Phonon sidebands and optical gain of light-emitting conjugated polymers
Hsin-Fei Meng*Institute of Physics, National Chiao Tung University, Hsinchu 300, Taiwan, Republic of China Vincent Chia-Hung Chang†
Department of Physics, National Taiwan Normal University, Taipei 116, Taiwan, Republic of China and National Center for Theoretical Sciences, Hsinchu 300, Taiwan, Republic of China
共Received 23 June 1999兲
We study the interaction of excitons and optical phonons in light-emitting conjugated polymers with the variational approach. The optical transition matrix elements for the phonon sidebands in the absorption and emission spectra are calculated. The electron-phonon coupling constant is determined by fitting the theoretical oscillator strengths of the emission sidebands with the experimental photoluminescence spectra for the various poly( p-phenylene vinylene兲 derivatives. Using this coupling constant, we predict that the absorption one-phonon sideband is as important as the zero-one-phonon sideband for the ideal systems. Taking the experimental data of the peak absorption coefficient as input, we obtain the optical gain coefficient for a wide range of exciton densities. The gain is found to be determined by the exciton density n0by a general expression 1.98 ⫻10⫺16n
0 cm2, which is in good agreement with experiments.关S0163-1829共99兲06243-8兴
I. INTRODUCTION
Conjugated polymer has been identified as a promising candidate for future optoelectronic applications1. Encouraged by the discovery of their electroluminescence,2an enormous amount of research has been conducted to further explore the possibilities of electro-optical devices based on conjugated polymers. A nature step forward is the search for lasing ac-tions. Preliminary optically pumped polymer lasers have been demonstrated.3–6The effort to develop these prototypes into realistic coherent light sources with preferably electrical pumping has, however, encountered major difficulties. In or-der to solve these difficulties, more works need to be done on not only the technical issues such as device fabrications, but also the basic physics of lasing mechanisms in conjugated polymers. This paper is motivated by the latter.
Lasing takes place when the gain overcomes the loss in an optical resonator. For gas lasers with typical three or four-level operation, gain is achieved by the population inversion between the two active levels. Due to the inversion, the rate for the absorption from the lower state is smaller than the rate of stimulated emission from the upper state. In the case of inorganic semiconductor lasers, gain occurs between two bands of extended states, i.e., the conduction band and the valence band, instead of two discrete active states. Upon cur-rent injection, the conduction-band bottom is occupied by the electrons and the valence-band top is occupied by holes. Ab-sorption near the band edge is then depleted, while stimu-lated emission is enhanced at the same time. In other words, the current injection causes a spectral shift between the ab-sorption and the emission, which grows with the carrier den-sity and therefore injection current. As a result, net gain arises. Though conjugated polymers are also direct band-gap semiconductors, which exhibit gain upon pumping, the sce-nario for gain seems to be quite different from above. One of the most important differences is that even before optical pumping or current injection, a significant spectral shift
be-tween the absorption and emission, i.e. Stokes shift, exists for conjugated polymers. Due to this shift, the emitted pho-ton has a reduced chance to be reabsorbed and the optical gain is possible without electronic population inversion. The Stokes shift, common in organic materials, is usually due to the electron-phonon coupling, which causes vibrational re-laxations after electronic excitations. With the Stokes shift, lasing can occur without electronic population inversion, in contrast to the conventional compound semiconductors.
In this paper, we study theoretically the phonon side-bands in the absorption and emission spectra and the result-ing Stokes shift due to electron-lattice couplresult-ing for one ide-alized infinite periodic chain. The significance of extrinsic factors like the inhomogeneity broadened density of states 共DOS兲 will be investigated afterwards. The theory developed by Pollmann, Buttner, and Matsurra7,8共PBM兲 for polar inor-ganic semiconductors is adapted for the description of con-jugated polymers. Besides dimensionality, one of the major differences between conjugated polymers and polar semicon-ductors is the type of the electron-phonon coupling. While the coupling for polar semiconductors is of the Fro¨hlich type, the coupling for the covalently bonded conjugated polymers is of deformation nature.
If the oscillator strength of the absorption is not domi-nated by the zero-phonon band, there will be a subsequent relaxation among the vibrational levels from the higher pho-non states to the zero-phopho-non state after optical excitation. Such a relaxation is sometimes referred to as the genuine Stokes shift. This shift reduces the overlap between the emis-sion and the absorption spectra, and favors the achievement of gain. We predict that for ideal systems the oscillator strength of the absorption is evenly shared by the one- and zero-phonon band, while the emission is dominated by the zero-phonon band. The mirror symmetry common for small organic molecules is therefore broken. Moreover, this result implies the possibility of a genuine Stokes shift of one opti-cal phonon energy, contrary to the interpretations of some PRB 60
site-selective experiments.9,10 We shall come back to this point in the Discussion.
Because our calculation is only for one single chain, the absorption and emission coefficient for a realistic solid-state sample can be determined only up to a common proportional constant, which includes factors like transition dipole matrix element and chain packing density. We determine the con-stant from fitting with the experimental absorption coeffi-cient, then calculate the gain coefficient for arbitrary exciton density. We found that near the one-phonon emission band there is a universal relation between the peak gain coefficient and the exciton volume density, independent of the chain packing density and geometry. This result gives a relation between the resonator loss and the threshold exciton density required for lasing in practical resonators. The agreement with the experimental estimates is quite reasonable.
This article is organized as follows: In Sec. II, we present our model Hamiltonian and the Lee-Low Pines 共LLP兲 uni-tary transformation. In Sec. III, the variational approach to derive the wave functions and energy is introduced. In Sec. IV, we calculate the optical transition matrix elements and present our results on the absorption and emission spectra with phononside bands. In Sec. V, we study the gain coeffi-cient for arbitrary exciton density and compare the theoreti-cal predictions with experiments. Sections VI and VII are the discussion and the conclusion, respectively.
II. THE EXCITON-PHONON HAMILTONIAN Poly( p-phenylenevinylene兲 共PPV兲 is idealized as an one-dimensional lattice. Each unit cell contains eight carbon at-oms, with one pzorbital at each carbon atom. The-electron bands are formed by the superposition of these pz orbitals. The total number ofelectrons is equal to the total number of carbon atoms. Simple tight-binding calculation shows that PPV is a direct band-gap semiconductor. Because optical absorption and emission near the band gap involve transi-tions between the valence and the conduction bands only, the problem can be simplified from eight bands to only two bands. The system then consists of an electron and a hole interacting with each other and with the longitudinal-optical phonons. It is convenient to write an effective Hamiltonian for the system
H⫽Hex⫹Hp⫹Hex⫺p, 共1兲 with Hex⫽ pe2 2me ⫹ ph 2 2mh⫹V共r e⫺rh兲 共2兲 and Hp⫽
兺
s⫽1 n兺
k s共k兲as,k † a s,k. 共3兲The three terms in H are for the electron-hole system of exciton, phonon, and their interaction, respectively. Hex⫺p will be derived below. pe(h) is the momentum operator for electron共hole兲. me,h are the effective mass of the conduction and valence bands, respectively. V(r) is the effective Cou-lomb attraction between the electron and the hole. s is the
phonon branch index, and k is the crystal momentum. The
-electron band edge is chosen to be at k⫽0. as,k †
and as,k are the phonon creation and annihilation operators, respec-tively.
