Activation of the ERK signal transduction pathway
by Epstein–Barr virus immediate-early protein Rta
Yu-Hsiu Lee,
1Ya-Fang Chiu,
2Wen-Hung Wang,
2Li-Kwan Chang
3and Shih-Tung Liu
2Correspondence Shih-Tung Liu [email protected]
1Institute of Microbiology and Immunology, National Yang-Ming University, 155 Linong Street
Section 2, Taipei 112, Taiwan, ROC
2Molecular Genetics Laboratory, Department of Microbiology and Immunology, Chang-Gung
University, 259 Wen-Hua 1st Road, Kwei-Shan, Taoyuan 333, Taiwan, ROC
3Institute of Microbiology and Biochemistry, National Taiwan University, 1 Roosevelt Road Section
4, Taipei 106, Taiwan, ROC
Received 7 May 2008 Accepted 9 June 2008
BRCA1-associated protein 2 (BRAP2) is known to interact with the kinase suppressor of Ras 1 (KSR1), inhibiting the ERK signal transduction cascade. This study found that an Epstein–Barr virus (EBV) immediate-early protein, Rta, is a binding partner of BRAP2 in yeast and confirmed the binding in vitro by a glutathione S-transferase pull-down assay and in vivo by
co-immunoprecipitation in 293(maxi-EBV) cells. Binding studies also showed that Rta and KSR1 interacted with the C-terminal 202 aa region in BRAP2. Additionally, Rta appeared to prevent the binding of KSR1 to BRAP2, activating the ERK signal transduction pathway and the transcription of an EBV immediate-early gene, BZLF1. Activation of the ERK signal transduction pathway by Rta may be critical for the maintenance of the lytic state of EBV.
INTRODUCTION
Epstein–Barr virus (EBV) is a human herpesvirus that infects
lymphoid and epithelial cells. Although EBV infection is
commonly asymptomatic, infection by this virus also causes
infectious mononucleosis (Diehl et al., 1968) and is closely
associated with many neoplastic disorders (Einhorn et al.,
1970; Gunven et al., 1970; Johansson et al., 1970; Klein et al.,
1970). Although EBV typically remains latent after infection
of B lymphocytes, the virus must enter a lytic cycle to produce
virus particles. During the onset of the lytic cycle, the virus
expresses the proteins Rta and Zta, encoded by BRLF1 and
BZLF1, respectively, to activate the genes required for the viral
lytic cycle (Chevallier-Greco et al., 1986; Chiu et al., 2007;
Feederle et al., 2000; Granato et al., 2006; Hardwick et al.,
1988; Lu et al., 2006). Although the exact means by which the
EBV lytic cycle is activated in vivo is unknown, activation in
vitro occurs after latently infected cells are exposed to
12-O-tetradecanoylphorbol-13-acetate (TPA), calcium ionophores,
transforming growth factor (TGF)-b1 or anti-IgG (Daibata
et al., 1990; Faggioni et al., 1986; zur Hausen et al., 1978).
TPA, anti-IgG and TGF-b1 are known to activate the ERK
signal transduction pathway (Fahmi et al., 2000; Fenton &
Sinclair, 1999; Gao et al., 2001; Satoh et al., 1999), which
ultimately activates transcription of BZLF1 and the EBV lytic
cycle (Borras et al., 1996; Flemington & Speck, 1990).
It is known that EBV must express Zta to activate its lytic
genes (Chevallier-Greco et al., 1986; Chiu et al., 2007; Feederle
et al., 2000). Earlier studies have established that Rta
upregulates transcription of the Zta gene, BZLF1 (Adamson
et al., 2000; Ragoczy et al., 1998; Zalani et al., 1996). This
activation is associated with activation of the p38 and JNK
signal transduction pathway, causing the phosphorylation of
ATF1/2 and the activation of transcription through an ATF1/
2 site in the ZII region of the promoter (Adamson et al.,
2000). However, the exact means by which Rta activates these
signal transduction cascades is unknown. In this study, we
used a yeast two-hybrid analysis to show that Rta interacts
with BRCA1-associated protein 2 (BRAP2, also known as
IMP), a protein that is known to interact with the kinase
suppressor of Ras 1 (KSR1) (Matheny et al., 2004). Mu¨ller et
al. (2001) demonstrated that KSR1 functions as a scaffold,
providing a platform for the phosphorylation of MEK1/2 and
ERK1/2 (Muller et al., 2001; Nguyen et al., 2002; Roy et al.,
2002). However, BRAP2 appears to prevent KSR1 from
interacting with the cytoplasmic membrane and
homo-oligomerization, thus inhibiting the ERK signal transduction
pathway (Chen et al., 2008; Matheny & White, 2006; Matheny
et al., 2004). This study demonstrated that Rta prevents the
binding of BRAP2 to KSR1, activating the ERK signal
transduction pathway and the transcription of BZLF1 to
influence the viral lytic cycle.
METHODS
Cell lines and EBV lytic induction.293 cells infected with maxi-EBV (a mutant maxi-EBV that contains an F replicon and can be
maintained in Escherichia coli)[293(maxi-EBV) cells] or its mutant derivative, MI-270, which contains a transposon insertion in BRLF1 (Chiu et al., 2007; Delecluse et al., 1998), were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10 % fetal calf serum. Cells were treated with 30 ng TPA ml21and 3 mM sodium butyrate to induce the EBV lytic cycle (Chang & Liu, 2000; Davies et al., 1991; Luka et al., 1979).
Plasmids.A DNA fragment containing the BRAP2 gene was PCR amplified using primers 59-CGCGGATCCGAATCCATGAGTGTG-TCACTGGTTGTTATCCG-39 and 59-CGGGGTACCAAGCTTTC-AGGGATGTCTGTTGCTCTGAAGG-39, and a human testis cDNA library (BD Clontech) as template. Plasmids pGEX-BRAP2 and pHA-BRAP2 were constructed by inserting this PCR fragment into the BamHI/SmaI sites in pGEX-4T1 (Amersham Biosciences) and BamHI/KpnI sites in pcDNA3-HA2, respectively. A plasmid expres-sing full-length BRAP2 fused to a glutathione S-transferase (GST) sequence at the N terminus (pGST-BRAP2) was constructed by inserting the PCR fragment into pENTR3C (Invitrogen) at the BamHI/EcoRI sites and transferring this fragment to pDEST27 using the Gateway system (Invitrogen). Plasmids that expressed deleted GST–BRAP2, including BN262, BC399 and B263/398, which contain the BRAP2 regions aa 1–262, 399–600 and 263–398, respectively, were constructed in a similar way. Plasmid pET-Rta contained BRLF1 transcribed from the T7 promoter (Chang et al., 2004). Plasmids pCMV-R and pCMV-Z contained BRLF1 and BZLF1, respectively, transcribed from the cytomegalovirus immediate-early promoter (Chang et al., 1998; Hung & Liu, 1999). Plasmid pCMV-3 is an empty vector that was used to construct pCMV-R (Chang et al., 1998). Plasmid pHA-Rta is a plasmid that expresses haemagglutinin (HA)-tagged Rta (Chang et al., 2004). Plasmids pHA-RN415, pHA-RN315, RN190, R190/315, RC255, R255/415, pHA-RC361, pHA-RC416 and pHA-191/415, which express the regions aa 1–415, 1–315, 1–190, 190–315, 255–605, 255–415, 361–605, 416–605 and 191–415 of HA–Rta (Chang et al., 2004), respectively, were constructed to map the regions in Rta that interact with BRAP2. Plasmids pCDNA3-Flag-KSR1 (Zhang et al., 1997) and pSG/RNLSm (Hsu et al., 2005) express Flag-tagged KSR1 (Flag–KSR1; obtained from Deborah K. Morrison, NCI-Frederick, MD, USA) and an Rta protein with a mutated nuclear localization signal (NLS; obtained from Tsuey-Ying Hsu, 1 Jen-Ai Rd Section 1, Taipei 100, Taiwan, ROC), respectively. Plasmids pZp-Luc and pNS3 were constructed by inserting the 2240 to+38 and 257 to +38 regions of the BZLF1 promoter into the HindIII/SmaI sites in pGL2-Basic (Promega), respectively.
