國 立 交 通 大 學
機 械 工 程 學 系
碩士論文
應用團塊理論分析一氧化碳毒化效應對
PEMFC 暫態性能之影響
Transient Analysis of CO Poisoning effect to Cell
Performance of PEMFC Using Agglomerate Model
研 究 生:洪家瑜
指導教授:曲新生 教授
應用團塊理論分析一氧化碳毒化效應對 PEMFC 暫態性
能之影響
Transient Analysis of CO Poisoning effect to Cell
Performance of PEMFC Using Agglomerate Model
研 究 生:洪家瑜 Student:Chia-Yu Hung 指導教授:曲新生 Advisor:Hsin-Sen Chu 國 立 交 通 大 學 機 械 工 程 學 系 碩 士 論 文 A Thesis
Submitted to Department of Mechanical Engineering National Chiao Tung University
In Partial Fulfillment of the Requirements For the Degree of
Master In
Mechanical Engineering June 2006
Hsinchu, Taiwan, Republic of China
應用團塊理論分析一氧化碳毒化效應對
PEMFC 暫態
性能之影響
研究生:洪家瑜 指導教授:曲新生 國立交通大學機械工程學系摘要
本研究主要,針對質子交換膜燃料電池中陽極觸媒層內之一氧化 碳毒化現象進行理論分析。使用之理論模式為包含團塊模式與毒化模 式之暫態傳輸行為,並給定邊界條件,求得一氧化碳毒化在觸媒層內 氫氣、一氧化碳、覆蓋率、電流密度的分佈情形與達穩態所需的時間。 研究中主要是改變不同的一氧化碳濃度、觸媒層厚度以及觸媒顆粒半 徑的改變,來探討不同條件對電池內部所產生之影響。 研究結果顯示,隨著一氧化碳濃度愈低,氫氣含量越高時,電池 系統啟動後,需較長的時間達到穩態。在相同20μm 觸媒層厚度下, Agglomerate 模式,探討氫氣、一氧化碳濃度和覆蓋率的分佈情況時, 仍可看出在20μm 厚的觸媒層處,有明顯擴散現象,但 Homogeneous 模式下,探討氫氣、一氧化碳濃度和覆蓋率的分佈情況時,只能看出 在 10μm 厚的觸媒層處,有明顯擴散現象。然而,在改變觸媒顆粒 半徑及觸媒層厚度情況下,觸媒半徑在10nm 和觸媒層厚度在 10μm 時,電流密度均達到一個最佳值。Transient Analysis of CO Poisoning effect to Cell
Performance of PEMFC Using Agglomerate Model
Student:Chia-Yu Hung Advisor:Hsin-Sen Chu Department of Mechanical Engineering
National Chiao Tung University
Abstract
The behavior of poisoning phenomena in the anode catalyst layer for polymer electrolyte fuel cells were considered in this study. According to boundary conditions, computational solutions of hydrogen, carbon monoxide concentration, preferentially adsorbing to the platinum surface and current density in catalyst layer and the time to reach stability were obtained by using agglomerate model and langmuir model. Our discussion focuses on the effects of different carbon monoxide concentration, catalyst layer thickness, and catalyst pellet radius on the performance of polymer electrolyte fuel cells.
The results show that a long steady state can be achieve using pure hydrogen at a relatively low CO content. At the same thickness of the anode catalyst layer for both model, this phenomena of hydrogen, carbon monoxide concentration and coverage rate are concentrated within the outermost 20μm of the active layer in the agglomerate model, diffusing and absorbing into the inner part of the active layer, but the homogeneous model are concentrated within the outermost 10μm of the active layer. However, in the situation of different catalyst layer thickness and catalyst pellet size, the thickness of 10μm and the pellet radius of 10nm has the maximum current density.
誌 謝
研究所裡求學的期間,感謝吾師 曲新生不辭辛勞地指導,不管 是在課業以及生活上,均讓學生受益良多,尤其是從事研究的過程 中,所應抱持的求學態度與解決問題的方法,讓學生得以順利完成論 文。 感謝陳發林教授、宋齊有教授、顏維謀教授以及鄭金祥教授,在 報告時所提出的建議以及平時在學業上的指正與教導。另外,感謝實 驗室裡學長姐,在課業與生活上的照顧。還有實驗室學弟們在課業上 的互相討論,在課業之餘一起運動,讓我在研究所的生活能更充實。 最後感謝我的家人,因為他們的支持,讓我能全心全意投入學業當中。目錄
中文摘要
... i
英文摘要
... ii
誌謝
...iii
目錄
... iv
表目錄
... vii
圖目錄
...viii
符號說明
... xii
第一章 緒論
... 1
1.1 前言 ... 1
1.2 燃料電池的發展 ... 2
1.3 燃料電池的應用 ... 3
1.3.1 燃料種類 ... 3
1.3.2 燃料電池的關鍵技術 ... 5
1.4 質子交換膜燃料電池(PEMFC)... 7
1.4.1 燃料電池的基本原理 ... 7
1.5 文獻回顧 ... 8
1.5.1 Pseudo-Homogeneous 觸媒層數學模式... 9
1.5.2 Heterogeneous 觸媒層數學模式 ... 11
1.5.3 陽極觸媒層毒化數學模式 ... 13
1.6 本文探討主題 ... 16
第二章 理論分析
... 22
2.1 基本假設 ... 22
2.2 統御方程式 ... 22
2.2.1 濃度分佈 ... 22
2.2.2 觸媒表面覆蓋率 ... 25
2.2.3 傳輸電流密度與離子相電位 ... 26
第三章 數值方法
... 29
3.1 有限差分法 ... 29
3.2 收斂條件 ... 30
第四章 結果與討論
... 31
4.1 數值方法之驗證 ... 31
4.1.1 格點測試 ... 31
4.1.2 實驗驗證 ... 32
4.2 比較兩種數學模式下一氧化碳毒化之暫態效應... 32
4.3 Agglomerate 模式下一氧化碳毒化之穩態效應... 35
第五章 結論與建議
... 65
5.1 結論 ... 65
5.2 建議 ... 66
表目錄
表
1-1 燃料電池的主要氫供給來源[3~7]
...17圖目錄
圖
1-1 能源趨勢圖[1]
...18圖
1-2 燃料電池示意圖
...19圖
1-3 燃料電池系統簡圖[2]
...20圖
1-4 Agglomerate 模式之質子交換膜半電池示意圖
...21圖
2-1 物理模型示意圖
...28圖
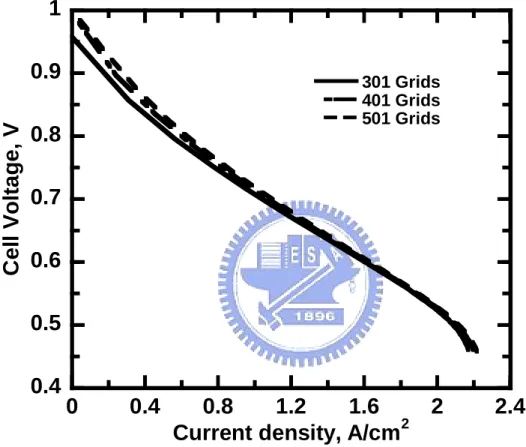
4-1 為陽極觸媒層中,極化曲線的格點測試結果圖。
