行政院國家科學委員會補助專題研究計畫
■ 成 果 報 告 □期中進度報告(計畫名稱)
缺血性腎衰竭於基因治療後之氧化壓力與細胞凋亡評估
計畫類別:■
個別型計畫 □
整合型計畫
計畫編號:NSC 92-2314- B -002 -331
執行期間:92 年 8 月 1 日至 93 年 7 月 31 日
計畫主持人:賴明坤
共同主持人:鄭劍廷
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):■精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計
畫、列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:台大醫學院醫學系
中 華 民 國 93 年 10 月 30 日
中文摘要
本研究以 adenoviral bcl-2 vector 前處理活體大白鼠兩側腎動脈阻斷四十五分鐘再遭 受不同時段的血液灌流造成急性腎衰竭作為研究模式,探討腎臟遭受缺血 再灌流 後之細胞凋亡的情形。我們利用三種方式評估細胞凋亡;in situ apoptosis assay (TUNEL reaction)的免疫分析,DNA ladder 的生化檢定及流式細胞儀來測定腎近端小 管之細胞凋亡數量。我們發現 adenoviral bcl-2 vector 前處理會明顯增加 Bcl-2 protein 的表現量而降低自由基 superoxide 在腎臟遭受缺血 再灌流後之數量(出現於近端位 置, 以 NBT 的方式來測定)及釋放量(以超感度冷光的方式來測定血液,組織即活體 腎臟)。也降低 apoptosis 與 BUN 與 Creatinine。以 Adv-bcl-2 vector 的研發可應用於 保存液的製造與改進將對於減輕臨床上因腎臟移除後之移植腎之延遲作用或是經腎 缺血後再灌流的傷害有相當大的幫助。
關鍵字:細胞凋亡,自由基,bcl-2 基因,腺病毒
Abstract
Ischemia/reperfusion induces oxidative injury to proximal and distal renal tubular cells. We hypothesize that Bcl-2 protein augmentation with adenovirus vector mediated bcl-2 (Adv-bcl-2) gene transfer may improve ischemia/reperfusion induced renal proximal and distal tubular apoptosis through the mitochondrial control of Bax and cytochrome C translocation. Twenty-four hours of Adv-bcl-2 transfection to proximal and distal tubular cells in vitro upregulated Bcl-2/Bax ratio and inhibited hypoxia/reoxygenation induced cytochrome C translocation, O2.- production and tubular apoptosis. Intrarenal arterial
Adv-bcl-2 administration with renal venous clamping augmented Bcl-2 protein of rat kidney
in vivo in a time-dependent manner. The maximal Bcl-2 protein expression appeared at 7
days after Adv-bcl-2 administration and the primary location of Bcl-2 augmentation was in proximal and distal tubules, but not in glomeruli. With a real-time monitoring O2-. production
and apoptosis analysis of rat kidneys, ischemia/reperfusion increased renal O2-. level,
potentiated proapoptotic mechanisms, including decrease in Bcl-2/Bax ratio, increases in caspase 3 expression and poly-(ADP-ribose)-polymerase fragments, and subsequent proximal and distal tubular apoptosis. However, Adv-bcl-2 administration significantly enhanced Bcl-2/Bax ratio, decreased ischemia and reperfusion induced O2-. amount and
inhibited proximal and distal tubular apoptosis. Our results suggest that Adv-bcl-2 gene transfer significantly reduces ischemia/reperfusion induced oxidative injury in the kidney. Keywords: reactive oxygen species, apoptosis, adenoviral vector, bcl-2
Introduction
Complete or partial cessation (ischemia) followed by restoration of blood flow (reperfusion) impairs many organs, such as the heart, brain, liver, and kidney (1-4). Ischemia/reperfusion injury induces burst release of reactive oxygen species (ROS) (1-4), which contribute to abnormal signal transduction or cellulardysfunction (5,6) and initiate the cascade of apoptosis/necrosis (2,7).
Mitochondrial dysfunction following oxidative injury is an early event in apoptotic cell death, since the apoptogenic factor, cytochrome C, is released into the cytoplasm (7). Once this translocation occurs, cytochrome C binds to another cytoplasmic factor, Apaf-1, and the formed complex activates the initiator caspase-9 that in turn activates the effector caspases, of which caspase-3 is a prominent member (8). Release of cytochrome C from the mitochondria can be triggered by the proapoptogenic Bax (9). While Bax has been shown to trigger cell death (10), the anti-apoptotic Bcl-2 can block cytochrome C release and caspase activation (11). In the kidney, ROS are produced in significant amounts in renal proximal rather than distal tubular epithelium under ischemia/reperfusionor hypoxia/reoxygenation conditions (2). The increased ROS production enhances Bax/Bcl-2 ratio (2,12), caspase 3 (CPP32) expression (2,13), and poly-(ADP-ribose)-polymerase (PARP) fragments (14), and subsequentlyresulted in severe apoptosis, including increases in DNA fragmentationand apoptotic cell number in renal tubules.
Antioxidant Bcl-2 (15) resides in the mitochondria and prevents activation of the effector caspases by mechanisms such as blockade of the mitochondria permeability transition pore (MTP) opening (16), or by functioning as a docking protein (17). Overexpression of Bcl-2 can block both apoptosis and necrosis (18,19), and protect ischemic tissue against reperfusion induced oxidative stress (20). Therefore, the application of local bcl-2 gene transfer into the kidney to augment Bcl-2 protein seems promising as a therapeutic strategy for reduction of renal ischemia/reperfusion injury.
