的多維度 FCF 計算公式[55]。除此
之外,Dotorov 等人使用同調態 (coherent state) 方法,得到一個重疊積分的
遞迴方程式[56,57],這是一個很容易使用的方法,所以被廣為採用。所謂 Dotorov 的方法,只要準確的算出原帶 0 0 0 的 FCF 數值,便可利用遞迴公式推 算出其它振動態的 FCF,但其缺點是可能會花費較長的計算時間,或是需要 大量的記憶體。因此,許多研究者發展了不同的演算法[58-64],以改善遞迴 法的計算效率。另外,Malmqvist 和 Forsberg 應用 LU 矩陣分解法計算 FCF, 他們的 L 和 U 矩陣也可是以遞迴法獲得[65]。然而,使用遞迴法計算 FCF,
無法避免的一個問題是:在某些情況下,會產生較大的誤差。例如當原帶 0 0
0 的 FCF 計算偏差較大時,此誤差會傳遞至後續的計算步驟,而使誤差不斷
的擴大。
另一方面,Faulkner 和 Richardson 提出兩種轉換方法 (transformation
method),用以計算多原子分子的 FCF[66,67],此方法並由 Kulander 加以延
伸[68]。因為計算的收斂速率較慢,所以他們的方法並沒有被廣泛的採用。
Chen 和 Pei 應用 Laguerre 多項式,導出一個二維 FCF 的公式,其展開式包
含了無限多的項次,所以計算效率也深受收斂速率的影響[69]。最後,Lin
與其同事[70,71]藉由複雜的數學技巧,對於多維度 FCF 的計算方法做出了
重要的貢獻; Mebel 等人使用厄米多項式 (Hermite polynomial) 的周道積分
(contour integral),獲得了一個四維 FCF 的閉合式 (closed-form formula)
[72];而 Islampour 等人採用生成函數的方法,發展出一個可推導任意維度 FCF 的通解,但他們只推導出四維的公式[73],更高維度的公式仍需要相當 複雜的數學推演。2005 年,Liang 等人使用厄米多項式一個展開公式,也推 導出一個計算 FCF 的公式[74],其方法較 Islampour 等人的方法簡易。然而, 不論是從理論或實用的觀點來看,由於杜辛斯基效應使得多原子分子的 FCF 複雜難解,所以我們致力於尋求一個新的、簡易的、而且是有效率的計算方 法。 截至目前我們可以藉由 2008 年 Chang 提 出 的 新 計 算 方 法 , 計 算 在
四 種 混 合 模 式 下,任 意 能 階 躍 遷 的 FCF,而 Chang 提 出 的 方 法 的 特 點 與 先 前 的 文 獻 相 較 下 , 我 們 可 以 發 現 Chang 的 計 算 方 法 優 勢 在 於 : (1) 所 推 導 的 公 式 是 一 個 封 閉 型 的 公 式 , 會 有 單 一 的 計 算 結 果 , 而 非 採 用 遞 迴 函 數 的 趨 近 , 因 此 計 算 結 果 的 準 確 性 相 對 的 提 高;(2) 計 算 過 程 巧 妙 運 用 了 厄 米 多 項 式 的 展 開 式,簡 化 了 繁 冗 的 計 算 過 程 , 縮 短 計 算 的 時 間 。 因 為 H2CS 和 H2CS - 的 光 電 子 光 譜,杜 辛 斯 基 效 應 很 小( 見 3.4 節 ),所 以 本 研 究 採 用 Chang 在 2005 年 所 提 出 的 一 維 諧 振 子 FCF 計 算 方 法,我們的計算方式是運用分子在躍遷前後的平衡結構差異∆d,與不 同振動模式的角頻率ω ,將分子的振動波函數重疊積分取平方,計 算 硫 甲 醛 H2CS ( ) ~ 1 1A X 游 離 至 H2CS+ ( ) ~ 2 2B X 的 FCF, 以 及 H2CS - ) ( ~ 1 2B X 脫 電 子 至 H2CS ( ) ~ 1 1 A X 的 FCF; 另 外 , 我 們 也 計 算 H2CS - 不 同 振 動 模 式 的 波 茲 曼 分 布 Pv[75],其中 Pv為居量 (population),藉由 FCF 與居量 Pv 兩者的乘積,模擬 H2CS - 分子的光電子光譜。
1.4 量 子 計 算 發 展 沿 革
量 子 計 算 發 展 沿 革
量 子 計 算 發 展 沿 革
量 子 計 算 發 展 沿 革
量子計算是理論化學的一個重要分支,在量子力學的理論基礎上,藉 由數學上的近似法以及運用電腦程序計算分子的量子力學方程式,可以求 得分子的基本結構、總能量、偶極矩、振動頻率…等,並用以解釋一些具 體的化學問題。傳統量子化學發展至 1930 年代已然成形,理論上分子的 電子結構皆可由薛丁格方程式表示,而對於少量原子的系統,藉由解薛丁 格方程式也確實計算出非常精確的結果,例如 1927 年 Heitler 和 London 應用量子力學分析氫原子軌域結構[76]與 1932 年 Mulliken 的分子軌域概念 [77],皆能精確計算氫原子的鍵長、解離能…等。 但多電子體系中,電子與電子間以及電子與原子核之間複雜的交互作 用,一直無法獲得具體解釋,直到 1930 年 Hartree 與 Fock 進一步提出可 將多電子體系中的電子,視為在其他電子構成的平均勢場中運動的粒子 [78-79],將多電子體系複雜的薛丁格方程式,簡化為單電子波函數的乘 積:Ψ(1,2,3,…,n)=Ψ(1)×Ψ(2)×Ψ(3)…Ψ(n) (1-2) 再利用變分法求得波函數的解。 至此,所有的化學基本量,理論上皆可以藉由解薛丁格方程式來獲 得,而 Hartree-Fock 理論也成為現代量子計算的重要基石。Hartree-Fock 理論牽涉到複雜與大量的計算,對於少量電子的體系 (N=20),尚能精確