In contrast to the Fro¨hlich type coupling in polar semicon-ductors, the electron-phonon coupling in conjugated poly-mers are of deformation nature. Its explicit form can be ob-tained as follows. Consider the interaction Hamiltonian He⫺p between conduction-band electron and phonon first. From the crystal momentum conservation, the matrix ele-ment of He⫺pbetween conduction-band Bloch states兩k
典
and 兩k⬘典
must be of the form具
k⬘
兩He⫺p兩k典
⫽兺
sq e s,q共a s,q⫹as,⫺q † 兲␦ k⬘,k⫹q. 共4兲 For deformation type of interaction, the coupling constante
s,q approaches to a finite value as q→0. Since the exciton is composed of Bloch states near the Brillouin zone center, we only need to consider electron scattering in that region, where es,q can be replaced by its value at q⫽0, denoted simply by es below. In fact, from the relation
具
k兩He⫺p兩k典
⫽es
(as,0⫹as,0 †
), es is identified to be the change of the conduction-band edge due to the q⫽0 mode of the lattice displacement with normal coordinates,q, which is equal to (as,q⫹as,q
†
)/
冑
2 in the second quantized form. The lattice displacement vector usi(R) for the ith carbon atom in the nth unit cell can be expressed in terms of the normal coordinates,qby ui共Rn兲⫽
冑
1 N兺
q,s冑
ប 2 Mcs共q兲⑀s i 共q兲eiqRn冑
2 s,q. 共5兲 N is the total number of unit cells in the chain. Rn is the location of the nth unit cell. Mc is the mass of the carbon atom. ⑀si(q) are the polarization vectors. i⫽1→8 label the carbon atoms in each unit cell. The dependency of the conduction-band edge c on s,0 is through the modulation of the hopping integral between neighboring carbon pz orbit-als by the lattice displacement. Assume that␣ is the change in the hopping integral per unit length of the bond-length change. The bond-length change is of the order of 兩ui兩, which is in turn of the order of冑
ប/2NMcs(q), so we havee s⫽ c s,0 ⬃␣
冑
2N Mប c0 , 共6兲assuming that all the phonon branches are dispersionless and share the same frequency 0. This relation gives only the correct order of magnitude for the electron-phonon coupling constant es. The exact expression for es depends on the polarization vectors ⑀s
i
(q) of the phonon modes. Instead of solving the complete lattice dynamic equation to obtain all the polarization vectors, we introduce a phenomenological dimensionless parameter e, and assume that the coupling constantes is independent of the branch index s, by
e s⫽
e⫽e␣
冑
បe represents the strength of the electron-phonon coupling. Similarly, for the valence band we can calculate the modu-lation of the valence-band edge by the lattice displacement of the normal phonon modes s,q, and obtain the correspond-ing hole-phonon couplcorrespond-ing constant h. The lattice structure of PPV is invariant under inversion respect to the center of the benzene ring. Therefore, the set of eight polarization vec-tors ⑀s,qi for each mode (s,q) must be either even or odd upon inversion. The energies of the electronic bands at q ⫽0 is actually the eigenvalues of a 8⫻8 symmetric matrix A, with Ai j equal to the hopping integral between carbon site i and j. A is invariant upon lattice inversion when the lattice is in the equilibrium configuration. When the lattice is dis-placed from the equilibrium configuration, the matrix is modulated A→A⫹⌬A. One can show that for even polar-ization vectors, ⌬A is odd upon lattice inversion. On the other hand,⌬A is even upon lattice inversion for odd polar-ization vectors. There is a bandedge modulation to the linear order in the displacement only when⌬A is even. Moreover, we found that for such⌬A, the modulation of the conduction band and the valence band are always exactly the same in size but opposite in sign. Therefore, we have the general relation⫺v⫽e⬅. However, because hole is the vacancy of the valence band, the matrix elements between the one-hole states are opposite in sign to the matrix elements of the corresponding valence-band states. In other words, we have h
具
k⬘
兩Hh⫺p兩k典
h⫽⫺v具
k⬘
兩Hh⫺p兩k典
v up to a constant. Here, 兩k典
v is the one-body valence-band Bloch state, while兩k典
h is the many-body Slater determinant with one hole in the oth-erwise filled valence band. So we haveh⫽⫺v⫽. Finally in the real-space representation, the exciton-phonon interac-tion Hamiltonian becomesHe p⫺p⫽He⫺p⫹Hh⫺p⫽
兺
sk 共eiqre⫹eiqrh兲共a
s,q⫹as,⫺q † 兲, 共8兲 with ⫽␣
冑
2N Mប c0 . 共9兲re,h are the one-dimensional coordinates of the electron and hole, respectively. In this interaction Hamiltonian, is the only fitting parameter.
Expressed in terms of the center-of-mass and relative co-ordinates of the exciton, the total Hamiltonian becomes
H⫽P 2 2 M⫹ p2 2⫹ប0
兺
s,k as,k† as,k ⫹兺
s,k 关e ikR k共r兲as,k⫹h.c.兴⫹V共r兲. 共10兲 Here M⫽me⫹mh is the total mass,⫽memh/ M is the re-duced mass. R⫽re⫹rh is the center-of-mass coordinate, and r⫽re⫺rh is the relative coordinate.k(r) is defined ask共r兲⬅exp共is2kr兲⫹exp共is1kr兲 共11兲
with the ratio s1⫽me/ M and s2⫽mh/ M . Next, we perform the Lee-Low-Pines 共LLP兲 transformation11 to remove the center-of-mass degree of freedom
H1⬅U1⫺1HU1,U1⫽exp
冋
i冉
Q⫺兺
s,kkas,k† as,k
冊
R册
. 共12兲 The transformed Hamiltonian becomesH1⫽ ប2Q2 2 M ⫹ p2 2⫹V共r兲⫹
兺
k 关k共r兲ak⫹H.c.兴 ⫹兺
k ប⍀˜ 共k,Q兲ak † ak⫹兺
k,k⬘ ប2kk⬘
2 M ak † ak†⬘akak⬘, 共13兲 with ប⍀˜ 共k,Q兲⬅ប0⫺ ប2kQ M ⫹ ប2k2 2 M . 共14兲Q is the total momentum of the system, which is conserved and taken as a c number here. Note that we have included the summation over the phonon branch index s into the summa-tion over wave number k implicitly.
This Hamiltonian is of similar form with the exciton-phonon Hamiltonian for conventional three-dimensional po-lar inorganic semiconductors studied by PBM.7,8In addition to dimensionality, there are two major differences between the Hamiltonians for conjugated polymers and polar semi-conductors. First the k dependency of the coupling constant is different. The coupling is a constant for the polymers, but proportional to 1/k for the polar semiconductors. Second, the electron-phonon coupling and hole-phonon coupling ink(r) have the same sign for polymers, but different in polar semi-conductors.