In this study, a replication-defective adenovirus vector-mediated gene transfer was used to augment Bcl-2 in rat kidney to obviate concerns regarding the purity of the enzyme and preparation or fluctuations in Bcl-2 protein delivery. Our results demonstrate that proximal and distal tubules enriched in Bcl-2 are more resistant to the damaging effects of ischemia/reperfusion by downregulation in ROS amount, Bax/Bcl-2 ratio, cytochrome C release, and subsequent reduction of apoptotic cell death.
Methods
Preparation of Recombinant adenoviral vector
A human bcl-2 cDNA containing the entire coding sequence was subcloned into the E1 and E3 deleted adenovirus shuttle plasmid, which contains a promotor of the human phosphoglycerate kinase (Adv-pgk). A recombinant adenovirus (Adv-bcl-2) was generated by homologous recombination and amplified in human embryonic kidney (HEK) 293 cells as previously described (21). The human bcl-2 cDNA was kindly provided by from Dr. J.Y. Yen at Institution of Biomedical Science, Academia Sinica (22). Viruses stocks were purified by CsCl density gradient centrifugation, aliquoted, and stored at -80°C. Viral titers, plaque-forming units (pfu), were determined by plaque-forming assay that HEK 293 cells were infected with serially diluted viral preparations and then overlaid with low melting-pointagarose after infection. Numbers of plaques formed were countedwithin 2
weeks.
Animals
Female Wistar rats (200-250 g) were housed at the Experimental Animal Center, National Taiwan University, at a constant temperature and with a consistent light cycle (light from 07:00 to 18:00 o'clock). Food and water were provided adlibitum. All surgical and experimental procedures were approved by the animal care and experimental protocols were in accordance with the guidelines of the National Science Council of the Republic of China (NSC 1997).
Gene transfer to renal proximal (PT) and distal tubules (DT)
Under sodium pentobarbital (40 mg/kg, i.p.) anesthesia, kidneys were flushed with 20 ml of ice-cold Krebs-Henseleit-saline buffer via an aortal catheter. Specific isolation of PT and DT was performed as previously described (2). Each of the two types of cells was identified and cultured (2,23). For infection, ~3×105
cells were seeded on a 10-cm2 Petri dish and treated with Adv-bcl-2 at the 107 pfu for 24 hours. Induction of hypoxia/reoxygenation of the renal tubule cells was performed as described (2). The cultures were first placed in an atmosphere of 95% O2/5% CO2 at 37°C for 30 min. Hypoxia was achieved by gassing with
95% N2/5% CO2 for 15 min, whereas reoxygenation was performed by reintroduction of
95% O2/5% CO2 for 30 min. For determinationof the number of apoptotic cells in culture,
the cells were fixed with 70% ethanol, stained with propidium, and counted with FACSCalibur (BectonDickinson, San Jose, CA). Cell viability was counted with a Trypan blue dye exclusion test. The amounts of ROS in PTand DT cells (106 cells/ml) were detected by the lucigenin-enhanced ROS test (2).
Intrarenal arterial gene delivery
For direct gene delivery, an intrarenal arterial catheter was performed via the left femoral artery (2). Under avertin anesthesia (400 mg/kg, ACROS ORGANICS, NJ, USA), a length of stretched PE10 tubing was introduced into the left renal artery from the left femoral artery via the aorta. Base on our preliminary results, 108 pfu of Adv permitted efficient gene delivery. Recombinant adenovirus of 108 pfu in 0.2 ml of saline was infused into the left kidney at a rateof 20 µL/min via the intrarenal arterial catheter with clamping of the renal vein for 10 min. Toascertain the transgene expression at the region of Adv infusion,we infused adenoviruses containing a green fluorescentprotein (GFP) gene, Adv-GFP, into the left kidney and examinedthe GFP expression in rat kidneys 3-28 days later. High levels of GFP were visualized under UV in the arterial lining cells and tubular cells in the left kidney sections but not in the right kidney. These results confirmed the uptake and expression of Adv by the left kidneys.
After Adv-bcl-2 injection into the left kidney, the incision was closed in layers with 3.0 suture (Ethicon), and the animals were allowed to recover. Rats were sacrificed by overdose of sodium pentobarbital at 3, 5, 7, and 28 days after Adv-bcl-2 administration. In some rats,
renal glomeruli were isolated by graded sieving (250, 150, and 75 µm) (24) and PT and DT cells were isolated as described above. The isolated proteins from separated glomeruli, PT and DT were used to determine Bcl-2 protein expression after Adv-bcl-2.
Induction of unilateral renal ischemia
On the day of the experiments, the rats were anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and were tracheotomized. The left kidney was exposed via a flank incision and dissection from the surrounding tissue. For induction of total ischemia in the kidney, the left renal artery was clamped 45 min with a small vascular clamp. Sham-operated (control) animals underwent similar operative procedures without occlusion of the renal artery. Reperfusion was initiated by removal of the clamp for 4-10 hours. After ischemia/reperfusion insults, the kidney was resected and divided into two parts. One part was stored in 10% neutral buffered formalin for routine histology and in situ apoptotic assay, and another was quickly frozen in liquid nitrogen and stored at –70°C for protein isolation.
