Novel Alternating Fluorene-Based Conjugated Polymers Containing
Oxadiazole Pendants with Various Terminal Groups
Hsiao-Hsien Sung and Hong-Cheu Lin*
Department of Materials Science and Engineering, National Chiao Tung University, Hsinchu, Taiwan, ROC
Received May 21, 2004; Revised Manuscript Received August 9, 2004
ABSTRACT: A series of soluble alternating fluorene-based copolymers containing symmetrical and asymmetrical 1,3,4-oxadiazole (OXD) pendants with various terminal groups are synthesized by the palladium-catalyzed Suzuki coupling reaction. These polymers possess higher glass transition temper-atures than that of the analogous dialkoxy-substituted polymer (PFPOC6) consisting of the same backbone without OXD pendants. The photophysical and electrochemical properties of these polymers are affected by the polar effect (electron-withdrawing group, -CN, and electron-donating group, -R or -OR) and the size effect (the size of the grafted side chain) of the OXD pendants. Owing to the large steric hindrance of OXD pendants, the aggregation of these polymers in solids is reduced, which results in almost identical PL emissions in both solution and solid states. The bulky OXD pendants on the polymer side chains can provide the polymer films with lower HOMO and LUMO energy levels and better electron injection property. Since only one emission peak is observed in both PL and EL spectra of these polymers, it is evidenced that effective energy transfer from the OXD pendants to the conjugated polymer backbones has occurred, thus eliminating the light emission from the OXD pendants. These asymmetrical OXD-substituted polymers have higher quantum yields and less aggregation in the solid state than the symmetrical OXD-substituted polymers. The symmetrical OXD-substituted polymer (P1) has a longer PL emission wavelength than the asymmetrical OXD-substituted polymers (P2-P8), which may be due to the improvement of the coplanarity between the polymer backbone and the symmetrical OXD pendants and/or the introduction of two electron-withdrawing OXD pendants.
Introduction
Polymer light-emitting diodes (PLEDs) have been studied intensively during the past decade because of their potential applications in a new generation of flat display and lighting technologies.1,2 Compared with inorganic electroluminescent (EL) materials, π-conju-gated polymers have many virtues such as ease of forming large area, mechanical flexibility, low fabrica-tion cost, and easy color tuning through molecular engineering.3 Therefore, it is generally expected that PLEDs will be a possible candidate of the next genera-tion display technology.
To obtain highly efficient PLED devices, a balance in the injection and transportation of both holes and electrons into the polymer emissive layer is necessary.4-8 However, most EL polymers inject and transport holes more efficiently than electrons due to the inherent richness of π-electrons in the EL polymers, which results in an unbalance of opposite carrier currents and a shift of the recombination zone toward the region near the interface of the polymer and the cathode. Thus, the synthesis of efficient electron-transporting polymers is needed to improve EL device performance. Polymers containing electron-withdrawing units in the main chains or side chains usually have large electron affini-ties. In recent years, polyquinolines, polyquinoxalines, polybenzobisazoles, and polypyridines have been dem-onstrated as electron-transporting (ET) polymers due to the good film quality and the excellent thermal and oxidative stability.9-11 Molecular and polymeric 2,5-diphenyl-1,3,4-oxadiazolediyl derivatives are the most
widely studied classes of electron-injection and/or hole-blocking materials12by virtue of many merits owned by 1,3,4-oxadiazole (OXD) moieties, such as prominent electron affinity, high photoluminescence (PL) quantum yield, and good thermal stability.13On the other hand, to achieve highly efficient and luminescent PLED devices, it is important to suppress the self-quenching process, which originates from the formation of excimers due to the intermolecular interactions in the solid state. Since large steric hindrance of lateral substitution will cause the decrease of crystallinity in the resulting polymers, the bulky substitution with symmetrical and asymmetrical structure on the side chains of the poly-mer can provide the polypoly-mer film with an amorphous property.14In addition, the pseudo-orthogonal arrange-ment of the rigid pendant groups can reduce the
π-stacking and thus increase the radiative decay
quan-tum efficiency and photostability of the polymers.12c,15 In this paper, we report a series of alternating fluorene-based copolymers containing symmetrical and asym-metrical substituted OXD pendants with different ter-minal groups (on the side chains of the polymers). The electrochemical, spectroscopic, and thermal properties of these polymers with respect to various terminal OXD groups are also investigated in this study.
Experimental Section
Measurements. 1H NMR spectra were recorded on a Varian Unity 300 MHz spectrometer using CDCl3 sol-vent. Elemental analyses were performed on a HERAEUS CHN-OS RAPID elemental analyzer. Transition temperatures were determined by differential scanning calorimetry (Perkin-Elmer Pyris 7) with a heating and cooling rate of 10 °C/min. Thermogravimetric analysis (TGA) was conducted on a Du Pont Thermal Analyst 2100 system with a TGA 2950 thermo-gravimetric analyzer under a heating rate of 20 °C/min. Gel * Author for correspondence: Tel 8863-5712121 ext 55305; Fax
8863-5724727; e-mail [email protected].