III. VARIATIONAL SCHEME
Following the work of PBM, the trial ground state兩⌿
典
of the whole system is chosen to be of the form兩⌿
典
⫽ex共r兲U2关Fk共r兲兴兩0典
ph. 共15兲ex(r) is the exciton trial wave function.兩0
典
ph is the phonon ground state. U2 is a displacement operator such that the phonon trial ground state is a coherent stateU2关Fk共r兲兴⫽exp
冋
兺
kFk*共r兲ak⫺Fk共r兲ak†
册
. 共16兲 Fk(r) is our variational function. We defineH2⬅U2⫺1关Fk共r兲兴H1U2关Fk共r兲兴, 共17兲 and
H0⬅ph
具
0兩H2兩0典
ph, 共18兲 such that the trial ground-state energy具
⌿兩H1兩⌿典
is equal to具
ex兩H0关Fk(r)兴兩ex典
. The variational ground state is deter-mined by the minimum conditions␦ ␦Fk共r兲
具
ex兩H0关Fk共r兲兴兩ex
典
⫽0 共I兲,␦
␦ex共r兲
具
ex兩H0关Fk共r兲兴兩ex典
⫽0 共II兲. 共19兲 Due to the translational symmetry of the system, the phonon variational function should be of the form12Fk共r兲⫽ ប0共 fk 1eis2kr⫺ f k 2eis1kr兲. 共20兲 So the first of the two minimum conditions is replaced by
␦
␦fki
具
ex兩H0兩ex典
⫽0. 共21兲 A. Phonon displacement trial functionsWe proceed with condition I in Eq. 共19兲 first. For given exciton trial wave function ex(r), condition I gives the phonon trial functions Fk(r) as functionals ofex. In terms of fk
i
, the minimum condition becomes
␦ ␦fki
具
ex兩H0兩ex典
⫽具
ex兩 ␦Fk共r兲 ␦fki ␦ ␦Fk共r兲 H0兩ex典
⫽0. 共22兲 Assuming the inversion symmetry of the exciton wave func-tion ex(r)⫽ex(⫺r), the equations can be solvedfk1⫽共1⫹Gk兲共1⫹R2 2k2兲⫺共1⫹G k兲Gk 共1⫹R1 2 k2兲共1⫹R22k2兲Gk2 , fk2⫽⫺共1⫹Gk兲共1⫹R1 2k2兲⫹共1⫹G k兲Gk 共1⫹R1 2 k2兲共1⫹R22k2兲⫺Gk2 , 共23兲 where Gk⬅
具
ex兩eikr兩ex典
, and R1,2⬅冑
ប/2me,h0 is the so called polaron radius. Note Gkis real due to the symmetry ofex(r). The dependency of the phonon trial function fk i
on the exciton trial wave function ex(r) is now only through the quantity Gk. We now substitute Eq. 共23兲 into
具
ex兩H0关Fk(r)兴兩ex典
, and express the expectation value in terms of Gk具
ex兩H0兩ex典
⫽具
ex兩Hf ree兩ex典
⫹R2␣¯2␦ 0冋
I1⫺ ប0 R I2⫹I3册
. 共24兲 Here, Hf ree⫽p2/2⫹V(r). I1,2,3 are certain dimensionless integrals defined below. aB⫽ប2/e2⑀ is the exciton Bohr radius.R is the exciton Rydberg ប2/2aB2. The lattice con-stant a is written as␦0aB. In addition, we define the dimen-sionless polaron radius R¯i as Ri/aB and the dimensionless electron-phonon coupling constant ␣¯ by
␣ ¯⫽ ␣ ប0
冑
ប 2 M0 . 共25兲For the explicit forms of the integrals I1, I2, and I3, the integration variable k is changed to a dimensionless variable t⬅kaB, such that f1,2(t)⫽ fk
1,2
. The function G(t) is defined as Gt/aB. The integrals are
I1⫽
冕
⫺⬁ ⬁ dt 2t 2兵关s 2f1共t兲兴2⫹关s1f2共t兲兴2 ⫹2s1s2f1共t兲f2共t兲G共t兲其, I2⫽冕
⫺⬁ ⬁ dt 2关 f 1共t兲⫺ f2共t兲兴关1⫹G共t兲兴, 共26兲 I3⫽冕
⫺⬁ ⬁ dt 2冋
ប0 R ⫹ 共1⫹兲2t 2册
⫻兵关 f1共t兲兴2⫹关 f2共t兲兴2⫺ f1共t兲f2共t兲G共t兲其. Here, is me/mh. Note the integrals I1,2,3 are expressed in terms of the function G(t), which depends only on the ex-citon trial wave function ex(r). In order to obtain具
ex兩Hf ree兩ex典
and the integrals I1,2,3 in具
ex兩H0兩ex典
, we need to chose a definite form of the variational exciton wave functionex(r).B. Exciton trial wave function
Without the electron-phonon coupling, an exciton in the polymer chain can be modeled as a one-dimensional hydro-gen atom with Coulomb interaction cut off at small dis-tances. In fact, the binding energy of the one-dimensional hydrogen atom is infinity without a cutoff.13 Because our two-band effective mass approach is valid only for Bloch states within the first Brillouin zone, the real-space cutoff for the Coulomb interaction is the lattice constant a, whose cor-responding momentum-space cutoff is at the Brillouin zone boundary. The corresponding Schro¨dinger equation is
⫺ ប 2 2
d2
dr2 ⫹V共r兲⫽E. 共27兲 The cutoff Coulomb potential is
V共r兲⫽
再
⫺ e 2 ⑀兩r兩, 兩r兩⬎a ⫺e 2 ⑀a, 兩r兩⬍a. 共28兲Here,⑀is the dielectric constant along the chain. We assume that the main effect of exciton-phonon interaction is to intro-duce a modified electron-hole Coulomb attraction. So we take the variational trial wave function for ex(r) as the solution of the Schro¨dinger equation for V(r) with ⑀ changed to ⑀v. The subscript ‘‘v’’ means variation. The
modified Bohr radius, denoted by a0 is then
a0⫽ ប2
We take a0 as our variational parameter to minimize the system total energy. Following the work on Loudon, we de-fine three dimensionless variables
␦⫽aa 0 , ⫽
冑
2a0 2 ប2 兩E兩, z⫽ 2r a0 , 共30兲and express the solution and energy eigenvalue in terms of them. ␦ is now our variational parameter. For the ground state, and␦ are related by the equation
ln
冉
2␦ 冊
⫹1
2 ⫽0. 共31兲
In terms of the new variable z, the cutoff length of the Cou-lomb potential is z0⫽2␦/, and the dimensionless ground-state wave function is
共z兲⫽
再
cos冋冉
⫺1 4⫹ a02 4a冊
1/2 z册
, z⬍z0 Ce⫺ 1 2z冉
z ln z⫺1 冊
, z⬎z0. 共32兲The constant C is determined by the continuity condition
C⫽ cos共⫺1⫹␦2兲1/2␦ e⫺␦/
冉
2␦ ln 2␦ ⫺ 1 冊
. 共33兲The actual wave function ex(r) is related to(z) by
ex共r兲⫽
冑
1 a0N共␦兲冉
2r a0冊
. 共34兲The dimensionless quantity N(␦) is equal to 兰0⬁兩(z)兩2dz, such that 兰⫺⬁⬁ 兩ex(r)兩2dr⫽1. Now we substitute the trial wave function ex with variational parameter␦ into the ex-pression for the quantity G(k) in Eq.共23兲
G共t,␦兲⫽ 1 2N共␦兲
冕
⫺⬁⬁
兩共z兲兩2eit␦z/2␦0dz, 共35兲 where␦0⫽a/aB as defined above. After substituting G(t,␦) into Eqs. 共23兲, 共26兲, and 共24兲, we obtain the variational ground-state energy as the function of a single variational variable␦. The ground state is then determined by minimiz-ing the function. The vibarional excited states can be ob-tained by applying U2 to states containing one or more phonons. Their energy can be obtained by calculating the expectation values of H2 for them. For example, for states with one phonon at k, the energy is ph
具
0兩akH2ak†兩0
典
ph. IV. OPTICAL ABSORPTION AND EMISSIONA. Oscillator strengths
The optical absorption of conjugated polymers occurs through the creation of an exciton. This process is character-ized by the oscillator strength f and the optical absorption coefficient␣, both of which are related to the corresponding exciton transition-matrix element:
f⫽cf
兺
m 兩Pm兩 2, 共36兲 h␣共h兲⫽c␣兺
m 兩Pm兩 2␦共E m⫺E0⫺h兲,where cf,c␣ are proportional constants and h is the energy of the photon. The subscript m characterizes the states of the phonon. E0 is the electronic band gap plus the ground-state energy of our Hamiltonian in Eq. 共1兲. Em is the energy of phonons in the final state of the optical transition. The exci-ton transition-matrix element for this process Pmis given by
Pm⫽⑀ជ•Mជc,*v
再
具
0兩exp冋
兺
k,s „Fk *共0兲ak,s ⫺Fk共0兲ak,s † …册
兩m典
冎
ex共0兲 ⫽⑀ជ•Mជc,*vexp共⫺g/2兲 ⫻具
0兩exp冋
兺
k,s Fk*共0兲ak,s册
兩m典
ex共0兲, 共37兲 where the factor g isg⫽
兺
k,s 兩Fk共0兲兩 2⫽n兺
k冉
ប0冊
2 共 fk 1⫺ f k 2兲2 ⫽n2兺
k冉
␣2 2បM03冊
共 fk 1⫺ f k 2兲2. 共38兲Here, n is the number of even optical phonon modes.⑀ជis the unit polarization vector of the electric field, Mជc,*v is the di-pole transition-matrix element between the valence- and conduction-band states. From the exciton transition-matrix element, we can calculate the total oscillator strength for the transition to states with a certain number of phonons. For transitions to the states with one exciton plus zero, one, and two phonons, the oscillator strengths are respectively given by
f(0)⫽AL兩ex共0兲兩2exp共⫺g兲,
f(1)⫽ f(0)g, 共39兲
f(2)⫽ f(0)g2/2.