....40圖
4-2 驗證 Chen et al. [42]在一氧化碳濃度 100ppm、氫氣濃
度
100%下,電池暫態極化曲線分佈圖。
...41圖
4-3 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,氫氣濃度隨觸媒層厚度之暫態
變化曲線。
...42圖
4-4 Homogeneous model 在 10ppm 一氧化碳和 75%氫
氣、觸媒層厚度
20μm 下,氫氣濃度隨觸媒層厚度之
暫態變化曲線。
...43圖
4-5 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化碳濃度隨觸媒層厚度之
暫態變化曲線。
...44圖
4-6 Homogeneous model 在 10ppm 一氧化碳和 75%氫
氣、觸媒層厚度
20μm 下,一氧化碳濃度隨觸媒層厚
度之暫態變化曲線。
...45圖
4-7 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,氫氣覆蓋率隨觸媒層厚度之暫
態變化曲線。
...46圖
4-8 Homogeneous model 探討在 10ppm 一氧化碳和 75%
氫氣、觸媒層厚度
20μm 下,氫氣覆蓋率隨觸媒層厚
度之暫態變化曲線。
...47圖
4-9 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化碳覆蓋率隨觸媒層厚度
之暫態變化曲線。
...48圖
4-10 Homogeneous model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化碳覆蓋率隨觸媒層厚度
之暫態變化曲線。
...49圖
4-11 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化電流密度隨觸媒層厚度
之暫態變化曲線。
...50圖
4-12 Homogeneous model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化碳電流密度隨觸媒層厚
度之暫態變化曲線。
...51圖
4-13 Agglomerate model 探討在 10ppm 一氧化碳和 75%氫
氣、觸媒層厚度
20μm 下,氫氣電流密度隨觸媒層厚
度之暫態變化曲線。
...52圖
4-14 Homogeneous model 探討在 10ppm 一氧化碳和 75%
氫氣、觸媒層厚度
20μm 下,氫氣電流密度隨觸媒層
厚度之暫態變化曲線。
...53圖
4-15 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化碳及氫氣覆蓋率對電流
密度之暫態變化曲線。
...54圖
4-16 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化碳及氫氣覆蓋率對電流
密度之暫態變化曲線。
...55圖
4-17 操作條件為理想電位 1.2V、10ppm 一氧化碳和 75%
氫氣,觸媒層厚度為
20μm 下的電池極化曲線圖。
56圖
4-18 操作條件為理想電位 1.2V、10ppm 一氧化碳和 75%
氫氣,觸媒層厚度為
20μm 下的功率曲線圖。
...57圖
4-19 操作條件為理想電位 1.2V、100ppm 一氧化碳和 75%
氫氣,觸媒層厚度為
20μm 下的電池極化曲線圖。
58圖
4-20 操作條件為理想電位 1.2V、100ppm 一氧化碳和 75%
氫氣,觸媒層厚度為
20μm 下的功率曲線圖。
...59圖
4-21 在 10ppm 一氧化碳 75%與 40%的氫氣和 100ppm 一
氧化碳
75%與 40%氫氣下達穩態時的電池極化曲線
圖。
...60圖
4-22 在 10ppm 一氧化碳 75%與 40%的氫氣和 100ppm 一
氧化碳
75%與 40%氫氣下達穩態時的功率曲線圖。
...61圖
4-23 在 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20μm、
觸媒顆粒半徑
100nm 及不同 Nafion Phase 厚度下,
電流密度隨位置之穩態變化曲線圖。
...62圖
4-24 在 10ppm 一氧化碳、75%氫氣、觸媒層厚度 20μm、
Nafion 半徑 1000nm 及不同觸媒顆粒半徑下,電流密
度隨位置之穩態變化曲線圖。
...63圖
4-25 在不同一氧化碳、75%氫氣、觸媒層厚度 20μm、Nafion
半徑
1000nm、觸媒顆粒半徑 100nm 及操作電壓 0.6V
下,不同觸媒層厚度與電流密度之穩態變化曲線圖。
...64符號說明
a :每單位體積觸媒的表面積(cm2/cm3) C :濃度(mole/cm3)
D :擴散係數(cm2/s)
F :法拉第常數(Faraday constant) (C/mol) i :電流密度(A/cm2)
n :電子數
R :理想氣體常數 (atm cm3/mol K) (式 2-3) ; (J/mol K) (式 2-5) T :溫度(K) m :觸媒質量(mg Pt/cm2) r :觸媒顆粒半徑(cm) H :亨利常數 L :觸媒層厚度(cm) t :時間(s) S :源項(mol/s cm3) V :Nafion 體積分率 T :溫度(K) k :反應速率常數(1/s) E :有效因子
X :莫耳分率 P :大氣壓力(atm) K :吸附速率常數 b :脫附速率常數 x :座標位置(μm)
希臘字母
α :傳輸係數 ε :孔隙率 η :離子相電位(V) θ :覆蓋率 δ :Nafion phase 厚度(cm) β :Thiele modulus ξ :觸媒表面的面積密度與法拉第常數的乘積 κ :離子傳導係數上標
g :氣體 Agg :團塊A :陽極 eff :有效值 ref :參考值 i :氫氣或一氧化碳 Pt :觸媒(白金) N :Nafion phase r :外部 CL :觸媒層
第一章 緒論
1.1 前言
邁入 21 世紀以來,科技進步帶動人類經濟活動的快速發展,各 種經濟活動製造出多樣產品,富裕人類之生活。目前人類所享受的生 活便利性,可以說是來自各種不同形式的能源消耗,這些人類生活中 使用的能源絕大部分是來自化石能源。如圖1-1 所示 AAPPS Bulletin[1] 在2003 年的報告指出,隨著人口的增加,化石能源從 2010 後開始逐 年遞減,預測石油蘊藏量可能僅可以再供人類使用半個世紀。因此如 何節約能源,提高能源使用效率,並善用再生能源即成為現今能源科 技發展的主流。 如今資源的濫用造成了地球環境的嚴重破壞,使得生態環境遭受 到極大威脅,其程度遠超過自然復原的能力,如此一來,不僅危害到 現代人類的生活,也會影響到後代子孫的福祉。舉例而言,二氧化碳 的過度排放,造成大氣的溫室效應,影響到氣候的變化。根據聯合國 1996 年氣候變化綱要公約(UNFCCC)之評估報告指出,人類若不對此 現象採取任何防制措施,全球平均地面氣溫於2100 年時,將比 1990 年時增加攝氏兩度,海平面因而上升 50 公分,此將會嚴重影響地球 之生態環境。 因此,新一代的潔淨能源技術在本世紀之後將非常蓬勃,例如, 潮汐、地熱、太陽能、風能等其發電技術均有顯著進步,不過最具發 展潛力的潔淨能源科技為氫能源(Hydrogen energy)。特別是燃料電池 的技術,很有可能成為二十一世紀最重要的能源科技之一。在 2000 年9 月號的「經濟學人雜誌(Economist)」更提到,燃料電池與資訊科技、生物科技並列為目前最重要的三大科技。所以,深受美國、歐洲 及日本等先進國家的重視,其均投入大量的經費及人力在燃料電池相 關研究上,使二十一世紀能成為氫能源經濟的時代。
1.2 燃料電池的發展
燃料電池於 1839 年由英國人發明,但直到 1960 年代美國才應 用於太空計畫(NASA)中,阿波羅號(Apollo)及雙子星(Gemini)太空船 即是利用燃料電池作為動力,NASA 最先是選定 PEFC 機組的燃料電 池,但是因當時的電解質壽命過短,因而改用 AFC。之後氣體研究 所(Gas Research Institute, GRI)開始支持 International Fuell Cell(IFC)公 司發展PAFC,經歷 1960~70 年代的 TARGET 計畫(12.5kW)及 1970~80 年代的GRI/DOE 計畫(40kW),並在 1980 年代發展 200kW 燃料電池, 而能源部(Department of Energy, DOE)和電力研究所(Electric Power Research Institute, EPRI)也相繼投入,並興建 4MW 及 11MW 展示電 廠,以及 1994 年 200KW 的初期商業化。能源部(DOE)、氣體研究所 (GRI)和電力研究所(EPRI)也於 1980 年開始研發 MCFC 與 SOFC。此 外運輸部(Department of Transport, DOT)也支持 PAFC 的推廣以及攜 帶型 PEFC 及 DMFC 的發展。近年來的燃料電池用途包括可攜帶式 小型電力、車輛動力及發電廠,而美國則積極發展在燃料電池車輛方 面。 反觀國內則是由經濟部能委會與工研院能資所開始,近年來,出 現了更多研究團隊,如中科院材料所、工研院材料所與原委會,且由 於台灣經濟研究院、經濟部、國科會熱流學門大力地支持,學術界關 於質子交換模燃料電池相關的基礎研究與應用相繼出現。這種結合產 官學的燃料電池研究風潮正逐漸蔓延出去。1.3 燃料電池的應用
