幽門螺旋桿菌抑制小鼠巨噬細胞感染模式抗菌一氧化氮之產生; Helicobacter pylori suppresses antimicrobial nitric oxide production in a mouse macrophage model
46
0
0
全文
(2) 致謝 時光荏苒,兩年的時光很快的過去了,在這段時間裡,不管是遇到了什麼樣的挫 折及困難,身邊總有許多老師、朋友的幫助,才使得我能順利度過難關。在這兩年內, 有太多太多的人要感謝,首先要感謝我的指導教授賴志河老師,剛進實驗室時的成員不 多,在學期中時更常常是只有老師與我兩人留守實驗室,也正因為如此,老師對我特別 照顧,不管是研究或課業上,老師都給予很多的關心、指導,老師也給了我很多待人處 事上的觀念,受益良多。另外要感謝我大學時期的指導老師,台灣體育大學競技運動學 系方世華老師,在她的推薦下才使我能進入賴老師實驗室學習。 再來要感謝同窗兩年的同學們,共同經歷了許多課業上的壓力,在大家的相互扶 持、照顧下,每個人都可以順利通過試驗,我無法忘記那年夏天我們在澎湖的畢業旅行 所留下的美好回憶,沒有你們,這些回憶不會這麼美麗。還有所辦秘書麗如姊,所有大 大小小的事,總是要麻煩她,而麗如姊也溫柔的守護著我們,幫助我們克服了許多困難。 也要感謝分子系統生物醫學所馬明琪老師,生物科技學系徐媛曼老師,醫學藥理學科湯 智昕老師以及清華大學分子與細胞生物研究所王雯靜教授,期間也受了他們及實驗室成 員們許多的照顧;更要感謝實驗室的助理及學弟妹們的陪伴,讓我們的實驗室總是洋溢 著愉悅的氣氛。 最後我要感謝我的父母,讓我可以自由的去做我想做的事,我的姑姑、兄嫂及姊 姊們,有他們的支持與鼓勵,我才能堅持下去。這一路上有太多太多的人要感謝,我會 永遠記得你們對我的幫助與照顧,並在此祝福大家平安喜樂。.
(3) Contents 中文摘要 (ABSTRACT IN CHINESE) ............................................................................................................. II ABSTRACT.........................................................................................................................................................III INTRODUCTION................................................................................................................................................. 1 MATERIALS AND METHODS .......................................................................................................................... 5 ANTIBODIES AND MATERIALS ............................................................................................................................ 5 BACTERIAL STRAIN AND CELL CULTURE .......................................................................................................... 5 PREPARATION OF MOUSE PERITONEAL EXCLUDED MACROPHAGES (PEMS).................................................. 6 WESTERN BLOT ................................................................................................................................................. 7 SEMI-QUANTITATIVE RT-PCR ANALYSIS.......................................................................................................... 7 BACTERIAL SURVIVAL ASSAY ............................................................................................................................. 8 DETERMINATION OF NITRIC OXIDE ................................................................................................................... 9 CELL VIABILITY ASSAY ...................................................................................................................................... 9 TRANSIENT TRANSFECTION OF NF-ΚB, INOS AND AP-1 REPORTER GENE ....................................................10 IMMUNOFLUORESCENCE LABELING OF PHOSPHORYLATED P65 TRANSLOCATION .........................................10 STATISTICAL ANALYSIS .....................................................................................................................................11 RESULTS..............................................................................................................................................................12 DISCUSSION .......................................................................................................................................................18 PROSPECTION...................................................................................................................................................25 REFERENCES.....................................................................................................................................................27 FIGURES..............................................................................................................................................................34 APPENDIX...........................................................................................................................................................39. I.
(4) 中文摘要 (Abstract in Chinese) 在幽門螺旋桿菌(Helicobacter pylori)存活於胃黏膜組織進而引發慢性胃發炎的反應 中,菌體必須具有能在免疫系統活化反應下存活的能力。在胃黏膜組織對於幽門螺旋桿 菌的免疫反應及相關的發炎反應中,一氧化氮(nitric oxide, NO)扮演著相當重要的角色。 病原體可能會活化巨噬細胞的誘發性一氧化氮合成酶(inducible nitric oxide synthase, iNOS)表現,而 iNOS 在不同細胞中的表現則會經由細菌的脂多醣(lipopolysaccharide, LPS) 刺激細胞所調控。有某些病原體具有避免巨噬細胞清除病原的能力,這些能力包括抑制 巨噬細胞的活化或引發急速的細胞死亡。在我們的研究中,我們使用了小鼠巨噬細胞的 實驗模式,最主要是為了確認幽門螺旋桿菌的感染是否會抑制由 LPS 所誘發產生的一氧 化氮及 iNOS 的表現。研究中我們分析了 iNOS 的 mRNA 及蛋白質的表現情形,而結果 也證實了幽門螺旋桿菌能抑制 iNOS 的轉錄及轉錄後的表現情形,而這些作用情形主要 是由活菌並具有正常功能的菌體所造成的。除此之外,這些現象還包括抑制裂殖原活化 蛋白激酶(mitogen-activated protein kinase, MAP kinase)傳遞路徑,而此一路徑最主要是影 響 nuclear factor (NF)-κB 活化並進入細胞核。而我們的結果也顯示,幽門螺旋桿菌具有 調控先天性免疫反應的能力,而這個調控機制對於菌體存活在宿主胃體並持續進行感染 是有益的。. 關鍵字:幽門螺旋桿菌,一氧化氮,巨噬細胞,NF-κB. II.
(5) Abstract The ability of the Helicobacter pylori to survive in the interaction of non-phagocytes and phagocytes is postulated to enhance the persistence of this pathogen in the gastric mucosa and then to cause chronic inflammation. Nitric oxide (NO) production plays an important role in the gastric mucosal immune response to H. pylori and the associated inflammation. Pathogens might activate macrophage inducible nitric oxide synthase (iNOS) expression. The expression of iNOS is regulated in various cell types and can be enhanced by stimulation of bacterial lipopolysaccharide (LPS). In some pathogens, the ability to evade macrophage killing involves inducing rapid death of macrophages or suppression of macrophage activities. In this study, we used a mouse macrophage infection model to demonstrate that H. pylori could inhibit lipopolysaccharide (LPS)-induced NO production and iNOS expression. Analysis of iNOS specific mRNA and protein expression levels after infection revealed that H. pylori inhibited iNOS expression at both transcriptional and post-transcriptional levels, and only live and functional bacteria would do. Furthermore, this phenomenon involved down-regulation of mitogen-activated protein (MAP) kinase pathway, which triggered translocation of active nuclear factor (NF)-κB into nucleus. Our data suggested a new mechanism for H. pylori regulating innate immune responses of host cells to benefit persistent infection in host stomachs.. III.
(6) Keywords: :Helicobacter pylori, macrophage, nitric oxide, NF-kappa B. Abbreviations: LPS, lipopolysaccharide; NO, nitric oxide; iNOS, inducible nitric oxide synthase; MAP, mitogen-activated protein; ERK, extracellular signal-related kinase; JNK, c-Jun-N-terminal kinase; NF-κB, nuclear factor (NF)-κB; IKK, IκB kinase; PEMs, peritoneal excluded macrophages; ODN, oligonucleotide dominate negative.. IV.
(7) Introduction Helicobacter pylori is the most commonly causative agent of duodenal and gastric diseases in human. Infection of the pathogen usually occurs in childhood and can persist in the stomach for a life-time (1-3). The most common outcome of infection is chronic gastritis, but some patients may develop peptic ulcers, gastric carcinoma, and gastric lymphoma. Persistent infection of H. pylori in the gastric mucosa results in release of interleukine (IL)-8, which attracts neutrophile infiltration and causes chronic gastritis. It has been demonstrated that CagA+ H. pylori can induce NF-κB activation and secrete of IL-8 in gastric epithelial cells (4). It has also been found that employing NOD1-deficient mice reduced control of H. pylori densities in the stomach (5). These findings suggest that H. pylori is essential for induction of proinflammatory responses in host stomach. Chronic gastritis is induced not only by epithelium secretion of IL-8 but also by mononuclear cell infiltrates, included of lymphocyte and macrophages. In the study of macrophages, which are major source of the IL-6 present in H. pylori induced chronic gastritis, are known to play a critical role in the pathogenesis of mucosal inflammations (6). It was also reported that macrophages secretion increased levels of IL-1β and TNF-α in H. pylori infected gastric tissues (7, 8). Analysis of IL-1 gene cluster polymorphisms indicated that IL-1β is associated with an increased risk of gastric cancer (9). Nevertheless, those immune responses is failed to eliminate H. pylori from gastric mucosa completely, suggesting. 1.