計算,但隨著電子數目的增加,待解的變分參數將以 10n倍數成長[47],例 如處理 N=100 的電子系統,待解的變分參數將有 10150個,因此 Hartee- Fock 理論在沉寂 20 年後,才在 1953 年由 Pople 等學者花費兩年時間使用 手搖計算器完成對氮氣分子的量子化學的計算,對於 HF 理論的驗證有重 要的劃時代意義,自此之後,隨著電腦運算技術的進步與理論發展臻備, 目前已經可以解決多電子體系龐大的量子計算,這樣的計算方法也稱為第
一原理方法 (Ab initio methods)。
本研究使用的密度泛涵理論 (density functional theory, DFT),則是建
立在 Hartree-Fock 方程的理論基礎上,相異之處在於,Hartree-Fock 理論計 算中有 3N 個變數(N 為電子數),因此方程式變得相當複雜;而 DFT 理 論是以電子密度分佈函數 ρ(r) 取代波函數 Ψ(1,2,3,…,n) 做計算,使得原本 有 3N 個變數(每個電子包含三個空間變數)轉變成只包含三種變數之電 子機率密度,電子密度與系統大小無關,問題顯得單純化許多,計算耗時 也相對較少;DFT 的另一項優點,就是對於分子平衡結構和振動頻率的計 算結果,通常與實驗值相當接近,因此近年來密度泛函理論可以說是解決 多電子系統波函數複雜之交互作用問題的方法之ㄧ。
1.5 論 文 架 構
論 文 架 構
論 文 架 構
論 文 架 構
本論文共分成四章,除了本章緒論外,第二章介紹本研究所採用的研 究方法,包括密度泛函理論、H2CS 分子、正離子和負離子的量子計算、 法蘭克-康登因子、馬克斯威爾─波茲曼分布函數、光譜模擬的計算方法 說明等,第三章為結果與討論,敘述 H2CS 分子以及正、負離子的計算結 果,包括平衡結構、振動頻率、絕熱游離能、電子親和力和法蘭克-康登 因子,以及模擬 H2CS 分子以及負離子的光電子光譜,並與實驗進行比較 分析。第四章為研究結論。第二章
第二章
第二章
第二章
理論計算方法
理論計算方法
理論計算方法
理論計算方法
本 研 究 利 用 密 度 泛 函 理 論 中 的 B3LYP 方 法 , 先 計 算 硫 甲 醛 分 子 、 正 離 子 與 負 離 子 的 平 衡 結 構 與 振 動 頻 率 , 再 以 本 研 究 室 發 展 的 方 法 計 算 法 蘭 克 - 康 登 因 子 , 進 而 模 擬 光 電 子 光 譜 , 將 量 子 計 算 結 果 與 模 擬 之 光 電 子 光 譜 , 與 實 驗 值 進 行 比 對 與 探 討 。 本 章 將 依 序 介 紹 研 究 所 使 用 的 理 論 計 算 背 景 與 方 法 。 本 章 共 分 為 四 節 , 第 一 節 介 紹 密 度 泛 函 理 論 以 及 選 用 的 基 組 , 第 二 節 介 紹 幾 何 優 選 與 正 規 振 動 模 式 的 計 算 方 法 與 流 程,第 三 節 介 紹 法 蘭 克 -康 登 因 數 的 假 設 、 推 導 和 計 算 方 法 。 第 四 節 介 紹 波 茲 曼 分 布 函 數 的 計 算 。 第 五 節 說 明 光 譜 模 擬 的 方 法 。
2.1 密
密
密 度 泛 函 理 論
密
度 泛 函 理 論
度 泛 函 理 論
度 泛 函 理 論 (density functional theory, DFT)
1920 年代量子力學蓬勃發展,許多物理學者認為,量子力學方程式已經包含所有的資訊,只要能夠解出波函數的解,便能求得所有的物理量。但
是在實際計算上,傳統量子力學的方法,只能處理少量電子系統的問題,對
於多電子系統而言,因電子與電子之間複雜的交互作用,使用薛丁格方程式
來求解波函數是相當困難的。目前在計算化學方法中,依其所使用的理論背
景可分成分子力學 (Molecular Mechanics)、半經驗法 (Semi-Empirical)、全
成熱、分子內的轉動活化能、以及振動光譜等,但是分子力學無法計算分子 軌域能階和分子激發態的行為;半經驗法是針對全始計算的方法加以簡化, 並加入實驗參數,才可以進行大分子的量子計算,但由於加入了實驗的參 數,使得計算結果的精確度難以評估,當有新型態的化學結構出現時,原先 加入的參數可能無法預測新化合物的性質,而影響計算的精確度。 本研究採用的計算方法為密度泛函理論法,其中的理論根據 1927 年波 恩-奧本海默近似法[80]假設出發,由於原子核的質量比電子大上許多,所以 與電子相互作用時可以視為固定不動,因此原子核的座標與動能皆可以視為 常數,而電子又都是電荷、質量、自旋等特徵完全相同的粒子,因此分子結 構問題的研究就化為 N 個全同粒子體系的研究,運用在研究多電子體系中, 將電子與原子核間的運動,視為分開的獨立事件,簡化了原來多粒子體系的 複雜度。 在全始計算中除了一些基本的常數(如蒲朗克常數、光速、原子核與電 子的質量等)外,不加入任何的實驗參數.故稱為全始或第一原理計算。但 因其計算量龐雜,早期受限於電腦運算效率,所以全始計算只能處理少量電 子的系統。 本研究採用的密度泛函理論,證明了分子基態的電子能量、波函數以及 其它的性質,可以由只包含三種變數的電子機率密度決定,也因此解決全始 計算方法在面臨多電子系統繁雜求解的問題,因為計算過程中的變數由原本