燃料電池的開發,最初是以運用於大型集中式發電裝置為主要重 點,但目前現場型、家用型發電機組之實用化與商業化,有後來居上 的傾向。適合裝設此種發電機組之場所,舉例而言,如一般住宅、旅 館、醫院、商店、辦公大樓、公寓、工廠等等,以供應全部或特定電 力需求。家用型燃料電池機組可以替代或補充一般電網提供之電力, 適合一般住宅、小型商用設備(譬如:冷凍櫃)及工業設施(譬如: 臨時工程用電)。一般而言,家用型燃料電池機組包括三部分如圖 1-2;第一部分是燃料處理器(Fuel processor),內含燃料電池重組器 (Fuel reformer)和一氧化碳清理器,燃料可用天然氣、甲醇等含氫 氣成份較高之燃料,經過重組後產生氫氣。一氧化碳清理器則是減低 一氧化碳之濃度(降至50ppm 以下),防止白金觸媒(Platinum catalyst ) 產生中毒現象。第二部分是質子交換模燃料電池組,主要是利用電化 學原理產生電力。第三部份是電力整流裝置(Power conditioner),即 將燃料電池產生之低電壓直流電轉換成高電壓交流電,方便電力之使 用。1.3.1 燃料種類
燃料電池以氫作為燃料,其來源包含從水之電解取得,以及可從 甲醇、乙醇、天然氣、汽油、柴油、煤油、煤等含氫能源,利用「燃 料重組器」來分離出氫氣,因此現有燃料電池之燃料來源非常多樣, 甚至還有用生物能或垃圾掩埋場、廢水處理場產生的沼氣(甲烷)來 重組氫。 用水電解取氫的方法大都是先利用太陽能、風力或其他可再生能源來發電,再用其電能來電解水。此種方法從純粹的能源利用觀點來 看,雖然不具任何效益,不過在沙漠、離島等偏遠地區為儲存光能、 風力,還是有其一定的作用。以下介紹各種燃料的特性如表1-1: 氫:平常為無色無味無臭的氣體,質氫、能源密度小,儲存、運 送不便,且具爆炸的危險性,不需重組器就可和空氣直接進行發電反 應。甲醇(Methanol):有機,有毒,可燃性無色液體,沸點64.5℃, 溶於水或酒精等,化學式為 CH3OH,石化業的中游原料,可用天然 氣為原料所產生的 CO 和由 H2 構成的合成氣體在觸媒的觸發作用 下,精製而成。戴姆勒和福特汽車等公司看好這種燃料,不過需要加 300℃以上的水蒸氣來使其產生反應,所以裝置就很難縮小。且具毒 性與腐蝕性,搬運與保管需要小心等缺點。汽油:揮發性的可燃性液 體,由含C5~C10的碳化氫組成。Royal Dutch Shell 與美國 Exxon Mobil
等石油公司主張採用此一燃料,因為可以利用現有供應設施,不過要 從其中取出氫比甲醇更為困難,一般需要900℃的水蒸氣,因此加溫 到能讓車子開始走動需要花十分鐘的時間。GM 嘗試以此為燃料,正 研究設法降低啟動速度,又豐田、日產與雷諾等也加重這方面的研 究。甲烷(Methane):有機,無臭無味無色氣體,較空氣輕,化學 式為CH4。天然氣中含有大量此物質,天然氣從採取到液化的過程, 必須先除去塵埃、硫磺、碳酸氣和水分等雜質,因此 LPG 要比汽、 煤油乾淨。從甲烷取出氫和汽油一樣困難,故家庭或辦公室的自備發 電機雖可利用現有的天然氣供應網,但用在汽車就有困難了。環己 烷:芳香族碳氫化合物,不用水蒸氣就可以用較低溫度取出氫,可利 用和汽油一樣的供應設備,不過具毒性。
1.3.2 燃料電池的關鍵技術
燃料電池的關鍵技術有:燃料儲存技術、燃料重組器、電解質材 料、觸媒技術、隔離版、反相器、起動技術(起動時間從一小時到半 小時縮短為一分鐘)、防凍技術、系統控制、熱交換系統、溫溼度控 制、空壓器、燃料多樣與廢熱利用等。 (一) 燃料儲存技術 燃料電池若要使用純氫時,雖然可不需燃料重組,但卻需考慮氫 氣的的供給方式。目前已知的氫氣儲存方式有高壓氫筒、液化氫和儲 氫合金等。 高壓氫筒普通使用150 氣壓,儲存同量的氫氣容積要較其他方式 大上許多,重量同時也隨之增加,若提高氣壓以增加儲存量,危險性 則又跟著提升。液化氫的儲氫量雖較高壓氫筒大上六倍,但溫度需降 到負253℃,並需要花費能源與隔熱、冷卻容器,不適用於一般家庭 與車輛,且具爆炸性。 儲氫合金是用鑭、鈰、釔、鋰、鈦、鋯、鎂等稀有吸氫金屬和鐵、 鈷、鎳、鋁、銅、錳等幾乎不吸氫金屬構成,儲氫量介於前兩者之間, 使用的氣壓低,安全性也就提高。問題是成本高、重量重,儲氫量達 不到理想標準,因此各國的科學家都在積極開發新配方,以提高氫的 重量儲存比。 此外新的儲氫材料開發方向,目前正朝石墨與碳奈米材料發展, 此外,含氫溶液的新研發最近也有大突破,以日本工學院大學須田精 二郎教授為首的集團開發出氟化氫溶液,含氫量高達10﹪左右,不 僅容易分離出氫,且可重複使用。 (二) 燃料重組技術:從天然氣或其他油品重組氫需經脫硫、重組、CO 轉換、CO 去 除等手續才能取得純淨的氫、重組的方式有水蒸氣重組法(Steam reformer)、部分氧化法(Partial oxidation reactor)等。
水蒸氣重組法由將水和燃料蒸發的氧化器、執行重組的重組器、 供應熱能給氧化器和重組器的觸媒燃燒器,以及在轉換反應時降低 CO 濃度的 CO 選擇氧化器等構成,其開發重點在簡單、耐用、小型 與低成本化等,而關鍵技術在觸媒的開發與系統的整合。大阪瓦斯是 這方面的技術領導者,其開發的觸媒去硫技術能降到現有的千分之一 左右,達十億分之一(1ppb)以下。 部分氧化法由美國業者所開發,方式是一面燃燒甲醇,一面重組 氫,熱效率較前法差,且溫度高達800℃,所以不能採用輕質的鋁材 料。不過直接燃燒甲醇的發熱和重組吸熱能夠平衡運作,因此具有能 迅速因應起動和負載變化的優點。 (三) 電解質材料技術 電解質是左右燃料電池性能的關鍵材料,最接近實用化的固體高 分子燃料電池所用的離子交換膜,目前是以杜邦開發的磺酸類氟膜 (Nafion 膜)為主,供應商有杜邦、W.L. Gore Associates、旭化成和 旭硝子等。離子交換膜主要注重的是使用強度,燃料電池用的交換膜 為增加離子的傳導性,除注重強度外,更注意膜厚,各家業者都正試 製更薄的產品,目前Core 開發的新產品有 10~20 微米厚的膜。另外, Nafion 膜使用溫度限制在 80~90℃,耐溫性差、壽命短,發電效率無 法大幅提高,因此許多研究機構正研發高溫用替代薄膜。日本積水化 學 與 產 業 技 術 總 研 等 已 開 發 出 以 辛 烷 和 己 烷 為 原 料 的 耐 熱 膜 (350℃),若能商品化,不僅可提升發電效率與廢熱的使用,而且可 直接燃燒分離氫時產生的 CO,減少觸媒的中毒。另外以陶瓷或玻璃
薄膜取代高分子薄膜的下一代技術也正由西門子西屋、BMW 和日本 學者分別開發中,加熱溫度需達 1000℃,發電效率雖高達 60%,但 需採用昂貴的耐熱材料。若能降低加熱溫度到600℃左右,則商業化 的可能性將大幅提高。
1.4 質子交換膜燃料電池(PEMFC)
基於能源與環境保護的觀點,燃料電池值得研究、運用與推廣, 因此其化學反應原理、電池特性以及應用層面,將探討如下:1.4.1 燃料電池的基本原理
燃料電池的運作原理,就是電池含有陰陽兩個電極,而兩個電極 間則為具有滲透性的薄膜所構成。氫氣由燃料電池陽極進入,氧氣(或 空氣)則由陰極進入燃料電池。經由催化劑的作用,使得陽極的氫原 子分解成兩個氫質子(Proton)與兩個電子(Electron),其中質子藉 由電解液傳遞到薄膜的另一邊,電子則經由外電路形成電流後到達陰 極。也就是說,燃料電池為一電化學裝置,可將反應物(燃料與氧化 物)中氫氧之化學內能通過電極反應直接轉化為電能、廢熱、水,因 此為一能量轉換裝置,而非能量儲存裝置。 故以質子交換膜燃料電池為例,以氟磺酸型固體聚合物為電解 質,純氫氣或者經重組器所得出的氫氣為燃料、空氣或者純氧為氧化 劑,Pt/C 或 Pt-Ru/C 為電催化劑。 薄膜兩側分別供應氫氣與氧氣,氫原子含有一個質子及一個電 子,質子被氧吸引形成水分子,因此燃料電池唯一液體是水,腐蝕性 問題相當小,同時其操作溫度在 80℃至 100℃的燃料電池,具有可在 室溫下快速啟動、無電解質液流失、易組裝生產、水易排出及操作壽命長的優點,目前是最受注目的一型燃料電池如圖 1-3。其陽極的半 反應式為: H2 → 2H+ +2e− (1-1) 其中氫離子經由電解質到達陰極端,而電子則是經由外部迴路傳 遞,兩者皆到陰極端的觸媒層與氧氣進行發生如下還原反應: O H H e O2 2 2 2 2 1 + − + + → (1-2) 其總反應式為: H2 O2 H2O 2 1 → + (1-3) 因此,在一大氣壓下,其化學反應之Gibbs 自由能的大小為: kJ s T h g = Δ − Δ = −237.2 Δ (1-4) 若化學能完全轉換成電能,則其電池的理想電壓為: V nF G E =−Δ =1.23 Δ (1-5) 但實際上電池電壓會受到活化、歐姆、濃度過電位的損失,所以 實際開迴路電壓值會小於理想電壓值。然而,觸媒白金價格昂貴,若 減少其使用量,電池性能必受到影響。再者,白金容易與一氧化碳反 應而發生中毒現象,因此比較不適合用在大型發電廠,而適合作為汽 車動力來源。