The dipole transition-matrix element, the polarization vec-tors, and the proportional constant are all absorbed in the new constant A. L is the chain length. It is clear that the relative strength among these transitions depends on the magnitude of g. To calculate g, we need to know the param-eter
冑
n, where is the dimensionless electron-phonon coupling constant. We extract this number from the experi-mental emission spectra of the conjugated polymers. The transition rate, as expected, is proportional to the system size L.Exciton radiative recombination occurs mainly after re-laxation from higher phonon states to zero-phonon state. It then emits a photon and transits to the electron-hole ground state 共no exciton兲 plus some phonons. We first assume that
the total momentum Q of the exciton in the initial state is zero. This is true for zero-temperature transition. The tem-perature dependence can be taken into account later. The corresponding exciton transition-matrix element for decay is similar to that of the absorption:
P ¯m⫽⑀ជ•Mជ c,v *
再
具
m兩exp冋
兺
k,s „Fk *共0兲ak,s ⫺Fk共0兲ak,s † …册
兩0典
冎
ex *共0兲 共40兲 ⫽⑀ជ•Mជc,*v再
exp共⫺g/2兲 ⫻具
0兩exp冋
兺
k,s Fk*共0兲ak,s册
兩m典
冎
*ex*共0兲. 共41兲 The total emission rate for the various phonon final states is proportional to the square of the above matrix element inte-grated over the energy of the emitted photon. For an exciton with strictly zero total momentum, the emission rate to the zero, one, and two phonon states are give byh(0)⫽AL兩ex共0兲兩2exp共⫺g兲, h(1)⫽AL exp共⫺g兲兩ex共0兲兩2n兩F0兩2, 共42兲 h(2)⫽AL exp共⫺g兲兩 ex共0兲兩2 n2 2
兺
k 兩Fk F⫺k兩2. Because of momentum conservation, only the phonon with zero momentum can be emitted in the one-phonon process, and the two phonons must be of opposite momenta k and ⫺k in the two-phonon process. To be selfconsistent, the emission rate should remain finite as the chain length L goes to infinity. As shown in Eqs. 共9兲 and 共15兲, the quantity Fk scales as L⫺1/2. The emission rates for the one and two pho-non processes, indeed, do not scale with L. However, the zero-phonon emission rate h(0) is proportional to L. This problem can be fixed only when we consider the case of finite temperature.At finite temperature, the exciton total momentum Q is allowed to thermally fluctuate and become nonzero. How-ever, the final state, electron-hole ground state with no pho-non, has a total momentum equal to zero. From momentum conservation, the rate is nonzero only for initial states with zero total momentum, which, as the length L of the polymer chain becomes large, occupies a phase space scaling with the inverse of L. The transition rate is the product of h(0)and the probability that the exciton momentum Q⫽0. Since the two factors have inverse scaling with respect to L, the physical rate stays finite as L goes to infinity. We assume that the probability distribution is determined by a Boltzman distri-bution P共Q兲⫽1 L
冑
2ប2 m* exp冉
⫺ ប2Q2 2m*冊
. 共43兲 By the periodic boundary condition, Q takes the discrete val-ues 2m/L, with m being an integer. The normalization of the probability distribution is determined by兺
QP共Q兲⫽ L 2
冕
⫺⬁⬁
P共Q兲dQ⫽1. 共44兲 Because of the momentum conservation, only excitons with Q⫽0 can decay radiatively. Thus the temperature-dependent zero-phonon emission rate ⌫0(T) is given by
⌫0共T兲⯝h (0)
兺
Q P共Q兲␦Q,0⫽h(0)冕
⫺⬁ ⬁ P共Q兲␦共Q兲dQ ⫽h(0)P共0兲⫽A兩 ex兩2e⫺g冑
2ប2 m* . 共45兲Now the zero-phonon emission rate is now finite as L be-comes infinity. The above formula Eq. 共45兲, however, im-plies strong temperature dependence of the zero-phonon emission rate, which is not observed experimentally. The reason is that the sharp Dirac␦ function␦(Q) above should be, in reality, replaced by a distribution function with finite width⌬K. The strict momentum selection rule is smeared by defects or environmental inhomogeneity, which break the discrete translational invariance. So, we replace the Dirac ␦ function in Eq. 共45兲 by a Lorentzian distribution L(Q;⌬K) ⫽(⌬K)⫺1关1⫹(Q/⌬K)2兴⫺1 with width ⌬K. The zero-phonon emission rate becomes
⌫0共T兲⯝h(0)
冕
⫺⬁ ⬁ P共Q兲L共Q;⌬K兲dQ ⫽h(0)2 L 1 ⌬Kr共T;⌬K兲 ⫽A兩ex共0兲兩 2e⫺g 2 ⌬Kr共T;⌬K兲. 共46兲 The dimensionless parameter r(T;⌬K) is equal tor共T;⌬K兲⫽ L 2
冕
⫺⬁ ⬁ P共Q兲 1 1⫹冉
Q ⌬K冊
2dQ ⫽ 1冑
冕
0 ⬁ e⫺⑀ 1冑
⑀ 1 1⫹ ⑀ ⌬E d⑀. 共47兲Here,⌬E is the corresponding energy range and we assume that it equals (⌬K)2/2M . r is basically the fraction of the thermal probability distribution that is within the range of ⌬K. When the temperature goes to zero, r approaches one accordingly.
While such replacement has significant effect on the tem-perature dependence of the zero-phonon emission rate, it does not affect other phonon bands. For one or more phonon emission, there is always a continuum of available final states with arbitrary phonon momentum. In addition, the transition rate h(1)is not sensitive to the exciton momentum Q. Thus, the one-phonon emission rate⌫(1)(T) at finite tem-perature can be written as
⌫(1)共T兲⯝h(1)
兺
K,Q P共Q兲␦K,Q⫽h(1)兺
Q P共Q兲 ⫻冕
⫺⬁ ⬁ ␦共K⫺Q兲dK⫽h(1). 共48兲 Here, Q is the momentum of the exciton共initial state兲 and K is the momentum of the emitted phonon共final state兲. The ␦ function above indicates that the momentum conservation is strictly enforced. Clearly, it does not matter whether we use a␦ function or a distribution with finite width. The formula also shows that the one-phonon rate is insensitive to T and ⌬K. The case for two-phonon emission rate is similar, i.e.,⌫(2)共T兲⯝h(2). 共49兲 Our model thus predicts a mild temperature dependence for zero-phonon emission rate while the multiple phonon emis-sion rates are temperature independent.