的 3N 個(N 為電子數目)變成 3 個維度的變數,變數減少了,所以計算速度 快,並且密度泛涵理論也考慮到電子之間的交換-相關能,因而提高計算的 精確性,在現代化學計算領域,是一個重要的理論。 密度泛函理論發展最早可追溯至 1927 年 Thomas 和 Fermi 發展的 Thomas-Fermi 模型[81-82],內容是利用電子密度分佈函數的泛函來計算原 子 動 能 的 理 論 。 其 中 的 原 理 是 先 以 局 部 密 度 近 似 (Local-density approximation, LDA )[83]求得以密度分佈函數 ρ(r)為變量的能量泛函 E[ρ(r)],進而計算原子的能量,在當時開創量子計算另一種新的思維,但是 Thomas-Fermi 理論忽略了電子彼此之間交互作用的交換能,限制了計算的 準確性,也因此未受重視。直到 1964 年 Hohenberg 和 Kohn 證明 DFT 兩個 重要的定理[84]: (1) 在 一 個 穩 定 的 量 子 力 學 系 統 中 每 一 個 可 觀 測 的 物 理 量( 包 括能量在內),都可藉由基態 (ground-state) 的電子密度來 計 算。 因 此 可 將 每 個 可 觀 測 的 物 理 量,以 基 態 電 子 密 度 的 泛 函 表 示。換 言 之,在 多 電 子 體 系 中,電 子 基 態 的 能 量 也 可 由 電 子密度函數 ρ(r)的泛函表示。 ( 2 ) 藉 由 變 分 法 , 計 算 電 子 密 度 函 數 ρ(r)為 變 數 的 能 量 泛 函 EG. S .[ρ(r)]的 最 小 值 , 亦 是 求 其 最 低 能 量 狀 態 , 便 能 得 到 基 態 能 量 EG. S .。
這二個定理,對於 DFT 有重要的貢獻,其原理簡述如下:我們將一個 粒子的總能量,以不含時薛定格方程式表示為式(2-1)
( )
r E( )
r Hψ = ψ (2-1) 以漢彌爾頓算子 H 表示為式(2-2) ψ ψ ψ V E m = + ∇ − 2 2 h (2-2) 其中 2 2 2 2 2 2 2 z y x ∂ ∂ + ∂ ∂ + ∂ ∂ = ∇ (2-3) 雙電子系統則可以表示為式 (2-4)(
)
(
)
(
)
2 1 2 1 2 1 2 2 2 1 , , , 2 2m m V r r r r E r r ψ ψ ψ ψ = + ∇ − ∇ − h h (2-4) 其中ψ為多電子波函數,(
)
2 1,r r V 為由原子核產生的位能 (external potential)。 在式(2-4)中(
)
2 1, r r V 是無法再分離變數的,因而使得薛丁格方程式,無法求 得實際的本徵值。為了解決上述的問題,Hohenberg-Kohn 的第一定理指出: 可將基態的總能 EG..S.寫成電荷密度函數的泛函,因此能量的計算簡化成式 (2-5),大大降低了計算過程的自由度。 EG. S .[Ψ(r1 ,r2 ,r3 … …,rN)]→EG. S[ρ(r)] (2-5) 第二,如果代入的數值不是基態的電荷密度函數 ρG.S.(r),則總能量 EG. S .可 表示為式(2-6): EG. S .[ρ(r)]≥EG. S .[ρG.S.(r)] (2-6) 式中 EG. S[ρ(r)]必定會大於或等於基態能量 EG. S .[ρG.S.(r)],而 Hohenberg-Kohn理論只證明了 EG. S .[ρG.S.(r)]的存在,並沒有明確的計算公式,因此在當時只 能採用變分法, 將不同的 ρ(r)代入 EG. S .[ρ(r)]直到求得最小值,進而獲得基 態能量的近似值,再將基態的總能量分成動能與位能兩部分,如式(2-7):
( )
[
r]
[
( )
r]
[
( )
r]
S G S Gρ
ρ
ρ
. . . G.S. U E = Τ + (2-7) 在式(2-7)中,我們可以藉由庫倫定律計算出位能[
( )
r]
S G. . ρ U ,但對於基 態的總動能[
( )
r]
S G. ρ Τ ,當時尚無任何物理定律可以計算出精確的結果,所 以 DFT 理論在當時仍待後續的發展。 2.1.1 2.1.1 2.1.1 2.1.1 Kohn-Sham方程式方程式方程式方程式1965 年由 Kohn 和 Sham 繼續發展 DFT 理論,提出了 Kohn-Sham 方法
[85],成功說明如何由電子密度函數 ρ(r)計算電子的能量 EG..S.,它提供另
一種新的計算模式,這樣的方法隨著科技的進步與電腦計算效率提升,不僅
節省計算時間,計算結果與實驗值非常接近,其理論推導如下:
以 ρ(r)為變數的能量泛函 EG. S .[ρ(r)]可表示為
EG. S .[ρ(r)]=Tm[ρ(r)]+Eee[ρ(r)]+Eext[ρ(r)] (2-8)
其中 Tm[ρ(r)]為多電子動能,可表示為
[ ]
(
)
(
, ,..)
2 ,.. , Tm 1 2 2 r1 r2 m r r iψ ψ ρ = −∑
∇ h (2-9) Eee[ρ(r)]是電子與電子間的交互作用能,可表示為[ ]
(
, ,..)
(
, ,..)