1.5 文獻回顧
燃料電池的研究中,一般主要可分為三種等級來探討:1.燃料電 池及週邊系統、2.燃料電池組、3.單電池,對於燃料電池系統而言, 主要是由巨觀觀點來探討每個系統的組成,包括:系統整合、系統設計等。對於燃料電池組以及單電池而言,則主要利用微觀的模式,來 探討在電池中更複雜的現象以及性能的好壞,尤其Wood et al. [8]年 針對燃料電池作兩千小時的實驗研究後,藉由循環伏安法量得結果發 現,造成電池性能下降的主要因素為觸媒層中特性的改變,因此,為 了對質子交換膜燃料電池中的觸媒層更加瞭解,本章將對於觸媒層方 面進行相關文獻的探討。
1.5.1 Pseudo-Homogeneous 觸媒層數學模式
1997 年 Broka and Ekdunge [9]比較 Film 數學模式和 Agglomerate 數學模式分別對陰極側的質子交換膜燃料電池的影響,結果顯示, Film 數學模式隨著反應層的增加,在高電流密度下,過電位也隨之增 加,其極限電流密度減少,因為質傳的障蔽提高且質子的傳導率也被 限 制 住 ; 而 用 Agglomerate 數 學 模 式 時 , 其 極 限 電 流 密 度 因 與 Agglomerate 的外表面有比例關係,所以,當反應層的厚度增加,反 而會增加了極限電流密度。
2001 年 You and Liu [10]探討質子交換膜燃料電池中,陰極觸媒 層各種參數間的影響,結果發現,當過電位(η)為 0.35 時,電流密度 達到極限電流密度,因為由氧氣的質傳所造成的,同時,氧氣濃度在 較靠近觸媒層與薄膜界面處即消耗完畢;當質子傳導率(κ)為 0.5Ωcm-1 時,在觸媒層一半處(ξ=0.5)即有 90%的電流產生,因此,判斷在 ξ=0.75、κ=0.5 時,可有較好的性能出現,另外,還探討孔隙度與觸 媒表面的反應面積對觸媒層性能的影響,當孔隙度(ε)為 0.35 時,可 使全部觸媒層均勻反應完全,而當反應面積(Av)為 1.4x105 時,也可 造成叫均勻的電化學反應。
2004 年 Song et al. [11]利用四種參數分別為 Nafion 含量、Pt 負 載、觸媒層厚度和孔隙度,探討陰極觸媒層之最佳化設計,數值分析 顯示當只改變四個參數中的一個時,厚度對觸媒層性能影響最大,其 最佳值為13.0972μm;當改變四個參數中的兩個時,厚度和 Pt 負載量 可 使 觸 媒 層 的 電 流 密 度 達 到 最 高 性 能 , 最 佳 值 為 2.2807μm 和 0.044mg/cm2。
2004 年 Wang et al. [12]則主要探討 Nafion 的含量對質子交換膜 燃料電池電極極化曲線的影響,結果得知,在 Nafion 含量均勻分佈 的情況下,其最佳值為 35wt%,另外,當靠近氣體擴散層與薄膜層 邊,Nafion 含量分別為 30wt%和 40wt%時,能夠減少在觸媒層中的 電位損失大約 3~5%,因為,較低的 Nafion loading 量減少了觸媒層 與氣體擴散層界面孔隙被阻塞的可能性,使產生的水容易排除,而較 高的Nafion loading 量增加了薄膜與觸媒層界面處的接觸面積,改善 了質子的傳輸阻抗,進而提升電池性能。 2004 年 Jeng et al. [13]利用無因次化法,描述質子交換膜燃料電 池陰極觸媒層的三個參數,分別為氧氣還原反阻抗(πI)、質子傳導阻 抗(πK)和氧氣擴散阻抗(πD),了解發生在觸媒層的複雜現象。模擬得 知,當 πD增加五倍的基本值時,表示氧氣通透率下降,使得在觸媒 層中間其還原反應即停止,氧氣濃度瞬間消耗完畢,而電流密度也迅 速達到穩態,但當 πD改為五分之一倍的基本值時,還原反應的量與 氧氣的濃度均勻分佈在觸媒層中,而電流密度則呈現平滑曲線達到穩 態。
1.5.2 Heterogeneous 觸媒層數學模式
1995 年 Bultel et al. [14]探討質子交換膜燃料電池中反應層的質 傳現象,結果發現,氫氣的氧化反應主要是由擴散所控制,而觸媒顆 粒的幾何形狀並不會對氧氣的還原反應有太大的影響,但對電化學反 應速率卻有很大的限制。在1997 年 Bultel et al. [15]延續 1995 年的模 式,加入了歐姆阻抗對反應層的性能影響,結果發現,當觸媒粒子間 距離少於 10 倍的觸媒直徑時,觸媒顆粒的影響只能從電化學中看 出,同時因為加入了歐姆阻抗效應,使傳統的數學模式更能產生精準 的結果。又於1998 年,Antoine et al. [16]延續 1995 年的數學模式, 加入了擴散效應,研究結果發現,當考慮氧氣的還原反應時,觸媒顆 粒的分佈或者如何排列影響並不大,反而是觸煤顆粒的有效反應面積 為主要因素,另外,當觸媒顆粒比較小的時候,氫氣會有不錯的氧化 反應。而在1999 年 Bultel et al. [17]所延伸的數學模式,只針對反應 層的濃度來探討,因此,其通用性稍嫌不足。後來在 2000 年,Bultel et al. [18]將文獻[16]的數學模式複雜化,探討質子交換膜燃料電池中 反應層濃度和過電位的分佈,研究結果發現,球形擴散的機制和歐姆 阻抗的下降,對於電流密度會有很大的影響。1998 年 Gloaguen et al. [19]所考慮的 Agglomerate 數學模式係沿 Bultel et al. [14]的數學模式,但不同的地方在於文獻[19]並非僅單一 考慮觸媒顆粒,而是針對整個觸媒層對電池性能的影響,因此探討質 子交換膜燃料電池中 Micro-homogeneous 和 Agglomerate 的數學模式 對 氧 氣 還 原 反 應 的 影 響 。 研 究 結 果 發 現 , 沒 有 孔 隙 存 在 的 Micro-homogeneous 數學模式,其擴散造成的過電位並不如預期的正 確,而有孔隙存在的 Agglomerate 數學模式,其得到較合理的過電位
值。 2002 年 Genevey et al. [20]探討熱、質量和電子傳遞在質子交換膜 燃料電陰極觸媒層的暫態分析,結果發現,在較高的孔隙率和觸媒顆 粒的含量可有利於提升電池性能,而較高的電池溫度對觸媒層性能有 負面的影響,且同時證明了當極限電流密度接近薄膜與觸媒層界面 時,氧氣濃度會趨近零。 2004 年 Wang et al. [21]在質子交換膜燃料電池的陰極觸媒層 中,考慮了氧氣還原反應、質子在聚合電解質傳遞、氧氣藉由擴散通 過 氣 孔 以 及 溶 融 態 的 氧 氣 在 電 解 質 中 擴 散 等 效 應 的 球 型 Flooded-agglomerate 數學模式,結果得知,靠近氣體擴散層邊的觸媒 層,若增加處其觸媒層孔隙度,碳含量和疏水性時,會增加氧氣和電 子的傳輸現象,而靠近薄膜邊的觸媒層,若減少區域的孔隙度,增加 Nafion 的含量及親水性時,會改善質子傳輸現象。
2004 年 Song et al. [22]利用多層 Agglomerate 數學模式探討質子 交換膜中,陰極觸媒層隨著不同 Agglomerate 尺寸,Agglomerate 孔 隙度和次層孔隙度的性能研究,結果發現,當Agglomerate 尺寸與次 層孔隙度由氣體擴散層向薄膜遞減時,會使陰極電位提升,因為 Agglomerat 半徑減少,會增加其在陰極觸媒層內的電流密度而增加了 電流生成量,另外,當次層孔隙度減少時,會增加反應面積,改善性 能,而當Agglomerate 孔隙度逐漸遞增時,也會使陰極電位提升,因 為增加了氧氣的擴散量。
2004 年 Wang et al. [23]建立了兩種不同成分球型的 Agglomerate 數學模式,一種是混合碳與觸媒粒子和 PFSI 的結構;另一種是混合 碳與觸媒粒子和水充滿孔隙的結構,探討質子傳遞深度、表面電位、 Agglomerat 的半徑以及氧氣濃度相互間的影響,結果發現,當從