B. Line shapes
Now we turn to the line shapes of the absorption and emission spectra. So far, we consider only a single chain, and the line shape of the spectra is give by Eq.共36兲. However in real sample the inhomogeniety broadening seems to domi-nate over the theoretical line shapes. We observe that the experimental linewidth is 70 meV at 20 K, which is much larger than the theoretical linewidth, i.e. the thermal energy kBT. This difference shows that the experimental broadening is of disorder origin, instead of intrinsic origins like thermal fluctuation and electronic bandwidth. Consequently, in order to make a reliable prediction on the gain profile, we choose not to use the theoretical line shapes. Instead, we attach Gaussian distributions with adjustable width to the phonon bands with the corresponding oscillator strength. In Sec. IV C, a fitting of our prediction with the observed emission spectra will be described, which shows almost perfect match and confirms our assumption of Gaussian distributions here. The absorption rate ␣c() as a function of the photon frequency can be written as
␣c共兲⫽␣c0共兲⫹␣c1共兲⫹␣c2共兲, 共50兲 with ␣ci共兲⫽ f(i)Fi共兲, Fi共兲⫽ 1
冑
i aexp冋
⫺冉
⫺i a i a冊
2册
. 共51兲 The subscript ‘‘c’’ denotes chain. f(i) is the absorption os-cillator strength for the ith sideband obtained in Eq.共39兲, andFi() the Gaussian distribution centered at the peak frequen-ciesiaof the sidebands in the absorption spectrum.ais the line broadening for the absorption spectrum. It is convenient to factor out the physical dimension and define a dimension-less line-shape function␣¯ ():
␣c共兲⫽ LA兩ex共0兲兩2e⫺g
冑
1 a ␣¯共兲, 共52兲 where␣¯ () is given by ␣ ¯共兲⫽冑
1 a关F 0共兲⫹gF1共兲⫹12g 2F 2共兲兴. 共53兲 Now, we consider the absorption coefficient ␣. For a real sample made out of a large number of chains, the absorption coefficient ␣() is proportional to the single-chain absorp-tion rate␣c() times the volumn density of chains, which is determined by chain packing geometry. We can combine the chain density, the factor LA兩ex(0)兩2e⫺gin Eq.共52兲 and the proportional constant into a common dimensionful constant B for all the phonon sidebands and write␣共兲⫽B␣¯共兲. 共54兲 B has the same unit of cm⫺1as the absorption coefficient␣. Similarly, the recombination rate␥c() for an exciton in a single chain can be written as
␥c共兲⫽␥c0共兲⫹␥c1共兲⫹␥c2共兲, 共55兲 with ␥ci共兲⫽⌫(i)Gi共兲, Gi共兲⫽ 1
冑
i eexp冋
⫺冉
⫺i e i e冊
2册
. 共56兲Gi() is the Gaussian distribution centered at the peak of the phonon sideband in the emission spectrum. Plugging in the oscillator strength⌫(i), we have
␥c共兲⫽
Aa兩ex兩2e⫺g
冑
1e ¯␥共兲. 共57兲 The dimensionless function␥¯ () is
␥ ¯共兲⫽
冑
1 e冋
2 ⌬Kar共T;⌬K兲G0共兲⫹ L an兩F0兩 2G 1共兲 ⫹L a n2 2兺
k 兩Fk F⫺k兩2G2共兲册
. 共58兲 When there are Nex excitons in the chain, the photon emis-sion rate becomes␥c共兲Nex⫽LA兩ex兩2e⫺g共nexa兲␥¯共兲, 共59兲 where nex⬅Nex/L is the chain exciton density. nexa is the number of excitons per unit cell. For the same reason as in absorption, the photon emission coefficient ␥(,nex) for a real sample can be written as
␥共,nex兲⫽Bnexa¯␥共兲. 共60兲 The factor B here is the same as the one in Eq.共54兲 since the absorption and emission processes do not differ in chain packing geometry and dipole matrix element. The optical spectra are thus determined up to a common factor B, which can only be fixed by comparing with the experiment. Com-bining the absorption spectrum and the stimulated emission spectrum, we can obtain the gain coefficient of the sample. This will be discussed in Sec. V.
C. Comparison with experiments and predictions We try to fit the recently observed14 emission spectra of the various conjugated polymers by three equally spaced Gaussian distributions, as suggested in Eq.共55兲. Figure 1 is the fitting for poly共dioctyloxy phenylene vinylene兲 共Pd0PV兲 film at 300 K. As clearly shown in the figure, the data perfectly match the sum of three Gaussian distributions. This is also true for Pd0PV film at 10 K, Pd0PV solution at 300 K and poly共2-methoxy,5-共2
⬘
ethyl 兲-hexyloxy-phenylene-vinylene兲 共MEH-PPV兲 solution at 300 K. From the fitting, we can determineie,ieand⌫(i). For simplicity, we assume that the absorption and emission roughly share the same broadening, i.e., ia⫽ie. From the ratio of ⌫(2) and ⌫(1), we can deduce the product of , the effective electron-phonon coupling constant, and冑
n. They always appear to-gether in the calculation. The wave functionex and associ-ated variables such as fk1,2are evaluated using the variational method described in Sec. III. We choose me⫽0.117m0 and mh⫽0.0658m0, here m0 is the free-electron mass.15 The di-electric constant⑀ is set to 3. The wave number of the non is deduced from the spacing between the emission pho-non sidebands, which corresponds to 1454 cm⫺1. With冑
n, we can calculate the factor g in Eq.共38兲, which deter-mines the oscillator strength of absorption. Thus we obtain a prediction for the absorption spectra. This prediction will be used in the next section to discuss the optical gain. Direct comparison of the predicted absorption spectrum with ex-periment is so far difficult, because the phonon structures are washed away by the chain length distribution and otherbroadening mechanisms. On the other hand, the ratio be-tween ⌫(1) and⌫(0) determines the parameter⌬K. The fit-ting for the 300 and 10 K Pd0PV film data gives⌬K equal to 0.17/a and 0.19/a, respectively. The fact that they are so close confirms our explanation of the temparature depen-dence. The results of
冑
n and⌬K for four kinds of conju-gated polymers are shown in Table I.The effective electron-phonon coupling constant may be different for other conjugated polymers. In Figs. 2 and 3 we plot the strength of the higher phonon sidebands relative to the zero-phonon band for the absorption and the emission spectra as functions of
冑
n. For larger冑
n, the phonon modes are more displaced in the excited state with respect to the ground state. The spectra are consequently more domi-nated by the higher phonon bands.V. OPTICAL GAIN
Excitons are created through either optical excitation or electron-hole current injection. In addition to the spontane-ous decay discussed in the previspontane-ous section, excitons can also decay radiatively through stimulated emission. There-fore, in the presence of excitons, an electromangetic wave propagating in the polymer sample may experience net gain and be amplified. Gain is achieved when stimulated emission FIG. 1. The observed emission spectrum of Pd0PV film at 300
K. The solid line indicates the theoretical fitting using three Gauss-ian distributions corresponding to zero-, one- and two-phonon emis-sion.
FIG. 2. The ratios of the one phonon共solid line兲 and two pho-non emission rates共dashed line兲 to the zero phonon emission rate as a function of the effective coupling parameter
冑
n.FIG. 3. The ratios of the one-phonon 共solid line兲 and two-phonon absorption rates共dashed line兲 to the zero-phonon absorption rate as a function of the effective coupling parameter
冑
n. TABLE I. The ratio among the experimental emission phononsidebands⌫i, the fitted electron-phonon couling constant
冑
n, thecorresponding phonon displacement factor g, and the fitted momen-tum smearing⌬K are shown for various samples and conditions. Material ⌫0/⌫1 ⌫1/⌫2
冑
n g a⌬KPd0PV film 300 K 0.91 3.33 2.1 0.94 0.17 Pd0PV film 10 K 1.14 3.23 2.1 0.94 0.19 Pd0PV solution 300 K 1.37 3.33 2.1 0.94 0.07 MEH-PPV solution 300 K 1.27 3.12 2.2 1.03 0.08
has a higher rate than absorption. It is more likely to occur in the emission spectral region where overlap with the absorp-tion spectrum is minimal. Our calculaabsorp-tion on the relative strength of the phonon sidebands shows that, due to vibra-tional relaxation, there is a significant redshift of the emis-sion spectrum relative to the absorption. Such shift reduces the spectral overlap and favors gain. Below, we first study the gain coefficient for arbitrary frequency and exciton den-sity, then compare our prediction with the experiment.
The gain coefficient g() is the difference between the emission coefficient and the absorption coefficient
g共兲⫽␥共,nex兲⫺␣共兲⫽Bg¯共兲, 共61兲 with
g
¯共兲⫽共nexa兲␥¯共兲⫺␣¯共兲. 共62兲 It depends only on the intrinsic properties of the polymer chain, i.e.,␥() and␣(), as well as the number of exciton Nex in the chain. With the dimensionful constant B pulled out, the function g¯ () is dimensionless. For a given frequncy
, net gain is achieved when g()⬎0. In other words, the threshold exciton density nexT is given by
nexTa⫽␥ ¯共兲
␣
¯共兲. 共63兲
In the emission spectral region where overlap with the ab-sorption spectrum is small, we have
␥
¯Ⰷ␣¯ , n ex
TaⰆ1. 共64兲
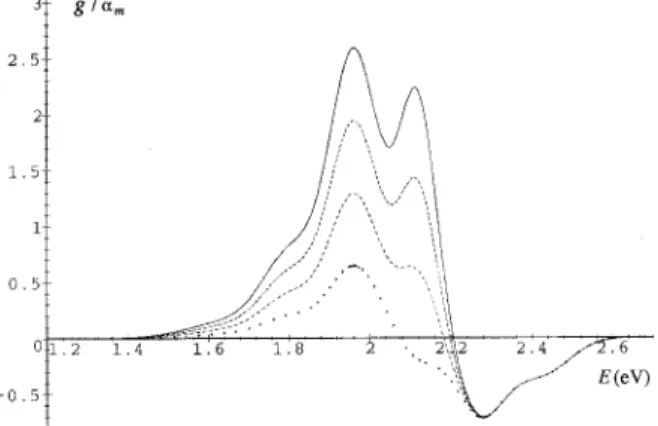
nexa is the fraction of the valence-band electrons that are excited to the conduction-band and form excitons. Therefore, in this case gain will occur even though only a small fraction of electrons in the valence band are excited共see Fig. 4兲.