2 1 2 2 1 r r r r e r r E ij i j ee ρ ψ∑
ψ − = (2-10)式(2-9)、(2-10)其密度泛函的形式是未知的。 Eext[ρ(r)]是外界施加的位勢可表示為:
[ ]
=∫
V( ) ( )
r r d r Eext ρ ext ρ 3 (2-11) 其 中V( )
r ext 是 已 知 的 函 數 , 可 隨 著 不 同 的 問 題 帶 入 不 同 的 波 函 數 。 而 Kohn-Sham 方法的貢獻則是從式(2-9)、(2-10)中分離出已知的物理量, 分別是電子獨立運動的總動能[ ]
ρ s T 與電子間的庫倫位能[ ]
ρ H E 。剩下的部份 則視為電子間的交換相干能,所以系統總能 EG.S.[ρ(r)]可重新表示成:( )
[
ρ]
[ ]
ρ[ ]
ρ[ ]
ρ[ ]
ρ ext XC H S S G r T E E E E = + + + . (2-12) 其中 ρ(r)公式如式(2-13)( )
r =∑
Ψ( )
2 i i r ρ (2-13) 並且 ρ(r)公式必須符合式(2-14)的歸一化條件: ( ) ( ) ( ) ∫ = ∑ ∫ Ψ Ψ = i i r i r d r r N ρ * 3 (2-14) 至此除了交換相干能[ ]
ρ XC E 之外,所有的泛函項皆有明確的公式可表 示,其中電子獨立運動的總動能[ ]
ρ S T 可表示成式(2-15)[ ]
=−∑ ∫
( )
∇( )
i i s r r d r m T * 2 3 2 2 ψ ψ ρ h (2-15) 而靜電庫倫位能可表示為式 (2-16)[ ]
∫
( ) ( )
=∫
( ) ( )
− = d r V r r d r r r r r EH 3 ' H 3 ' ' ρ ρ ρ ρ (2-16) 最後,Kohn-Sham 方法中,雖然無法求得相干交換能[ ]
ρ XC E 精確值,但可定 義趨近的位能[
( )
r]
XC ρ表示成:
[ ]
[
( )
r]
( )
r d r EXC ρ εXC ρ ρ 3∫
= (2-17) 再其中針對交換相干能再以變分法取得近似,表示為下列式 (2-18)。( )
[
r]
LDA[
( )
r]
XC XC ρ ε ρ ε ~ (2-18) 這代表著原本需要知道整個 ρ(r)函數分布,才能知道各點εXC的大小,但現 在近似成只要將某定點位置 r0,代入 ρ(r),便可以得到該位置的 ρ0,也就可 以得到該位置的 LDA XC ε 值。也就是 XC ε 的大小只跟該位置的電荷密度大小有關。 最後我們在式(2-12)總能泛函中,藉由變分原理求得各泛函數的導數, 並將各個波函數在滿足歸一化條件下,將 Kohn-sham 方程式表示成式(2-19): i i i xc ext H r V r V r E V m ψ ψ = + + + ∇ − ( ) ( ) ( ) 2 2 2 h (2-19) 而計算過程則是利用迭代法,先藉由數個正交基底函數展開數個Ψi後,解 一組 1 i Ψ ,並求出 ρ1,解一組 2 i Ψ ,並求出 ρ2 再一一的求出相對應的局部 EG.S.[ρ(r)]。因此在多電子的系統中,藉由初始電荷密度 ρin(r),得到有效位 能Vext(r),再代入 Kohn-sham 方程式求得各能階及對應的軌域,而算出新的 電荷密度 ρout(r)。若新的電荷密度與初始電荷密度不相同,則經由混合方法 (Mixing scheme),重新計算一個新的電荷密度,計算方式如式(2-20)( )
(
)
( )
i out i in i in r α ρ r αρ ρ+1 = 1− + (2-20) 重複這樣的運算過程,直到計算結果收斂,差異小於設定的條件,這樣的方 法亦稱為自洽場計算[86]。2.1.2 2.1.2 2.1.2
2.1.2 泛函數泛函數泛函數泛函數(Functio(Functio(Functional)(Functional)nal) nal)
在量子化學計算上,我們選用「基組」與「方法」進行計算,DFT 計
算方法的名稱是以交換-相關泛函數的廣義公式代替,亦即 DFT 的名稱來源
是由交換泛函數及相關泛函數的名稱組合而成。
本研究所採用的 B3LYP 是指使用 Becke 三參數交換泛函數[88]與 LYP
相關泛函數[88,89]做計算,其形式如下: LYP C c VWN C c B X x exact X LSDA X x LYP B XC a a E a E a E a E a E E =(1− − ) + + 88+(1− ) + 0 0 3
其 中 LYP 是 描 述 由 Lee、 Yang、 Parr 三 人 所 提 出 的 相 關 泛 函 數 ,
而 Becke 的 主 要 貢 獻 在 於 交 換 泛 函 數 的 常 數 決 定,透 過 與 一 些 分 子 的 化 學 能 相 對 照 而 決 定 出a0=0.20、 =0.72 x a 、 =0.81 c a ,將 常 數 帶 入 其 公 式 如 下 : exact x VWN c LYP c LSDA x B x LYP B XC E E E E E E 3 =0.72× 88+0.08× +0.81× +0.19× +0.2× (2-21) 其中,B88[88]是 Becke1988 泛函數,LSDA 是[89]局部自旋交換作用, LYP[90]是相關泛函數,VWN 是局部自旋密度(LSD)相關作用泛函數。Becke 三參數交換泛函數是 Beck 利用 Hatree-Fork (HF) 的交換函數與 DFT 理論的 交換─相關能函數進行混合所得的新函數方法,其進行計算所得到的修正能 量為 HF 交換能與 DFT 能混合後的結果,因此使用 B3LYP 方法在計算上比 HF 方法更能夠準確的計算出能量值。 而理論化學家也發現採用 B3LYP 方法計算所得的結果,與其他高層次之計