Agglomerate 表面往內部移動時,局部電位增加、電化學反應減少、 氧氣濃度降低外,另外,當Agglomerate 的半徑縮短時,其局部電位、 氧氣濃度、電化學反應均有提升的效果,同時,針對 PFSI 與水的 Agglomerat 兩種模式作比較,得知後者具有較大的有效因子,表示氧 氣反應均勻。 2004 年 Lin et al. [24]建立了一為穩態,兩相流及等溫的質子交換 膜燃料電池陰極半電池模式,其中觸媒層是利用 Thin-film-agglomerat 數學模式的方法表示,探討在氣體擴散層和觸媒層所產生的Flooding 現象對電池性能的影響,結果得知,在觸媒層厚度約13μm 時,電池 性能最好,因為其厚度薄,所以水排除的速度快,不易累積而產生 Flooding 影響性能,但當厚度大於 13μm 時,除了造成更多水產生外, 離子的過電位、Nafion phase 的阻抗均會增加,而使電池型能下降。 2002 年 Siegel et al. [25]利用 CFDesign 和 FEMAP 探討二維質子 交換膜燃料電性能的研究,結果發現,當過電位在 0.5V 時,觸媒層 孔隙度有最佳值為 0.04,而若觸媒層較薄時,會造成擴散阻抗的增 加。2003 年 Siegel et al. [26]延續 2002 年的研究,再加入了液態水的 傳輸,結果發現,有20%~40%的水會從陰極傳遞到薄膜內,使得電 池性能並不如未考慮液態水時來的高,換句話說,考慮液態水的傳輸 是必要的,否則燃料電池的性能研究並不準確。
1.5.3 陽極觸媒層毒化數學模式
由於質子交換膜燃料電池適合低溫情況下操作,即使一氧化碳濃 度很低(10~100ppm)仍然對陽極觸媒層氫氣的氧化反應及電池的輸出 功率有很嚴重的影響。因此,1987 年 Dhar et al. [27]利用簡單的 Temkinmodel 及實驗,探討一氧化碳毒化對陽極電位的影響,結果發現,一 氧化碳的表面覆蓋率隨著不同溫度會和一氧化碳及氫氣的比值取自 然對數會有線性關係,再與實驗數據作比對後其結果趨近吻合。另 外,還計算出一氧化碳吸附時的標準自由能以及標準墒分別為-14.5 到-12.1kcal/mol 和-39cal/molK,故一氧化的覆蓋的確會造成氫氣氧化 反應的困難。到1996 年 Bellows et al. [28]把二氧化碳效應一起考慮進 去,探討在使用高效率的純氫燃料下對燃料電池的影響,得知一氧化 碳的電氧化反應對於白金(Pt)的一氧化碳容忍度有很重要的影響且會 隨溫度增加而電氧化反應跟著增加,而同時發現一氧化碳會比二氧化 碳更快吸附在白金上,所以造成極化損失的量也遠大於二氧化碳吸附 時嚴重。 2001 年 Springer et al. [29]才首先提出一氧化碳完整的數學模 式,模式包括一氧化碳的吸附、脫付及電化學反應的項,描述了當陽 極氣體同時存在氫氣及一氧化碳時,進入到陽極觸媒層的物理機制及 化學反應,造成了電流密度的改變,同時也影響到整體的電池性能。 同年,Baschuk and Li [30]整合 1995 到 1998 年的文獻和結果 [27,31~33],了解到一氧化碳吸附在白金上的反應是很複雜的,尤其 在低溫下為電位和一氧化碳濃度的函數,另外還提到主要有三種方法 可減緩一氧化碳的影響(1)使用合金的白金觸媒,(2)較高的電池 操作溫度,(3)加入氧化物氣體或者空氣。然而第一種方法價格無 法有效降低,第二種方法則必須考慮到 Nafion 膜乾化及耐熱的問題, 因此第三種方法目前看來是較為實際。因此在2001 年 Murthy et al. [34] 設定電池操作溫度70℃、反應面積 25cm2、500 和 3000ppm 的一氧化 碳含量、PRIMEA Series 5561 的 MEA 膜以及兩種不同 GDM 的操作 情況下,利用空氣吹入法(Air-bleed)探討對質子交換膜燃料電池的影
響。結果得知,通入5%的空氣、500ppm 的一氧化碳毒化達穩態時, 造成電流密度少於 0.6A/cm2,而 3000ppm 的一氧化碳毒化時,即使 通入 15%的空氣其電流密度仍無法有效改善,大約在操作電壓為 0.6V,電流密度只達到 200mA/cm2。
2003 年 Chan et al. [35]將 Springer et al. [29]的一氧化碳模式加入 Bernardi et al. [36,37]的全電池模式,探討穩態、不同一氧化碳濃度 下、過電位與覆蓋率的分不情形,以及極化性能曲線圖。同年 Baschuk and Li et al. [38]利用 Langmuir-Hinshelwood 機制探討陽極毒化半電池 模式,改善了文獻[29,36,37]所使用較簡化的模式來描述的缺點,且 結合了氧氣輸入法(O2-bleed)的模式。研究結果發現,當 100ppm 一氧 化碳情況下,陽極的過電位不超過 0.2V 而加入 O2-bleed 後可很明顯 看出陽極過電位大幅提升,最高達0.55V。因此,可知陽極過電位對 一氧化碳的毒化是很明顯的。另外提到,為了降低價格,就必須減低 白金的使用量,因此若能有效利用 O2-bleed 或者 Air-bleed 而改善電 池因毒化造成的性能下降時,觸媒的及電池整體的價格就有機會下 降。同年,Baschuk et al. [39]更進一步利用非等溫、多維的模式加入 吸附、脫附及電化學的反應探討一氧化碳、氫氣和氧氣的全電池模 式,結果發現溫度的改變對電池電流密度的影響不大,反而是當一氧 化碳濃度在 20ppm 下,得知增加操作壓力會增加低電流密的值,但 相對在高電流密度的地方就會降低。
到了2004 年 Zhou and Liu [40]利用 3-D 模式,探討 CO 毒化的問 題,其中提到了藉由三維模式所發現的新現象無法容易的從一般過去 一維、二維的模式來解釋。在 50ppm 下,隨著通入足夠的氫氣,電 池性能會因為陽極流率的減少而增加,且在一個合理的範圍下,電池 性能會隨著陽極氣體擴散層的孔隙比減少而增加,因為孔隙比的減少
降低了質傳損失而增加電池性能。
1.6 本文探討主題
由以上的文獻回顧,我們可知質子交換膜燃料電池中,利用 Agglomerate 的模式如圖 1-4,探討關於觸媒層毒化的現象非常缺乏, 所以本文特別針對這部分加以研究,更進而探討暫態下 Agglomerate 的 毒 化 現 象 對 陽 極 觸 媒 層 的 影 響 。 文 獻 中 所 提 到 的 Thiele’s modulus[41]以及有效反應因子,最原始推導是由單一顆觸媒粒子而 來,因此,本文中針對Thiele’s modulus[41]的推導過程中,加入了均 勻毒化的項,來代表陽極觸媒毒化現象。表
1-1 燃料電池的主要氫供給來源[3~7]
類別 優點 缺點 氫氣 有害物質的排放量少,不需要重組器 可達到輕量化,且可迅速啟動,發電 效率高 氫氣較難處理,需要龐大 儲氫桶。建設供氫的基礎 設施需花巨大成本。 甲醇 原料較豐富,容易廉價取得。 較其他燃料容易分出氫。 具毒性,處理時需要小心 重組時會產生CO 等副產 物、啟動需花 4~5 分鐘 汽油、煤油 可利用既有的汽油加油站,容易普 及,處理也較方便 重組溫度高,較難分離出 氫。需要去除硫磺等雜 質。啟動費時,效率較低。 甲烷(天然氣) 可從食品、農業廢棄物來製造 重組溫度和汽、煤油一樣 高。也必須除去硫磺等雜 質。 氫和金屬的化 合物水溶液 可常溫下分離出氫。可重複使用。可 將全套系統小型化。 必須先設置供應設備及建 立生產體制。容器需要耐 腐蝕加工。 環己烷(六氫化 苯) 可在較低溫度下分離出氫。 可利用汽油的供應設備 具毒性圖
1-1 能源趨勢圖[1]
(年)
1750 1800 1850 1900 1950 2000 2050 2100再生能源
核能
石油
煤
人口
世界人口數︵十億
︶
能源消耗量
( 十億噸
/ 年
)
1 2 3 4 5 6 7 8 8 7 6 5 4 3 2 1流道 電極板 薄膜 電極板 流道
圖
1-3 燃料電池示意圖[2]
陽極 陰極
H+ H+ H+ O2 O2 O2 O2 H+ H+H
2O
2H
2O
e -e -e -e -O2 H+ H+H
2O
圖
1-4 Agglomerate 模式之質子交換膜半電池示意圖
H2 Carbon薄膜
觸媒層
氣體擴散層
陽極
碳顆粒
觸媒顆粒
H2 H2 H2第二章 理論分析
2.1 基本假設
本文利用團塊理論探討陽極觸媒層內部一氧化碳毒化現象,對質 子交換膜燃料電池暫態性能的影響。此裝置只探討一層陽極觸媒多孔 性材質,如圖2-1 所示。故本文先作以下的基本假設來簡化問題: 1.空間為一維直角座標系。 2.不考慮兩相流的影響。 3.在 Nafion 相裡的離子傳導為常數。 4.陽極觸媒層為均勻等溫 5.電子在碳顆粒的傳導遠高於離子在 Nafion 相中的傳導。 6.空間內的氣體為理想氣體。 7.觸媒顆粒為均勻分佈。 8.在觸媒顆粒上為均勻毒化。2.2 統御方程式
探討陽極觸媒層之座標系統。如圖 2-1 所示。本文將座標原點設 於陽極氣體擴散層與觸媒層的界面處,原點到質子交換膜厚度為L, 則其各物質傳輸以及毒化之統御方程式如下:2.2.1 濃度分佈