In vivo chemiluminescence recording for ROS activity
The ROS generation in response to ischemia/reperfusion injury was measured from the kidney surface by a chemiluminescence detection method (2). The measurement of ROS from the kidney was started by intrarenal arterial infusion of a superoxide anion probe, 2-Methyl-6-(4-methoxyphenyl)-3,7-dihydroimidazo-[1,2-a]-pyrazin- 3-one-hydrochloride (MCLA) (0.2 mg/ml/h, TCI-Ace, Tokyo Kasei Kogyo Co. Ltd., Tokyo, Japan) throughout the experiment by use of a Chemiluminescence Analyzing System (CLD-110, Tohoku Electronic In. Co., Sendai, Japan). The MCLA-enhanced chemiluminescence counts were continuously recorded every 10 sec. The real-time displayed chemiluminescence signal was recognized as ROS level from the kidney surface.
In situ demonstration of superoxide generation and apoptosis formation
A nitroblue tetrazolium (NBT) perfusion method was used for localizing de novo ROS generation in the insulted kidney (2). Rats (n=3 in each group) were sacrificed at the end of ischemia/reperfusion injury. An 18-gauge needle connected to an infusion pump (Infors AG, CH-4103, Bottmingen, Switzerland) was inserted into the lower abdominal aorta just below the level of the renal artery. The kidneys were perfused with 37°C Hanks' balanced salt solution (flow rate, 10 ml/min; pH 7.4), and the perfusate was allowed to drain from the inferior vena cava. Once blood had been removed, NBT (1 mg/ml) was added to the solution, and the kidney was perfused for an additional 10 min at a flow rate of 5 ml/min. All unreacted NBT was removed from the kidneys by perfusion with Hanks’ solution. The NBT-perfused kidney was removed and fixed in zinc/formalin (1% ZnSO4 in 10% formalin)
for histologic examination for formazan deposits.
The method for the terminal deoxynucleotidyl transferase-mediated nick-end labeling method (TUNEL) was performed as previously described (2,12,13). Sections of the kidney were prepared, deparaffinized, and stained by the methyl green and
TUNEL-avidin-biotin-complex method. Twenty high-power (×400) fields were randomly selected for each bladder section, and the value of apoptotic cells/(apoptotic cells and methyl green stained cells) was counted. The number of apoptotic cells was expressed per 100 of the tubular cells in each section.
Immunostaining for Bax and Bcl-2
Localization of Bax and Bcl-2 was detected in 10% neutral-buffered formalin fixed, paraffin-embedded 4-µm sections by immunohistochemistry using a standard avidin-biotin peroxidase complex technique. After deparaffinization and hydration, the sections were heated at 95°C in pH=7.0 10 mmol/L citrate acid buffer for Bax or Bcl-2. The activity of endogenous peroxidase was quenched by 3% (vol/vol) H2O2 in methanol. The sections were
then incubated with the primary antibody overnight at 4°C in a humid atmosphere. Bax and Bcl-2 (human Bcl-2 peptide, a.a. 49-179, Transduction, Bluegrass-Lexington, KY, USA) was diluted at 1:400. The sections were stained by an avidin-biotinylated horseradish-peroxidase procedure using a commercially available kit (ABC Elite; Vector Laboratories). Finally, the color was developed by the 3'-Amino-9-ethylcarbazole substrate (Vector Laboratories) and counter stained with hematoxylin. Control sections were incubated with the same species normal IgG or serum at the same protein concentration as the primary antibody.
Bax, Bcl-2, cytochrome C, CPP32, and PARP expression
Cytosolic Bax translocation to mitochondria and mitochondrial leakage of cytochrome C to cytosol are required for triggering apoptotic pathway (25,26). PT and DT cells were subjected to differential centrifugation to obtain the mitochondrial and cytosolic fractions. Protein was determined by the BioRad Protein Assay (BioRad Laboratories, Hercules, CA, USA). Ten µg of protein was electrophoresed as described below. The primary antibody was a polyclonal rabbit antihuman cytochrome C (Santa Cruz Biotechology, Inc., Santa Cruz, CA, USA) used at 1:1000 or Bax (human Bax synthetic peptide, a.a. 44-62, Chemicon, Temecula, CA, USA).
The expression of Bax/Bcl-2, caspase 3, and PARP of kidney tissue was evaluated by western immunoblotting (2). Briefly, the total proteins were homogenized with a prechilled mortar and pestle in extraction buffer, which consisted of 10 mM Tris-HCl (pH 7.6), 140 mM NaCl, 1 mM phenylmethyl sulfonyl fluoride, 1% Nonidet P-40, 0.5% deoxycholate, 2% β-mercaptoethanol, 10 µg/ml pepstatin A, and 10 µg/ml aprotinin. The mixtures were homogenized completely by vortexing and kept at 4°C for 30 min. The homogenate was centrifuged at 12,000 ×g for 12 min at 4°C, the supernatant was collected, and the protein concentrations were determined by BioRad Protein Assay (BioRad Laboratories).
Antibodies raised against Bax (Chemicon), Bcl-2 (human Bcl-2 peptide, a.a. 49-179, Transduction, Bluegrass-Lexington, KY, USA), the activation fragments of caspase 3 (CPP32/Yama/Apopain, Upstate Biotechnology, Lake Placid, NY, USA), PARP (N-terminal
peptide from the p85 fragment, Promega, Madison, WI, USA), and β-actin (Clone AC-74, Sigma, Saint Louis, MI, USA) were used. All of these antibodies cross-react with the respective rat antigens.
Statistical analysis
All values were expressed as mean ± SE. Differences within groups were evaluated by paired t-test. One-way analysis of variance was used for establishing differences among groups. Intergroup comparisons were made by Duncan's multiple-range test. Differences were regarded as significant if P < 0.05 was attained.