算結果非常接近,而計算時間卻減少許多,因此 B3LYP 為本研究採用的交 換相關泛函數。 2.1.3 2.1.3 2.1.3 2.1.3 基底函數組基底函數組基底函數組基底函數組((((basis set)))) 在進行密度泛函理論時,需要選擇一個基底函數組 (basis set 簡稱基組) 來表示分子軌域之波函數。所謂基底函數組是由ㄧ群基底函數組合而成,而 基底函數則是由許多高斯函數 (primitive Gaussin) 組合而成,主要藉由高斯 函數的線性組合 (linear combination) 來描述分子軌域。因此基組是用數學 式的方式來描述一個系統中,每個電子可能存在的軌域,也可進一步結合近 似全部的電子波函數進行理論計算。 基組必須符合每個電子的邊界條件(亦即每個電子只能出現在空間中的 特定區域)。較大的基組群對電子的空間所在限制條件較少,更能符合量子 理論中電子在空間中任何位置皆可能出現的假設,因此,如欲對分子的波函 數做良好的描述,基組的選擇是非常重要的,以下針對本研究所選的基組, 進行介紹: ( ( (
(1))))分 離 價 殼 層 基 組分 離 價 殼 層 基 組分 離 價 殼 層 基 組分 離 價 殼 層 基 組 (((( Split valence basis sets))))
本 研 究 計 畫 所 使 用 的 基 底 函 數 是 Pople[91] 等 人 所 提 出 的
6-311++G(d,p) 、 6-311++G(2d,p) 、 6-311++G(3d,p) 、 6-311++G(3df,p) 、
6-311++G(3df,pd)[92-95] , 我 們 以 本 研 究 中 6-311++G(d,p) 為 例 , 其 中
態的基底函數組為 Pople 所提出,其形式如下:k-nlmG,k 是指使用了 k 個
初 始 高 斯 函 數 (primitive Gaussian) 來 組 合 內 殼 層 軌 域 (inner shell
orbital);而價殼層軌域則由三個基底函數分別由 n、l、m 個初始高斯函數 所組成。故 6-311G 基組表示,內殼層之每個原子軌域以一個基底函數來表 示,此基底函數是由 6 個初始函數線性組合(linear combination)而成。價 殼層(valence shell)則由三個基底函數組合而成,每一的基底函數則分別 是由 3、1 及 1 個初始函數(primitives)所構成。 ( 2) 擴 散 函 數擴 散 函 數擴 散 函 數 (擴 散 函 數((( Diffuse functions )))) 為了求得更精確的計算結果,除了上述的函數組合外,必須再加入擴散 函數 (diffuse function) [106]來涵蓋距離原子核較遠處的電子,以本研究中使 用的 6-311++G(d,p)為例,6-311++G(d,p)中的「+」號表示加入擴散函數,擴 散函數與標準的價層軌域函數的大小相比而言,為一個擁有 s 軌域型態或 p 軌域型態的低指數函數。因此,加入擴散函數之後,軌域會擁有更大的空間, 可以供給電子佔據。同樣的,兩個「+」號表示除了重原子 (C 和 S) 之外, 對氫原子也加上擴散函數。 ( 3) 極 化 性 基 底 函 數極 化 性 基 底 函 數極 化 性 基 底 函 數 (極 化 性 基 底 函 數(( Polarization functions )( )) ) 在分子結構的計算中,為了使每個原子可調變的位置更具彈性,可在基 組中加入極化函數 (polarization functions) [107]。極化性基底函數的特色, 是在對每個原子軌域的描述中,加入較其在基態所需要,含有較高角動量 (angular momentum) 的軌域型態,使整個基組更能接近真正的分子軌域性
質。以 6-311++G(d,p)為例,6-311++G(d,p)中的「d,p」表示加入極化函數, 第一個 d 號表示重原子中加入更高階的角動量函數,在 H2CS 分子中,重原 子為碳和硫原子,因此對其加入一組 d 形式的極化函數。第二個 p 號代表在 每一個氫原子中加入一組 p 形式的極化函數,若是 6-311++G(2d,2p)中,2d 則表示對重原子加入二組 d 形式的極化函數,2p 代表在每一個氫原子中加 入二組 p 形式的極化函數。6-311++g(3df,pd) 則是對重原子加上三個 d 軌域 函數和一個 f 軌域函數外,對氫原子也加上一個 p 軌域函數與 d 軌域函數組。 為了比較不同基組的結果,除了上述基組外,另外我們也採用 Dunning 等 人 [108-112] 所 提 的 基 底 函 數 組 AVDZ 與 AVTZ 。 這 兩 組 指 的 是 aug-cc-pVDZ 和 aug-cc-pVTZ,其中的基底函數也包含了極化函數和擴散函 數,我們以下表一列出 Dunning 基底函數組中各原子所包含的極化函數類 型:
表一、Dunning 的基底函數包含的極化函數類型 Atoms cc-pVDZ cc-pVTZ H 2s,1p 3s,2p,1d He 2s,1p 3s,2p,1d B-Ne 3s,2p,1d 4s,3p,2d,1f Al-Ar 4s,3p,1d 5s,4p,2d,1f Ga-Kr 5s,4p,1d 6s,5p,3d,1f 從上表一中可看出,基組 cc-pVDZ 包含對氫原子加入 s、p 軌域函數, 對重原子(在本研究指的是碳與硫原子)加入 s、p、d 函數,基組 cc-pVTZ 則包含對氫原子加入 s、p、d 函數,對重原子加入 s、p、d、f 函數,皆已 包含極化函數。另外在這些基底函數關鍵字之前加上 aug-前置字串,指示增 加擴散函數,使基組更能接近真正的分子軌域的性質。
2.1.4 2.1.4 2.1.4
2.1.4 耦合簇理論耦合簇理論耦合簇理論耦合簇理論 ( ( ( (CoupledCoupled-CoupledCoupled---cluster theorycluster theorycluster theorycluster theory))))
本研究對硫甲醛中性分子、正離子、負離子,以 DFT 的 B3LYP 的方法,
搭配 2.1.3 節的七種基組,進行幾何優選和振動頻率的計算。在獲得 H2CS
分子與相關正負離子的平衡結構後,再以這七個基組,搭配耦合叢集
CCSD(T)(Coupled-c1uster single, doubles and perturbative triples) 方法,計算
分子與離子的單點能量。 耦合簇理論[83-84]常運用在多電子系統,在進行 Hartree-Fock 方法計算 後,對於多電子的相關能進行更精確的計算,計算多運用在中小型分子。耦 合簇理論是由 Kümmel 等人[89]在 1980 年提出,本來運用在核物理光譜, 經修正後,運用在分子和原子對電子相關能計算。本研究為了求得更精確的 能量,因此採用 CCSD(T)的方法進行計算。CCSD(T)的方法是指對單重激發 態能量和雙重激發態能量的耦合簇計算,並且加入微擾理論 (perturbation theory) [90]用以計算三重激發的能量。以下簡述本研究利用耦合簇理論,計 算游離能和電子親和力的方法。 藉由上述的 B3LYP 方法,搭配七種基組,理論計算硫甲醛分子與正、 負離子,所得六種模式的振動頻率ν1~ν6,可分別求得中性分子、正離子與
負離子的零點能 (zero-point energy, ZPE),計算方式為
=
∑
i i ν 2 1 ZPE 。 然後用 CCSD(T)方法,計算 B3LYP 搭配七種基組所得的平衡結構的能 量,若中性分子的能量為 E2、負離子的能量為 E1,而正離子的能量為 E3,則將分子與負離子的能量差 E2-E1,加上分子與負離子的零點能之差
(ZPE2-ZPE1),所得能量便是電子親和力 (electron affinity)。接著將正離子與
分子的能量差 E3-E2,加上兩者零點能之差 (ZPE3-ZPE2),所得的能量,便
2.2 中 性 分 子
中 性 分 子
中 性 分 子 、
中 性 分 子
、
、
、 正 離 子
正 離 子
正 離 子 、
正 離 子
、
、
、 負 離 子 的 平 衡 結 構 計 算
負 離 子 的 平 衡 結 構 計 算
負 離 子 的 平 衡 結 構 計 算
負 離 子 的 平 衡 結 構 計 算
本研究選用 Gassion03 套裝軟體中含電子相關之 DFT(B3LYP)方法並配 合 6-311++G(d,p)、6-311++G(2d,p)、6-311++(3d,p)、6-311++g(3df,p)、 6-311++g(3df,pd)和 AVDZ 與 AVTZ 等七種基組進行計算。首先進行硫甲醛 分子(圖一)、正離子、負離子的幾何優選 (geometry optimization),進行幾 何優選的目的在尋找最穩定能量的分子結構,計算過程中會不斷調整分子的 幾何形狀,包括鍵長、鍵角、二面角等,直到達成位能曲面上的穩定點為止。 所謂幾何優選可採用的策略分為(1)完全幾何優選 (full oprimization) 及(2)部分幾何優選 (partial optimization) 計算。完全幾何優選,是對所 有指定的變數進行優選計算,以找出能量最低的結構,但由於變數比較多, 因此計算所耗費的時間會更多;部分幾何優選則只針對其中的一些變數進 行幾何優選。 本研究採用部分幾何優選的策略進行計算,原因在於,研究目的是要探 討藉由理論計算所模擬出的光電子光譜與實驗數據的差異性,而 H2CS 分子 的實驗數據已經相當完整,因此,可以確定分子與離子的幾何結構形狀,對 於某些變數可以給予固定,以縮短計算時間,提高計算的效率。 此外,我們也依據幾何優選的計算結果,計算硫甲醛 (H2CS) 分子、正 離子與負離子的振動正規模式的頻率。由於,位能曲面上的穩定點除了可能point),因此可以藉由計算結果進行判斷,若振動正規模式的頻率皆為正值,
則代表此種分子結構為位能曲面上的穩定結構。反之,若振動正規模式的頻
率,其中之ㄧ為虛數,則代表此種結構,為位能曲面上的鞍點,無法穩定存
2.3 法 蘭 克
法 蘭 克
法 蘭 克 -康 登 因 子
法 蘭 克
康 登 因 子
康 登 因 子
康 登 因 子