10.1021/ma0489927 CCC: $27.50 © 2004 American Chemical Society Published on Web 09/23/2004
permeation chromatography (GPC) analysis was conducted on a Waters 1515 separation module using polystyrene as a standard and THF as an eluant. UV-vis absorption spectra were recorded in dilute chloroform solutions (10-5M) on a HP G1103A spectrophotometer, and fluorescence spectra were obtained on a Hitachi F-4500 spectrophotometer. Fluorescence quantum yields were determined by comparing the integrated PL density of a reference 9,10-diphenylanthracene in toluene with a known quantum yield (ca. 5× 10-6M, quantum yield ) 1.0). Cyclic voltammetry (CV) was performed at a scanning rate of 100 mV/s on a BAS 100 B/W electrochemical analyzer, which was equipped with a three-electrode cell. Pt wire was used as a counter electrode, and an Ag/AgCl was used as a reference electrode in the CV measurement. The polymer thin film was cast onto a Pt disk as a working electrode with ferrocene as a standard in actonitrile, and 0.1 M tetrabutyl-ammonium hexafluorophosphate (TBAPF6) was used as a supporting electrolyte. Polymer thin films were spin-coated on a quartz substrate from chloroform solution with a concentra-tion of 10 mg/mL (this condiconcentra-tion consists with the electrolu-minescent device).
Materials. Chemicals and solvents were reagent grades and purchased from Aldrich, ARCROS, TCI, and Lancaster Chemi-cal Co. Dichloromathane and THF were distilled to keep anhydrous before use. The other chemicals were used without further purification.
The synthetic routes of monomers M1-M8 are shown in Scheme 1. First, 2,5-dibromo-p-xylene was oxidized with potassium permanganate in pyridine to afford 2,5-dibromo-terephthalic acid and 2,5-dibromo-4-methylbenzoic acid. After then, the acid group was further esterified to give 2,5-dibromoterephthalic acid dimethyl ester (1) and 2,5-dibromo-4-methylbenzoic acid methyl ester (2), which were synthesized by the modified procedure of ref 16. After the ester groups had been converted to benzoyl hydrazides (3 and 4), they were reacted with various aryl chloride to form compounds 5 and 6. Finally, monomers M1-M8 were produced by the dehydra-tive cyclization of hydrazides in POCl3.
2,5-Dibromoterephthaloyldihydrazides (3). A mixture of 3.19 g (9.07 mmol) of methyl-2,5-dibromoterephthalate and 4.53 g (90.70 mmol) of hydrazine monohydrate was dissolved in 35 mL of methanol and then refluxed for 24 h. After cooling, the product was poured into water and recrystallized from methanol. Yield: 83%.1H NMR (ppm, DMSO-d
6): 4.51 (s, 4H), 7.58 (s, 2H), 9.69 (s, 2H).
2,5-Dibromo-4-methylbenzoic Acid Hydrazide (4). Yield: 78%.1H NMR (ppm, DMSO-d
6): 2.33 (s, 3H), 7.52 (s, 1H), 7.68 (s, 1H), 9.57 (s, 1H).
Compound 5. 0.81 g (3.02 mmol) of 4-octyloxybenzoyl chloride was added into a solution containing 0.53 g (1.51 mmol) of 1 and 0.25 mL (3.02 mmol) of pyridine in 10 mL of NMP. The reaction mixture was stirred for 12 h and then Scheme 1. Synthetic Routes of Monomers
poured into water. The product was filtered and crystallized from methanol. Yield: 73%.1H NMR (ppm, DMSO-d
6): 0.83 (t, 6H), 1.21-1.80 (m, 24H), 4.04 (t, 4H), 7.02 (d, J ) 9 Hz, 4H), 7.78 (s, 2H), 7.89 (d, J ) 8.7 Hz, 4H), 10.55 (s, 2H), 10.58 (s, 2H).
Benzoic Acid N-(2,5-Dibromo-4-methyl-benzoyl)hy-drazide (6-2). Yield: 81%.1H NMR (ppm, DMSO-d
6): 2.38 (s, 3H), 7.49-7.62 (m, 3H), 7.68 (s, 1H), 7.77 (s, 1H), 7.91 (d, J ) 8.4 Hz, 2H), 10.38 (s, 1H), 10.44 (s, 1H).
4-Methylbenzoic Acid N′ -(2,5-dibromo-4-methylben-zoyl)hydrazide (6-3). Yield: 66%.1H NMR (ppm,
DMSO-d6): 2.37 (s, 3H), 2.38 (s, 3H), 7.30 (d, J ) 8.1 Hz, 2H), 7.68 (s, 1H), 7.76 (s, 1H), 7.84 (d, J ) 7.5 Hz, 2H), 10.40 (s, 1H), 10.58 (s, 1H).
4-Methoxybenzoic Acid N′ -(2,5-dibromo-4-methylben-zoyl)hydrazide (6-4). Yield: 88%.1H NMR (ppm,
DMSO-d6): 2.38 (s, 3H), 3.82 (s, 3H), 7.03 (d, J ) 8.7 Hz, 2H), 7.68 (s, 1H), 7.76 (s, 1H), 7.90 (d, J ) 8.7 Hz, 2H), 10.36 (s, 1H), 10.51 (s, 1H).
4-Octyloxybenzoic Acid N′ -(2,5-dibromo-4-methylben-zoyl)hydrazide (6-5). Yield: 79%.1H NMR (ppm,
DMSO-d6): 0.83 (t, 3H), 1.25-1.79 (m, 12H), 2.38 (s, 3H), 4.03 (t, 2H), 7.00 (d, J ) 8.7 Hz, 2H), 7.68 (s, 1H), 7.76 (s, 1H), 7.87 (d, J ) 9 Hz, 2H), 10.37 (s, 1H), 10.50 (s, 1H).
4-Cyanobenzoic Acid N′ -(2,5-Dibromo-4-methylben-zoyl)hydrazide (6-6). Yield: 82%.1H NMR (ppm,
DMSO-d6): 2.38 (s, 3H), 7.67 (s, 1H), 7.77 (s, 1H), 8.00 (d, J ) 8.4 Hz, 2H), 8.05 (d, J ) 8.1 Hz, 2H), 10.59 (s, 1H), 10.97 (s, 1H).