Results
Adv-bcl-2 transfection augmented Bcl-2 on PT and DT
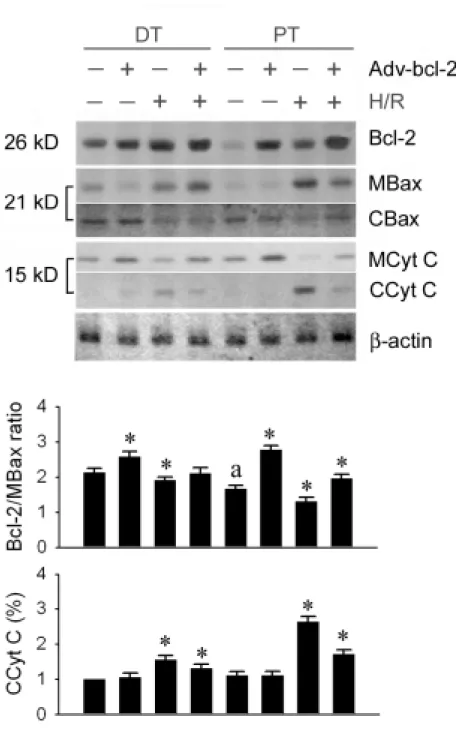
To examine the role of the induction of Bcl-2 protein by Adv-bcl-2, primary PT and DT cultures that overexpressed Bcl-2 proteins were established. Adv-bcl-2 was transfected into the PT and DT cultured cells for 24 hours. In the basal level without Adv-bcl-2, the Bcl-2 protein expression of DT was 4.7±0.8 times higher (P<0.05) than that of PT. Adv-bcl-2 administration augmented 1.7±0.3 folds of DT and 6.5±1.4 folds of PT Bcl-2 expression. Hypoxia/reoxygenation enhanced Bcl-2 expression, and activated Bax translocation to mitochondria and cytochrome C translocation to cytosol in both PT and DT cells (Figure 1 upper panel). Overexpression of Bcl-2 protein by Adv-bcl-2 inhibited cytochrome C translocation, but not Bax translocation in both DT and PT cultures. Increased Bcl-2/mitochondrial Bax ratio by Adv-bcl-2 transfection was efficiently inhibited cytochrome C release to cytoplasm (Figure 1 middle and lower panels)
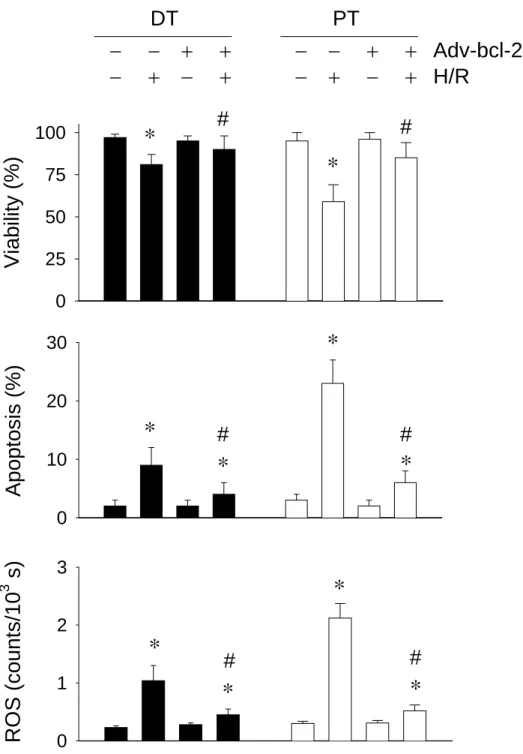
Bcl-2 augmentation inhibits DT and PT apoptosis and ROS amount
In correlation with the level of Bcl-2 proteins expressed, PT and DT apoptosis and ROS release induced by hypoxia/reoxygenation was inhibited by Adv-bcl-2 administration (Figure 2).Theaugmented Bcl-2 present in DT and PT cells was sufficient to antagonize the apoptotic and oxidant action of hypoxia/reoxygenation.
Intrarenal arterial Adv-bcl-2 increased Bcl-2 expression in the kidney in vivo
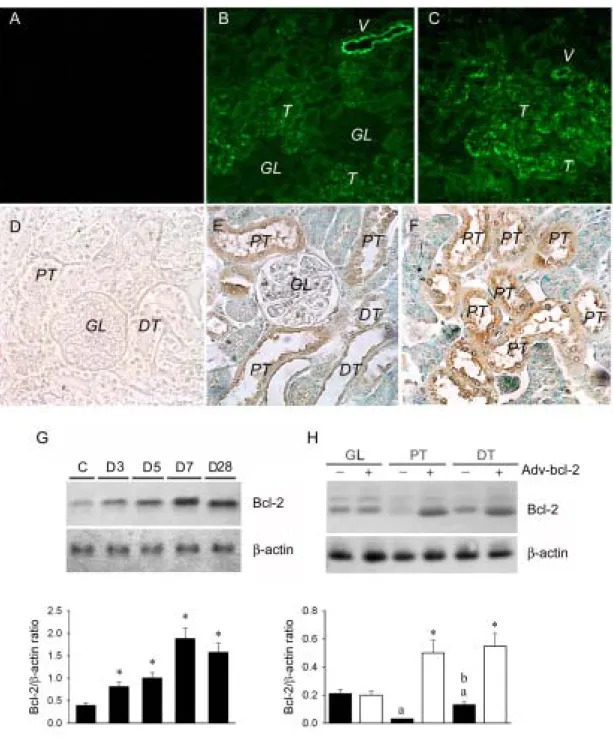
To evaluate the efficiency of adenovirus vector administration via the intrarenal arterial route, we infused Adv-GFP into the left kidney of normal rats. After 3-28 days of administration, the green fluorescent intensity was found in the renal vessels and tubules (Figures 3B & 3C) of Adv-GFP kidney, but not found in the Adv-pgk treated kidney (Figure 3A).