實際上,觸媒層內部是以團塊的方式緊密靠在一起,團與團之間 可能會有裂縫以提供氧氣的傳輸,因此,當氧氣到達團塊周圍時,其 必先溶融在觸媒層內的 Nafion 相後,再經由擴散的機制,到達白金表面產生電化學反應,其表示式如下: i i g i eff CL i CL S x C D x t C − ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ ∂ ∂ ∂ ∂ = ∂ ∂ ,
ε
ε
(2-1)
其中,有效擴散係數可由下列表示之: 5 . 1 , [(1 CL) N] g i g i eff D V D = −ε
(2-2)
其中,Ci為氫氣或一氧化碳濃度、 g i D 為氫氣或一氧化碳的擴散係 數、ε
CL為觸媒層孔隙度、VN 為 Nafion 的體積分率。而式(2-1)中 的Si為電化學反應的源項其可表示為: ⎟⎟ ⎟ ⎟ ⎟ ⎠ ⎞ ⎜⎜ ⎜ ⎜ ⎜ ⎝ ⎛ + = A i A g i Agg r N i i i E k D a H RTC S , 1δ
(2-3)
其中,R為理想氣體常數、T 為電池操作溫度、δ
N 為 Nafion phase 的厚度、arAgg為每單位團塊體積的外表面積、kA,i為氫氣或一氧化碳 的反應速率常數、EA為有效因子。其中 Agg r a 、kA,i 及EA分別可由式 (2-4)、(2-5)及(2-6) ) 1 ( ) ( 2 CL N Pt Agg r r aε
δ
− + =(2-4)
) (exp( , , i A ref i ref Agg Pt i A RT F nFC i a k =
α
η
(2-5)
) 1 ) tanh( 1 ( 3β
β
β
θ
− = i A E(2-6)
其中,rPt為觸媒顆粒半徑、 Agg Pt a 為每單位團塊體積的觸媒表面積、iref 為參考電流密度、F 為法拉第常數、Ci,ref 為氫氣或一氧化碳的參考 濃度、η
i為氫氣或一氧化碳離子相電位、θ
i為氫氣或一氧化碳覆蓋 率、n為電子數、α
A為陽極傳輸係數、β
為Thiele modulus。而 Agg Pt a 及β
可分別由式(2-7)及(2-8)表示成: ) 1 ( CL Pt Pt Agg Pt L m a aε
− =(2-7)
g i eff i i A Pt D k r , , 3θ
β
=(2-8)
Pta
為每單位觸媒質量的表面積、m
Pt為每單位面積的觸媒質量、L
為 觸媒層厚度。 擴散方程式的邊界條件如下: g i g i C C = x = 0(2-9a)
0 = dx dC D g i i x = L(2-9b)
初始條件如下:
( )
x,0 =0 Cig t = 0(2-10c)
2.2.2 觸媒表面覆蓋率
當陽極觸媒層同時存在氫氣與一氧化碳時,氫氣與一氧化碳為了 進行化學反應,會與空出來的白金表面進行吸附、脫附以及電化學反 應產生電流,因此,對傳統的 Bulter-volmer 方程式導入 Agglomerate 模式中,並修正毒化對觸媒顆粒覆蓋的效率因子,故影響到白金表面 氫氣與一氧化碳的覆蓋率其表示式如下:(
)
⎟ ⎟ ⎟ ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎜ ⎜ ⎜ ⎝ ⎛ + − − − − = A H A Agg r g H N H H H H H H CO H H H H E R a D H RTC F n K b P X K dt d 2 2 2 2 2 2 2 2 2 2 2 2 , 1δ
θ
θ
θ
θ
ξ
(2-11)
(
)
⎟⎟ ⎟ ⎟ ⎟ ⎠ ⎞ ⎜⎜ ⎜ ⎜ ⎜ ⎝ ⎛ + − − − − = A co A Agg r g co N CO CO CO CO CO CO CO H CO CO CO E R a D H RTC F n K b P X K dt d , 2 1δ
θ
θ
θ
θ
ξ
(2-12)
ξ
為觸媒表面的面積密度與法拉第常數的乘積,K、b分別代表吸附 和脫附速率常數, X 為莫耳分率,P為大氣壓力。等式左邊代表覆附項、脫附項與電化學反應造成之消耗項。 則覆蓋率的初始條件如下: 0 ) 0 ( 2 = H
θ
(2-13a)
0 ) 0 ( = COθ
(2-13b)
2.2.3 傳輸電流密度與離子相電位
在觸媒層中,由於受一氧化碳毒化的影響,觸媒表面覆蓋氫氣與 一氧化碳兩種氣體,又因為本文假設Agglomerate 中均勻充滿 Nafion 相,因此只考慮離子造成的傳輸電流密度和離子相電位為主而未考慮 電子的影響,所以氫氣與一氧化碳電化學反應所產生的傳輸電流密度 統御方程式如下: ⎟⎟ ⎟ ⎟ ⎟ ⎠ ⎞ ⎜⎜ ⎜ ⎜ ⎜ ⎝ ⎛ + + ⎟⎟ ⎟ ⎟ ⎟ ⎟ ⎠ ⎞ ⎜⎜ ⎜ ⎜ ⎜ ⎜ ⎝ ⎛ + = + = A co A g co Agg r N CO CO CO A H A g H Agg r N H H H CO H E k D a H RTC F n E k D a H RTC F n dx di dx di dx di , , 1 1 2 2 2 2 2 2δ
δ
(2-14)
其中第一項代表氫氣反應產生的傳輸電流密度,第二項則代表一氧化 碳的傳輸電流密度。而離子相電位的統御方程式如下:κ
η
i dx d i =其中
κ
為離子傳導係數。 則觸媒層的電流和相電位邊界條件如下: 0 = ix = 0 (2-15a)
0 = iη
x = 0 (2-15b)
圖
2-1 物理模型示意圖
氣
體
擴
散
層
觸
媒
層
x
L0
薄
膜
第三章 數值方法
由於上述之統御方程式中具有非線性項,不易獲得解析解,所以 本文藉由數值計算來加以求解。一般解常微分方程式的數值方法多採 用有限差分法,許多計算流體力學 (Computational fluid dynamics) 和 數值熱傳 (Numerical heat transfer) 的書籍都有介紹一些有限差分法 來解常微分方程式。
3.1 有限差分法
首先我們先來介紹有限差分法如何的使用在一個二階常微分方 程式:( )
b( )
x dx df x a dx f d + = 2 2 for x1≦ ≦x x2 (3-1) 其中 a(x)與 b(x)是一已知的函數。我們將 x 區域分成(m+1)個間隔, 接著定義下列標記法: f i = f (xi) for i = 1 , 2,…., m Δxi = xi+1 - xi for i = 1 , 2,…., m-1 (3-2) 其中xi,i = 1 , 2,…., m,是在 x 區域中連續的 m 個點。 所以經過有限中央差分法之後,一個二階常微分方程式可以表示為) ( 2 ) ( 2 ! 1 2 1 1 x b x f f x a x f f f i i i i i i i = Δ − ⋅ + Δ + − − + − + (3-3) 移項整理過後可以得到 2 1 1 ( ) 2 ) ( 1 2 2 ) ( 1 a x x fi fi a x x⎟fi =b x ⋅Δx ⎠ ⎞ ⎜ ⎝ ⎛ ⋅Δ − + − ⎟ ⎠ ⎞ ⎜ ⎝ ⎛ ⋅Δ + + − (3-4) 由上述步驟,我們得到 m-1 個聯立的代數方程式,只需再求解三角對 角矩陣系統便可得到(3-4)式的數值解。
3.2 收斂條件
在需要處理許多方程式、矩陣時,而又受陷於儲存及運算空間不 足時,疊代法 (Iterative method) 的是最佳的選擇,其利用第一次運 算結果去解第二次的值,在利用第二次運算結果求得第三次的值…, 如此不斷重複疊代直達到所要求的收斂條件為止,其流程如圖 xx 所 示。疊代法收斂與否的取決,可由下列式子來做判定 * * error a a a − = ε (3-5) 其中a 為運算後的值,a*為先前的值。接下來依照使用者所需條件 γ, 做為收斂的條件: error ε < γ (3-6)第四章 結果與討論
本文數值計算乃依據前述章節之基本假設、理論分析及邊界條 件,建立數值計算模型及格點劃分,並且以指定的數值方法及收斂條 件當作計算方法。求解 Agglomerate 模式下陽極觸媒層毒化時,氫氣 與一氧化碳濃度場、覆蓋率、電流密度與極化曲線圖的分佈情形。4.1 數值方法之驗證
以數值方法計算物理模型,通常必須經過實驗量測的數據或引用 曾發表在公認的學術期刊的論文,來支持個人的研究方法。Chen et al. [42]探討在質子交換膜燃料電池中,受到一氧化碳毒化且加入了 Air-bleed 的效應後,對電池性能曲線的影響。因此,本文將針對此篇 論文一氧化碳毒化的部分來做驗證,在數值模擬方面,採用 Laasonen implicit 方法來求解。4.1.1 格點測試