The optical gain depends on the constant B. Though we cannot calculate B, it can be inferred from the experimental data. Especially the maximum of the absorption coefficient
␣m can be measured directly. Let us discuss Pd0PV film at room temparature as an example. According to our model, in this case the maximum of the absorption occurs at the center of the zero-phonon band0e, i.e.,␣m⫽␣(0
a
)⫽B. Hence the calculable dimensionless function g¯ is equal to the gain co-efficient g(;nex) divided by␣m. In Figs. 5, 6, and 7, we plot the dimensionless function g()/␣mfor various exciton densities nexa. As shown in Fig. 5, gain indeed occurs for the spectral region around the one-phonon emission band FIG. 4. The ratio of⌫0over⌫1as a function of temparature T.
FIG. 5. Optical gain coefficients g() of Pd0PV film at 300 K with exciton densities nex equal to 0.004共solid line兲, 0.002, 0.001 共dashed line兲 and 10⫺5 共crossed line兲, normalized so that the peak absorption rate␣mis equal to 1.
FIG. 6. Optical gain coefficients g() of Pd0PV film at 300 K with exciton densities nexequal to 0.4共solid line兲, 0.3, 0.2 共dashed
line兲, and 0.1 共crossed line兲, normalized so that the peak absorption rate␣mis equal to 1.
FIG. 7. The gain coefficient g(), emission rate and absortion rate 共dashed lines兲 of Pd0PV film at 300 K with exciton densities
nexequal to 0.1, all normalized so that the peak absorption rate␣m
once the exciton density is above 10⫺4 per unit cell. The maximum absorption␣mfor PPV film has been estimated
14 to be about 2.3⫻105 cm⫺1. Since ␣m⫽B, we have B ⫽2.3⫻105 cm⫺1. We denote this value as B
0 for reference. We concentrate now on the gain profile near the maximal gain frequency 1e. According to our model calculation,
␥
¯ (1
e)⫽6.4 and␣¯ ( 1
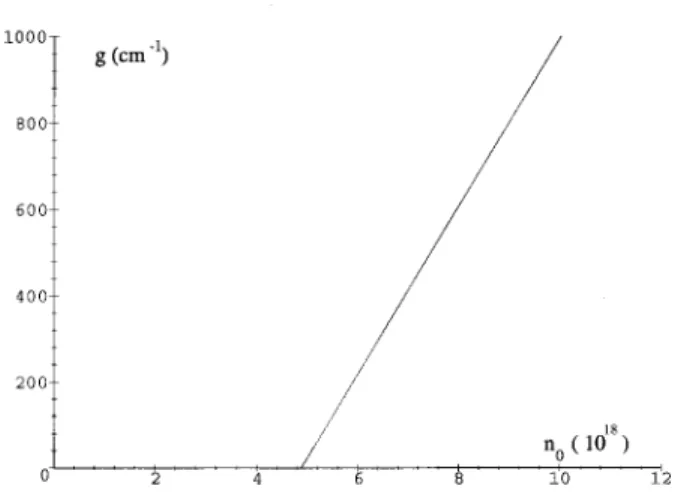
e)⫽0.004. For comparison with experi-ments, it is convenient to use the volume density of exciton, n0, instead of the chain density nex. n0 is related to nex by n0⫽nexa/v0, where v0 is the primitive cell volume. The density of the unsubstituted PPV unit cell has been estimated16to be 7.5⫻1021 cm⫺3, corresponding to a primi-tive cell volume v0 of 1.3⫻10⫺22 cm3. In Fig. 8, we plot the gain coefficient as a function of the exciton volume den-sity n0 at 1e. The onset of gain at the peak of the one-phonon emission band1e is at nexa⫽6.3⫻10⫺4. The corre-sponding threshold exciton volume density n0T for gain is therefore 4.8⫻1018 cm⫺3.
So far, we considered only the optical gain due to the active medium of conjugated polymers. In order to make a practical estimate on the exciton density required for lasing action in a resonator, we also need to know the resonator loss coefficient ␣r(), which arises from the loss mechanisms such as free-carrier absorption, scattering from optical inho-mogeneities, and imperfect mirror reflection. ␣r() is, of course, sensitive to material and mirror quality and is ex-pected to vary from case to case. The actual threshold of lasing is determined by the competition between the active medium gain g() considered above and the resonator loss
␣r(). Lasing occurs only when the former is larger than the latter.
If this actual threshold chain density nexa is much larger than the ratio between¯ and␥ ␣¯ , the formula can be simpli-fied significantly. For frequencies close to1
e
,␥¯⬃103␣¯ . It is hence a good approximation to ignore ␣ in Eq. 共61兲 if nexaⰇ10⫺3, i.e.,
g共兲⯝B0nexa␥¯共兲⫽B0v0n0␥¯ . 共65兲 With this approximation, our prediction for the gain coeffi-cient can be easily generalized to polymers of different
struc-tures. For PPV polymer samples with other packing geom-etry, the primitive cell volume v and the constant B are
different from v0and B0. However, B is proportional to the density of chains. Therefore, the product of B and the primi-tive cell volume is a constant, i.e., Bv⫽B0v0, assuming that one chain contains roughly identical numbers of primitive cells. The gain coefficient for a sample with an arbitraryv is
then
g⫽Bvn0¯␥⫽B0v0n0¯␥⫽3.1⫻10⫺17␥¯ n0 cm
2, 共66兲 where n0⫽nexa/v is the exciton volume density for this sample. Note that this formula is independent of v. It thus
applies to samples with arbitrary packing geometry and den-sity, including even polymer blends for which the active polymer共PPV兲 is dispersed in an inert matrix. The equation above gives a relation between the resonator loss␣rand the actual threshold exciton volume density n0T
␣r⫽3.1⫻10⫺17␥¯ n0
T cm2. 共67兲
For Pd0PV at room temperature, ¯␥⫽6.4 at the peak gain. Thus,
␣r⫽1.98⫻10⫺16n0 T
cm2. 共68兲
This treatment can be easily generalized to other PPV deriva-tive that are similar. The above relation holds for room-temparature Pd0PV samples of any form.
To check the relation Eq.共68兲, we obtain the correspond-ing resonator loss coefficient ␣r(theo) from the threshold density n0T estimated by the various experimental groups.
␣r(theo) is then compared with the experimental values
␣r(exp). The results are shown in Table II for the experi-mental data by Tessler et al.共Ref. 4兲, Frolov et al. 共Ref. 17兲 and Wegmann et al. 共Ref. 5兲. The difference between the theoretical predictions and the experiment data on the reso-nator loss coefficient are within 50%. No adjustable param-eter is introduced in this comparison. This check confirms the reliability of the relation Eq.共67兲. The values of n0T(exp) for Refs. 4 and 17 are estimated from the excitation laser pulse energy, focus area, and the ratio between exciton life-time and the pulse duration. For Ref. 5, it is given by the author. The condition nexa⭓10⫺3 is satisfied in all cases. We estimate the values of ␣r(exp) by separating it into two parts:␣r⫽␣s⫹␣m, where the former is due to inhomogene-ity scattering and the latter imperfect mirror reflection.␣mis equal to (ln R)/2d, where R is the mirror reflectance and d is the resonator length.␣s is assumed to take the usual magni-tute of 10⬃100 cm⫺1.18 In Ref. 4, ␣m is estimated to be 1.8⫻104 cm⫺1with d⫽100 nm and R⫽0.7. The scattering FIG. 8. The gain coefficient as a function of the exciton
vol-umne density n0at the maximal gain frequency of1
e
.