Yeast two-hybrid screen.Proteins that interacted with Rta were identified using a yeast two-hybrid screen with a bait plasmid, pR476, and a human testis cDNA library, according to a method described elsewhere (Chang et al., 2004).
Binding of Rta to BRAP2 in vitro.An E. coli BL21(DE3)(pGEX-BRAP2) lysate was prepared and a GST pull-down assay was performed as described previously (Chang et al., 2004). Glutathione–Sepharose 4B beads (Amersham Biosciences) were then added to the lysate to allow the binding of GST–BRAP2 to the beads. The beads (30 ml) were then added to an E. coli BL21(DE3)(pET-Rta) lysate (500 ml) or a lysate prepared from 293T cells transfected with a plasmid expressing Rta or its deletion derivatives. The reaction mixture was incubated on ice for 1 h. After the beads had been washed in RIPA buffer, electrophoresis sample buffer was added to elute the proteins from the beads by heating at 95uC for 5 min. Rta was finally detected by immunoblotting. His–Rta was purified from E. coli BL21(DE3)(pET-Rta) and bound to Ni-NTA agarose beads (Qiagen). The beads were added to the E. coli BL21(DE3)(pGEX-BRAP2) lysate. The binding of GST–BRAP2 to the beads was detected by immunoblotting with anti-GST antibody.
Competitive binding of Rta and KSR1 to BRAP2. 293T cells (56106) were transfected with 4 mg pGST-BRAP2, pCMV-R (0, 3, 6 or 12 mg) and 4 mg pCDNA3-Flag-KSR1. After 24 h of culture, lysates (500 ml) were prepared from the cells using a RIPA buffer without Triton X-100 and sodium deoxycholate. Glutathione–Sepharose beads (30 ml) were then added to the lysate prepared from cells that had been transfected with pGST-BRAP2. The mixture was mixed at 4uC for 1 h. The beads were washed in RIPA buffer and then mixed with lysate from cells that had been transfected with pCMV-R. After they had been mixed and washed, the beads were finally mixed with lysate from cells that had been transfected with pCDNA3-Flag-KSR1. Glutathione–Sepharose beads were also added to a mixture that contained 500 ml each of the lysates from 293T cells transfected with 4 mg pGST-BRAP2 and pCDNA3-Flag-KSR1. His–Rta, purified from E. coli BL21(DE3)(pET-Rta), was then added to the lysate mixture. Proteins that were bound to the beads were eluted with electrophor-esis sample buffer and analysed by immunoblotting.
Immunoprecipitation.293(maxi-EBV) cells (56106) were treated with TPA and sodium butyrate to induce the EBV lytic cycle. Co-immunoprecipitation of Rta and BRAP2 was performed with anti-Rta (1 : 500 dilution) (Argene) and mouse polyclonal anti-BRAP2 antibody (1 : 5000 dilution) as described previously (Chang et al., 2004). Immunoblotting was subsequently conducted to identify the co-immunoprecipitated proteins.
Indirect immunofluorescence analysis.293(maxi-EBV) cells were transfected with pCMV-Z or treated with TPA and sodium butyrate for 24 h to induce expression of Rta. Cells were collected by centrifugation, plated on poly-L-lysine (Sigma)-coated coverslips and fixed with 4 % paraformaldehyde in PBS at 4uC for 30 min. The cells were then incubated with Rta monoclonal antibody (mAb) and rabbit anti-BRAP2 polyclonal antibody for 1 h, followed by Alexa Fluor 488-conjugated goat anti-mouse IgG polyclonal antibody and Alexa Fluor 594-conjugated goat anti-rabbit IgG polyclonal antibody (Molecular Probes). After 1 h of incubation, cells were stained with 49,6-diamidino-2-phenylindole (DAPI). Finally, cells were washed in PBS, mounted in Citifluor (Agar Scientific) and examined under a confocal laser-scanning microscope (model LSM 510 META; Zeiss). The rabbit anti-BRAP2 antibody was produced using synthesized pepticle (KLPSRKGRSKRGK).
Activation of the ERK signal transduction pathway by Rta.293T cells transfected with pCMV-R were lysed using electrophoresis sample buffer 24 h after transfection. Proteins in the lysate were detected by immunoblotting. U0126 (10 mM; Cell Signalling) was added 4 h prior to lysate preparation to inhibit the ERK signal transduction pathway.
Immunoblot analysis.Proteins were detected by immunoblotting as described previously (Chang et al., 2004). Proteins on the membrane were detected using primary antibodies and horseradish peroxidase-conjugated secondary antibodies, and visualized using SuperSignal West Pico Chemiluminescent substrate (Pierce). Anti-HA was purchased from Roche and anti-a-tubulin and anti-Flag mAbs from Sigma. Anti-b-actin mAb was purchased from Novus Biologicals. Rabbit ERK1/2, phospho-ERK1/2 (Thr202/Tyr204), anti-MEK1/2 and anti-phospho-anti-MEK1/2 (Ser217/221) antibodies were purchased from Cell Signaling Technology. Rabbit anti-GST antibody was purchased from Santa Cruz Biotechnology. Mouse anti-Zta mAb was purchased from Argene. Anti-EA-D mAb was purchased from Millipore. Anti-BRAP2 antibody was produced in mice with bacterially expressed GST–BRAP2.
Transient transfection assay.Plasmids (800 ng) were transfected into 1.56105 293T cells using Lipofectamine 2000 (Invitrogen).
Luciferase activity was measured using a luminometer (model LB593; Berthod) as described previously (Chang et al., 1998). Each sample
was prepared in duplicate and each transfection experiment was repeated three times. The copy number of plasmids transfected into cells was determined by real-time PCR using a set of primers specific for the ampicillin-resistance gene (Chang & Liu, 2000). Luciferase activity was normalized to the copy numbers of plasmids.