為了使內部網格區分的大小、多寡,不至於影響到最後結果的正 確性。因此,往往在實行數值模擬方法之前,格點測試的工作有其必 要性。而網格空間分佈主要有準確性、數值穩定性和花費時間等三大 考量。通常而言,網格區分越細密,則所計算出來的值也會越精確, 相對地花費時間也較多。但在某些特殊情況下,網格區分太細密,會 因數值上四捨五入誤差(Round-off error)和網格變形的緣故,反而會導 致結果的不正確及數值的不穩定現象。因此可藉由格點測試的工作, 在有限的電腦資源中尋得網格分佈的最佳化:即為利用最少量的網格數,且其存在著最小的變形量,以獲得相同結果的正確解。 圖4-1 所示為陽極觸媒層中,極化曲線的格點測試結果圖,本文 分別做三組不同格點的極化曲線,以其找出最佳化的格點分佈,由圖 中可看出,401 與 501 格點分佈所呈現的極化曲線誤差 1%以下,而 301 格點分佈的電流密度卻與其他兩組格點分佈所呈現出的相差較 多,而再基於計算時間上的考量,本文在此採用每層401 個格點作為 物理模型格點取決依據。
4.1.2 實驗驗證
圖 4-2 驗證 Chen et al. [42]在一氧化碳濃度 100ppm、氫氣濃度 100%下,電池暫態極化曲線分佈圖,從圖中可知,因為受到一氧化 碳毒化的影響,在操作電壓為 0.6V 時,電流密度從第一秒大約 1.6 A/cm2到達穩態為1508 秒左右時降至 0.2 A/cm2。此驗證結果與 Chen et al. [43]在 100ppm 一氧化碳與 100%的氫氣濃度相較之下趨勢相吻 合。4.2 比較兩種數學模式下一氧化碳毒化之暫態效應
圖4-3、4-4 探討在 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20 μm 下 , 氫 氣 濃 度 隨 觸 媒 層 厚 度 之 暫 態 變 化 曲 線 。 圖 4-3 為 Agglomerate 模式下的曲線分布,可知氫氣濃度隨厚度的方向因與觸 媒反應產生電流密度而遞減,但隨時間達到穩態時,因觸媒顆粒受到 一氧化碳吸附減少了可和氫氣反應的面積,使氫氣濃度必須更往薄膜 方向的觸媒顆粒擴散,尋找反應而延遲了氫氣濃度的消耗。圖 4-4 為 Homogeneous 模式下的曲線分佈圖,其物理現象與圖 4-3 相同,但氫氣濃度隨厚度方向改變大約 10μm 以內時,濃度即被消耗完畢,而 圖4-3 卻持續消耗到 20μm 厚度。從實際情況來探討,圖 4-3 的現象 代表氫氣有能力擴散到觸媒層深處而使得在10~20μm 的觸媒沒有浪 費掉,但圖 4-4 的情況剛好相反,在 10μm 以後觸媒似乎完全用不到 而浪費掉。若從數學模式上來討論,因 Homogeneous 模式缺少考慮 在觸媒層內部實際上的複雜機制,例如:氣-固相的界面化學、液-氣 象的傳輸現象等,因此其氫氣的消耗只限於 Bulter-Volmer 電化學方 程式,所以其表現出的現象也較為簡化,而Agglomerate 模式有把觸 媒層內部複雜機制較完整的描述,所以其結果會與實際接近。 圖4-5、4-6 探討在 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20 μm 下,一氧化碳濃度隨觸媒層厚度之暫態變化曲線。圖 4-5 為 Agglomerate 模式下的曲線分佈圖,可知一氧化碳濃度隨厚度方向因 脫附、電化學反應而遞減。但隨時間達穩態的過程中,第一秒因一氧 化碳的分子量大於氫氣,所以擴散較慢,使得吸附造成毒化現象還沒 開始,當到達 2000 秒時,很明顯的看出,因一氧化碳吸附在白金上 後,強走了氫氣的反應面積且形成很強的鍵結不容易脫附而造成一氧 化碳濃度的累積。圖4-6 為 Homogeneous 模式下的曲線分佈圖,其物 理現象與圖 4-5 雷同,但時間從 10 秒開始,一氧化碳的脫附現象很 明顯而圖4-5 的脫附現象並不明顯。若從數學模式上來討論,在相同 的操作條件下,圖 4-6 因一氧化碳濃度只單純受 Bulter-Volmer 電化學 方程式的影響,而在一氧化碳脫附時並不像使用Agglomerate 模式來 模擬時,有多考慮了Nafion phase 的介質、觸媒尺寸等因素的阻礙, 所以脫附現象明顯,反之,Agglomerate 模式極不明顯。 圖4-7、4-8 探討在 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20 μm 下,氫氣覆蓋率隨觸媒層厚度之暫態變化曲線。圖 4-7 為
Agglomerate 模式下的曲線分佈圖,可知氫氣的覆蓋率隨厚度方向因 吸附及電化學反應而遞減,但隨時間達穩態的過程中,一氧化碳毒化 越來越嚴重,使氫氣的覆蓋率逐漸降低。圖4-8 為 Homogeneous 模式 下的曲線分佈圖,其物理現象和圖4-7 相似,但其氫氣覆蓋率最後只 覆蓋到 10μm 左右即無法在繼續覆蓋,代表觸媒層只被利用到 10μ m 以下,而 Agglomerate 模式卻可覆蓋到 20μm 且覆蓋率最後趨近平 衡狀態。 圖 4-9、4-10 探討在 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20μm 下,一氧化碳覆蓋率隨觸媒層厚度之暫態變化曲線。圖 4-9 為 Agglomerate 模式下的曲線圖,可知一氧化碳的覆蓋率隨厚度方向因 一氧化碳的脫附不易而逐漸累積到達平衡狀態,且隨時間達穩態時, 覆蓋因累積而慢慢上升到 0.9 左右。圖 4-10 為 Homogeneous 模式下 的曲線分佈圖,隨時間愈達穩態時,愈可看出大約在 5μm 的觸媒層 厚度處,有一個峰值接著即產生所謂脫附,使覆蓋率有些微的減少, 其結果和圖4-6 現象吻合且原因相同。圖 4-11、4-12 與前相同的操作 條件下,可看出兩種模式計算出的一氧化碳電流密度均很小,大約在 10-8~10-10之間。又圖4-13。4-14 為氫氣的電流密度圖均都在 1 A/cm2 以上,因此往後的討論即忽略一氧化碳所產生的電流密度,只以氫氣 的電流密度為主。 圖 4-15,4-16 探討在 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20μm 下,一氧化碳及氫氣覆蓋率對電流密度之暫態變化曲線。圖 4-15 為 Agglomerate 模式下,氫氣覆蓋率隨電流密度改變的曲線,由 圖中可看出往電流密度增加的方向氫氣的覆蓋率會降低,以時間為 300 秒來看,大約電流密度在 1.25 A/cm2處,氫氣覆蓋率急速下降且 能產生出的電流密度也已經不多,其原因由前途 4-7 可知氫氣覆蓋率
在觸媒層厚度大約 15μ時,變化趨勢開始平緩表示氫氣的吸附和脫 附達到一個接近平衡的狀態。另外,氫氣覆蓋率隨時間達穩態越來越 低,同時電流密度也越來越低,其最主要的原因還是由於一氧化碳的 吸附,減少了氫氣的電化學反應,由圖4-16 即可明顯看出來。 圖4-17,4-18 操作條件為理想電位 1.2V、10ppm 一氧化碳和 75% 氫氣,觸媒層厚度為20μm 下的電池極化曲線圖,以操作電壓為 0.6V 為準,當開始時間為一秒時,毒化可以說還沒開始,其原因在圖 4-5 有說明,此時電流密度大約在1.4 A/cm2,經過 4692.316 秒後達穩態, 其電流密度大約為 0.8 A/cm2。而圖 4-18 是功率與電流密度的關係 圖,由圖中到達穩態那條曲線來看,雖然電流密度最大到 1.1 A/cm2, 但實際使用電流密度時,最好的範圍應該是在高電流密度區的百分之 75%才是最恰當,因此大約在 0.8 A/cm2左右。 圖 4-19,4-20 操作條件為理想電位 1.2V、100ppm 一氧化碳和 75%氫氣,觸媒層厚度為 20μm 下的電池極化曲線圖,以操作電壓為 0.6V 為準,在時間為 1 秒時,其電流密度大約 1.4 A/cm2,經過1660.176 秒到達穩態情況時,其電流密度只剩0.1 A/cm2不到,由此看出100ppm 的一氧化碳毒化對電池性能的影響真的很大,所以目前質子交換膜燃 料電池一氧化碳的容忍度已經有辦法小於 50ppm。從圖 4-20 的功率 圖可更明顯看出電池幾乎沒有電流量可用了。
4.3 Agglomerate 模式下一氧化碳毒化之穩態效應