(8) that this bacteria has the ability to evade host immune elimination. Another bactericidal agent of macrophages, nitric oxide (NO) is generated by nitric oxide synthase (NOS)-mediated conversion of L-arginine to L-citrulline. It has been reported that H. pylori can activate iNOS expression in macrophage (10). H. pylori-infected gastritis was shown to have higher expression of iNOS than H. pylori-negative patients in gastric epithelium and lamina propria (11). Furthermore, H. pylori urease was demonstrated to stimulate of iNOS expression and NO production in a mouse macrophage infection system (12). It is important to know that H. pylori can also survive despite marked induction of inducible NOS (iNOS) in macrophages. Gobert et. al. reported that H. pylori prevents NO production by the bacterial cell envelope gene rocF, which encoded an arginase and competed with NOS (13). Mutation of the rocF gene results in efficient killing of H. pylori in an NO-dependent manner, suggesting that arginase might be important for protection of H. pylori from. macrophages attack. These evidences suggested that H. pylori has a delicate. mechanism for the balance between activation of macrophages and protection of the bacteria from immune attack. However, the interaction between macrophages and H. pylori to inhibit innate immunity has not been extensively studied. Although H. pylori is known to produce arginase to compete with iNOS for their substrate and regulate NO synthesis (14), the molecular mechanism of H. pylori to suppress NO production in macrophages has not been well defined. To address the question of how H.. 2.
(9) pylori evades antimicrobial activities by macrophage, we established an in vitro and ex vivo mouse model system to examine whether this bacteria could suppress LPS-induced NO production. Our study reveals that H. pylori infection in such a model does inhibit LPS-stimulated iNOS expression and NO production in macrophages. We also demonstrate that H. pylori triggers the increase of phophorylated p38, activation of Erk1/2 and NF-κB, subsequently suppresses LPS-stimulated macrophage responses. Thus, study with this model system reveals an important aspect that H. pylori has the ability to trigger macrophage activation and subsequently evading of early host immune responses. Although there is an innate response to the bacteria, one study has shown a attenuated uptake of bacteria into macrophages followed by the formation of megasomes as a result of phagosome fusion (15). These megasomes protect intracellular bacteria from efficient killing. In adaptive immunity, H. pylori can specifically block antigen-dependent proliferation of T-cells. This effect is mediated by the virulence factor VacA, which could act as an immunomodulator by interfering with the IL-2 signalling pathway in T-cells by blocking Ca2+ mobilization and the activity of the Ca2+/calmodulin-dependent phosphatase calcineurin (16). There were some findings support the possibility that VasA is immunosuppressive, but the mechanism involves a direct reaction on T cells rather than in antigen-presenting cells. The toxin inhibits the activation and proliferation of T cells (17). Even there are so many studies suggest that H. pylori could evade host immune. 3.
(10) responses by these mechanisms, we hope our team could find some new mechanisms about immune evasion of H. pylori.. 4.
(11) Materials and methods Antibodies and materials Polyclonal rabbit antibodies specific for iNOS, p-JNK antibody, α-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific for p38 MAP kinase, SAPK/JNK, p44/42 (Erk 1/2) were purchased from Cell Signaling (Danvers, MA). Phosphorylated p38 MAP kinase, mouse anti-actin antibody, phosphorylated MAP kinase 1/2 (Erk1/2) (Thr185/Tyr187) were purchased from Upstate (Lake Placid, NY). pSV-β-galactosidase vector and luciferase assay kit were purchased from Promega (Madison, MA). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO).. Bacterial strain and cell Culture H. pylori 26695 (ATCC700392) reference strain was recovered from frozen stocks on Brucella agar plates (Becton Dickinson) containing 10 % sheep blood, 6 µg/ml vancomycin and 2 µg/ml amphotericin B under microaerophilic conditions for 2–3 days as described previously (18). To develop H. pylori derivative extracts, the following manipulation was performed. Heat-killed H. pylori was obtained by boiling the bacteria suspension in PBS (1× 109/ml) for 30 min. H. pylori crude extracts were prepared by sonication on ice for 5 min. Crude extracts were then subjected to centrifugation at 16,000 ×g for 5 min at 4°C, the supernatant was. 5.
(12) filtrated through 0.22µm filter and used for the further analysis. RAW 264.7 cells (ATCC TIB-71, murine macrophage cell line) were cultured in RPMI 1640 (Invitrogene, Grand island, NY) medium. Ten percent of de-complement Fetal bovine serum (Hyclone, UT) was added in culture medium. Penicillin and streptomycin (Invitrogene) were used if needed. In bacteria infection system, cell culture medium was not supplemented with antibiotic reagents.. Preparation of mouse peritoneal excluded macrophages (PEMs) C57BL/6JNarl male mice were purchased from National Laboratory Animal Center in Taiwan and maintained in the animal center of China Medical University at Taichung. The animal room was maintained on a 12 hr light and 12 hr dark cycle with a standard temperature and humidity. All mice were grown at 8 weeks old, sacrificed under anesthesia, and used to obtain cells from peritoneal exudates. All procedures adhered to the “Guide for the Care and Use of Laboratory Animals” (NRC, USA) and were approved by the animal experiment committee of China Medical University. Mouse peritoneal excluded macrophages (PEMs) were obtained from mice by lavage with 10 ml of cold PBS per mouse at 3 days after intraperitoneal injection of 2 ml 3% thioglycollate (Sigma-Aldrich) in PBS. The PEMs were then seeded in culture plates and incubated at 37°C in humidified 5% CO2 and 95% air to allow macrophages adherence. After 2 hr, the non-adherent cells were removed by washing. 6.
(13) with warmed PBS and the remaining cells (90% macrophages, judged by non-specific esterase stain) were used for further experiments.. Western blot H. pylori infected cells were washed three times with PBS and then boiled with sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) sample buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 0.05% brilliant blue R) at 95°C for 10 min. The samples were then resolved by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). The membranes were blocked with 5% skim milk in PBS buffer containing 0.1% Tween 20 at room temperature for 1 h and then incubated overnight with first antibodies as described in materials (dilution, 1:1,000) at 4°C. The membranes were washed with PBS containing 0.1% Tween 20 and then incubated with HRP-conjugated secondary antibodies (Invitrogen) at a dilution of 1:5,000. The proteins of interest were visualized by using the ECLTM Western Blotting Detection Reagents (GE Healthcare, Little Chalfont, UK) and were detected by exposure the autoradiograph to X-ray films (Kodak, Rochester, NY).. Semi-quantitative RT-PCR analysis PEM cells were homogenized with Cyclo-PrepTM Total RNA Purification Kit. 7.
(14) (Amresco, Solon, OH) and reverse-transcription assay was performed using a Fast-RunTM HotStart RT-PCR (AMV) kit (Protech Technology Enterprise Co., Ltd, Taiwan). The oligonucleotide. primers. used. corresponded. to. murine. iNOS,. forward:. 5’-GCCTCGCTCTGGAAAGA-3’ and reversed: 5’- TCCATGCAGACAACCTT -3’; GAPDH,. forward:. 5’-ACTCCCACTCTTCCACCTTC-3’,. and. reversed:. 5’-TCTTGCTCAGTGTCCTTGC-3’. All oligonucleotide primers were synthesized by Invitrogen. The experiment was performed as following program: For 1st cDNA synthesis: RNA denaturation for 5 min, 58 °C, 1 cycle; cDNA synthesis for 30 min, 42°C, 1 cycle; AMV RT inactivation & RNA/cDNA/primer denaturation for 2 min, 94°C. For 2nd PCR reaction: denaturation for 30 sec, 94°C; annealing for 1.5 min (iNOS) or 40 sec (GAPDH), 57°C; extension for 1 min, 72°C and the PCR reactions total run 35 cycles. The final elongation at 72°C for 5 min. PCR products were analyzed on 2% agarose gels. The mRNA of GAPDH served as the internal control for sample loading and mRNA integrity.. Bacterial survival assay Mouse PEMs were cultured in the bottom layer of Trans-well plate (Corning, Lowell, MA) and infection with H. pylori at MOI of 0, 50, and 100. After 48 hr, a total of 1 × 106 H. pylori were added on the insert membrane (0.1 µm pore size) and co-incubated for a further 6 hr. After which, the bacteria on the insert membrane were suspended and cultured by serial. 8.