本研究採用 2005 年 Chang 提出的公式來計算法蘭克─康登因子,其理 論計算方法如下: 已知諧振子的波函數可表示為:(
)
− = 2 2 1 exp x x H N v v v α α (2-22)(
)
− = 2 ' ' ' ' 2 1 exp ' ' ' N H x x v v v α α (2-23) 其中 2 1 ! 2 = π α v N v v 為規一化常數,Hv( )
x 為 Hermite 多項式。 而 h ω α = ,其中ω是諧振子的角頻率, π 2 h = h ,h為 Planck’s constant。 將基態與激發態的振動波函數的重疊積分取平方,即為法蘭克-康登因子。( )( )
(
)
(
)
2 2 ' ' ' ' ' 0 ' 0 ' 2 ' ! ! 1 2 ) ' 2 ( ) 2 )( ' ( ) ( ) ! ' ! 2 ( ' ' + − Η Η = − − = = + −∑
∑
α α α α ν ν ν ν ν ν ν ν ν ν K b b Ae v v k k k k k k k k s (2-24) 其中:v 代表基態的振動量子數,v´代表激發態的振動量子數。 計算過程中,令 S 因子為: ' ' 2 α α αα + = d S (2-25) d x x'= + (2-26) d代表兩電子態之間平衡結構的改變量(正規改變量),可由下列公式求得: q L d = '∆ (2-27)L為位移矩陣 (displacement matrix) ' ' α α α α + − = d b (2-28) ' ' ' α α α α + − = d b (2-29)
(
2n−1)
!!=1×3×5×L×(
2n−1)
(2-30) ' ' 2 α α αα + = A (2-31) 而 2 ' k k K = + ,其中因為奇函數的積分為零,因此k +k'必須為偶數。 以上的計算過程雖然使用兩點假設: (1)基態和激發態的位能面皆為諧振子。 (2)忽略不同振動模式之間混合 (mode-mixinging) 的可能,亦即忽略杜辛 斯基效應 (Duschinsky effect)。 但是在分子躍遷的能量在低能量區時,分子真實的位能面和我們假設的 諧振子位能面是相當趨近的,因此這樣的計算結果很接近實際值。2.4 馬 克 斯 威 爾
馬 克 斯 威 爾
馬 克 斯 威 爾 ─
馬 克 斯 威 爾
─
─
─ 波 茲 曼 分 布 函 數
波 茲 曼 分 布 函 數
波 茲 曼 分 布 函 數
波 茲 曼 分 布 函 數
在模擬光電子光譜,我們除了考慮電子躍遷的機率(法蘭克─康登因 子),仍需考慮每個振動態的居量 Pv (population)。當我們計算法蘭克─康 登因子,會模擬電子從不同的振動態躍遷(本研究模擬 H2CS -從基態的ν= 0、 ν3=1、ν4=1、ν4=2 躍遷光譜),因而在同一個分子會模擬出數個不同模 式躍遷的光電子光譜,再以居量 Pv與光譜的強度(I)成正比的原理,將數個 光電子光譜,合成ㄧ個統整的光譜,也就是研究最終的模擬光電子光譜, 以下將簡述波茲曼分布函數的計算過程。 藉由馬克斯威爾-波茲曼分布函數[75],我們可以得知在絕對溫度 T 的 狀態下,電子在能量 E 的分布的最可能的方式。因此我們可以計算出在某 個系統中,能量 E1的居量為 n1,能量 E2的居量為 n2…等以此類推,其公 式如式 (2-32): kT E v ve
c
P
−×
=
( 2-32) 其中Pv為居量 (population) 常數 c 為歸一化常數,與系統的粒子數有關,T為絕對溫度。 k 為波茲曼常數=0.69503563 cm-1K-1 。 因此在計算電子躍遷的機率時,除了計算 FCF,仍需計算光譜中的居 量Pv並乘上 FCF,才是真正電子躍遷的機率,可將光譜的強度以 I 表示,寫成式(2-33) 2 ' v P I ∝ ×
ν
ν
(2-33) 為了模擬 H2CS -光電子光譜,我們必須計算 H2CS - 在不振動能階的居量 Pv,由式(2-33)我們可以知道居量 Pv正比於光譜線的強度除以 FCF,如式 (2-34) 2 ' vI
P
ν
ν
∝
(2-34) 以實驗室光電子光譜為基準[43],量取已知的 0 1 0 0 3 0 , 的譜線高度為強度 I1、 I2,將光譜強度I除以該激發態的 FCF,便可求得相對居量 Pv。 另外我們也可以用於計算絕對溫度 T,這個方法廣泛應用於天文學 上,對於已經被偵測的光譜,我們可以藉由波茲曼分布與 FCF,計算外太 空星體的溫度。其計算過程如下: 我們將光譜的相對居量 Pv取自然對數,並將式(2-32)整理成式(2-35)c
T
P
T
E
v vln
ln
k
+
×
=
−
(2-35) 由上式(2-35)可得知,溫度T是該式的斜率,因此我們用已知的能量 Ev除 以波茲曼常數 k,得到 k Ev − 為 Y 軸,以及相對居量的自然對數 lnPv為 X 軸, 可求得溫度 T。 藉由波茲曼分布函數與 FCF,不但可運用於計算外太空所發現的星體的 溫度,在本研究中,當求得絕對溫度 T 時,更可計算個別激發態的電子居量Pv,再將所有激發態的光電子光譜成乘上相對應的居量 Pv,並將這些不同振 動態的光電子光譜合併為一,即為本研究對於 H2CS 以及 H2CS - 兩者的模擬光 電子光譜。
2.5 光 譜 模 擬
光 譜 模 擬
光 譜 模 擬
光 譜 模 擬