Next, we infused Adv-bcl-2 into the left kidney of normal rats and determined Bcl-2 protein levels 3 to 28 days after administration. With Bcl-2 immunocytochemical analysis, the PT and DT cells (Figure 3E), especially in PT cells (Figure 3F), are highly stained by Bcl-2. With western blotting analysis, compared with Adv-pgk control, Adv-bcl-2 augmented Bcl-2 in a time-dependent manner (Figure 3G). Maximal augmentation of 4-5 fold of renal Bcl-2 was noted at 7 days after administration. The augmented Bcl-2 expression
was primarily located in the DT (4.5±0.7 folds) and PT (12.0±2.1 folds) tubules, not glomeruli by western blots (Fig. 3H).
Adv-bcl-2 reduced ischemia- and reperfusion-induced kidney ROS levels
Previous studies indicate bcl-2 transfer confers antioxidant and antiapoptotic potential against oxidative stress (20). We therefore evaluated the effects of Adv-bcl-2 administration on O2¯· levels and apoptosis cell death. Continuous infusionof MCLA into a control kidney
displayed a basal O2¯· level at 500-800 counts/10 s. In ischemic kidneys, an increase in O2¯·
value (1740±280 counts/10 s) was observed and wasmaintained at the increased level for 45 min (Figure 4, trace 1). In reperfused kidneys, the O2¯· level was further enhanced to
4120±570 counts/10 s and maintained at the high level for 4-6 hours. In the kidneys treated with Adv-bcl-2 (Figure 4, trace 2), the level of O2¯· in the baseline control, ischemic periods
and reperfusion stages were 550±90, 1120±190, and 1490±245 counts/10 s, respectively. Adv-bcl-2 augmented Bcl-2 protein significantly inhibited 55% and 75% of O2¯· production
in the ischemia and reperfusion stages.
In situ localization of O2¯· formation and proapoptotic Bax expression
We used an NBT staining method for localization of O2¯· productionin renal tissue (2).
NBT is reduced to an insoluble formazan derivative upon exposure to O2¯· and the
blue-coloredformazan is readily detectable in tissue by light microscopy. O2¯· generation was
not seen in controlkidneys. There was significant O2¯· production (blue deposits) in ischemic
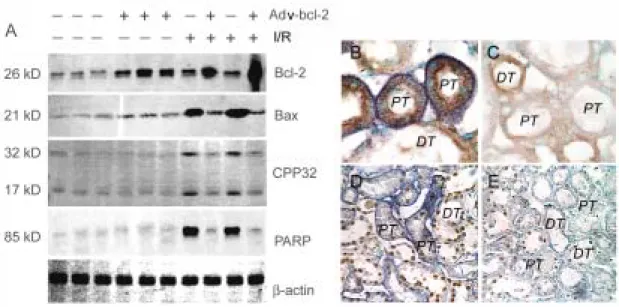
kidneys after4 hours of reperfusion (Figures 5 B&D). The NBT deposits were located mainly in the PT and sparsely in DT. In rats treated with Adv-bcl-2, significant decreases in NBT deposits in PT cells were observed (Figures 5 C&E). With double staining of proapoptotic Bax expression in the NBT treated kidneys, Bax was not present in the control kidneys (data not shown), but was highly stained in the PT and DT cells after ischemia/reperfusion injury (Figure 5B). Both NBT and Bax stains were highly expressed in the insulted PT. Adv-bcl-2 reduced NBT deposits and Bax expression, indicating a reduction of oxidant and apoptotic action by augmented Bcl-2 (Figure 5C).
Adv-bcl-2 gene transfer reduced ischemia/reperfusion-induced kidney apoptosis
The expression of Bax, Bcl-2, CPP32, and PARP in the kidney samples after ischemia/reperfusion was assessed by immunoblotting (Figure 5A). The expressionof Bcl-2 was detected in control renal tissues and was apparently enhanced after 7 days of Adv-bcl-2 administration. Bcl-2 expression was mildly increased by ischemia/reperfusion. Bcl-2 expression was significantly augmented by ischemia/reperfusion in the Adv-bcl-2 treated kidneys. Expression of Bax but not of CPP32 and PARP was detected in Adv-pgk and Adv-bcl-2 treated kidneys. The expression of three proteins was significantly increasedafter ischemia/reperfusion. The enhanced Bax, CPP32, and PARP expression by ischemia/reperfusion was significantly inhibited by Adv-bcl-2 treatment.
were not detected or were only rarelypresent in sections from Adv-pgk control or Adv-bcl-2 treated kidney. In kidneys subjected to ischemia/reperfusion, increased numbers of apoptotic cells were detectedin both PT (22.0±4.1%) and DT (7.3±1.8%) (Figure 5D). After 7 days of Adv-bcl-2 administration, augmented Bcl-2 expression significantly (P<0.05) reduced the numberof apoptotic cells in both PT (3.6±0.6) and DT (1.4±0.3%) cells (Figure 5E).