(15) dilution onto Brucella blood agar plate and colonies were counted after 4–5 days. Colony forming units (CFUs) were enumerated for anti-bacterial effects. Experiments were performed at least three times in triplicates.. Determination of nitric oxide The production of NO was estimated from the accumulation of nitrite (NO2−), a stable end product of NO metabolism, in the medium using the Griess reagent (Sigma-Aldrich) as described previously (19). Briefly, cells were incubated with medium containing various MOI of H. pylori wild type strain, in the presence or absence of LPS (2µg/ml) for the indication time. Equal volumes of culture supernatant and Griess reagent were mixed and incubated for 15 min at room temperature. The absorbance was measured at 570 nm on a spectrophotometer, and referred to a nitrite standard curve to determine the nitrate concentration in supernatants.. Cell viability assay The MTT assay was used to measure the effects of LPS and H. pylori to induce death of macrophage cells (20). The RAW 264.7 and PEMs were exposed to various MOI of H. pylori during 24 or 48hr incubation periods. Cell viability was measured by the ability of viable cells to reduce MTT (Sigma-Aldrich) to formazan based on the ability of living cells to utilize Thiazolyl Blue and convert it into purple formazan, which absorbs light at 570 nm and could. 9.
(16) be analyzed spectrophotometrically. Measurement was performed in triplicate. The absorbance was measured using the BioRad spectrophotometer. The mean OD value of the content of four wells was used for assessing the cell viability expressed as percentage of control.. Transient transfection of NF-κB, iNOS and AP-1 reporter gene RAW 264.7 cells were grown to 90% confluence in 12-well plate and was transfected NF-κB-Luc, iNOS-Luc and AP-1-Luc by using Lipofectamine 2000 (Invitrogen). After 24 hr incubation, transfection was complete, and cells were incubated with or without LPS and then infected with H. pylori strain for 24 hr. To prepare cell lysates, 100 µl of reporter lysis buffer (Promega) was added to each well, and cells were scraped from dishes. An equal volume of luciferase substrate was added to all samples, and luminescence was measured in a microplate luminometer. The value of luciferase activity was normalized to transfection efficiency monitored by the co-transfected β-galactosidase expression vector obtained from Promega (Madison, MA).. Immunofluorescence labeling of phosphorylated p65 translocation To visualize the H. pylori inhibits phosphorylated p65 translocated into epithelial cells, RAW cells were seeded onto cover-slips for 2hr and treated with or without LPS and H. pylori. 10.
(17) for a further 1hr at 37°C. Cells were washed and then fixed in 3.7% (wt/vol) paraformaldehyde for at least one hour at 4°C and permeabilized in PBS contained 0.5% (vol/vol) Triton X-100 for 2 minutes. To label p65, cell preparations were then incubated for 30 minutes with NF-κB p65 (H-286) rabbit polyclonal antibody (Santa Cruz Biotechnology), and DAPI (4',6-diamidino-2-phenylindole). The secondary antibody was fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (Chemicon). Then, cells were fixed in paraformaldehyde. Preparations were mounted and observed with a confocal laser scanning microscope (Zeiss LSM 510).. Statistical analysis Student's t test was used to calculate the statistical significance of the experimental results for two groups; a P value of <0.05 was considered significant.. 11.
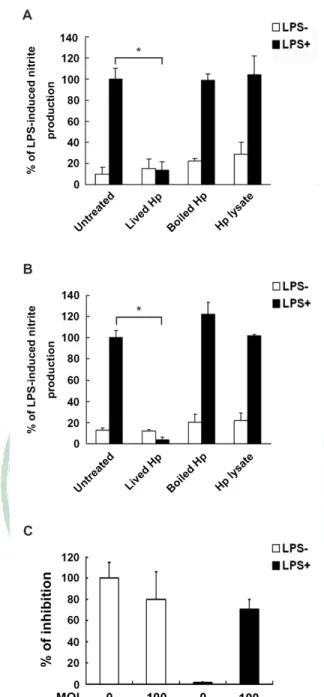
(18) Results 1. H. pylori inhibits LPS-mediated nitric oxide production in macrophages To assess whether H. pylori can inhibit LPS-induced nitric oxide production in macrophages, mouse macrophage RAW 264.7 cells were cultured with LPS (2µg/ml) and infected with H. pylori at MOI of 0-200 for 24 hr. As shown in Figure 1A, nitric oxide production was in an un-effective level when cells were treated with LPS and infected with H. pylori at low MOI (0-50). However, NO production in LPS-induced RAW 264.7 cells was suppressed at higher MOI of H. pylori infection. When cells were infected with H. pylori at MOI of 100, the LPS-induced NO production was reduced near 50%. The NO-production was diminished to basal line which without LPS-induced level after infection with H. pylori at MOI of 150 to 200 (Figure 1A). To further determine the effects of H. pylori suppresses LPS-induced NO production in murine primary macrophage, peritoneal excluded macrophages (PEMs) were prepared as described in material and methods and followed by co-incubated with LPS and H. pylori for a further 48 hr. The result of ex vivo study showed that while LPS treatment effectively induced NO production in uninfected cells (MOI=0). When PEMs were infected with H. pylori at MOI of 10 to 100, the LPS-induced NO production were reduced gradually (Figure 1B). Thus, these results demonstrated that not only H. pylori inhibits LPS-induced NO production in a dose-dependent manner in RAW 264.7 cells, but have the same effects on murine primary peritoneal excluded macrophages.. 12.
(19) 2. Live H. pylori is essential for inhibition of LPS-mediated nitric oxide production To determine the role of functional H. pylori can inhibit LPS-induced NO production in RAW 264.7 cells, we analyzed the effects of lived, killed bacteria, and bacterial crude extracts. The results showed that neither heat killed H. pylori nor H. pylori crude extracts inhibited LPS-induced NO production, in contrast to both live bacteria and water extract (Figure 2A). We also used murine primary macrophage to investigate the effects of H. pylori in the inhibition of LPS-stimulated NO production. In consistent with RAW 264.7 cells, the results also showed that live bacteria from H. pylori have the effects on inhibition of LPS-induced NO production in PEMs (Figure 2B). Thus, these data suggested that H. pylori can inhibit LPS-induced macrophage NO production in a manner that is dependent on a functional live bacteria. To mimic bacteria infection in a NO-containing condition, we tested whether H. pylori-inhibited LPS-induced NO production would enhance the survival of bacteria in a liquid culture environment. The experimental design was described in materials and methods. As shown in Figure 2C, our data showed that the higher MOI of H. pylori infection of PEMs, the lower anti-microbial activity was found. These data reveal that H. pylori have their potential on inhibition of NO production and thus reduced antimicrobial activity, subsequently enhance H. pylori survival after countered macrophage attack.. 13.
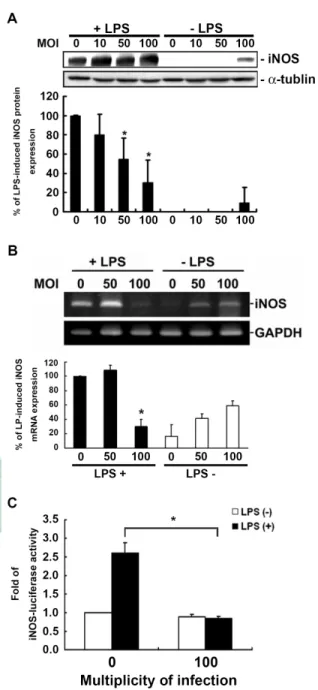
(20) 3.. H.. pylori. inhibits. LPS-induced. iNOS. expression. at. transcriptional. and. post-transcriptional levels To investigate the effects of H. pylori on the cytosolic protein levels of iNOS, PEMs were treated with LPS or LPS plus various MOI of H. pylori for 48hr and the protein levels of iNOS were analyzed by Western blot. The results showed that infection with H. pylori led to a significant decrease in LPS-induced expression of iNOS in a MOI-dependent manner (Figure 3A). When cells were infected with H. pylori at a MOI of 100, the decrease of LPS-induced iNOS expression approached to 80%. We further detected the effects of H. pylori on LPS-induced mRNA expression of iNOS using semi-quantitative RT-PCR analysis. Total RNA were extracted from PEMs after co-cultured with or without LPS and various MOI of H. pylori for 48hr. Our results showed that mRNA expression of iNOS was also reduced by H. pylori infection at a MOI of 100 (Figure 3B). To directly determine LPS-stimulated iNOS promoter activity was suppressed after H. pylori infection, RAW 264.7 macrophages were transiently transfected with p-iNOS-luciferase as an indicator of iNOS promoter activation. The result showed that LPS-induced macrophage iNOS luciferase activity was suppressed with H. pylori infection (Figure 3C). Thus, these data suggest that H. pylori may inhibit iNOS promoter activity and subsequently influence NO production in macrophages.. 14.