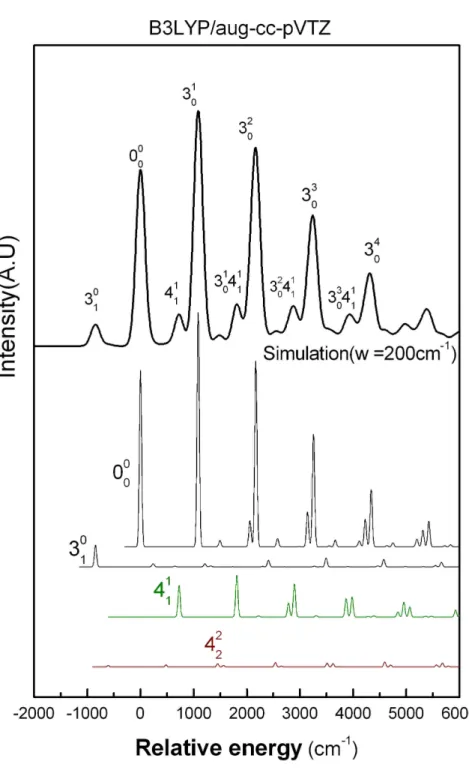
在計算中,我們以法蘭克-康登因子為每個譜線的強度,並假設每一 個躍遷的譜線,近似於一個羅倫茲函數 (Lorentzian function) 2 0) ( 4 1 ) ( w g ν ν κ ν − + = (2-36) 其中ν為頻率,ν0為躍遷的中心頻率,w 為其半高寬 (full width at halfmaximum),w 可在計算中任意調整。我們將每個躍遷的貢獻加起來, 即可得到整個分子躍遷的模擬光譜。在模擬光譜中,其半高寬預設為50 cm-1;若要和實驗值相比對,半高寬再視實驗的解析度加以調整。本研究 與低解析的實驗光電子光譜比對時,分子的光電子光譜設定半高寬為300 cm-1,硫甲醛負離子的光電子光譜設定半高寬為200 cm-1。 本研究的 H2CS - ,在四個振動態的法蘭克─康登因子,經由羅倫茲函 數所模擬出的光譜後,會產生四個光電子光譜,分別為ν=0、ν3=1、ν4 =1、ν4=2 躍遷的光電子光譜,經由對實驗光譜的分析,算出ν=0、ν3 =1、ν4=1、ν4=2 的相對居量分別為 1、0.2522、0.07189、0.0636。最後, 再將四個光譜結合成最後的 H2CS - 模擬光電子光譜。第三章
第三章
第三章
第三章
研究結果與討論
研究結果與討論
研究結果與討論
研究結果與討論
本章共分六節來呈現硫甲醛分子、正離子、負離子的計算結果,並且將 模擬光譜與實驗光譜做比對。第一節探討硫甲醛分子、正離子與負離子的計 算結果,其中包括分子、正離子、負離子的平衡結構。第二節探討振動模式 和頻率。第三節探討硫甲醛的游離能與電子親和力,第四節介紹硫甲醛分子 的法蘭克-康登因子。第五節探討 H2CS -的波茲曼分布計算結果。第六節呈 現 H2CS 與 H2CS - 的光電子光譜與實驗光譜的比對結果。由於計算資料眾 多,在本章僅列出部分基組資料進行解說,其餘基組的計算資料,則建置於 附錄。
3.1 硫
硫
硫 甲 醛 分 子
硫
甲 醛 分 子
甲 醛 分 子
甲 醛 分 子 、
、 正 離 子
、
、
正 離 子
正 離 子 、
正 離 子
、
、 負 離 子 平 衡 結 構 的 計 算 結 果
、
負 離 子 平 衡 結 構 的 計 算 結 果
負 離 子 平 衡 結 構 的 計 算 結 果
負 離 子 平 衡 結 構 的 計 算 結 果
3.1.1 3.1.1 3.1.1 3.1.1 平衡結構計算值與實驗值差距比較結果平衡結構計算值與實驗值差距比較結果平衡結構計算值與實驗值差距比較結果 平衡結構計算值與實驗值差距比較結果 根據 1998 年 Kuchitsu[44]整理實驗數據,硫甲醛 (H2CS) 分子的平衡結 構,屬於 C2V點群的平面分子,在分子平衡狀態下,S=C 鍵為雙鍵,結構的 實驗值分別為:rS-C=1.6108 Å,rC-H=1.0925 Å,∠SCH=121.7°、∠HCH=116.5°[44]。 在正離子平衡結構方面,目前並沒有實驗值可供比對,負離子有實驗值 rS-C=172±2 pm,我們也將各個離子的計算結果與分子的平衡結構進行比較。 以 七 種 基 組 6-311++G(d,p) 、 6-311++G(2d,p) 、 6-311++(3d,p) 、 6-311++g(3df,p)、6-311++g(3df,pd)和 AVDZ 與 AVTZ,計算硫甲醛的分子、正離子、負離子的平衡結構,並將結果與實驗值整理至表二~四。 硫甲醛分子七種基組的計算結果,所有鍵長的計算值與實驗值差距都很 小 ; SC 鍵 長 的 計 算 結 果 介 於 160.63~162.26 pm 之 間 , 以 最 大 基 組 aug-cc-pVTZ 計算值最接近實驗值,由下表二可得知,差距約 0.01%;CH 鍵計算鍵長介於 108.72~109.71 pm 之間,同樣以最大的基組 aug-cc-pVTZ 計算值最接近實驗值,兩者差距為 0.03 pm。整體而言,實驗值與我們採計 七種基組計算結果的比值在 0.9972~1.0094 之間,可證實分子的計算結果與 實驗值的鍵長相近,從表二顯示,H2CS 分 子 鍵 角 的 計算結果都很一致, ∠SCH 計算結果介於 121.9°~122.2°;∠HCH 計算結果在 115.6°~116.2°,兩 者計算結果與實驗值的差距小於 1°,實驗值與計算結果的比值在 0.99~1.004 之間。
表二、H2CS 不同計算基組之幾何優選與實驗值之比較a a 計算方法為 DFT 之 B3LYP。 b 參考文獻[44]。 鍵長單位為 pm,角單位為度。 基組 S=C C-H ∠SCH ∠HCH 6-311++G(d,p) 161.55 109.03 122.1 115.9 6-311++G(2d,p) 161.07 108.89 122.1 115.9 6-311++G(3d,p) 160.80 108.97 122.1 115.8 6-311++G(3df,p) 160.64 108.97 122.2 115.6 6-311++G(3df,pd) 160.63 108.89 122.2 115.7 aug-cc-pVDZ 162.26 109.71 121.9 116.2 aug-cc-pVTZ 161.11 108.72 122.1 115.8 實驗值b 161.08 108.69 121.7 116.5