RESULTS
Interaction between Rta and BRAP2 in yeast and
in vitro
A yeast two-hybrid screen was performed using a bait
plasmid, pR476 (Chang et al., 2004), to screen a human
testis cDNA library. A total of 6610
5transformants was
screened and 10 cellular proteins that interacted with Rta
were identified. Sequencing analysis revealed that one of
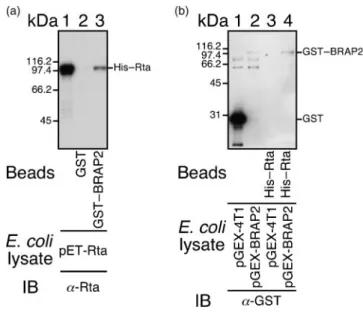
these proteins was BRAP2. A GST pull-down assay was
subsequently performed using bacterially expressed GST
and GST–BRAP2. GST or GST–BRAP2 bound to
glu-tathione–Sepharose beads was mixed with lysate prepared
from E. coli BL21(DE3)(pET-Rta) to confirm the
inter-action. After washing, proteins bound to the beads were
eluted and analysed by immunoblotting with anti-Rta
antibody. The results revealed that His–Rta in the lysate
(Fig. 1a, lane 1) was pulled down by GST–BRAP2–
glutathione–Sepharose beads (Fig. 1a, lane 3) but not by
GST–glutathione–Sepharose beads (Fig. 1a, lane 2). In
another set of experiments, His–Rta bound to Ni-NTA
agarose beads was mixed with E. coli
BL21(DE3)(pGEX-4T1) or E. coli BL21(DE3)(pGEX-BRAP2) lysate.
Immuno-blot analysis indicated that His–Rta bound to Ni-NTA
agarose beads retained GST–BRAP2 in the E. coli
BL21(DE3)(pGEX-BRAP2) lysate (Fig. 1b, lane 4). The
binding was not caused by an interaction between GST–
BRAP2 and the His tag in His–Rta, as an unrelated
His-tagged protein, His–FenB (Lin et al., 1998), did not interact
with GST–BRAP2 (data not shown). A negative control
also indicated that the His–Rta–Ni-NTA agarose beads did
not pull down GST (Fig. 1b, lane 3).
Interaction between BRAP2 and Rta in vivo
293(maxi-EBV) cells were treated with TPA and sodium
butyrate to induce the expression of Rta. Immunoblot
analysis indicated that Rta in the lysate (Fig. 2a, lane 1) was
immunoprecipitated by anti-Rta antibody (Fig. 2a, lane 3)
and co-immunoprecipitated with BRAP2 by anti-BRAP2
antibody (Fig. 2a, lane 4) but not by anti-Flag antibody
(Fig. 2a, lane 2). Additionally, BRAP2 in the lysate (Fig. 2b,
lane 1) was immunoprecipitated by anti-BRAP2 antibody
(Fig. 2b, lane 3) and co-immunoprecipitated with Rta by
anti-Rta antibody (Fig. 2b, lane 4) but not by anti-Flag
antibody (Fig. 2b, lane 2). These results showed that
BRAP2 interacts with Rta in 293(maxi-EBV) cells.
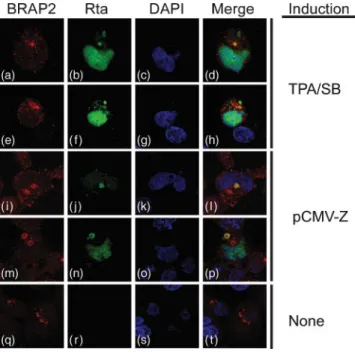
Subcellular localization of Rta and BRAP2
Immunofluorescence analysis of 293(maxi-EBV) cells was
performed with confocal microscopy to locate Rta and
BRAP2. Following lytic induction with TPA and sodium
Fig. 1. Interaction between Rta and GST–BRAP2 in vitro. (a) An E. coli BL21 DE3(pET-Rta) lysate was added to GST and GST– BRAP2 bound to glutathione–Sepharose beads. Proteins bound to the beads were extracted and analysed by immunoblotting (IB) with anti-Rta antibody. Lane 1 was loaded with the E. coli (pET-Rta) lysate. (b) Lysates prepared from E. coli BL21(DE3)-(pGEX4T-1) (lanes 1 and 3) and E. coli BL21(DE3)(pGEX-BRAP2) (lanes 2 and 4) were added to His–Rta–Ni-NTA agarose beads. Proteins bound to the beads were extracted and detected by IB using anti-GST antibody. Lane 1 in (a) and lanes 1 and 2 in (b) were loaded with 0.05 % of the cell lysate.
Fig. 2. Co-immunoprecipitation of Rta and BRAP2. Anti-BRAP2, anti-Rta and anti-Flag antibodies were added to the lysate prepared from 293(maxi-EBV) cells treated with TPA and sodium butyrate. Proteins immunoprecipitated (IP) by the antibody were analysed by immunoblotting (IB) with anti-Rta antibody (a) or anti-BRAP2 antibody (b). Lane 1 in (a) was loaded with 0.1 % of the cell lysate, whilst lane 1 in (b) was loaded with 2 % of the cell lysate.
butyrate, Rta was detected in the cytoplasm and nucleus
(Fig. 3b and f). However, BRAP2 was detected only in the
cytoplasm (Fig. 3a and e). Merged images showed that Rta
and BRAP2 were co-localized in the cytoplasm (Fig. 3d and
h). Similar co-localization was also observed in cells
transfected with pCMV-Z (Fig. 3i–p), but not in cells that
were latently infected by EBV (Fig. 3q–t).
Mapping the interaction domains in BRAP2, KSR1
and Rta
Plasmids expressing HA–Rta and its deletion derivatives
(Fig. 4a) were transfected into 293T cells and lysates were
prepared. Immunoblot analysis of these lysates confirmed
that these proteins were expressed (Fig. 4b). GST–
BRAP2–glutathione–Sepharose beads were then added
to the lysates. Immunoblot analysis using anti-HA
antibody indicated that the beads pulled down HA–Rta,
HA–RN415, HA–RN315, HA–RN190, HA–RC255 and
HA–RC416 (Fig. 4c, lanes 1–4, 6 and 8), indicating that
the regions aa 1–190 and 416–605 in Rta interacted with
BRAP2. However, the beads did not pull down proteins
that contained the Rta regions aa 191–315 (HA–R191/
315) and 255–415 (HA–R255/415) (Fig. 4c, lanes 5 and
7). Additionally, as expected, GST–glutathione–Sepharose
beads did not pull down any of these Rta proteins (data
not shown). A similar experiment with a GST fusion
protein containing full-length or a segment of BRAP2
(Fig. 4e, lanes 2–5) revealed that Rta was pulled down by
GST–BRAP2 (Fig. 4d, e, lane 8) and GST–BC399 bound
to glutathione–Sepharose beads (Fig. 4d, e, lane 11).