Discussion
The current study indicates that intrarenal arterial infusion of Adv-bcl-2 is effective in augmenting Bcl-2 expression in kidney tissues and in reducing tubular apoptosis by ischemia/reperfusion injury. In DT and PT cultures in vitro, augmented Bcl-2 can increase Bcl-2/Bax ratio, inhibit cytochrome C translocation to cytoplasm, and reduce ROS production and apoptotic cell death in hypoxia/reoxygenation injury. In rat kidney in vivo, the augmented Bcl-2 protein highly expressed in DT and PT cells, not glomeruli of rat kidney in vivo confers a selective protection against ischemia/reperfusion induced tubular oxidative stress and apoptosis.
One of the most common causes of acute tubular epithelial injury is ischemia/reperfusion and associated oxidative stress or ROS injury. Sensitivity to ischemia/reperfusion-induced injury differes along the renal nephron (27,28). Apoptotic cell death was recorded previously in both PT and DT cell populations in ischemic renal injury (2,13,29). The PT straight tubule was often seen to be acutely sensitive to ischemia/reperfusion, undergoing extensive necrosis and /or apoptosis. The DT was less sensitive to ischemia/reperfusion, with the dying cells being predominantly apoptotic (29,30). Therefore, correcting nephron dysfunction should be accomplished by transferring a particular molecule to the targeted nephron segments. In the current study, a specific tubular cell-targeted gene transfer techniques by a replication-defective Adv-bcl-2 could selectively transfect both PT and DT cells in the rat kidney without early local toxicity and CD4+/CD8+-mediated immune response as assessed by renal histology and function (data not shown). Furthermore, we performed intrarenal arterial administration technique and 10 min of renal venous clamping, which resulted in bcl-2 transfer in the cortico-medullary area without renal damage.
Mitochondria are the target and source of ROS (31), which play an important role in physiologic signaling mechanisms and in regulation of apoptotic pathway (9,32). Mitochondrial dysfunction caused by inappropriate MTP opening disrupts mitochondrial membrane potential for ATP synthesis and triggers oxidative and anoxic cell death (33). The outer mitochondrial voltage-dependent anion conductance (VDAC) channel is involved in cytochrome C release and is regulated by ROS and Bcl-2 family (16,34,35). O2¯· but not
H2O2 induces VDAC-dependent permeabilization of the outer mitochondrial membrane in
HepG2 cells (34). Bcl-2 family are able to regulate the status of MTP; Bax, a channel-forming protein, can open it (9,36), and Bcl-2 and Bcl-xL are able to stabilize and
inhibit its opening (37). Our data showed that Bax and O2¯· are coexpressed in the
ischemia/reperfusion kidney. Release of cytochrome C caused by increased MTP opening is a proximate trigger for evoking caspase 3 mediated apoptosis (8,16,35,36). Therefore, an increase in ROS and a reduction in Bcl-2/Bax ratio enhance MTP opening, cytochrome C release, and caspase 3 mediated renal tubular apoptosis (9,32). According to our in vitro and in vivo data, the decreased ratio of Bcl-2/Bax by hypoxia/reoxygenation or ischemia/reperfusion injury triggers O2¯· production, Bax translocation to mitochondria,
cytochrome C release to cytoplasm, CPP32 activation, and increases PARP fragments initiated apoptosis. Bcl-2/Bax ratio in mitochondria plays a critical role in regulation of apoptosis pathway.
Antioxidant Bcl-2 resides in the mitochondria and prevents activation of the effector caspases by mechanisms such as blockade of MTP opening (16,37), by functioning as a docking protein (17), and by decreasing cellular ROS (15,19,38). With an in situ vascular NBT perfusion technique, our recent (2) and current data showed thatthe cellular source of ROS synthesis was mainly the PT epithelialcells in the ischemia/reperfusion kidney. The increased Bax expression, apoptotic cell death as well asthe ROS production in PT cells can be ameliorated by superoxide dismutase (2) or Adv-bcl-2 administration. We also noted that, in culture, the number of apoptotic cells and the amounts of ROS formed are more evident in PT cells than in DT cells subjected to hypoxia/reoxygenation insult. Upon exposureto transient ischemia, the DT of the kidney often escapes thesevere damage that afflicts the PT. The resistance to ischemia/reperfusion injuryby DT cells may be attributed to increases of Bcl-2 protein expression and/or Bcl-2/Bax ratio in DT, whereas low levels of expression/ratio were detected in PT. This could also explain the generallylow level of ROS formation in DT cells and Adv-bcl-2 treated kidney or cells. Overexpression of the Bcl-2, which largely localizes to the outer mitochondrial membrane, protects cells not only from apoptotic but also from oxidative cell death (38). Adv-bcl-2 administration significantly augmented Bcl-2 protein expression in DT and PT tubules in vitro and in vivo. Increased Bcl-2/Bax ratio by Bcl-2 augmentation inhibited O2¯· production, cytochrome C
translocation to cytoplasm, CPP32 activation, and PARP fragments initiated apoptosis formation, but had no effect on Bax translocation to mitochondria. We implicate that augmented Bcl-2 protein by Adv-bcl-2 gene transfer confers renal DT and PT cells protection against hypoxia/reoxygenation or ischemia/reperfusion injury via the inhibition of MTP opening and cytochrome C release.
In summary, our results demonstrate that the rat kidney can be efficiently and selectively transduced with Adv-bcl-2 (108 pfu/kidney) without structural or functional side effect. We provide evidence that gene delivery to the kidneys is feasible, and Bcl-2 augmentation in both PT and DT tubules protects against ischemia/reperfusion injury via the antioxidant and antiapoptotic actions.
Acknowledgements
This work was supported by the National Science Council of the Republic of China (NSC 92-2320-B002-078, NSC 92-2314-B002-331, and NSC 92-2314-B002-163).