(21) 4. Involvement of ERK1/2 and MAPK-signaling pathways in H. pylori inhibits LPS-mediated macrophage NO production As LPS-induced NO production in macrophages has been shown several signaling pathways, including mitogen-activated protein kinase (MAPK), JNK, p42 and p44 (ERK 1/2). We performed Western blot analysis to elucidate the signal transduction mechanisms involved in H. pylori-inhibited LPS-induced NO production in PEMs from 0 to 60 min. As shown in figure 4A, the phophorylation of p38 induced by LPS was reduced within 0-5 min after H. pylori infection. The other signaling pathway, phosphorylation of p42 and p44 (p-ERK1/2), was also inhibited by infection with H. pylori during 0-5 min. In contrast to p38 and ERK1/2, phophorylation of JNK1/2 in LPS-stimulated macrophage was not influenced by H. pylori infection until 60 min. We further confirmed the inhibition of phosphorylation of p38 and ERK1/2 in LPS-induced macrophages after H. pylori infection at MOI of 0 and 100 within 5 min. The data showed that both phosphorylation of p38 and ERK1/2 were decreased when infection with H. pylori (Figure 4B). These results supported that H. pylori-inhibited LPS-stimulated NO production and iNOS expression in macrophages was regulated by p38 and ERK1/2 signaling pathway, while JNK1/2 was not involved in those effects.. 15.
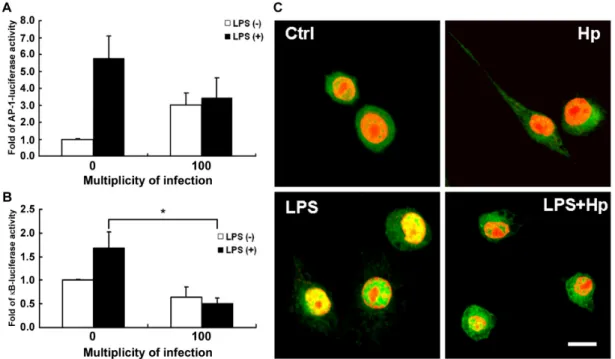
(22) 5. Suppression of NF-κB activation in H. pylori inhibits LPS-mediated macrophage NO production Consideration of the inhibition of iNOS transcription, we next investigated the effects of H. pylori on two essential transcription factors, NF-κB and AP-1, which have been demonstrated to play an essential role in the iNOS protein expression. We then examined the effects of H. pylori on transcription factor, NF-κB. The activity of NF-κB was determined by the luciferase assay in RAW 264.7 macrophages transfected with κB-lucifease plasmid. As shown in figure 5A, LPS-stimulated a significant activation of NF-κB promoter activity which was inhibited by infection of H. pylori. We further examined p65 localization, which showed that p65 was primarily located in the cytosol before LPS-treatment. In response to LPS for 2 hr, the p65 protein was translocated in nuclei. However, the p65 protein was even major in the cytosol when co-cultured with LPS and H. pylori. Another transcription factors AP-1 was also analyzed. In parallel, reporter assay driven by AP-1 transactivation in RAW 264.7 macrophages was also revealed the effect of H. pylori in inhibition of AP-1. These data suggested that H. pylori triggered not only the distribution of p65 but AP-1 transactivation in LPS-stimulated NO production in macrophage. To evaluate LPS-indcued iNOS promoter activity, the cells were co-treated with p38 ODN (oligonucleotide dominate negative), ERK ODN, IKK α/β ODN or scramble ODN. After transfection, incubated the cells with LPS (2µg/ml) and H. pylori (MOI=100) for 24 hr.. 16.
(23) The data showed even the cells mutant with P38, ERK or IKK α/β, the iNOS activity could be activated by LPS but not by H. pylori. And H. pylori had a significant inhibition of LPS-induced iNOS activity (data not show).. 17.
(24) Discussion Expression of iNOS and production of NO is well known to be an important effector molecule in macrophage for responses to bacteria due to antimicrobial activity (21). It has been reported that H. pylori can induce iNOS and NO production in macrophages (10, 12, 22). In vitro study also revealed that H. pylori are killed in an NO-dependent manner (22). However, this response maybe ineffective, as H. pylori generally persistent infects for the life of host and results in clinical outcomes. In this study we used a mouse macrophage model system to demonstrate that infection of H. pylori can attenuate LPS-induced macrophage expression of iNOS and suppress the production of NO. Our data also revealed that bacteria survived in the environment of co-cultured with H. pylori and LPS-induced macrophage in a trans-well model system. This finding was consistent with previous study that H. pylori arginase suppresses NO production and leading to immune evasion (13). In addition, H. pylori can also activate innate immune responses and induces apoptosis both in macrophage (23) as well as T cells (24). In the discovery of animal infection model, they found that macrophages are the mediators of gastritis of acute H. pylori infection in mice (25). Another report indicated that the ineffectiveness of NO-mediated antimicrobial activity maybe absence of local high NO concentration at the site of bacteria infection (26). In order to produce higher level of NO, we used LPS at the higher concentration of 2µg/ml to elevate NO production in macrophage. After treatment of LPS in RAW 264.7 cells for 24 hr or mouse PEMs for 48 hr,. 18.
(25) NO concentration was approached to ca. 20µM and 120µM, respectively. However, in the infection model system, co-cultured with H. pylori and LPS-induced macrophage still showed that the bacteria has the ability to inhibit NO production at higher bacterial load and resulted in bacteria survived (Figure 1 and 2C). Thus, these results reveal that H. pylori has a delicate mechanism to regulate the activation of macrophage and suppression of NO production. In our current study, using Western blot and RT-PCR analysis revealed that H. pylori can inhibit iNOS expression in LPS-induced macrophage at the transcriptional and post-transcriptional levels. Accordingly, the effectiveness of inhibitory activity was correlated with increasing of bacterial load (Figure 3). The expression of iNOS mRNA was reduced in LPS-induced macrophages infected with H. pylori at a MOI of 100. However, H. pylori also stimulated macrophage iNOS mRNA expression in the absence of LPS-induced condition. This may be the effectiveness of iNOS mRNA expression in H. pylori infection with macrophage for a long period (10). In this study, H. pylori suppression of NO production in LPS-induced macrophage was found that dependent on live bacteria (Figure 2A and 2B). Heat-killed and crude extract of H. pylori showed absence of LPS-induced NO production, this result was in consistent with previous finding (13), suggesting that this effect is mediated through direct interaction of H. pylori and macrophage. Although H. pylori has been considered as an extracelluar pathogen, several study revealed that this bacteria has the ability to invade into gastric epithelial cells. 19.
(26) (27, 28). In addition, H. pylori was found to stimulate megasome formation and leading to delay phagocytosis in macrophage (15). Moreover, a recent report showed that using transmission electron microscopy visualized that H. pylori was directly contacted with immune cells of lamina propria (29). Collectively, these findings may explain that the live H. pylori have the ability to invade into macrophage and presumably induction of ineffectiveness of immune cells. There were many research in nitric oxide inhibition, because of nitric oxide over production may cause shocks even lead to death (30, 31). It has well known that LPS stimulate iNOS gene expression and NO production mediated positively regulation of NF-κB, which is normally bound to its inhibitor IκB. Phosphorylation of IκB by IκB kinase (IKK) results in its degradation and leading to NF-κB activation, which then removes from cytosol to the nucleus. In addition to NF-κB, LPS also enable to activate MAP kinase pathway in macrophage, including Erk-1/2, JNK-1/2, and p38 [review in (32)]. During infection, nitric oxide produced is controlled by iNOS, and it could be regulated by TLR4 pathway, included NF-κB, IκB, IKKα/β and MAPK family (22, 33-38). Some research in Salmonella found it has similar inhibition of nitric oxide (34, 39, 40). Yersinia inhibits MAPK family activation by using Yop protein family (41-49). Here we found the ability of H. pylori to suppress LPS-induced NO production in macrophage, this was triggered by the inhibition of p65 nuclear translocation and NF-κB activation. Our current data also demonstrated that. 20.