However, Rta was not pulled down by GST–BN262 or
GST–B263/398 bound to glutathione–Sepharose beads
(Fig. 4d), which contained the regions aa 1–262 (Fig. 4e,
lane 9) and 263–398 (Fig. 4e, lane 10), respectively.
Furthermore, KSR1, which was expressed from
pCDNA3-Flag-KSR1 in 293T cells (Fig. 4f, lane 6), was pulled down
by GST–BRAP2 and GST–BC399 bound to glutathione–
Sepharose beads (Fig. 4f, lanes 8 and 11) but not by GST–
BN262
or
GST–B263/398
bound
to
glutathione–
Sepharose beads (Fig. 4f, lanes 9 and 10). These results
demonstrated that both Rta and KSR1 interacted with
GST–BC399.
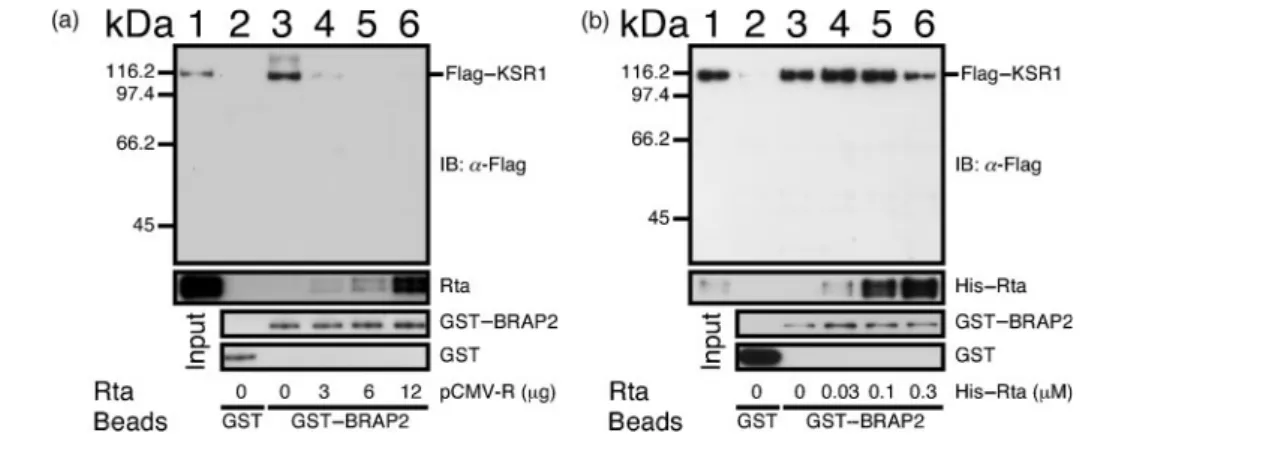
Competitive binding of Rta and BRAP2 to KSR1
As Rta and KSR1 both interact with the C-terminal 202 aa
region in BRAP2, we investigated whether Rta and KSR1
compete for the same binding sites in BRAP2. GST–
BRAP2–glutathione–Sepharose beads were incubated with
293T lysates from cells that had been transfected with
various amounts of pCMV-R to enable the binding of Rta
to the beads. The beads were then incubated with a 293T
lysate that contained Flag–KSR1 to determine how Rta
affected the binding of Flag–KSR1 to BRAP2. Immunoblot
analysis
indicated
that
GST–BRAP2–glutathione–
Sepharose beads pulled down KSR1 (Fig. 5a, lane 3).
However, GST–glutathione–Sepharose beads did not pull
down KSR1 (Fig. 5a, lane 2), confirming that KSR1
interacts with BRAP2. The binding of Flag–KSR1 to GST–
BRAP2 was reduced by pre-incubating the GST–BRAP2–
glutathione–Sepharose beads in lysate from cells
trans-fected with 3 mg pCMV-R (Fig. 5a, lane 4). Pre-incubating
the beads in lysate from cells transfected with 6 or 12 mg
pCMV-R further reduced the binding of Flag–KSR1 to
BRAP2 to a level that was undetectable by immunoblotting
(Fig. 5a, lanes 5 and 6), indicating that Rta impeded the
binding of Flag–KSR1 to BRAP2. Meanwhile, in a similar
study, two lysates prepared from 293T cells that had been
transfected separately with
pCDNA3-Flag-KSR1 and
pGST-BRAP2 were mixed in equal volumes. After adding
glutathione–Sepharose beads, His–Rta purified from an E.
coli lysate was then added to the lysate mixture.
Concentrations up to 0.1 mM bacterially expressed His–
Rta did not affect the interaction between Flag–KSR1 and
GST–BRAP2 and binding of the proteins to glutathione–
Sepharose beads (Fig. 5b, lanes 3–5), whilst adding 0.3 mM
His–Rta decreased the amount of Flag–KSR1 that was
retained by the beads (Fig. 5b, lane 6), showing that Rta
dislodges KSR1 from BRAP2.
Fig. 3. Indirect immunofluorescence analysis. 293(maxi-EBV) cells were treated with TPA and sodium butyrate (SB) (a–h) or transfected with pCMV-Z (i–p) to activate the EBV lytic cycle. Cells that were not treated with TPA and sodium butyrate or were not transfected with pCMV-Z are shown in (q)–(t). Cells were incubated with rabbit anti-BRAP2 polyclonal antibody (a, e, i, m and q; red) and anti-Rta mAb (b, f, j, n and r; green). DAPI staining (c, g, k, o and s; blue) revealed the positions of the nucleus. Cells were examined under a confocal laser-scanning microscope. Merged images are also shown (d, h, l, p and t).
Activating the ERK signal transduction pathway
by Rta
As Rta prevents BRAP2 from binding to KSR1, Rta may
eliminate the inhibition of the ERK signal transduction
pathway by BRAP2. Immunoblot analysis indicated that
transfecting 0.5–2 mg pCMV-R into 293T cells did not
change the overall level of MEK1/2 and ERK1/2 in the cell
(Fig. 6a, lanes 2–4, 6 and 8), but promoted the
phosphorylation of MEK1/2 and ERK1/2 (Fig. 6a, lanes
1–4). Additionally, adding U0126, which inhibits the
activity of MEK1/2, prevented the phosphorylation of
ERK1/2 activated by Rta and the Rta NLS mutant (Fig. 6a,
lanes 6 and 7). We also found that transfecting
pHA-BRAP2 inhibited the phosphorylation of MEK1/2 and
ERK1/2 activated by Rta and the Rta NLS mutant (Fig. 6a,
lanes 8 and 9). A similar study was also performed using
BJAB, Akata, EBV-negative Akata and P3HR1 cells; the
activation of ERK1/2 phosphorylation by Rta in these cell
lines was less obvious than that in 293T cells (data not
shown).