TABLE II. Based on the experimental threshold exciton density
n0, our predicted resonator loss␣r共theo兲 is compared with the
ex-perimental estimate␣r共exp兲.
Ref. 4 Ref. 17 Ref. 5
n0
T共exp兲/cm⫺3 1.4⫻1020 7.4⫻1017 ⬃1017
␣r共theo兲/cm⫺1 2.7⫻104 146 ⬃20
loss is much smaller and can be neglected in this case. For the case of Ref. 17,␣m is estimated to be 0.18 cm⫺1 with d⫽4.5 cm and R⫽0.2. It is much smaller than the scatter-ing loss and can be ignored. There is no mirror used in Ref. 5 and the surface reflectance in unknown; we assume that the loss is also dominated by the scattering just like Ref. 17.
VI. DISCUSSION
We present in this article a model to study the interaction of the excitons and the optical phonons in conjugated poly-mers. In our model, there is only one adjustable parameter, the strength of the electron-phonon coupling. The value of is different for different kinds of polymers since the lattice dynamic equation depends on the backbone structure as well as the side groups of the polymers. is a combination of the phonon polarization vectors ⑀si(k), and is, in principle, cal-culable. should be of order one, because the polarization vectors are all properly normalized. However, it is too com-plicated to calculate this way. For PPV, each unit cell contains eight carbon atoms. There are a total of sixteen degrees of freedom even if we consider only the motion of the atoms within the benzene plane. The normal modes will include fourteen optical phonon branches plus two acoustic branches. The lattice dynamic equation is thus quite in-volved. Instead of calculating it microscopically from the lattice structure, we determine the value of by fitting the experimental data of the relative oscillator strength of the photoluminescence phonon sidebands. However, it turns out that the strength of the zero-phonon band has an observable temperature dependence. In order to fit the temperature de-pendence of the zero phonon emission oscillator strength, we introduce another parameter ⌬K, which is the smearing of the momentum selection rule. The optical transition is al-lowed as long as the difference between the initial and final crystal momenta is within⌬K. The microscopic origin of the smearing includes, for example, lattice distortion, which breaks the discrete translational symmetry, and the emission of acoustic phonons in the process. As discussed in section IV, the oscillator strength of the one and two phonon side-bands in the luminescence spectrum are both independent of ⌬K, while the strength for the zero-phonon band depends on ⌬K mildly. The electron-phonon coupling constantis thus determined by the ratio between the one phonon and the two phonon luminescence sidebands, since this ratio is indepen-dent of the extra parameter⌬K. In other words, even though the value of ⌬K may vary slightly from sample to sample, this uncertainty has no effect on the determination of the intrinsic adjustable parameter. In Sec. IV, for two PPV derivatives共MEH-PPV and PdOPV兲, both in film and solu-tion, are determined. Their values differ by at most 7%. We, therefore, consider this value quite reliable. By fitting the experimental ratio between the strengths of the one- and zero-phonon sidebands, we determine the momentum smear-ing parameter ⌬K. For our samples, it is about 0.17/a, which is about 6% of the size of the first Brillouin zone. This is also quite reasonable. One of our main results is that once the electron-phonon coupling constant is determined by fitting the luminesence spectrum, we can use it to predict the
relative phonon sidebands of the absorption spectrum. For the absorption spectrum, none of the sidebands depend on the momentum smearing ⌬K. They depend only on one single parameter through the factor g(), as seen in Fig. 3. As shown in Sec. IV, the one-phonon sideband is as im-portant as the zero-phonon band, if the material quality can be improved to suppress the imhomogeniety broadening. The large weighting of the one-phonon band reduces the overlap between the absorption and the emission spectra and gives rise to gain in the emission spectral region. In Sec. V we use the calculated absorption spectrum together with the experi-mental emission spectrum to make quantitative predictions on the gain spectrum of the polymer sample.
The existence of a genuine Stokes shift has been ad-dressed by some site-selective photoluminescence 共PL兲 ex-periments recently.9,10In those experiments, the excitation is chosen to be deeply inside the low energy tail of the absorp-tion spectrum to ensure that only long chains are excited, so that no subsequent exciton migration to other chains occurs before emission. The work by Heun et al. shows that for PPV films the zero-phonon band of the PL is quite close to the excitation energy,10whereas Samuel et al. found that the energy of the zero-phonon band is unresolvable but the one-phonon band is about one optical one-phonon energy below the excitation energy. Those authors, thus, concluded that the site-selective absorption is dominated by the zero-phonon band in the absorption spectrum, implying there is no excited state relaxation among the vibrational levels, i.e., a vanishing genuine Stokes shift. However, such interpretation is some-what over simplified, because in the site-selective experi-ments the polymers are most likely to be excited to the zero-phonon states even if the oscillator strength of the higher phonon band is larger than the zero-phonon band. The reason is that the excitation is in the lower energy tail of the DOS, where the population of chains is supposed to increase expo-nentially with their electronic transition energy. Therefore, for a given excitation energy, the population of the chains whose zero-phonon band matches the excitation is much larger than the population of the chains whose higher phonon bands matches the excitation. This is simply because the electronic energy of the former is one or more optical pho-non energy above the latter. For PPV film, the DOS is esti-mated to be a Gaussian distribution with variance about 46 meV.19Since the optical phonon energy is about 0.2 eV, for a given excitation energy the population of the chains excited to the one-phonon states is about only one percent of the population of chains excited to the zero-phonon states. Therefore, unless the oscillator strength of the one-phonon band is one hundred times larger than the zero-phonon band, the site-selective absorption is always dominated by the zero-phonon band. Such dramatic contrast in the oscillator strengths is, of course, very unlikely. In other words, the interpretation of such experiments is complicated by the sharp DOS decay in the energy tail, and the lack of excited state energy relaxation cannot be used to rule out the possi-bility of dominant higher-phonon oscillator strength for one single chain.
Even though conjugated polymers behave like semicon-ductors in many aspects, the gain mechanism is quite differ-ent from the convdiffer-entional semiconductor lasers based on III-V compounds. In compound semiconductors there is
ba-sically no Stokes shift due to excited state lattice relaxation. Gain is achieved only when the pumping is so strong that the initial states are depleted and the final states are filled, i.e., when the quasi-Fermi level for the electrons is pushed up into the conduction band and the quasi-Fermi level for the hole is pulled down into the valence band. Under such pump-ing level, the absorption and emission spectra are displaced with respect each other to allow a net gain. In other words, the intensity of the pumping beam must be in the nonlinear regime where the absorption starts to saturate. In conjugated polymers, no such depletion is required because the vibra-tional relaxation in the excited state automatically provides the redshift of emission respect to absorption, even when the pumping intensity is still low in the linear regime, where the absorption remains basically the same as the ground state. This situation is actually similar to the case of excimer laser, for which population inversion is automatically realized be-cause excimers form only in the excited state. The only dif-ference is that for excimers the Stokes shift is due to the intermolecular distance relaxation, while for conjugated polymers it is due to the intrachain lattice relaxation involv-ing extended phonon modes.