Thiophene-2-carboxylic Acid N′ -(2,5-dibromo-4-meth-ylbenzoyl)hydrazide (6-7). Yield: 63%. 1H NMR (ppm, DMSO-d6): 2.38 (s, 3H), 7.20 (t, J ) 5.1 Hz, 1H), 7.65 (s, 1H), 7.77 (s, 1H), 7.86 (d, J ) 9.3 Hz, 1H), 7.88 (d, J ) 8.4 Hz, 1H), 10.47 (s, 1H), 10.71 (s, 1H).
Naphthalene-1-carboxylic Acid N′ -(2,5-dibromo-4-meth-ylbenzoyl)hydrazide (6-8). Yield: 71%. 1H NMR (ppm, DMSO-d6): 2.39 (s, 3H), 7.56-7.78 (m, 6H), 8.00 (d, J ) 7.2 Hz, 1H), 8.06 (d, J ) 7.2 Hz, 1H), 8.36 (d, J ) 8.7 Hz, 1H), 10.58 (s, 1H), 10.64 (s, 1H).
Monomer 1 (M1). 0.8 g (1.02 mmol) of 4a was dissolved in 100 mL of POCl3. The reaction mixture was heated to 130°C overnight and then cooled to room temperature. Most POCl3 in reaction mixture was removed at reduced pressure and poured into water. The product was filtered and crystallized from methanol and chloroform several times to get a yellow solid. Yield: 74%.1H NMR (ppm, CDCl
3): 0.88 (t, 6H), 1.30-1.86 (m, 24H), 4.05 (t, 4H), 7.03 (d, J ) 9 Hz, 4H), 8.08 (d, J
) 8.7 Hz, 4H), 8.51 (s, 2H). Anal. Calcd for C38H44Br2N24O4: C, 58.47; H, 5.68; N, 7.18. Found: C, 58.20; H, 5.79; N, 7.07. 2-(2,5-Dibromo-4-methylphenyl)-5-phenyl-[1,3,4]oxa-diazole (M2). Yield: 56%.1H NMR (ppm, CDCl
3): 2.46 (s, 3H), 7.52-7.65 (m, 3H), 8.14-8.17 (m, 2H), 8.24 (s, 1H). Anal. Calcd for C15H10Br2N2O: C, 45.72; H, 2.56; N, 7.11. Found: C, 46.06; H, 2.87; N, 6.87.
2-(2,5-Dibromo-4-methylphenyl)-5-p-tolyl-[1,3,4]oxadi-azole (M3). Yield: 68%.1H NMR (ppm, CDCl
3): 2.45 (s, 3H), 2.46 (s, 3H), 7.33 (d, J ) 7.8 Hz, 2H), 7.64 (s, 1H), 8.02 (d, J ) 8.4 Hz, 2H), 8.22 (s, 1H). Anal. Calcd for C16H12Br2N2O: C, 47.09; H, 2.96; N, 6.86. Found: C, 47.07; H, 3.23; N, 6.80.
2-(2,5-Dibromo-4-methylphenyl)-5-(4-methoxyphenyl)-[1,3,4]oxadiazole (M4). Yield: 44%.1H NMR (ppm, CDCl
3): 2.46 (s, 3H), 3.90 (s, 3H), 7.02 (d, J ) 9 Hz, 2H), 7.64 (s, 1H), 8.07 (d, J ) 8.7 Hz, 2H), 8.22 (s, 1H). Anal. Calcd for C16H12 -Br2N2O2: C, 45.31; H, 2.85; N, 6.61. Found: C, 45.09; H, 3.09; N, 6.51. 2-(2,5-Dibromo-4-methylphenyl)-5-(4-octyloxyphenyl)-[1,3,4]oxadiazole (M5). Yield: 81%.1H NMR (ppm, CDCl 3): 0.90 (t, 3H), 1.20-1.51 (m, 10H), 1.77 (m, 2H), 2.46 (s, 3H), 4.04 (t, 2H), 7.00 (d, J ) 8.7 Hz, 2H), 7.64 (s, 1H), 8.05 (d, J ) 8.7 Hz, 2H), 8.22 (s, 1H). Anal. Calcd for C23H26Br2N2O2: C, 52.89; H, 5.02; N, 5.36. Found: C, 53.13; H, 5.34; N, 5.30.
4-[5-(2,5-Dibromo-4-methylphenyl)-[1,3,4]oxadiazol-2-yl]benzonitrile (M6). Yield: 88%.1H NMR (ppm, CDCl
3): 2.48 (s, 3H), 7.67 (s, 1H), 7.84 (d, J ) 8.4 Hz, 2H), 8.25 (s, 1H), 8.26 (d, J ) 8.1 Hz, 2H). Anal. Calcd for C16H9Br2N3O: C, 45.86; H, 2.16; N, 10.03. Found: C, 45.82; H, 2.49; N, 10.07. 2-(2,5-Dibromo-4-methylphenyl)-5-thiophen-2-yl-[1,3,4]oxadiazole (M7). Yield: 88%.1H NMR (ppm, CDCl 3): 2.45 (s, 3H), 7.20 (t, J ) 5.1 Hz, 1H), 7.59 (d, J ) 5.1 Hz, 1H), 7.63 (s, 1H), 7.85 (d, J ) 5.1 Hz, 1H), 8.19 (s, 1H). Anal. Calcd for C13H8Br2N2OS: C, 39.03; H, 2.02; N, 7.00. Found: C, 39.37; H, 2.34; N, 6.92.