Activation of ERK1/2 by the BRAP2-interacting
domains in Rta
We also studied whether the expression of two Rta
fragments that bind to BRAP2, HA–RN190 (Fig. 4a) and
HA–RC361 (a protein that contains the C-terminal region
of Rta from aa 361–605), influenced the phosphorylation
of ERK1/2. Immunoblot analysis indicated that, although
transfecting a plasmid expressing HA–R191/415, which
does not interact with BRAP2, had little effect on the
phosphorylation of ERK1/2 (Fig. 6b, lane 4), transfecting a
plasmid that expressed HA–RC361 increased the
phos-phorylation level of ERK1/2 (Fig. 6b, lane 5). Similar
activation was also observed upon transfection of a plasmid
that expressed HA–RN190 (Fig. 6b, lane 3). Co-expressing
HA–RN190 and HA–RC361 also increased the
phosphor-ylation levels of MEK1/2 and ERK1/2 (Fig. 6b, lane 6).
Furthermore, similar activation was also observed after
co-expressing HA–RN190 and HA–RN416 (Fig. 4a and data
not shown). The addition of U0126 did not prevent the
phosphorylation of MEK1/2, but inhibited the
phosphor-Fig. 4. Mapping of the interaction domains in Rta, BRAP2 and KSR1. Plasmids expressing HA-tagged Rta (HA–Rta) and its deletion derivatives (a) were transfected into 293T cells. Proteins in the lysates were analysed by immunoblotting with anti-HA antibody (b). Proteins that were pulled down by GST–BRAP2–glutathione–Sepharose beads were analysed by immunoblotting with anti-HA antibody (c). The regions in BRAP2 that interacted with Rta and KSR1 were analysed in a similar way. GST fusion proteins containing full-length or a segment of BRAP2 (d) bound to glutathione–Sepharose beads were added to lysates prepared from 293T cells that had been transfected with pCMV-R (e) or pCDNA3-Flag-KSR1 (f). Lanes 1 and 6 in (e) and (f) were loaded with the cell lysate.
Fig. 5. Inhibition of binding of BRAP2 to KSR1 by Rta. (a) Glutathione–Sepharose beads were added to the lysate prepared from 5¾106293T cells transfected with 4 mg pGST–BRAP2 (lanes 3–6) to enable the binding of GST–BRAP2 to the beads. The beads were washed and mixed with 500 ml of a lysate prepared from the same number of 293T cells transfected with 0, 3, 6 or 12 mg pCMV-R (lanes 3–6). The beads were finally mixed with a lysate prepared from 5¾106293T cells transfected with pCDNA3-Flag-KSR1. (b) Lysates (500 ml) containing GST–BRAP2 and Flag–KSR1 were mixed. Glutathione–Sepharose beads and His–Rta were subsequently added to the lysate mixture as indicated. In (a) and (b), Flag-tagged KSR1 bound to the beads was extracted and examined by immunoblotting (IB) with anti-Flag antibody. GST–glutathione–Sepharose beads were also used to demonstrate the lack of binding between GST and KSR1 (lane 2). Lane 1 was loaded with 0.02 % of the lysate. Flag–KSR1 and Rta bound to GST–BRAP2–glutathione–Sepharose beads were detected with anti-Flag and anti-Rta antibody, respectively; GST and GST–BRAP2 were detected with anti-GST antibody.
Fig. 6. Activation of the ERK signal transduction pathway by Rta and its fragments. Lysates were prepared from 293T cells transfected with 0–2 mg pCMV-R (a) or plasmids expressing HA-tagged Rta and its deletion derivatives (b). Cells transfected with an empty vector, pcDNA3-HA2, were used as a negative control. MEK1/2 (MEK), phosphorylated MEK1/2 (pMEK), ERK1/ 2 (ERK), phosphorylated ERK1/2 (pERK), Rta, BRAP2 and b-actin in the lysate were detected by immunoblotting with their respective antibodies. U0126 in DMSO was used to inhibit the function of MEK1/2. Plasmid pSG/RNLSm expressed a mutant Rta protein lacking the NLS. Expression of HA–Rta, HA–RC361, HA–RN190 and HA–RN191/415 was examined by immunoblotting using anti-HA antibody.
ylation of ERK1/2 activated by HA–RN190 and HA–RC361
(Fig. 6b, lanes 6 and 7). This study also found that expressing
BRAP2 inhibited MEK and ERK phosphorylation activated
by HA–RN190 and HA–RC361 (Fig. 6b, lane 8).
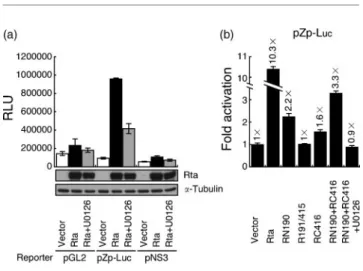
Activation of the BZLF1 promoter by Rta
Previous studies have established that BZLF1 transcription
is closely associated with activation of the ERK signal
transduction cascade (Fahmi et al., 2000; Fenton & Sinclair,
1999; Satoh et al., 1999). To determine whether Rta
activates BZLF1 transcription via the ERK signal
transduc-tion pathway, pCMV-R and a BZLF1 reporter plasmid,
pZp-Luc, were co-transfected into 293T cells. The BZLF1
promoter was activated by pCMV-R at a level about
10-fold higher than that achieved by pCMV-3 (Fig. 7a and b).
Adding U0126 reduced the promoter activity by 56 % (Fig.
7a), suggesting that activation of the ERK signal
transduc-tion pathway by Rta is important for the transcriptransduc-tion of
BZLF1. Additionally, pNS3, in which the four
TPA-response elements (TREs) and the ZII region in the
BZLF1 promoter were deleted, was not activated by
pCMV-R, indicating that TREs may be associated with
the activation. However, transfecting a plasmid expressing
HA–RN190 or HA–RC416 also activated BZLF1
transcrip-tion from pZp-Luc by 2.2-fold or 1.6-fold, respectively
(Fig. 7b). Transfecting plasmids that expressed both
fragments
increased
the
promoter
activity
3.3-fold
(Fig. 7b). Adding U0126 inhibited transcription to the basal
level, indicating the importance of Rta-induced ERK signal
transduction in activating the transcription of BZLF1.
Activation of EBV lytic genes by Rta via the ERK
signal transduction pathway
To demonstrate that activation of the ERK signalling
pathway by Rta is critical to the EBV lytic activation, we
transfected pCMV-R into 293 cells infected by a maxi-EBV
mutant strain, MI-270, containing a mutated BRLF1 (Chiu
et al., 2007). Immunoblotting revealed that the transfection
increased the expression levels of Zta and diffused early
antigen (EA-D) (Fig. 8, lanes 2 and 3). Adding U0126,
however, significantly lowered the level of Zta and EA-D
expression, indicating that activation of the ERK signal
transduction pathway by Rta is crucial for activation of the
EBV lytic cycle.