References
1. Bilzer M, Paumgartner G, Gerbes AL. Glutathione protects the rat liver against reperfusion injury after hypothermic preservation. Gastroenterology 1999; 117:200-210. 2. Chien CT, Lee PH, Chen CF, Ma MC, Lai MK, Hsu SM. De novo demonstration and co-localization of free-radical production and apoptosis formation in rat kidney subjected to ischemia/reperfusion. J Am Soc Nephrol 2001;12:973-982.
3. Plesnila N, Zhu C, Culmsee C, Groger M, Moskowitz MA, Blomgren K. Nuclear translocation of apoptosis-inducing factor after focal cerebral ischemia. J Cereb Blood Flow Metab 2004 24:458-466.
4. Rajesh KG, Sasaguri S, Suzuki R, Maeda H. Antioxidant MCI-186 inhibits mitochondrial permeability transition pore and upregulates Bcl-2 expression. Am J Physiol Heart Circ Physiol 2003; 285:H2171-2178.
5. Hunter T. Protein kinase and phosphatase: The yin and yang of protein phosphorylation and signaling. Cell 1995;80 : 225-236.
6. Stamler JS. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994;78:931-936.
7. Salahudeen AK, Huang H, Joshi M, Moore NA, Jenkins JK. Involvement of the mitochondrial pathway in cold storage and rewarming-associated apoptosis of human renal proximal tubular cells. Am J Transplant. 2003;3:273-80.
8. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997;91:479-489.
9. Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 2000;345:271-278.
10. Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J 1998;17:3878-3885. 11. Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science.
1998;281:1322-1326.
12. Basile DP, Liapis H, Hammerman MR: Expression of bcl-2 and bax in regenerating rat renal tubules following ischemic injury. Am J Physiol 1997; 272:F640-F647.
13. Kaushal GP, Singh AB, Shah SV: Identification of gene family of caspases in rat kidney and altered expression in ischemia-reperfusion injury. Am J Physiol 1998;274:F587 -F595.
oxygen intermediate(s) (ROI): Common mediator(s) of poly(ADP-ribose)polymerase (PARP) cleavage and apoptosis. FEBS Lett 1996;39: 299-303.
15. Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci USA. 2003 100:15035-15040.
16. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999; 399:483-487.
17. Reed JC. Double identity for proteins of the Bcl-2 family. Nature 1997; 387:773-776. 18. Israels LG, Israels ED. Apoptosis. Stem Cells 1999;17:306-313.
19. Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science 1993;262:1274-1277.
20. Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J Neurochem 2003;85:1026-1036.
21. Shyue SK, Tsai MJ, Liou JY, Willerson JT, Wu KK. Selective augmentation of prostacyclin production by combined prostacyclin synthase and cyclooxygenase-1 gene transfer. Circulation 2001;103:2090-2095.
22. Huang, HM., Huang CJ and Yen, JJY. Mcl-1 is a common target of stem cell factor and interleukin 5 for apoptosis prevention activity via MEK/MAPK and PI-3K/Akt pathways. Blood 2000;96:1764-1771.
23. Guder WG, Ross BD. Enzyme distribution along the nephron. Kidney Int 1984;26:101-111.
24. Chen YM, Chien CT, Hu-Tsai MI, Wu KD, Tsai CC, Wu MS, Tsai TJ. Pentoxifylline attenuates experimental mesangial proliferative glomerulonephritis. Kidney Int 56: 932-943, 1999.
25. Cai J, Yang J, Jones DP. Mitochondrial control of apoptosis: the role of cytochrome C. Biochem Biophys Acta 1998;1366-139-149.
26. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998; 281-1309-1312.
27. Cuttle L, Zhang XJ, Endre ZH, Winterford C, Gobe GC. Bcl-xL translocation in renal tubular epithelial cells in vitro protects distal cells from oxidative stress. Kidney Int 59:1779-1788, 2001.
28. Lieberthal W, Nigam SL. Acute renal failure. I. Relative importance of proximal vs distal tubular injury. Am J Physiol 1998;275:F623-F631.
29. Gobe G, Zhang XJ, Willgoss DA, Schoch E, Hogg NA, Endre ZH. Relationship between expression of Bcl-2 genes and growth factors in ischemic acute renal failure in
the rat. J Am Soc Nephrol 2000;11:454-67.
30. Brezis M, Shanley P, Silva P, Spokes K, Lear S, Epstein FH, Rosen S. Disparate mechanisms for hypoxic cell injury in different nephron segments. Studies in the isolated perfused rat kidney. J Clin Invest 1985;76:1796-806.
31. Richter C, Schweizer M, Cossarizza, Franceschi C. Control of apoptosis by the cellular ATP level. FEBS Lett 1996;378:107-110.
32. Saikumar P, Dong Z, Patel Y, Hall K, Hopfer U, Weinberg JM, Venkatachalam MA. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene 1998;17:3401-3415.
33. Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols: Increase of the gating potential by oxidants and its reversal by reducing agents. J Bio Chem 1994;269:16638-16642.
34. Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol 2001;155:1003-1015.
35. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997;275:1132-1136.
36. Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA 1998;95:14681-14686.
37. Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie ZH, Reed JC, Kroemer G. The permeability transition pore complex: A target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med 1998;187:1261-1271. 38. Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in
Figure Legends
Figure 1: Adv-bcl-2 augmented Bcl-2 blocks cytochrome C translocation without inhibiting Bax translocation in the PT and DT cultures. Ten µg of total protein (Bcl-2 and
β–actin), cytosolic Bax (CBax) and cytochrome C (CCyt C) and mitochondrial Bax (MBax) and cytochrome C (MCytc C) was analyzed by SDS-PAGE followed by immunoblotting. In baseline level, DT Bcl-2 level is significantly higher than PT Bcl-2. Adv-bcl-2 increased both DT and PT Bcl-2 expression. Hypoxia/reoxygenation (H/R) enhances Bcl-2, but also activates Bax translocation to mitochondria and cytochrome C translocation to cytosol. Overexpression of Bcl-2 protein by Adv-bcl-2 gene transfer can inhibit cytochrome C
translocation, but not Bax translocation in both DT and PT cultures. H/R injury decreases the Bcl-2/MBax ratio and increases the CCyt C in both DT and PT cells. Adv-bcl-2 transfection, however, increases the Bcl-2/MBax ratio in baseline level and preserves the Bcl-2/MBax ratio after H/R injury and inhibits cytosolic cytochrome C translocation. * P<0.05 when compared to control level of each culture cells. A P<0.05 PT vs. DT cells.
Viability (%)
0 25 50 75 100Apo
p
to
sis (%)
0 10 20 30RO
S (
c
ount
s
/10
3s)
0 1 2 3*
*
*
*
*
*
*
*
*
*
#
#
#
#
#
#
DT PT
− − + + − − + + Adv-bcl-2
− + − + − + − + H/R
Figure 2. Effect of hypoxia/reoxygenation (H/R) on renal proximal (PT) and distal (DT)
cells. Isolated PT and DT cells were subjected to 15 min of hypoxia (95% N2/5% CO2)
followed by 30 min of reoxygenation (95% O2/5% CO2). Cell viability and tubular apoptosis
were measured by Trypan blue exclusion and flow cytometry. Results are mean ± SEM for four cell preparations. H/R to PT and DT cells produced a statistically significant (P < 0.05) decrease in cell viability and an increase in tubular apoptosis and ROS generation. Adv-bcl-2 administration to PT and DT cells greatly protected cells from H/R injury in both PT and DT cells. *, P < 0.05 when compared with cells without Adv-bcl-2 and H/R treatment. #, P < 0.05 when compared with cells with H/R insult.
Figure 3. Effect of Adv-bcl-2 gene transfer in the kidney. After 3 (B) or 7 days (C) of
intrarenal arterial administration of Adv-GFP, renal tubules (T) and some renal vessel (V) but not glomeruli (GL) display high fluorescence intensity. There is no fluorescent intensity in the control kidney without Adv-GFP treatment (A). With immunohistochemical analysis for confirmation of successful transfection and subsequent protein transcription of bcl-2 gene, the renal proximal tubules (PT, E&F) and distal tubules (DT, E) display a positive brownish color in the kidney section. The kidney with Adv-pgk transfer does not display any brownish-colored stain (D). Bcl-2 protein levels in kidney tissues of rats receiving Adv-pgk vector, or Adv-bcl-2. Time course of Bcl-2 protein levels in kidney tissues transduced with
Adv-pgk control (C) at a representative time point and Adv-bcl-2 at 3-28 days (D3-D28) after treatment (G). Adv-bcl-2 increases the Bcl-2 protein expression in the rat kidney and a maximal expression is found at day 7. (H): Adv-bcl-2 enhances Bcl-2 protein expression primarily in the PT and DT, but not in GL. The lower panel shows densitometry of blots. The error bars are mean±SE of 3 experiments. *
, P < 0.05 when compared with control stage (C) or with Adv-pgk. a, P < 0.05 vs. GL without Adv-bcl-2 treatment. b, P < 0.05 vs. DT without Adv-bcl-2 treatment.
Figur e 4. O2-. detection from rat kidney surface in vivo. (A) Typical recordings of MCLA (1.0
mM)-enhanced O2-. from the surface of ischemia/reperfusion kidney (1, IR-Control),
ischemia/reperfusion kidney with pretreatment of Adv-bcl-2 gene transfer (2, IR-Adv-bcl-2), sham-operated kidney with Adv-bcl-2 pretreatment (3, Sham Adv-bcl-2), and sham-control kidney with saline (4, Sham Control). (B) Mean value of maximal O2-. amount detected from
the kidney surface during the 15 min-control (C), 45 min-ischemia, and 4 h-reperfusion periods is displayed. Ischemia and reperfusion enhance O2-. production in the insulted kidney,
however, the increased O2-. amounts are significantly attenuated by 7 days of Adv-bcl-2
pretreatment. *, P < 0.05 when compared with control stage. #, P < 0.05 when compared with IR-control group.
Figure 5. Effect of Adv-bcl-2 gene transfer on O2-., apoptosis-related proteins and
apoptotic cell death of rat kidney subjected to ischemia/reperfusion injury. (A): In the
baseline level, the Bcl-2 and Bax protein expression can be detected in the control kidneys. Adv-bcl-2 gene transfer significantly enhances renal Bcl-2 protein expression. In response to 45 min-ischemia/4 h-reperfusion (I/R), a mild increase in renal Bcl-2 protein and a marked enhancement in renal Bax protein are found to increase CPP32 and PARP expression. I/R enhanced Bax (brownish color) and O2-. expression (blue deposits) is primarily expressed in
the proximal (PT) but not in the distal tubules (DT) (B). The decreased Bcl-2/Bax ratio by the I/R insult leads to O2-. production and severe apoptosis (brownish nuclei) in the insulted
kidney (D). Adv-bcl-2 significantly decreases Bax and O2-. co-expression in the I/R kidney