2-(2,5-Dibromo-4-methylphenyl)-5-naphthalen-1-yl-[1,3,4]oxadiazole (M8). Yield: 68%.1H NMR (ppm, CDCl
3): 2.41 (s, 3H), 7.56-7.68 (m, 4H), 7.88-8.23 (m, 4H), 9.30 (d, J ) 8.1 Hz, 1H). Anal. Calcd for C19H12Br2N2O: C, 51.38; H, 2.72; N, 6.31. Found: C, 51.32; H, 3.01; N, 6.28.
Polymerization. The synthetic routes of polymers P1-P8 are shown in Scheme 2. The general procedure of polymeri-zation is proceeded through the Suzuki coupling reaction. A mixture of 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dioctylfluorene (1 equiv), dibromo compounds M1-M8 (1 equiv), and tetrakis(triphenylphosphine)palladium (1.0 mol %) Scheme 2. Synthetic Route of Polymers
was added in a degassed mixture of toluene ([monomer] ) 0.2 M) and aqueous 2 M potassium carbonate (3:2 in volume). The mixture was vigorously stirred and reacted at 87 °C for 72 h. After the mixture was cooled to room temperature, it was poured into 200 mL of methanol. A fibrous solid was obtained by filtration, and the solid was sequentially washed with methanol, water, and methanol.
P1. Yield: 51%.1H NMR (ppm, CDCl
3): 0.61-1.41 (m, 46H), 1.69-2.00 (m, 10H), 3.88 (br, 4H), 6.73 (d, J ) 8.4 Hz, 4H), 7.37-7.55 (m, 8H), 7.81 (d, J ) 7.2 Hz, 2H), 8.37 (s, 2H). Anal. Calcd for C63H76N4O4: C, 79.37; H, 8.04; N, 5.88. Found: C, 78.10; H, 7.97; N, 5.60.
P2. Yield: 23%.1H NMR (ppm, CDCl
3): 0.66-1.20 (m, 22H), 1.91 (br, 4H), 2.46 (s, 3H), 7.22-7.59 (m, 10H), 7.76-7.81 (m, 2H), 8.14 (s, 1H). Anal. Calcd for C40H42N2O: C, 84.77; H, 7.47; N, 4.94. Found: C, 83.26; H, 7.45; N, 4.83.
P3. Yield: 40%.1H NMR (ppm, CDCl
3): 0.66-1.23 (m, 22H), 1.92 (br, 4H), 2.28 (s, 3H), 2.46 (s, 3H), 7.03 (d, J ) 7.8 Hz, 2H), 7.36-7.50 (m, 7H), 7.74-7.82 (m, 2H), 8.12 (s, 1H). Anal. Calcd for C41H44N2O: C, 84.79; H, 7.64; N, 4.82. Found: C, 83.44; H, 7.55; N, 4.62.
P4. Yield: 35%.1H NMR (ppm, CDCl
3): 0.74-1.26 (m, 22H), 2.1 (br, 4H), 2.48 (s, 3H), 3.77 (s, 3H), 6.75 (br, 2H), 7.41-7.84 (m, 9H), 8.14 (s, 1H). Anal. Calcd for C41H44N2O2: C, 82.51; H, 7.43; N, 4.69. Found: C, 81.42; H, 7.50; N, 4.48.
P5. Yield: 78%.1H NMR (ppm, CDCl
3): 0.68-1.41 (m, 35H), 1.74-2.07 (m, 6H), 2.47 (s, 3H), 3.90 (br, 2H), 6.77 (d, J ) 8.4 Hz, 2H), 7.41-7.83 (m, 9H), 8.15 (s, 1H). Anal. Calcd for C48H58N2O2: C, 82.95; H, 8.41; N, 4.03. Found: C, 82.03; H, 8.27; N, 3.71.
P6. Yield: 54%.1H NMR (ppm, CDCl
3): 0.68-1.26 (m, 22H), 1.95 (br, 4H), 2.50 (s, 3H), 7.35-7.86 (m, 11H), 8.21 (s, 1H). Anal. Calcd for C41H41N3O: C, 83.21; H, 6.98; N, 7.10. Found: C, 82.26; H, 6.91; N, 6.73.
P7. Yield: 88%.1H NMR (ppm, CDCl
3): 0.75-1.20 (m, 22H), 2.04 (br, 4H), 2.48 (s, 3H), 6.95 (br, 1H), 7.37-7.49 (m, 7H), 7.76-7.85 (m, 2H), 8.11 (s, 1H). Anal. Calcd for C38H40N2OS: C, 79.68; H, 7.04; N, 4.89. Found: C, 78.47; H, 7.14; N, 4.69.
P8. Yield: 63%.1H NMR (ppm, CDCl
3): 0.48-1.20 (m, 22H), 1.92 (br, 4H), 2.48 (s, 3H), 7.12 (br, 1H), 7.39-7.55 (m, 8H), 7.80-7.87 (m, 4H), 8.20 (s, 1H), 9.04 (br, 1H). Anal. Calcd for C44H44N2O: C, 85.67; H, 7.19; N, 4.54. Found: C, 84.20; H, 7.31; N, 4.08.