(27) phosphorylation of p38 and Erk1/2 was attenuated by H. pylori infection, while JNK-1/2 was not involved in such interaction. This phenomenon might contribute to the inhibition of p65 phosphorylation and NF-κB activation, subsequently suppress both iNOS expression and NO production. Our result suggested that the upstream cellular target of H. pylori is more directly interacted with p38 and Erk1/2 but not JNK-1/2. A study in Drosophila model improve that nitric oxide production contributes to the induction of innate immune responses to gram-negative bacteria (50). The importance of iNOS in gastric epithelium cells is the regulation of apoptosis in H. pylori-infected cells, this reaction could prevent preneoplastic transformation (35). During pulmonary infection, flagellin from gram-negative bacteria is related to the immune response which was activated by toll like receptor (TLR) 5 (51). There were many studies suggest that lipopolysaccharide (LPS) from gram-negative bacterium could induce nitric oxide production and iNOS expression of macrophage. Such reactions could be considered as inflammatory responses as well as through TLR 4 pathway (52, 53). H. pylori is a gram-negative bacterium, also there were LPS on its’ outer membrane. Some reports have been discovered that LPS from H. pylori could active NF-κB through TLR2 but not TLR4 (54, 55). In additional, it is thought that LPS from H. pylori might be an antagonist to TLR4 (56). Previous report also suggested that inhibition of nitric oxide by H. pylori infection of macrophage would enhance bacterial viability (13). When H. pylori. 21.
(28) infected with macrophage, the inflammation responses was reduced and the inflammatory regulators also down-regulated (22). In gastric epithelium cells, H. pylori infection could induce a chronic inflammation by inducing nitrite production (57). Both in human and animal model, nitric oxide synthase expression in epithelium were up-regulated by H. pylori infection, and these cause the chronic gastritis (58). In human stomach epithelium, the infection by H. pylori would up-regulate endothelial nitric oxide synthase (eNOS) expression and induces angiogenesis in gastric mucosa of dyspeptic patients (59). Helicobacter pylori infects to stomach may cause chronic gastritis, gastric ulcer, duodenal ulcer even cause cancer (60). However, H. pylori is a pathogen, the host innate immunity may clean the bacterium, but in fact, it could dodge the immune response then survive in tissues (61, 62). Many studies focus on the pathogenesis of H. pylori and they confirmed it go through type IV secretion and cag pathogenicity island (63-65). Innate immunity could clean the pathogens then protect body from illness. But during H. pylori infection, the innate immunity has lost its’ functions (66, 67). Some agreed that H. pylori has an immune invasion because of the cholesterol glucosylation (68). Also in other bacterium have the ability of immune evasion, some could escape from immune response by reducing nitric oxide (69). In the infection of Staphylococcus aureus, it secretes proteins to inhibit complement activation and neutrophil chemotaxis or that lyse neutrophils, neutralizes antimicrobial defensin peptides, and its cell surface is modified to reduce their effectiveness.. 22.
(29) The organism can survive in phagosomes, express polysaccharides and proteins that inhibit opsonization by antibody and complement, and its’ cell wall is resistant to lysozyme. Besides, S. aureus expresses several types of superantigen that corrupt the normal humoral immune response, resulting in anergy and immunosuppression. In contrast with Staphylococcus epidermidis, S. epidermidis must rely primarily on cell-surface polymers and the ability to form the biolfilm to survive in the host (70). In a early discovery, they found H. pylori could fuse the phagosomes into megasomes, then proliferated in the megasomes (15). Recently finding is the urease of H. pylori could help the organism escape form megasomes (71). Above these discussions, we know that bacterium have some mechanisms to escape from immune system. The ability of immune evasion is important to pathogens. When pathogens dodge the attack of immune system, they have more chances to survive, proliferate and pathogenesis. H. pylori was already known which could survive in host tissues and induce a chronic inflammation. But the mechanisms how H. pylori escape from immune system still not clear. The ability to interfere NF-κB activation is a strategy for pathogen to enhance immune evasion. Several studies have been demonstrated that pathogens can exploit the NF-κB to manipulate cellular responses (72). Chlamydia sp. was found to cleavage of p65 protein, subsequently suppress host inflammatory immune response (73). Another report of enteropathogenic E. coli was also found to trigger NF-κB activation and result in inhibit iNOS. 23.
(30) expression (74). Our current results are consistent with previous finding that bacteria can trigger NF-κB activation, subsequently manipulate immune mediators. This general effect on host signaling may support the hypothesis that H. pylori enable to modulate host signaling events and protect bacteria from immune elimination. In conclusion, in this study we demonstrate that infection with live H. pylori can inhibit LPS-induced iNOS gene transcription and NO production in a mouse macrophage model system. H. pylori inhibits LPS-induced MAP kinase pathway results in reduced NF-κB transactivation. This phenomenon including suppress antimicrobial mediators in innate immune responses of H. pylori. Collectively, our study reveals a new mechanism whereby H. pylori protects macrophage inflammation and supports the hypothesis that bacteria modulate host signaling to exert its beneficial effects.. 24.
(31) Prospection In our results, we confirmed H. pylori could inhibit LPS-induced nitric oxide production. And the inhibitions of nitric oxide were through iNOS expression inhibition; the phosphorylation inhibition of ERK1/2, P38; and the down-regulation of NF-κB activation. One of our experiments, we did bacteriocied assay (Fig. 2C), the result showed us when H. pylori inhibits nitric oxide producing, there were more bacterium survive. This result demonstrates our contention, inhibit nitric oxide production could help more bacterium surviving. We already know H. pylori inhibit nitric oxide producing to raise the survival rate of bacterium, but there still have many questions need to answer. In previously discussions, some suggest H. pylori is an antagonist to TLR4, but these discovers were in gastric cancer, vascular endothelial cells and human embryonic kidney cells (54-56). We would like to know how H. pylori interact with macrophages. In our research, we used LPS which from E. coli to induce the inflammatory reactions of macrophages. When cocultured with H. pylori could down-regulate the inflammatory reactions. If it is possible when infect with H. pylori could prevent the immune response activating by other bacterium infection? Also, we would like to define the mechanism pathway which is used by H. pylori to escape form the attack of immune system. And the interaction of H. pylori with other pathogens.. 25.
(32) As we showed in introduction, H. pylori could cause gastric ulcer, duodenal ulcer even cause cancer. Once infection with H. pylori, the bacterium could survive in the host for a long life time. Commonly infection with pathogens could cause the host immune response to clean the pathogens. But in the infection of H. pylori, the host response is mild chronic inflammation. And which is the regulating pathway to control the chronic inflammation still unclear. We also have great interest in the H. pylori inducing chronic gastric inflammation. To our proposal, we hope to demonstrate the activation of inflammatory factor Interleukin-8 (IL-8) during chronic inflammation if is regulated by Src/p110β PI3K/Akt pathway. This proposal is the beginning to my research direction in the future. After we find out the mechanism of chronic gastric inflammation, I expect I could study more deeply in how H. pylori could prevent the attack of immune systems then survive in host, and the mechanisms that cause the chronic gastric inflammation. In the future, I wish I could study broadly, not only in the pathogenesis of H. pylori, but also in other pathogens.. 26.