DISCUSSION
Rta is known to interact with cellular proteins to affect its
own functions or those of the cell. For instance, the
interaction with Ubc9 and PIAS1 causes Rta sumoylation,
which enhances the transcription activity of Rta (Chang et
al., 2004; Liu et al., 2006). The interaction between Rta and
Rb releases E2F1 from Rb to affect cell-cycle progression
(Swenson et al., 1999; Zacny et al., 1998). Rta also interacts
with MCAF1, an binding protein, to enhance
Sp1-mediated transcription (Chang et al., 2005). This study
found that Rta interacts with a KSR1-binding protein,
BRAP2. The interaction was confirmed in vitro using a GST
Fig. 7. Activation of the BZLF1 promoter by Rta and its domains that interact with BRAP2. (a) Reporter plasmids pGL2-Basic (pGL2), pZp-Luc and pNS3 were co-transfected with pCMV-3 (vector) or pCMV-R (Rta) into 293T cells. The amount of Rta in the lysate was determined by immunoblotting (bottom panel). (b) pZp-Luc was co-transfected with pcDNA3-HA2 (vector), pCMV-R, pHA-RN190, pHA-R191/415 and pHA-RC416 into 293T cells. The copy number of plasmids that were transfected into the cells was determined by real-time PCR using a set of primers specific for the ampicillin-resistance gene. The luciferase activity was normalized to the copy number of the plasmids to determine the fold activation. U0126 was added to inhibit the ERK signal transduction cascade. Luciferase activity was measured at 24 h after transfection. Each sample was prepared in duplicate and each experiment was repeated three times. RLU, Relative light units.
Fig. 8. Inhibition of Rta-induced Zta and EA-D expression by U0126. Plasmid pCMV-R was transfected into 293T cells infected by a mutant maxi-EBV strain, MI-270, which contained a mutated BRLF1. Expression of Rta, Zta, EA-D and b-actin was determined by immunoblot analysis 24 h after transfection. U0126 was added to inhibit the ERK signal transduction pathway.
pull-down assay (Fig. 1) and in vivo by
co-immunopreci-pitation (Fig. 2). Confocal microscopy showed that Rta and
BRAP2 co-localized in the cytoplasm (Fig. 3), which is
consistent with the knowledge that BRAP2 is a cytoplasmic
protein (Asada et al., 2004; Li et al., 1998).
As BRAP2 interacts with the NLS in BRCA1 and p21 to
retain these two proteins in the cytoplasm (Asada et al.,
2004; Li et al., 1998), we investigated whether expression
of BRAP2 also retained Rta in the cytoplasm. To
accomplish this, 293(maxi-EBV) cells were transfected
with pHA-BRAP2 and treated with TPA and sodium
butyrate. Confocal microscopy revealed that, although
BRAP2 was expressed abundantly in the cell, the
expression did not affect the localization of Rta and its
ability to transactivate a promoter containing an
Rta-response element (data not shown), showing that BRAP2
probably does not retain Rta in the cytoplasm to affect its
nuclear functions. BRAP2 is also a ubiquitin E3 ligase
(Matheny et al., 2004; Pai et al., 2007). However, we found
that Rta, although conjugated by SUMO-1 (Chang et al.,
2004), was not conjugated by ubiquitin (unpublished
results), suggesting that BRAP2 does not influence the
function of Rta via ubiquitination. Another important
function of BRAP2 is its ability to bind to KSR1 to inhibit
ERK signal transduction (Chen et al., 2008; Matheny et al.,
2004). KSR1 is a scaffolding protein that facilitates the
phosphorylation of MEK1/2 and ERK1/2 in the ERK
signal transduction cascade (Muller et al., 2001; Nguyen et
al., 2002; Roy et al., 2002). However, BRAP2 prevents
KSR1 homo-oligomerization and prevents it from
asso-ciating with the cytoplasmic membrane to inhibit ERK
signal transduction (Chen et al., 2008; Matheny et al.,
2004). Therefore, we investigated whether Rta influenced
the capacity of BRAP2 to inhibit ERK signal transduction.
A mapping study found that Rta and KSR1 both
interacted with the C-terminal 202 aa region in BRAP2
(Fig. 4). Thus, we suggest that Rta binds to BRAP2 to
prevent the interaction between BRAP2 and KSR1, which
activates
the
ERK
signal
transduction
cascade.
Furthermore, Rta activates the BZLF1 promoter through
an ATF2 site in the ZII region (Adamson et al., 2000).
Therefore, activation of the ERK signal transduction
pathway by Rta may ultimately influence the transcription
of BZLF1 through the activation of ATF2, a downstream
target of the ERK signal transduction pathway (Morton
et al., 2004; Ouwens et al., 2002). The first piece of
evidence in support of this hypothesis is the fact that the
binding capacity of GST–BRAP2–glutathione–Sepharose
beads to KSR1 declined substantially when the beads were
pre-incubated with cell lysates containing Rta (Fig. 5a);
adding bacterially expressed His–Rta to a lysate mixture
containing Flag–KSR1 and GST–BRAP2 also dislodged
Flag–KSR1 from the GST–BRAP2–glutathione–Sepharose
beads (Fig. 5b), indicating that Rta prevents the
interaction between BRAP2 and KSR1. Secondly, the
results of the immunoblot analysis showed that
transfect-ing 293T cells with pCMV-R increased the degree of
phosphorylation of MEK1/2 and ERK1/2 (Fig. 6a).
Finally, previous studies have established that TPA,
anti-IgG and TGF-b1 activate BZLF1 transcription
through the four TREs and the ZII region in the BZLF1
promoter (Adamson et al., 2000; Fenton & Sinclair, 1999;
Ragoczy et al., 1998; Satoh et al., 1999). A transient
transfection study revealed that the capacity of Rta to
activate the mutant BZLF1 promoter without these sites
(pNS3) was reduce substantially (Fig. 7a). Additionally,
the fact that Rta-activated BZLF1 transcription is
inhibited by U0126 (Fig. 7a) also supports the suggestion
that Rta activates transcription via activation of the ERK
signal transduction pathway. Notably, transient
transfec-tion analysis indicated that U0126 did not completely
repress Rta-activated BZLF1 transcription (Fig. 7a),
which is probably due to the fact that Rta also activates
BZLF1 transcription via the p38 and JNK pathway
(Adamson et al., 2000), and activation via these routes
was unaffected by the U0126 treatment. We also found
that expressing the two domains in Rta that interact with
BRAP2 sufficiently increased the degree of
phosphoryla-tion of MEK1/2 and ERK1/2 (Fig. 6b) and activated the
BZLF1 promoter (Fig. 7b). Unlike intact Rta,
transactiva-tion of the BZLF1 promoter was completely inhibited by
U0126 (Fig. 7b), implying that, unlike the full-length Rta,
these two Rta fragments are not involved in activating the
p38 and JNK signalling pathway to activate BZLF1
transcription. Previous studies have demonstrated that
activation of ERK signal transduction by TPA, anti-IgG
and TGF-b1 is crucial to activation of BZLF1
transcrip-tion and the EBV lytic cycle (Fahmi et al., 2000; Fenton &
Sinclair, 1999; Satoh et al., 1999). The fact that U0126
reduced the capacity of Rta to activate expression of Zta
and EA-D (Fig. 8) indicates that activation of the ERK
signal transduction pathway by Rta is critical to EBV
reactivation. Our results also explain why an NLS mutant
of Rta was found previously to activate the EBV lytic cycle
(Hsu et al., 2005).