Results and Discussion
Synthesis and Characterization. The synthetic routes of monomers M1-M8 and polymers P1-P8 are shown in Schemes 1 and 2. All these polymers were easily dissolved in common organic solvents such as chloroform, THF, dichloromethane, and toluene to form good transparent films on glass substrates. The average molecular weights were measured by gel permeation chromatography (GPC) with polystyrene as a standard and THF as an eluting solvent. The related data obtained from GPC are given in Table 1. The number-average molecular weights (Mn) of polymers are between
4900 and 25 200 g/mol, and the polydispersity indexes (PDI) are found between 1.2 and 2.8. The thermal stability of the polymers in nitrogen was evaluated by thermogravimetric analysis (TGA), and their corre-sponding data are also summarized in Table 1. The TGA analysis indicates that the degradation temperatures (Td) of 5% weight loss in nitrogen are between 398 and 446 °C. The results of TGA reveal that the polymers are quite stable in nitrogen, and compared with the other polymers (P2-P8) containing asymmetrical OXD pendants, P1 containing symmetrical OXD pendants has the lowest Td. The glass transition temperatures (Tg) and melting temperatures of these polymers were determined by differential scanning calorimetry (DSC) in a nitrogen atmosphere at a scanning rate of 10 °C/ min. Their glass transition temperatures are shown between 85 and 148 °C, whereas their melting temper-atures are not observed even up to 280 °C. The long flexible alkoxy-substituted OXD polymer, P5, has the lowest Tg, and the polar cyano-substituted OXD poly-mer, P6, demonstrates the highest Tg. In contrast to P1 containing symmetrical OXD pendants, analogous P5 containing asymmetrical OXD pendants possesses a lower Tg(34 °C lower). Comparing P8 with P2, when the phenyl group is replaced with bulky a naphthyl group in the terminal group of OXD pendants, an increase of 23 °C in Tgis observed in P8. In comparison with the analogous structure without oxadiazole pen-dants, i.e., poly(fluorene phenlyene) (PFP),17 the deg-radation temperatures of these novel polymers are higher than that of PFP (Tdis 398 °C), and the glass transition temperatures of these polymers are also higher than that of PFPOC6(Tgis 72 °C).
Optical Properties. The photophysical characteris-tics of the monomers and polymers were studied by PL and UV-vis absorption in both dilute chloroform solu-tions and thin films. The optical properties of the monomers and polymers are summarized in Tables 2 and 3 and Figures 1-5 serially. It is noted that similar absorption patterns between 250 and 400 nm are observed in various substituted monomers, except M1.
Table 1. Molecular Weights and Thermal Properties of Polymers
polymer yield (%) Mn PDI Tda(°C) Tgb(°C)
P1 51 4 900 1.2 398 119 P2 23 6 700 1.7 418 109 P3 40 14 800 1.5 421 125 P4 35 26 600 2.8 414 128 P5 78 24 500 2.7 410 85 P6 54 25 200 2.6 422 148 P7 88 12 200 1.9 407 120 P8 63 6 500 1.7 446 132
aTemperature of 5% weight loss was measured by TGA in nitrogen.bThe glass transition temperature (T
g) was measured on the first cooling cycle after the sample had been heated to 280 °C.
As expected, both of the absorption and PL spectra are affected by the substituted terminal groups. Compared with those of M2, the maximum absorption and PL wavelengths of M3-M8 increase as the polar uents and π-excessive thiophene (naphthalene) substit-uents are attached to the OXD rings. Interestingly, compared with that of M4 (λPL,sol) 370 nm) containing a OMe pendant terminal group, the emission wave-length of M6 (λPL,sol) 361 nm), in which the polar cyano group is attached to the OXD segment, is not efficiently enlarged. This phenomenon is contrary to our previous study, where the cyano group attached to the chro-mophoric core will elongate the conjugation and result in a longer emission wavelength.18 Whereas, the PL emission spectra of the polymers have shown that the cyano-substituted polymer P6 has a longer emission wavelength (λPL,sol) 430 nm) than that of the alkoxy-substituted polymer P4 (λPL,sol) 405 nm).
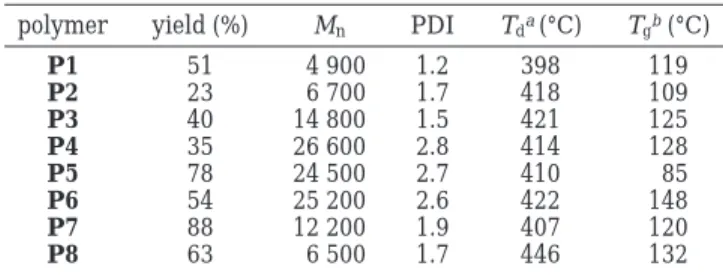
The absorption spectra of synthesized polymers in dilute solutions (chloroform) are depicted in Figure 1.
It is noticed that the polymers with asymmetric OXD pendants exhibit two absorption peaks, except P8. The absorption spectrum of P8 shows only one peak, which may be due to the overlap of absorption peaks in both OXD pendants and polymer main chains. The shorter absorption ranging at 290-305 nm is originated from the absorption of the conjugated OXD segments, which is confirmed by the same maximum absorption of
Table 2. Absorption and PL Emission Spectral Data of Monomers in Chloroform monomer λabsa (nm) λPL,solb (nm) monomer λabsa (nm) λPL,solb (nm) M1 337 418 M5 299 372 M2 283 347 M6 290 361 M3 286 352 M7 302 367 M4 297 370 M8 320 379
aWavelength of the maximum absorbance.bWavelength of the maximum PL emission.
Table 3. Absorption and PL Emission Spectral Data of Polymers in Chloroform and in Thin Solid Films
polymer λabs,sola (nm) λabs,filma (nm) band gap b (eV) λPL,sola (nm) λPL, film (nm) ΦPL,solc fwhmsol(nm) P1 321 322 3.14 445 447 0.15 70 P2 327 327 3.33 405 411 0.30 60 P3 329 326 3.32 402 407 0.39 58 P4 325 323 3.32 405 412 0.36 58 P5 322 321 3.33 404 409 0.31 58 P6 323 322 3.32 430 430 0.17 68 P7 323 324 3.33 409 415 0.26 61 P8 328 328 3.34 410 410 0.38 62
aWavelength of the maximum absorbance or PL emission. bBand gaps were calculated from the onsets of UV-vis absorption spectra of polymer solutions.cPL quantum yield in CHCl
3.