(33) References 1.. Miyaji, H., T. Azuma, S. Ito, Y. Abe, F. Gejyo, N. Hashimoto, H. Sugimoto, H. Suto, Y. Ito, Y. Yamazaki, Y. Kohli, and M. Kuriyama. 2000. Helicobacter pylori infection occurs via close contact with infected individuals in early childhood. Journal of gastroenterology and hepatology 15:257-262.. 2.. Goodman, K. J., and P. Correa. 2000. Transmission of Helicobacter pylori among siblings. Lancet 355:358-362.. 3.. Rothenbacher, D., G. Bode, G. Berg, U. Knayer, T. Gonser, G. Adler, and H. Brenner. 1999. Helicobacter pylori among preschool children and their parents: evidence of parent-child transmission. The Journal of infectious diseases 179:398-402. Brandt, S., T. Kwok, R. Hartig, W. Konig, and S. Backert. 2005. NF-kappaB activation. 4.. 5.. 6. 7.. 8.. 9.. 10.. 11.. and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proceedings of the National Academy of Sciences of the United States of America 102:9300-9305. Viala, J., C. Chaput, I. G. Boneca, A. Cardona, S. E. Girardin, A. P. Moran, R. Athman, S. Memet, M. R. Huerre, A. J. Coyle, P. S. DiStefano, P. J. Sansonetti, A. Labigne, J. Bertin, D. J. Philpott, and R. L. Ferrero. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nature immunology 5:1166-1174. Sartor, R. B. 1994. Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology 106:533-539. Harris, P. R., L. E. Smythies, P. D. Smith, and A. Dubois. 2000. Inflammatory cytokine mRNA expression during early and persistent Helicobacter pylori infection in nonhuman primates. The Journal of infectious diseases 181:783-786. Yamaoka, Y., M. Kita, T. Kodama, N. Sawai, and J. Imanishi. 1996. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology 110:1744-1752. El-Omar, E. M., M. Carrington, W. H. Chow, K. E. McColl, J. H. Bream, H. A. Young, J. Herrera, J. Lissowska, C. C. Yuan, N. Rothman, G. Lanyon, M. Martin, J. F. Fraumeni, Jr., and C. S. Rabkin. 2000. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404:398-402. Wilson, K. T., K. S. Ramanujam, H. L. Mobley, R. F. Musselman, S. P. James, and S. J. Meltzer. 1996. Helicobacter pylori stimulates inducible nitric oxide synthase expression and activity in a murine macrophage cell line. Gastroenterology 111:1524-1533. Fu, S., K. S. Ramanujam, A. Wong, G. T. Fantry, C. B. Drachenberg, S. P. James, S. J. Meltzer, and K. T. Wilson. 1999. Increased expression and cellular localization of 27.
(34) inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology 116:1319-1329. 12.. Gobert, A. P., B. D. Mersey, Y. Cheng, D. R. Blumberg, J. C. Newton, and K. T. Wilson. 2002. Cutting edge: urease release by Helicobacter pylori stimulates macrophage inducible nitric oxide synthase. J Immunol 168:6002-6006.. 13.. Gobert, A. P., D. J. McGee, M. Akhtar, G. L. Mendz, J. C. Newton, Y. Cheng, H. L. Mobley, and K. T. Wilson. 2001. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proceedings of the. 14.. 15.. 16.. 17. 18.. 19.. 20.. 21. 22.. National Academy of Sciences of the United States of America 98:13844-13849. McGee, D. J., F. J. Radcliff, G. L. Mendz, R. L. Ferrero, and H. L. Mobley. 1999. Helicobacter pylori rocF is required for arginase activity and acid protection in vitro but is not essential for colonization of mice or for urease activity. Journal of bacteriology 181:7314-7322. Allen, L. A., L. S. Schlesinger, and B. Kang. 2000. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. The Journal of experimental medicine 191:115-128. Boncristiano, M., S. R. Paccani, S. Barone, C. Ulivieri, L. Patrussi, D. Ilver, A. Amedei, M. M. D'Elios, J. L. Telford, and C. T. Baldari. 2003. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med 198:1887-1897. Gebert, B., W. Fischer, E. Weiss, R. Hoffmann, and R. Haas. 2003. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099-1102. Lai, C. H., C. H. Kuo, P. Y. Chen, S. K. Poon, C. S. Chang, and W. C. Wang. 2006. Association of antibiotic resistance and higher internalization activity in resistant Helicobacter pylori isolates. The Journal of antimicrobial chemotherapy 57:466-471. Chi, D. S., M. Qui, G. Krishnaswamy, C. Li, and W. Stone. 2003. Regulation of nitric oxide production from macrophages by lipopolysaccharide and catecholamines. Nitric Oxide 8:127-132. Rao, Y. K., S. H. Fang, and Y. M. Tzeng. 2007. Evaluation of the anti-inflammatory and anti-proliferation tumoral cells activities of Antrodia camphorata, Cordyceps sinensis, and Cinnamomum osmophloeum bark extracts. Journal of ethnopharmacology 114:78-85. Fang, F. C. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nature reviews 2:820-832. Bussiere, F. I., R. Chaturvedi, Y. Cheng, A. P. Gobert, M. Asim, D. R. Blumberg, H. Xu, P. Y. Kim, A. Hacker, R. A. Casero, Jr., and K. T. Wilson. 2005. Spermine causes loss of innate immune response to Helicobacter pylori by inhibition of inducible nitric-oxide synthase translation. J Biol Chem 280:2409-2412. 28.
(35) 23.. Gobert, A. P., Y. Cheng, J. Y. Wang, J. L. Boucher, R. K. Iyer, S. D. Cederbaum, R. A. Casero, Jr., J. C. Newton, and K. T. Wilson. 2002. Helicobacter pylori induces. 24.. macrophage apoptosis by activation of arginase II. J Immunol 168:4692-4700. Zabaleta, J., D. J. McGee, A. H. Zea, C. P. Hernandez, P. C. Rodriguez, R. A. Sierra, P. Correa, and A. C. Ochoa. 2004. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta). J Immunol. 25.. 173:586-593. Kaparakis, M., A. K. Walduck, J. D. Price, J. S. Pedersen, N. van Rooijen, M. J. Pearse,. 26.. O. L. Wijburg, and R. A. Strugnell. 2008. Macrophages are mediators of gastritis in acute Helicobacter pylori infection in C57BL/6 mice. Infect Immun 76:2235-2239. Vallance, B. A., W. Deng, M. De Grado, C. Chan, K. Jacobson, and B. B. Finlay. 2002.. 27.. 28.. 29.. 30.. 31. 32.. 33.. 34.. Modulation of inducible nitric oxide synthase expression by the attaching and effacing bacterial pathogen citrobacter rodentium in infected mice. Infection and immunity 70:6424-6435. Amieva, M. R., N. R. Salama, L. S. Tompkins, and S. Falkow. 2002. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cellular microbiology 4:677-690. Semino-Mora, C., S. Q. Doi, A. Marty, V. Simko, I. Carlstedt, and A. Dubois. 2003. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. The Journal of infectious diseases 187:1165-1177. Necchi, V., M. E. Candusso, F. Tava, O. Luinetti, U. Ventura, R. Fiocca, V. Ricci, and E. Solcia. 2007. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology 132:1009-1023. Zapelini, P. H., G. T. Rezin, M. R. Cardoso, C. Ritter, F. Klamt, J. C. Moreira, E. L. Streck, and F. Dal-Pizzol. 2008. Antioxidant treatment reverses mitochondrial dysfunction in a sepsis animal model. Mitochondrion 8:211-218. Singer, M. 2008. Cellular dysfunction in sepsis. Clin Chest Med 29:655-660, viii-ix. Dobrovolskaia, M. A., and S. N. Vogel. 2002. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes and infection / Institut Pasteur 4:903-914. Kuo, C. L., F. M. Ho, M. Y. Chang, E. Prakash, and W. W. Lin. 2008. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem 103:931-940. Lahiri, A., P. Das, and D. Chakravortty. 2008. Arginase modulates Salmonella induced nitric oxide production in RAW264.7 macrophages and is required for Salmonella 29.