圖 4-21,4-22 探討在 10ppm 一氧化碳 75%與 40%的氫氣和 100ppm 一氧化碳 75%與 40%氫氣下達穩態時的電池極化曲線圖,由 圖 4-21 中可知,操作電壓固定在 0.6V,一氧化碳濃度越高且氫氣濃度愈高時,電流密度越大且一氧化碳濃度高氫氣濃度低時,愈容易達 到穩態,換句話說電池很快就被毒化而減少壽命,反之。圖 4-22 可 看出毒化後的結果,其性能的確很差! 圖4-23 探討 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20μm、 觸媒顆粒半徑100nm、不同 Nafion phase 厚度下,電流密度隨位置之 穩態變化曲線圖。由圖中可看出,當Nafion phase 厚度在 10000nm 時, 電流密度只剩下0.2A/cm2,主要原因是因為當Nafion phase 過厚時, 氫氣傳輸的阻抗增加不容易與白金表面產稱化學反應,而使電流密度 值很小。隨著厚度減少到 100nm 時,可看出電流密度逐漸上升,其 電流密度值大約在1.3A/cm2左右。 圖4-24 探討 10ppm 一氧化碳和 75%氫氣、觸媒層厚度 20μm、 Nafion 半徑 1000nm、不同觸媒顆粒半徑下,電流密度隨位置之穩態 變化曲線圖。由圖中可看出,當觸媒顆粒半徑尺寸在 1000nm 時,電 流密度約為0.9A/cm2左右,隨著半徑的減少到10nm 時,可看出電流 密度升高到約 1.3A/cm2 其主要原因在於觸媒顆粒越小表反應面積增 加。但當半徑持續減小至 1nm 時,發現電流密度反而降至約 1.1 A/cm2。因此,本研究結果發現觸媒顆粒尺寸半徑在 10nm 時,有最 高的電流密度。主要原因在於觸媒尺寸變小時,會增加與氫氣反應產 生電流的表面積,但當尺寸太小或者太大時,因為相對於 Nafion 的 阻抗過大以及反應面積量不夠的情況下,而造成電流密度的下降。 圖 4-25 探討
在不同一氧化碳、75%氫氣、觸媒層厚度 20
μm、Nafion 半徑 1000nm、觸媒顆粒半徑 100nm 及操作電
壓
0.6V 下,不同觸媒層厚度與電流密度之穩態變化曲線圖。
由圖中可看出,以
10μm 的觸媒層厚度為準,隨著觸媒層厚
度增加的方向電流密度持續減少,主要因為歐姆阻抗的增
加,而隨著觸媒層厚度減少的方向電流密度仍減少,主要的
原因是因為觸媒層的總反應量減少了。所以本研究結果發
表
4-1 基本操作參數
參數 符號 值 觸媒層厚度 L 0.002 cm 電池溫度 T 358 K 陽極壓力 P 3 atm 觸媒層孔隙度 εCL 0.06 [24] 傳輸係數 α 0.5 氫氣反應之電子數 n 2 氫氣擴散係數 g H D 2 1.9 x 10 -5 cm2/s 一氧化碳擴散係數 g CO D 1.5 x 10-7 cm2/s 觸媒顆粒半徑 rPt 1 x 10 -6 cm Nafion 膜厚度 δN 1 x 10-4 cm 觸媒顆粒內的Nafion 體積分率 VN 0.39 [24] 觸媒表面積 aPt 8000 1/cm 參考電流密度 iref 3 x 10 -3 A/cm2 [26] 參考氫氣濃度 CH2,ref 5.66 x 10 -6 參考一氧化碳濃度 CCO,ref 3.45 x 10 -3 離子導電度 κ 0.556 x 10-1 S/cm 觸媒表面的面積密度與法拉第 ξ 0.5 C/cm2常數的乘積 氫氣吸附速率常數 KH2 100 A/cm 2atm 氫氣脫附速率常數 bH2 0.5 atm [29] 一氧化碳吸附速率常數 KCO 10 A/cm 2atm [29] 一氧化碳脫附速率常數 bCO 2.75 x 10 -9 atm
0.4
0.5
0.6
0.7
0.8
0.9
1
0
0.4
0.8
1.2
1.6
2
2.4
301 Grids 401 Grids 501 GridsC
el
l
V
o
lt
ag
e,
V
Current density, A/cm
20.4
0.5
0.6
0.7
0.8
0.9
1
0
0.5
1
1.5
2
2.5
1S 1500S EXP VOL AT 1S S.S EXP VOLCe
ll
V
o
lt
a
g
e
, V
Current density, A/cm
2圖
4-2 驗證 Chen et al. [42]在一氧化碳濃度 100ppm、氫氣濃
度
100%下,電池暫態極化曲線分佈圖。
Experiments [42]
Present results
100% H2, 0ppm CO ○ 100% H2, 100ppm CO □0
0.2
0.4
0.6
0.8
1
0
5
10
15
20
t = 1 s t = 2000 s t = 3500 s tss = 4692.3 sC
H 2/ C
H 2 , inx, μm
圖
4-3 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、觸
媒層厚度
20μm 下,氫氣濃度隨觸媒層厚度之暫態變化曲
線。
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0
5
10
15
20
t = 1 s t = 300 s t = 500 s tss = 612.7 sC
H 2/
C
H 2 , inx, μm
圖
4-4 Homogeneous model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,氫氣濃度隨觸媒層厚度之暫態變化曲
線。
0
0.2
0.4
0.6
0.8
1
0
5
10
15
20
t = 1 s t = 10 s t = 2000 s t = 3500 s t = 4692.3 sC
co/ C
co , inx, μm
圖
4-5 Agglomerate model 在 10ppm 一氧化碳和 75%氫氣、觸
媒層厚度
20μm 下,一氧化碳濃度隨觸媒層厚度之暫態變化
曲線。
0
0.2
0.4
0.6
0.8
1
0
5
10
15
20
t = 1 s t = 10 s t = 200 s t = 500 s tss = 612.7 sC
co/
C
co , inx, μm
圖
4-6 Homogeneous model 在 10ppm 一氧化碳和 75%氫氣、
觸媒層厚度
20μm 下,一氧化碳濃度隨觸媒層厚度之暫態變
化曲線。
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0 5 10 15 20 t = 1 s t = 2000 s t = 3500 s tss = 4692.3 s H 2 Co ve ra ge x, μm
![表 1-1 燃料電池的主要氫供給來源[3~7] 類別 優點 缺點 氫氣 有害物質的排放量少,不需要重組器可達到輕量化,且可迅速啟動,發電 效率高 氫氣較難處理,需要龐大儲氫桶。建設供氫的基礎設施需花巨大成本。 甲醇 原料較豐富,容易廉價取得。 較其他燃料容易分出氫。 具毒性,處理時需要小心重組時會產生CO 等副產 物、啟動需花 4~5 分鐘 汽油、煤油 可利用既有的汽油加油站,容易普及,處理也較方便 重組溫度高,較難分離出氫。需要去除硫磺等雜 質。啟動費時,效率較低。 甲烷(天然氣)](https://thumb-ap.123doks.com/thumbv2/9libinfo/8460369.183148/37.892.130.768.351.862/有害物質較其他燃料容易分出氫具毒性處理時需要小心重組時會CO.webp)
![圖 1-1 能源趨勢圖[1] (年) 1750 1800 1850 1900 1950 2000 2050 2100 再生能源 核能 石油 煤 人口 世界人口數︵十億︶能源消耗量( 十億噸/ 年) 1 2 3 4 5 6 7 8 8 7 6 5 4 3 2 1](https://thumb-ap.123doks.com/thumbv2/9libinfo/8460369.183148/38.892.168.788.392.836/能源趨勢再生能源核能石油煤人口世界人口億源消耗量年872.webp)



![表 4-1 基本操作參數 參數 符號 值 觸媒層厚度 L 0.002 cm 電池溫度 T 358 K 陽極壓力 P 3 atm 觸媒層孔隙度 ε C L 0.06 [24] 傳輸係數 α 0.5 氫氣反應之電子數 n 2 氫氣擴散係數 D Hg 2 1.9 x 10 -5 cm 2 /s 一氧化碳擴散係數 D COg 1.5 x 10 -7 cm 2 /s 觸媒顆粒半徑 r Pt 1 x 10 -6 cm Nafion 膜厚度 δ N 1 x](https://thumb-ap.123doks.com/thumbv2/9libinfo/8460369.183148/58.892.126.797.200.1124/壓力觸媒層孔εC傳輸係數α之電子數氫氣一氧化觸媒顆粒半徑.webp)
![圖 4-2 驗證 Chen et al. [42]在一氧化碳濃度 100ppm、氫氣濃 度 100%下,電池暫態極化曲線分佈圖。 Experiments [42] Present results 100% H2, 0ppm CO ○ 100% H2, 100ppm CO □](https://thumb-ap.123doks.com/thumbv2/9libinfo/8460369.183148/61.892.144.761.267.792/一氧化碳濃度氫氣度下電池暫態極化曲線分佈圖ExperimentsPresentH○H□.webp)