Figure 1. UV-vis spectra of polymer solutions in dilute chloroform.
Figure 2. (a) UV-vis spectra of M5 and P5. (b) UV-vis and PL emission spectra of M1 and P1.
polymer P5 and monomer M5 (Figure 2a). The identical absorption maximum of the OXD segment in dilute solutions before and after the polymerization suggests that the electronic interactions between the conjugated oxadiazole segments and the polymer backbones are quite limited.19The result also implies that part of the conjugated OXD units (the phenyl-oxadiazole-phenyl conjugated unit) in the asymmetrical side-chain struc-tures twist away from the plane of the conjugated polymer backbone. The longer wavelength peak in the region of 310-370 nm is attributed to the electron transition of π-π* along the conjugated backbone of the polymer. Comparing Figures 1-3, the polymer main-chain absorption peaks of the solution and film samples (Figure 2a) are almost identical. This result indicates that the conformation of the polymer chains is very similar in both solution and solid states; in addition, the polymer assembly is in a rather disordered state and refrains from aggregation by this molecular design even in the solid state.
The maximum absorption value of P1 (321 nm) comes from the fluorene backbone, which can be proved by similar maximum absorption values of all polymers P1-P8 (321-329 nm), regardless of various oxadiazole pendant groups in P1-P8. The absorption strength of
OXD segments in P1 (containing symmetrical OXD pendants) does not appear as a peak but only shows a shoulder around 337 nm (see Figure 2b), which corre-sponds to the absorption peak of M1. This phenomenon illustrates that the fluorene and both symmetrical OXD segments of P1 do not twist away from the conjugated plane of the polymer backbone and induce an elongated conjugation. In addition, the absorption strength of P1 does not show any absorption peak in the region of 270-320 nm (which corresponds to M5 absorption). It implies the conjugated OXD units of P1 keep better coplanarity and do not twist away from the conjugated plane. We also observe the solid-state absorption spectrum of P1 (as shown in Figure 2b) has a tail in the low-energy region around 370-600 nm; it also proves that P1 has a better coplanarity between the symmetrical OXD pendants and the polymer backbone.
Compared with analogous PFP (λabs) 370 nm), our polymers exhibit blue-shifted absorption maxima (41-49 nm). The blue shifts of the absorption maxima may be ascribed to two reasons. One is that the coplanarity of the polymer backbone is lost due to the asymmetrical bulky substituents, which cause the steric hindrance and induce the distortion of the backbone. Therefore, the conjugation of polymer backbone is interrupted and induces a blue shift of the absorption maximum. The other is that as mentioned previously the OXD pendants act as electron-withdrawing substituents on the side of the polymer backbone. The electron-withdrawing sub-stituted groups decrease the electron density of the polymer backbone and reduce the absorption maxi-mum.17
The optical band gap energies obtained from UV-vis absorption in solutions are also given in Table 3. The band gap energies of polymers were determined by the intersection of tangents through the turning point of the lower energy side of the spectrum and the lengthened baseline for each polymer. The optical band gaps of asymmetrical OXD-substituted polymers from the edge absorption of solution samples are almost identical (3.32-3.34 eV), regardless of terminal functional groups (P2-P8) of OXD pendants. Nevertheless, the sym-metrical OXD-substituted polymer (P1) has a red-shifted absorption curve and a smaller band gap (3.14 eV). The smaller band gap may be due to the introduc-tion of two electron-withdrawing OXD moieties and/or a better coplanarity between the polymer backbone and the symmetrical substituents in P1. In comparison with PFP, all band gap energies of these polymers are larger than that of PFP (2.92 eV).
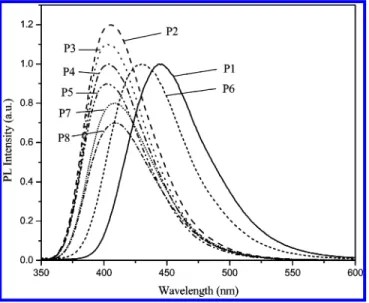
The fluorescence quantum yield (ΦPL), the PL emis-sion maxima (λPL), and the full width at half-maximum (fwhm) values are also presented in Table 3. All emis-sion data given here were obtained by the excitation at the maximum absorption peaks. In dilute solutions, all these polymers only show one emission peak ranging from 402 to 445 nm (Figure 4). Emissions from the OXD pendants are not observed, even when polymers are excited at the absorption peak of the OXD pendants. This indicates the existence of efficient energy transfer from the OXD pendants to the polymer backbone. Except P6, all asymmetrical OXD-substituted polymer solutions and films have PL emission in the purple region (Figures 4 and 5). Among these purple-emitting polymers, the electron-donating substituted groups (CH3, OCH3, and OC8H17) in P3-P5 do not seem to affect the emission spectra significantly as compared
Figure 4. PL spectra of polymer solutions in dilute chloro-form.
with that of P2. However, in contrast to P3-P5, when the electron-withdrawing cyano group is introduced into the terminal group of OXD pendants in P6, an obvious red shift (about 25 nm) can be observed in the PL spectrum. The symmetrical OXD-substituted polymer (P1), apparently, has a longer PL emission wavelength than the other asymmetrical OXD-substituted polymers (P2-P8). This result may be due to the improvement of the coplanarity between the polymer backbone and the symmetrical OXD pendants and/or the introduction of two electron-withdrawing OXD pendants.