(36) pathogenesis in mice model of infection. Microbes Infect 10:1166-1174. 35.. Miyazawa, M., H. Suzuki, T. Masaoka, A. Kai, M. Suematsu, H. Nagata, S. Miura, and H. Ishii. 2003. Suppressed apoptosis in the inflamed gastric mucosa of Helicobacter pylori-colonized iNOS-knockout mice. Free Radic Biol Med 34:1621-1630.. 36.. Suh, S. J., T. W. Chung, M. J. Son, S. H. Kim, T. C. Moon, K. H. Son, H. P. Kim, H. W. Chang, and C. H. Kim. 2006. The naturally occurring biflavonoid, ochnaflavone, inhibits LPS-induced iNOS expression, which is mediated by ERK1/2 via NF-kappaB. 37.. 38.. 39.. 40.. 41. 42.. 43.. 44.. 45.. 46.. regulation, in RAW264.7 cells. Arch Biochem Biophys 447:136-146. Sharma, R. K., A. Sodhi, H. V. Batra, and U. Tuteja. 2005. Phosphorylation of p42/44 MAP kinase is required for rF1-induced activation of murine peritoneal macrophages. Mol Immunol 42:1385-1392. Sharma, R. K., A. Sodhi, and H. V. Batra. 2005. Involvement of c-Jun N-terminal kinase in rF1 mediated activation of murine peritoneal macrophages in vitro. J Clin Immunol 25:215-223. Bourret, T. J., S. Porwollik, M. McClelland, R. Zhao, T. Greco, H. Ischiropoulos, and A. Vazquez-Torres. 2008. Nitric oxide antagonizes the acid tolerance response that protects Salmonella against innate gastric defenses. PLoS ONE 3:e1833. Foster, N., S. D. Hulme, and P. A. Barrow. 2005. Inhibition of IFN-gamma-stimulated proinflammatory cytokines by vasoactive intestinal peptide (VIP) correlates with increased survival of Salmonella enterica serovar typhimurium phoP in murine macrophages. J Interferon Cytokine Res 25:31-42. Mukherjee, S., and K. Orth. 2008. In vitro signaling by MAPK and NFkappaB pathways inhibited by Yersinia YopJ. Methods Enzymol 438:343-353. Hao, Y. H., Y. Wang, D. Burdette, S. Mukherjee, G. Keitany, E. Goldsmith, and K. Orth. 2008. Structural requirements for Yersinia YopJ inhibition of MAP kinase pathways. PLoS ONE 3:e1375. Sweet, C. R., J. Conlon, D. T. Golenbock, J. Goguen, and N. Silverman. 2007. YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell Microbiol 9:2700-2715. Lindner, I., J. Torruellas-Garcia, D. Kolonias, L. M. Carlson, K. A. Tolba, G. V. Plano, and K. P. Lee. 2007. Modulation of dendritic cell differentiation and function by YopJ of Yersinia pestis. Eur J Immunol 37:2450-2462. Autenrieth, S. E., I. Soldanova, R. Rosemann, D. Gunst, N. Zahir, M. Kracht, K. Ruckdeschel, H. Wagner, S. Borgmann, and I. B. Autenrieth. 2007. Yersinia enterocolitica YopP inhibits MAP kinase-mediated antigen uptake in dendritic cells. Cell Microbiol 9:425-437. Thiefes, A., A. Wolf, A. Doerrie, G. A. Grassl, K. Matsumoto, I. Autenrieth, E. Bohn, 30.
(37) H. Sakurai, R. Niedenthal, K. Resch, and M. Kracht. 2006. The Yersinia enterocolitica effector YopP inhibits host cell signalling by inactivating the protein kinase TAK1 in 47.. the IL-1 signalling pathway. EMBO Rep 7:838-844. Mukherjee, S., G. Keitany, Y. Li, Y. Wang, H. L. Ball, E. J. Goldsmith, and K. Orth. 2006. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312:1211-1214.. 48.. Bliska, J. B. 2006. Yersinia inhibits host signaling by acetylating MAPK kinases. ACS Chem Biol 1:349-351.. 49.. Zhang, Y., A. T. Ting, K. B. Marcu, and J. B. Bliska. 2005. Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J Immunol 174:7939-7949.. 50.. Foley, E., and P. H. O'Farrell. 2003. Nitric oxide contributes to induction of innate immune responses to gram-negative bacteria in Drosophila. Genes Dev 17:115-125. Liaudet, L., C. Szabo, O. V. Evgenov, K. G. Murthy, P. Pacher, L. Virag, J. G. Mabley, A. Marton, F. G. Soriano, M. Y. Kirov, L. J. Bjertnaes, and A. L. Salzman. 2003. Flagellin from gram-negative bacteria is a potent mediator of acute pulmonary inflammation in sepsis. Shock 19:131-137. Ryan, K. A., M. F. Smith, Jr., M. K. Sanders, and P. B. Ernst. 2004. Reactive oxygen and nitrogen species differentially regulate Toll-like receptor 4-mediated activation of NF-kappa B and interleukin-8 expression. Infect Immun 72:2123-2130. Thomas, K. E., C. L. Galligan, R. D. Newman, E. N. Fish, and S. N. Vogel. 2006. Contribution of interferon-beta to the murine macrophage response to the toll-like receptor 4 agonist, lipopolysaccharide. J Biol Chem 281:31119-31130. Yokota, S., T. Ohnishi, M. Muroi, K. Tanamoto, N. Fujii, and K. Amano. 2007. Highly-purified Helicobacter pylori LPS preparations induce weak inflammatory reactions and utilize Toll-like receptor 2 complex but not Toll-like receptor 4 complex.. 51.. 52.. 53.. 54.. 55.. 56.. 57.. FEMS Immunol Med Microbiol 51:140-148. Triantafilou, M., F. G. Gamper, P. M. Lepper, M. A. Mouratis, C. Schumann, E. Harokopakis, R. E. Schifferle, G. Hajishengallis, and K. Triantafilou. 2007. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol 9:2030-2039. Lepper, P. M., M. Triantafilou, C. Schumann, E. M. Schneider, and K. Triantafilou. 2005. Lipopolysaccharides from Helicobacter pylori can act as antagonists for Toll-like receptor 4. Cell Microbiol 7:519-528. Shiotani, A., H. Iishi, M. Kumamoto, and Y. Nakae. 2004. Helicobacter pylori infection and increased nitrite synthesis in the stomach. Inflammation and atrophy 31.
(38) connections. Dig Liver Dis 36:327-332. 58.. Obonyo, M., D. G. Guiney, J. Fierer, and S. P. Cole. 2003. Interactions between inducible nitric oxide and other inflammatory mediators during Helicobacter pylori infection. Helicobacter 8:495-502.. 59.. Lazaraki, G., J. Kountouras, S. Metallidis, E. Vrettou, V. Tzioufa, G. Germanidis, D. Chatzopoulos, C. Zavos, K. Giannoulis, and P. Nikolaidis. 2008. Helicobacter pylori infection upregulates endothelial nitric oxide synthase expression and induces angiogenesis in gastric mucosa of dyspeptic patients. Eur J Gastroenterol Hepatol. 60. 61.. 62. 63. 64.. 65.. 66.. 67.. 68.. 69. 70.. 20:441-449. Armuzzi, A., A. Gasbarrini, M. Gabrielli, F. Cremonini, M. Anti, and G. Gasbarrini. 2001. Helicobacter pylori and gastric carcinoma. Ann Ital Chir 72:5-11. Cooke, C. L., J. L. Huff, and J. V. Solnick. 2005. The role of genome diversity and immune evasion in persistent infection with Helicobacter pylori. FEMS Immunol Med Microbiol 45:11-23. Lee, S. K., and C. Josenhans. 2005. Helicobacter pylori and the innate immune system. Int J Med Microbiol 295:325-334. Backert, S., Y. Churin, and T. F. Meyer. 2002. Helicobacter pylori type IV secretion, host cell signalling and vaccine development. Keio J Med 51 Suppl 2:6-14. Galgani, M., I. Busiello, S. Censini, S. Zappacosta, L. Racioppi, and R. Zarrilli. 2004. Helicobacter pylori induces apoptosis of human monocytes but not monocyte-derived dendritic cells: role of the cag pathogenicity island. Infect Immun 72:4480-4485. Hohlfeld, S., I. Pattis, J. Puls, G. V. Plano, R. Haas, and W. Fischer. 2006. A C-terminal translocation signal is necessary, but not sufficient for type IV secretion of the Helicobacter pylori CagA protein. Mol Microbiol 59:1624-1637. Robinson, K., R. H. Argent, and J. C. Atherton. 2007. The inflammatory and immune response to Helicobacter pylori infection. Best Pract Res Clin Gastroenterol 21:237-259. Grebowska, A., A. P. Moran, A. Matusiak, L. Bak-Romaniszyn, E. Czkwianianc, T. Rechcinski, M. Walencka, I. Planeta-Malecka, W. Rudnicka, and M. Chmiela. 2008. Anti-phagocytic activity of Helicobacter pylori lipopolysaccharide (LPS)--possible modulation of the innate immune response to these bacteria. Pol J Microbiol 57:185-192. Wunder, C., Y. Churin, F. Winau, D. Warnecke, M. Vieth, B. Lindner, U. Zahringer, H. J. Mollenkopf, E. Heinz, and T. F. Meyer. 2006. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med 12:1030-1038. Shin, H., M. Mally, M. Kuhn, C. Paroz, and G. R. Cornelis. 2007. Escape from immune surveillance by Capnocytophaga canimorsus. J Infect Dis 195:375-386. Foster, T. J. 2005. Immune evasion by staphylococci. Nat Rev Microbiol 3:948-958. 32.