This investigation found that Rta did not promote the
phosphorylation of ERK1/2 in three B-lymphocyte cell
lines (Akata, BJAB and P3HR1) as much as in 293T cells
(data not shown). There is a possibility that the ERK signal
transduction pathway in these B lymphocyte cells is less
responsive to BRAP2 inhibition, so Rta does not
signific-antly activate the pathway in such cells. Earlier studies have
demonstrated that Zta also activates the BRLF1 promoter
by activating the ERK signal transduction cascade in
epithelial cells (Chang et al., 2006), showing that both Rta
and Zta influence ERK signal transduction to influence the
EBV lytic cycle. As the EBV particles produced by epithelial
cells exhibit a tropism towards B lymphocytes (Borza &
Hutt-Fletcher, 2002; Guerreiro-Cacais et al., 2004), in
which EBV latency is established, activation of the BZLF1
promoter by Rta through the ERK signal transduction
pathway may be critical to viral lytic replication and to the
infection of B lymphocytes in the B-lymphocyte/epithelial
cell infection cycle.
ACKNOWLEDGEMENTS
The authors would like to thank Deborah K. Morrison and Tsuey-Ying Hsu for providing the plasmids used in this study. This research was supported by the National Science Council of the Republic of China (NSC96-3112-B-182-002 and NSC95-2320-B-182-041-MY3), the National Health Research Institute of the Republic of China (NHRI-EX96-9417BI) and the Chang-Gung Molecular Medicine Research Center (CMRPD160111).
REFERENCES
Adamson, A. L., Darr, D., Holley-Guthrie, E., Johnson, R. A., Mauser, A., Swenson, J. & Kenney, S. (2000).Epstein–Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J Virol 74, 1224–1233.
Asada, M., Ohmi, K., Delia, D., Enosawa, S., Suzuki, S., Yuo, A., Suzuki, H. & Mizutani, S. (2004).Brap2 functions as a cytoplasmic retention protein for p21 during monocyte differentiation. Mol Cell Biol 24, 8236–8243.
Borras, A. M., Strominger, J. L. & Speck, S. H. (1996).
Characterization of the ZI domains in the Epstein–Barr virus BZLF1 gene promoter: role in phorbol ester induction. J Virol 70, 3894–3901.
Borza, C. M. & Hutt-Fletcher, L. M. (2002).Alternate replication in B cells and epithelial cells switches tropism of Epstein–Barr virus. Nat Med 8, 594–599.
Chang, L. K. & Liu, S. T. (2000).Activation of the BRLF1 promoter and lytic cycle of Epstein–Barr virus by histone acetylation. Nucleic Acids Res 28, 3918–3925.
Chang, L. K., Lee, Y. H., Cheng, T. S., Hong, Y. R., Lu, P. J., Wang, J. J., Wang, W. H., Kuo, C. W., Li, S. S. & Liu, S. T. (2004).Post-translational modification of Rta of Epstein–Barr virus by SUMO-1. J Biol Chem 279, 38803–38812.
Chang, L. K., Chung, J. Y., Hong, Y. R., Ichimura, T., Nakao, M. & Liu, S. T. (2005). Activation of Sp1-mediated transcription by Rta of Epstein–Barr virus via an interaction with MCAF1. Nucleic Acids Res 33, 6528–6539.
Chang, P. J., Chang, Y. S. & Liu, S. T. (1998). Role of Rta in the translation of bicistronic BZLF1 of Epstein–Barr virus. J Virol 72, 5128–5136.
Chang, Y., Lee, H. H., Chen, Y. T., Lu, J., Wu, S. Y., Chen, C. W., Takada, K. & Tsai, C. H. (2006).Induction of the early growth response 1 gene by Epstein–Barr virus lytic transactivator Zta. J Virol 80, 7748–7755.
Chen, C., Lewis, R. E. & White, M. A. (2008).IMP modulates KSR1-dependent multivalent complex formation to specify ERK1/2 pathway activation and response thresholds. J Biol Chem 283, 12789–12796.
Chevallier-Greco, A., Manet, E., Chavrier, P., Mosnier, C., Daillie, J. & Sergeant, A. (1986).Both Epstein–Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J 5, 3243–3249.
Chiu, Y. F., Tung, C. P., Lee, Y. H., Wang, W. H., Li, C., Hung, J. Y., Wang, C. Y., Kawaguchi, Y. & Liu, S. T. (2007).A comprehensive library of mutations of Epstein–Barr virus. J Gen Virol 88, 2463–2472.
Daibata, M., Humphreys, R. E., Takada, K. & Sairenji, T. (1990).
Activation of latent EBV via anti-IgG-triggered, second messenger pathways in the Burkitt’s lymphoma cell line Akata. J Immunol 144, 4788–4793.
Davies, A. H., Grand, R. J., Evans, F. J. & Rickinson, A. B. (1991).
Induction of Epstein–Barr virus lytic cycle by tumor-promoting and
non-tumor-promoting phorbol esters requires active protein kinase C. J Virol 65, 6838–6844.
Delecluse, H. J., Hilsendegen, T., Pich, D., Zeidler, R. & Hammerschmidt, W. (1998). Propagation and recovery of intact, infectious Epstein–Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A 95, 8245–8250.
Diehl, V., Henle, G., Henle, W. & Kohn, G. (1968).Demonstration of a herpes group virus in cultures of peripheral leukocytes from patients with infectious mononucleosis. J Virol 2, 663–669.
Einhorn, N., Klein, G. & Clifford, P. (1970).Increase in antibody titer against the EBV-associated membrane antigen complex in Burkitt’s lymphoma and nasopharyngeal carcinoma after local irradiation. Cancer 26, 1013–1021.
Faggioni, A., Zompetta, C., Grimaldi, S., Barile, G., Frati, L. & Lazdins, J. (1986).Calcium modulation activates Epstein–Barr virus genome in latently infected cells. Science 232, 1554–1556.
Fahmi, H., Cochet, C., Hmama, Z., Opolon, P. & Joab, I. (2000).
Transforming growth factor beta 1 stimulates expression of the Epstein– Barr virus BZLF1 immediate-early gene product ZEBRA by an indirect mechanismwhichrequirestheMAPKkinasepathway.JVirol74,5810–5818.
Feederle, R., Kost, M., Baumann, M., Janz, A., Drouet, E., Hammerschmidt, W. & Delecluse, H. J. (2000). The Epstein–Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J 19, 3080–3089.
Fenton, M. & Sinclair, A. J. (1999).Divergent requirements for the MAPK(ERK) signal transduction pathway during initial virus infection of quiescent primary B cells and disruption of Epstein– Barr virus latency by phorbol esters. J Virol 73, 8913–8916.
Flemington, E. & Speck, S. H. (1990).Identification of phorbol ester response elements in the promoter of Epstein–Barr virus putative lytic switch gene BZLF1. J Virol 64, 1217–1226.