The PL spectra of polymer thin films are shown in Figure 5. These asymmetrical and symmetrical OXD-substituted polymers emit purple and blue color light, respectively. Compared with the spectra of asymmetri-cal OXD-substituted polymers (P2-P8) in solutions, the PL emission peaks of solid films only show a small red shift (the maximum red-shifted value is 7 nm). In contrast to the asymmetrical OXD-substituted poly-mers, the thin film PL peak of P1 is just red-shifted for 2 nm, but the emission spectrum shows slight tails in the low-energy ends, which may be due to the aggrega-tion of polymer chains in the solid state. The PL quantum yield will be decreaed when pendant groups (side chains) are conjugated with the polymer main chains.12c,19,20It may be due to the multifarious conju-gated forms which cannot confine the excitons ef-ficiently; hence, the PL quantum yield decreases in the OXD-substituted polymer. It is interesting to note that the PL quantum yield of the symmetrical OXD-substi-tuted polymer (P1) is lower than those of asymmetrical OXD-substituted polymers (P2-P8), which may be due to the different degree of conjugation between the OXD pendants and the backbone in the symmetrical and asymmetrical structures, respectively.
Compared with PFP, side-chain conjugated PPV (OPO-PPV), and cross-conjugated PPE (PPE-PPV),14c,20b these OXD-substituted polymers (P1-P8) exhibit al-most identical spectra in the solution and thin film samples. This result is distinct from PFP, OPO-PPV, and PPE-PPV, which have significant red-shifted spec-tra in the thin film samples as compared with the solution samples. The results demonstrate that the configuration of the OXD-substituted polymers in our study is not affected by the variation of the state in these polymers. It can be understood by the enhanced steric effect between the OXD pendant groups and the poly-mer backbone, which consequently increases interchain distances and suppresses the formation of aggregation efficiently. In comparison with PFP, the PL spectra of all our polymers (no matter in solution or solid states) exhibit structureless emission spectra; it is different from PFP, which possesses a well-resolved vibronic band in the emission spectrum. This phenomenon suggests that the OXD pendants can efficiently enhance the amorphism of the polymers and reduce the vibronic structures in the solid state.
Klemm and co-workers reported that the full width at half-maximum (fwhm) values of alkoxy-substituted PPV-PPE hybrid polymers strongly depend on the length of the attached side chains.21 The fwhm value increases from 36 nm in PPV-PPE hybrid polymers containing longer side chains (-OC18) to 67 nm in PPV-PPE hybrid polymers containing shorter side chains (-OC8). They explain that the smaller fwhm value of the long side-chain polymers is due to the emission coming from isolated main-chain fluorophores; however,
the larger fwhm value of the short side-chain polymers is a contribution of isolated main-chain fluorophores and a certain degree of aggregation. In our synthetic poly-mers, the fwhm values of the emission curves of the polymers in the solution and solid states are almost independent of the terminal groups (P2-P5 and P7-P8) of the OXD pendants. Only about 10 nm of increase in the fwhm value was observed in symmetrical P1 and asymmetrical P6 (with a strong electron-withdrawing cyano substituent attached to the end of the OXD pendant). The strong polar groups of the cyano termi-nals in the pendants of P6 may induce more or less intramolecular and/or intermolecular interactions and consequently broaden the fwhm value. Otherwise, com-paring P5 with P2-P4 of our polymers containing OXD pendants, the fwhm value of the long terminal side-chain polymer (P5) is almost identical with those of short terminal side-chain polymers (P2-P4). From this result, we can generally conclude that the asymmetrical OXD pendants can provide good steric hindrance and reduce aggregation, except OXD pendants containing strong polar cyano groups (such as P6). In general, the lowest quantum yields of P1 (due to the symmetrical pendant structure) and P6 (due to the strong polar cyano substituent) are also correlated to their largest fwhm values.
In summary of the absorption and PL results, we can conclude the photophysical properties are strongly af-fected by the OXD pendants. In asymmetrical substi-tuted polymers, the absorption maximum peaks and optical band gaps are almost independent of the elec-tronic effect (withdrawing group, -CN, electron-donating group, -R or -OR) and size effect (the size of the terminal functional group) of the substituted OXD pendants. However, the PL emission peaks and the quantum yields are affected by the electronic effect. When the strong electron-withdrawing cyano group is attached to the terminal phenyl group of the OXD pendant, the PL emission peak is red-shifted and the quantum yield is reduced. Except P6, these polymers have similar PL emission peaks as that of PFP (λabs,sol ) 408 nm, λabs,film ) 422 nm), but they show larger Stokes shifts than that of PFP. As a symmetrical OXD-substituted polymer, P1 has an almost identical absorp-tion maximum value as those of asymmetrical substi-tuted polymers, but relatively it possesses a longer PL emission peak and a lower quantum yield.
Electrochemical Properties. To fit the energy band structural scheme of PLED device, it is necessary to determine the energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of each component, which was carried out by CV to investigate the redox behavior of the polymer thin films. The potential estimated here was based on the reference energy level of ferrocene (4.8 eV below the vacuum level) according to the following equation:21EHOMO/ELUMO) [-(Eonset- 0.45) - 4.8] eV. The onset potentials were determined from the intersec-tion of two tangents drawn at the rising and background currents of the cyclic voltammogram. A crude estimation of the LUMO levels of reduction polymers were deduced from the HOMO values and the optical band gaps.