(39) 71.. Schwartz, J. T., and L. A. Allen. 2006. Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J Leukoc Biol 79:1214-1225.. 72.. Tato, C. M., and C. A. Hunter. 2002. Host-pathogen interactions: subversion and utilization of the NF-kappa B pathway during infection. Infection and immunity 70:3311-3317.. 73.. Lad, S. P., J. Li, J. da Silva Correia, Q. Pan, S. Gadwal, R. J. Ulevitch, and E. Li. 2007. Cleavage of p65/RelA of the NF-kappaB pathway by Chlamydia. Proceedings of the National Academy of Sciences of the United States of America 104:2933-2938.. 74.. Maresca, M., D. Miller, S. Quitard, P. Dean, and B. Kenny. 2005. Enteropathogenic Escherichia coli (EPEC) effector-mediated suppression of antimicrobial nitric oxide production in a small intestinal epithelial model system. Cellular microbiology 7:1749-1762.. 33.
(40) Figures. Figure 1. Inhibition by H. pylori of LPS-induced NO production in RAW 264.7 cell line and mouse primary peritoneal excluded macrophage. Cells were treated with or without LPS (2µg/ml) and infected with H. pylori at various MOI of 0 to 200. After incubation for 24 hr and 48 hr, the culture supernatant of RAW 264.7 cell line (A) and mouse primary PEMs (B) were then collected for the assay of nitric oxide production, respectively. The data are the means and standard deviations of at least three independent experiments performed in triplicate. Statistical significance was evaluated using Student's t test (*, P < 0.05).. 34.
(41) Figure 2. Live H. pylori is essential for inhibition of LPS-induced nitric oxide production by macrophage. Cells were treated with or without LPS (2µg/ml) and were left un-infected or infected with lived, heat-killed and crude extract derived from H. pylori. After 24 hr and 48 hr, the culture supernatant of RAW 264.7 cell line (A) and mouse primary PEMs (B) was then collected for the assay of nitric oxide production, respectively. (C) Mouse primary PEMs grown on Trans-well bottom layer were infected with H. pylori at MOI of 0 to 100 for 48 hr, and then co-incubated with H. pylori in the trans-well insert membrane (0.1µm) for a further 6 hr, after which, the CFUs were counted. The bactericidal activity was expressed as the means of at least three independent experiments performed in triplicate. Statistical significance was evaluated using Student's t test (*, P < 0.05). 35.
(42) Figure 3. Inhibition by H. pylori of expression of protein and gene transcription for iNOS in LPS-induced macrophage activation. Mouse primary PEMs were treated with or without LPS (2µg/ml) and infected with H. pylori at different MOI for 48 hr. Cell lysates were prepared for the determination of iNOS in protein levels (A) and mRNA levels (B). The data of protein and mRNA expression were quantified with densitometric analysis and normalized with α–tubulin and GAPDH, respectively. (C) RAW 264.7 cells transfected with reporter gene iNOS and β-gal-lacZ (each at 1µg) were treated with or without LPS (2µg/ml) and infection with or without H. pylori. Luciferase activity was normalized to the transfection efficiency with β-gal-lacZ. The data was expressed as the means of at least three independent experiments performed in triplicate. Statistical significance was evaluated using Student's t test (*, P < 0.05). 36.
(43) Figure 4. Signal transduction mechanisms involved in attenuation by H. pylori for LPS-induced macrophage activation. (A) Mouse primary PEMs were incubated with LPS (2µg/ml) and infection with or without H. pylori for indicated time intervals, and p-p38, p-ERK, and p-JNK expression were determined by Western blot analysis. H. pylori inhibited LPS-induced p38 and extracellular signal-related kinase 1/2 (ERK1/2) pathway, as evidenced by the decrease in both phosphorylated p38 and phosphorylated ERK at 0-5 min, but not phosphorylated JNK1/2. (B) Mouse primary PEMs were treated with or without LPS (2µg/ml) at MOI of 0 and 100 for 5 min, p-p38 and p-ERK expression were determined by Western blot analysis. Infection of cells with H. pylori at a MOI of 100 results in suppression of LPS-induced p38 and ERK1/2 phosphorylation. 37.
(44) Figure 5. Inhibition of LPS-induced NF-kappaB activation by H. pylori. RAW 264.7 cells transfected with reporter gene AP-1 (A), NF-kappaB (B), and β-gal-lacZ (each at 1µg) were treated with or without LPS (2µg/ml) and infection with or without H. pylori. Luciferase activity was normalized to the transfection efficiency with β-gal-lacZ. The data was expressed as the means of at least three independent experiments performed in triplicate. Statistical significance was evaluated using Student's t test (*, P < 0.05). (C) The nuclear translocation of p65 after 1 hr treatment with or without LPS and H. pylori was shown by immunofluoresence, which was inhibited by infection with H. pylori. Scale bar = 10 µm. 38.
(45) Appendix LPS LBP. CD14. TLR4 TLR4. M D-2. Macrophage. M yD88 PI3K. Cytoplasm. Nitrite release. IRAK. Citrulline. Akt. TRAF6. Ras c-Raf. PKR. MEKK1/4. MEK1/2. TAB2. ECSIT. TAK1. MEKK1. P. ERK1/2. p38. JNK. ?. IKKβ β. MKK3/6. MKK4/7. L-Arginine. iNOS. P. Iκ κB P p50 p65 Translation MSK1. Nucleus. P. P P. P. P. P. P. P. P. iNOS mRNA. P. Elk1 SRF. c-Jun c-Fos. c-Jun AT F-2. CREBATF-1. p50 p65. SRE. AP-1. AP-1. CRE. NF-κ κB. iNOS gene. Transcription. I. The activation of TLR4 and the down-stream pathway. [This figure was adapted from the article which was published in Cellular signaling (2001), Mausumee G et al.]. 39.
(46) LPS LBP. CD14. TLR4 TLR4. M D-2. Macrophage. M yD88 PI3K. Cytoplasm. Nitrite release. IRAK. Citrulline. Akt. TRAF6. Ras c-Raf. PKR. MEKK1/4. MEK1/2. TAB2. ECSIT. TAK1. MEKK1. P. ERK1/2. p38. JNK. ?. IKKβ β. MKK3/6. MKK4/7. L-Arginine. iNOS. P. Iκ κB P p50 p65 Translation MSK1. Nucleus. P. P P. P. P. P. P. P. P. iNOS mRNA. P. Elk1 SRF. c-Jun c-Fos. c-Jun AT F-2. CREBATF-1. p50 p65. SRE. AP-1. AP-1. CRE. NF-κ κB. iNOS gene. Transcription. II. Helicobacter pylori infect to macrophage could suppress the inflammatory factors activation. To our finding, H. pylori could inhibit the nitrite production, iNOS expression. H. pylori also suppressed the activation of NF-κB and AP-1. Thus, H. pylori could attenuate the phosphorylation of p38 and ERK1/2.. 40.
(47)
數據


+2
相關文件
• 由於細胞代謝、紫外線的電離輻射、不良的生 活習慣 ( 煙、酒、肥胖 ) 、各種感染物所引起 的中性粒細胞、巨噬細胞的激活, 會催化分子 氧發生單價還原產生的 "
Seals, if any, essential for sealing the pressure sensing element, and in direct contact with the process medium, made of or protected by aluminum, aluminum alloy, aluminum
而在後續甲烷化反應試驗方面,以前段經厭氧醱酵產氫後之出流水為進流基 質。在厭氧光合產氫微生物方面,以光合作用產氫細菌中產氫能力最好的菌株 Rhodopseudomonas palustris
評估以 S-649266 或最佳現有療法進行治療罹患抗 Carbapenem 革蘭氏陰性菌感染的患者的臨床結果 (包括詴驗用藥 S-649266
一、「透明導電氧化物玻璃」之種類主要為「氧化銦錫導電玻璃(Indium Tin Oxide Coating Glass, ITO)」、「氧 化鋅透明導電玻璃(Al-doped Zinc Oxide
和富蘭克林·史達(Franklin Stahl)於 1958 年進行研究而得以證實。 1952 年赫希與蔡斯利用噬菌 體與細菌進行研究,證明 DNA 在噬菌體可作為遺傳物質(Hershey
Thus any continuous vector function r defines a space curve C that is traced out by the tip of the moving vector r(t), as shown in Figure 1.... The curve, shown in Figure 2,
Then we can draw a right triangle with angle θ as in Figure 3 and deduce from the Pythagorean Theorem that the third side has length.. This enables us to read from the