In Sec. V, we check our theoretical prediction on the re-lation between the lasing threshold and the resonator loss with experiment data. Our prediction of the resonator mirror loss agrees with the experiment within 50% 共Ref. 4兲. In the case of the scattering loss, accurate comparison with the ex-periment is impossible because the optical characteristics of the polymer films used in the experiment are not specified. Nonetheless the predictions are all in the right order of mag-nitude 共Refs. 17 and 5兲. We also estimate the minimum threshold exciton density for lasing to be realized in PPV samples. In the experiment of the Cambridge group,4one end of the 100-nm resonator containing the polymer film is a distributed Bragg reflector with reflectance of about 70%. The corresponding loss is about 1.8⫻105 cm⫺1. The total resonator loss is expected to be dominated by the mirrors. We found that to overcome a resonator loss of the order of 104 cm⫺1, a volume exciton density of the order of 1020 cm⫺3 is required. This is much larger than the typical lasing threshold of about 1018 cm⫺3 for conventional com-pound semiconductor laser. However, if the DBR is replaced by a high-reflectance mirror, the resonator loss can be re-duced to the residual loss from mechanisms like light scat-tering by optical inhomogeneity and defect absorption. The loss is usually of the order of only 10⫺100 cm⫺1. The vol-ume exciton density required to overcome this loss is pre-dicted to be only of the order of 1017 cm⫺3. This threshold density is then smaller than that of the conventional semi-conductor lasers. The corresponding exciton number per PPV monomer nexa, is as low as 10⫺4. This is the range shown in Fig. 5. So in principle gain can be realized in high-reflectance resonators when there is only one exciton per ten thousand PPV monomers. Unfortuantely, when a polymer sample is sandwiched between two mirrors of high reflectance, it can no longer be optically excited from outside easily. The question now is whether it is possible to generate excitons to the density of the order of 1017 cm⫺3 through electric current injection. This corresponds to the level of excitations required for electrically pumped polymer laser. The obstacle to electric pumping of conjugated polymer laser
is twofold. One is the difficulties of carrier injection. Another is related to the fact that, even if current can be easily in-jected, the electrons and holes form both singlet and triplet excitons, with a rough ratio of 1:3 based on spin statistics. In most cases, the triplet exciton energy is actually lower than the singlet. The singlet ratio is thus even lower than 25%. Only singlet excitons will undergo optical transitions. The corresponding total exciton density required for electric pumping polymer laser with residual loss of 10 ⫺100 cm⫺1is therefore around 1018 cm⫺3, comparable to the typical density for compound semiconductor lasers. Still, as discussed above, the corresponding total number of exci-ton per PPV monomer is only less than 10⫺3. There is no fundamental reason preventing the realization of such exci-ton density, if the injection problem can be solved.
Photoinduced absorption due to interchain excited state, which spectrally overlaps with the stimulated emission, has been reported to be an important factor to diminish the gain for some polymer samples. It has been argued that interchain species, identified as excimers or polaron pairs, have a domi-nating quantum yield.20,21Such claims are, however, contra-dicted by other works on the absolute PL quantum efficiency,22 and some controversies remain. Even though interchains species do seem to appear in high-quantum yield for some polymers such as CN-PPV,23 their polulation can be controlled by the side groups. For example, large and bulky side groups will presumably isolate the polymer back-bones from each other and prevent the formation of inter-chain species. In this paper, we do not attempt to include the effect of such species.
The wavelength at which the gain coefficient is maximal depends slightly on the side groups of PPV. In general, the emission spectrum of unsubstituted PPV is blueshifted with respect to other soluble derivatives. The possible reason is that the former is polymerized only after coating on the sub-strate, while the latter can be directly spin coated in the poly-mer form. The effective conjugation length of unsubstituted PPV film is therefore expected to be smaller than films made of other derivatives. This explains its higher band gap and emission spectrum. Our theoretical calculations do not as-sume any particular value of the band gap. Therefore, the predictions on the gain coefficient around the one-phonon emission peak does not depend on where it actually is. For example, the peak is at 2.35 eV共547 nm兲 for PPV, and 1.95 eV 共631 nm兲 for MEH-PPV.
VII. CONCLUSION
Starting from a model Hamiltonian for the exciton-phonon coupling, we study the absorption and luminescnece spectra including the phonon side bands. The electron-phonon coupling constant is the only fitting parameter, de-termined by fitting the experimental ratio between the one-and two-phonon sidebone-ands in the photoluminescence spec-trum. We then calculate the absorption spectrum and predict that the one phonon band is as important as the zero-phonon band. This is contrary to the case of luminescence, which is dominated by the zero-phonon band for the samples we con-sidered. The gain coefficient is calculated based on the re-sults. We derive a general relation between the resonator loss
and the volume exciton density required for lasing threshold, applicable to PPV samples with arbitrary packing geometry and density. Our result is in good agreement with the experi-ments for both high- and low-loss resonators. For low-loss resonator with loss of 10⫺100 cm⫺1, the threshold singlet exciton density required for lasing action is of the order of 1017 cm⫺3. This is, to our knowledge, the first theoretical work that makes quantitative predictions on the gain spec-trum of such systems. The implication on the feasibility of polymer laser based on PPV is discussed.
ACKNOWLEDGMENTS
共H.F.M.兲 is grateful to W.S. Fann and J.H. Hsu for useful discussions and for providing experimental data prior to pub-lication.共C.H.C.兲 would like to thank M. C. Chang for dis-cussions. The authors also thank the National Center for Theoretical Sciences of Taiwan, R.O.C., for hospitality and support. This work was supported by the National Science Council of Taiwan, R.O.C., under Contract No. NSC 88-2112-M-009-006 共H.F.M.兲 and No. NSC 88-2112-M-003-017 共C.H.C兲.
*Electronic address: [email protected]
†Electronic address: [email protected]
1For review, see S. Greenham and R. Friend, in Solid State Physcs,
edited by H. Ehrenreich and F. Spaepen共Academic Press, Bos-ton, 1995兲.
2J. Burroughes, D. Bradley, A. Brown, R. Marks, K. Mackay, R.
Friend, P. Burns, and A. Holmes, Nature 共London兲 347, 539
共1990兲.
3D. Moses, Appl. Phys. Lett. 60, 3215共1992兲.
4N. Tessler, G. Denton, and R. Friend, Nature共London兲 382, 539 共1996兲.
5G. Wegmann, H. Giessen, A. Greiner, and R. Mahrt, Phys. Rev.
B 57, R4218共1998兲.
6M. McGehee, R. Gupta, S. Veenstra, E. Miller, M. Diaz-Garcia,
and A. Heeger, Phys. Rev. B 58, 7035共1998兲.
7J. Pollmann and H. Bu¨ttner, Phys. Rev. B 16, 4480共1977兲. 8M. Matsuura and H. Bu¨ttner, Phys. Rev. B 21, 679共1980兲. 9N. Harrison, D. Baigent, I. Samuel, R. Friend, A. Grimsdale, S.
Moratti, and A. Holmes, Phys. Rev. B 53, 15 815共1996兲.
10S. Heun, R. Mahrt, A. Greiner, U. Lemmer, H. Bassler, D.
Hal-liday, D. Bradley, P. Burn, and A. Holmes, J. Phys.: Condens. Matter 5, 247共1993兲.
11T.D. Lee, F. Low, and D. Pines, Phys. Rev. B 90, 297共1953兲.
12H. Haken, Quantum Field Theory of Solids: An Introduction 共North-Holland, New York, 1976兲.
13R. Loudon, Am. J. Phys. 27, 649共1959兲.
14W.S. Fann and J.H. Hsu, J. Phys. Chem. A 103, 2375共1999兲. 15
P. Gomes da Costa and E. Conwell, Phys. Rev. B 48, 1993
共1993兲.
16P. Gomes da Costa, R. Dandra, and E. Conwell, Phys. Rev. B 47,
1800共1993兲.
17S. Frolov, W. Gellermann, M. Ozaki, K. Yoshino, and Z.
Vard-eny, Phys. Rev. Lett. 78, 729共1997兲.
18B. Saleh and M. Teich, Fundamentals of Photonics共Wiley, New
York, 1991兲.
19R. Kersting, U. Lemmer, R. Mahrt, K. Leo, H. Kurz, H. Ba¨ssler,
and E. Go¨bel, Phys. Rev. Lett. 70, 3820共1993兲.
20M. Yan, L. Rothberg, F. Papadimitritrakopoulos, M. Galvin, and
T. Miller, Phys. Rev. Lett. 72, 1104共1994兲.
21J. Hsu, M. Yan, T. Jedju, and L. Rothberg, Phys. Rev. B 49, 712 共1994兲.
22N. Greenham, I. Samuel, G. Hayes, R. Phillips, Y. Kessener, S.
Moratti, A. Holmes, and R. Friend, Chem. Phys. Lett. 241, 89
共1995兲.
23I.D.W. Samuel, G. Rumbles, and C.J. Collison, Phys. Rev. B 52,