Gao, X., Ikuta, K., Tajima, M. & Sairenji, T. (2001). 12-O-tetradecanoylphorbol-13-acetate induces Epstein–Barr virus reactiva-tion via NF-kB and AP-1 as regulated by protein kinase C and mitogen-activated protein kinase. Virology 286, 91–99.
Granato, M., Farina, A., Gonnella, R., Santarelli, R., Frati, L., Faggioni, A. & Angeloni, A. (2006).Regulation of the expression of the Epstein–Barr virus early gene BFRF1. Virology 347, 109–116.
Guerreiro-Cacais, A. O., Li, L., Donati, D., Bejarano, M. T., Morgan, A., Masucci, M. G., Hutt-Fletcher, L. & Levitsky, V. (2004).Capacity of Epstein–Barr virus to infect monocytes and inhibit their development into dendritic cells is affected by the cell type supporting virus replication. J Gen Virol 85, 2767–2778.
Gunven, P., Klein, G., Henle, G., Henle, W. & Clifford, P. (1970).
Epstein–Barr virus in Burkitt’s lymphoma and nasopharyngeal carcinoma. Antibodies to EBV associated membrane and viral capsid antigens in Burkitt lymphoma patients. Nature 228, 1053–1056.
Hardwick, J. M., Lieberman, P. M. & Hayward, S. D. (1988).A new Epstein–Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J Virol 62, 2274–2284.
Hsu, T. Y., Chang, Y., Wang, P. W., Liu, M. Y., Chen, M. R., Chen, J. Y. & Tsai, C. H. (2005).Reactivation of Epstein–Barr virus can be triggered by an Rta protein mutated at the nuclear localization signal. J Gen Virol 86, 317–322.
Hung, C. H. & Liu, S. T. (1999).Characterization of the Epstein–Barr virus BALF2 promoter. J Gen Virol 80, 2747–2750.
Johansson, B., Klein, G., Henle, W. & Henle, G. (1970).Epstein–Barr virus (EBV)-associated antibody patterns in malignant lymphoma and leukemia. I. Hodgkin’s disease. Int J Cancer 6, 450–462.
Klein, G., Geering, G., Old, L. J., Henle, G., Henle, W. & Clifford, P. (1970). Comparison of the anti-EBV titer and the EBV-associated
membrane reactive and precipitating antibody levels in the sera of Burkitt lymphoma and nasopharyngeal carcinoma patients and controls. Int J Cancer 5, 185–194.
Li, S., Ku, C. Y., Farmer, A. A., Cong, Y. S., Chen, C. F. & Lee, W. H. (1998).Identification of a novel cytoplasmic protein that specifically binds to nuclear localization signal motifs. J Biol Chem 273, 6183–6189.
Lin, G. H., Chen, C. L., Tschen, J. S., Tsay, S. S., Chang, Y. S. & Liu, S. T. (1998).Molecular cloning and characterization of fengycin synthetase gene fenB from Bacillus subtilis. J Bacteriol 180, 1338–1341.
Liu, S. T., Wang, W. H., Hong, Y. R., Chuang, J. Y., Lu, P. J. & Chang, L. K. (2006).Sumoylation of Rta of Epstein–Barr virus is preferentially enhanced by PIASxb. Virus Res 119, 163–170.
Lu, C. C., Jeng, Y. Y., Tsai, C. H., Liu, M. Y., Yeh, S. W., Hsu, T. Y. & Chen, M. R. (2006). Genome-wide transcription program and expression of the Rta responsive gene of Epstein–Barr virus. Virology 345, 358–372.
Luka, J., Kallin, B. & Klein, G. (1979).Induction of the Epstein–Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 94, 228–231.
Matheny, S. A. & White, M. A. (2006).Ras-sensitive IMP modulation of the Raf/MEK/ERK cascade through KSR1. Methods Enzymol 407, 237–247.
Matheny, S. A., Chen, C., Kortum, R. L., Razidlo, G. L., Lewis, R. E. & White, M. A. (2004).Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature 427, 256–260.
Morton, S., Davis, R. J. & Cohen, P. (2004).Signalling pathways involved in multisite phosphorylation of the transcription factor ATF-2. FEBS Lett 572, 177–183.
Mu¨ller, J., Ory, S., Copeland, T., Piwnica-Worms, H. & Morrison, D. K. (2001). C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell 8, 983–993.
Nguyen, A., Burack, W. R., Stock, J. L., Kortum, R., Chaika, O. V., Afkarian, M., Muller, W. J., Murphy, K. M., Morrison, D. K. & other authors (2002).Kinase suppressor of Ras (KSR) is a scaffold which facilitates mitogen-activated protein kinase activation in vivo. Mol Cell Biol 22, 3035–3045.
Ouwens, D. M., de Ruiter, N. D., van der Zon, G. C., Carter, A. P., Schouten, J., van der Burgt, C., Kooistra, K., Bos, J. L., Maassen, J. A. & van Dam, H. (2002).Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras–MEK– ERK pathway and of Thr69 through RalGDS–Src–p38. EMBO J 21, 3782–3793.
Pai, M. T., Tzeng, S. R., Kovacs, J. J., Keaton, M. A., Li, S. S., Yao, T. P. & Zhou, P. (2007).Solution structure of the Ubp-M BUZ domain, a highly specific protein module that recognizes the C-terminal tail of free ubiquitin. J Mol Biol 370, 290–302.
Ragoczy, T., Heston, L. & Miller, G. (1998).The Epstein–Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J Virol 72, 7978–7984.
Roy, F., Laberge, G., Douziech, M., Ferland-McCollough, D. & Therrien, M. (2002).KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev 16, 427–438.
Satoh, T., Hoshikawa, Y., Satoh, Y., Kurata, T. & Sairenji, T. (1999).
The interaction of mitogen-activated protein kinases to Epstein–Barr virus activation in Akata cells. Virus Genes 18, 57–64.
Swenson, J. J., Mauser, A. E., Kaufmann, W. K. & Kenney, S. C. (1999).The Epstein–Barr virus protein BRLF1 activates S phase entry through E2F1 induction. J Virol 73, 6540–6550.
Zacny, V. L., Wilson, J. & Pagano, J. S. (1998).The Epstein–Barr virus immediate-early gene product, BRLF1, interacts with the retinoblas-toma protein during the viral lytic cycle. J Virol 72, 8043–8051.
Zalani, S., Holley-Guthrie, E. & Kenney, S. (1996).Epstein–Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc Natl Acad Sci U S A 93, 9194–9199.
Zhang, Y., Yao, B., Delikat, S., Bayoumy, S., Lin, X. H., Basu, S., McGinley, M., Chan-Hui, P. Y., Lichenstein, H. & Kolesnick, R. (1997).Kinase suppressor of Ras is ceramide-activated protein kinase. Cell 89, 63–72.
zur Hausen, H., O’Neill, F. J., Freese, U. K. & Hecker, E. (1978).
Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature 272, 373–375.