These polymers have the same backbone structure of poly(fluorene-co-alt-phenylene) with different substit-uents on the OXD pendants which are attached to the phenylene groups. It is interesting to evaluate that how do these OXD pendants modulate the electrochemical
behavior of these polymers. As shown in Figures 6 and 7, most of the polymers possess two anodic peaks, which can be attributed to two different blocks in the OXD pendants and polymer backbones of the copolymers, respectively. Since the reduction potentials are difficult to be measured, only P1 and P6 can be determined, which may be due to their strong electron-withdrawing property. The asymmetrical substituted polymers (P2-P5) show oxidized peaks irreversibly at potentials ranging from 1.72 to 1.98 V (Table 4) and the onset potentials of oxidation between 1.57 and 1.62 V in the anodic scan. Comparing P3-P5 with P2, it seems that the electron-donating groups slightly reduce the overall electron-withdrawing effect of the OXD pendants. Com-pared with P2, the oxidized onset potentials of P7 and P8 are not remarkably changed by the replacement of the phenyl group with the π-excessive thiophene and naphthalene groups.
As for P6 with cyano-substituted OXD pendants, the oxidation onset potential is 1.39 V and reduction onset
potential is -1.51 V. It is obvious that P6 is much easier to be reduced than the other polymers; this is probably due to the strong electron-withdrawing cyano group which increases the overall electron-withdrawing effect of OXD pendants. Compared with P2, the HOMO energy level of P6 can be easily adjusted for 0.4 eV by changing the terminal functional groups of OXD pen-dants with CN terminals. As for the symmetrical OXD-substituted polymer (i.e., P1), the oxidation onset potential is 1.74 V and reduction onset potential is -1.65 V. The band gap obtained from the difference of onset potentials between the reduction and oxidation processes is estimated to be 3.20 V, which is close to that obtained from the absorption edge in the UV-vis spectrum (3.14 V).
The OXD pendants act as electron-withdrawing sub-stituents on the backbones of poly(fluorine-co-alt-phen-ylene)s and thus decrease the LUMO levels (-2.60 to -2.84) of all polymers as compared to that of PFP (HOMO ) -5.36, LUMO ) -2.47). The lower LUMO potentials of these polymers indicates that the incorpo-ration of electron-withdrawing OXD pendants increases the electron affinities and reduces the LUMO levels of wide band gap polymers (such as PFP) to match the Figure 6. Cyclic voltammetry of polymers during the oxidation process.
Figure 7. Cyclic voltammograms of P1 and P6.
Table 4. HOMO and LUMO Energies and Electrochemical Properties of Polymers polymer Ered/onset (V) Ered/peak (V) Eox/onset (V) Eox/peak (V) EHOMO (eV) ELUMO (eV) P1 -1.65 -1.79 1.55 1.74 -5.90 -2.70a P2 1.62 1.90 -5.97 -2.64b P3 1.57 1.72 -5.92 -2.60b P4 1.58 1.98 -5.93 -2.61b P5 1.61 1.73 -5.96 -2.63b P6 -1.51 -1.72 1.39 1.50 -5.74 -2.84a P7 1.69 1.87 -6.04 -2.71b P8 1.60 1.77 -5.95 -2.61b
aLUMO Energies were obtained from the cyclic voltammetry. bLUMO Energies were deduced from HOMO values and optical band gaps.
work function of the cathode. From these results, we can conclude that both HOMO and LUMO energy levels can be adjusted not only by the OXD pendants of the polymer backbone but also by the attached functional groups of the OXD pendants.
Electroluminescent (EL) Properties of PLED Devices. The CV and UV-vis results show that both HOMO and LUMO energy levels of our new polymers do not match the work functions of indium-tin oxide (ITO) anode and Al cathode. Therefore, we choose PEDOT and Ca as the anode and cathode, respectively, to overcome these large energy barriers. A series of double-layer EL devices with the configuration of ITO/ PEDOT:PSS/Polymer/Ca/Al were made by spin-coating the polymer solutions (with a concentration of 10 mg/ mL) onto PEDOT-coated glass substrates, and their EL data are demonstrated in Table 5.
The current-voltage and luminescence-voltage, i.e., EL response, curves of one typical PLED device (ITO/ PEDOT:PSS/P3/Ca/Al) are displayed in Figure 8. All these devices show turn-on voltages for current from 6.5 to 8.5 V and turn-on voltages for light from 7 to 9 V (shown in Table 5). Compared with P2 without flexible ends on OXD pendants, P5 with OC8terminals on OXD pendants has a higher turn-on voltage. This result may be due to the intrinsic insulation property of the long alkoxy side chains in P5 which consequently increases the turn-on voltage. The attainable maximum lumi-nance is 462 cd/m2at 15 V for P8 as an emitter. These similar turn-on voltages for current and light illustrate that a matched balance of injection and transportation in charge is achieved. The balanced injection and transportation of both charge carriers may be due to the limited electronic interactions between the
cross-conjugated OXD pendants and the cross-conjugated polymer backbone.15c
As shown in Figure 9, these devices of asymmetrical OXD-substituted polymers give purple to blue emission colors, and the EL emission peaks are between 406 and 452 nm (Table 5). The EL emission peaks, general speaking, are well matched with the corresponding PL emission peaks of the thin films (Table 4). This indicates that the similar radiative excited states are involved in both EL and PL processes. In contrast to all asym-metrical OXD-substituted polymers, the emission spec-tra (Figure 10) of P1 at different voltages are slightly voltage-dependent. Since the absorption and fluores-cence data reveal that P1 forms excimers due to the aggregation in the solid film, the EL spectra of P1 are red-shifted and become broader by increasing voltage. Conclusion
A series of soluble alternating fluorene-based copoly-mers with OXD pendants were synthesized by the palladium-catalyzed Suzuki coupling reaction. These polymers exhibit good thermal stability up to 400 °C and possess higher glass transition temperatures than
Table 5. EL Data of PLED Devicesa
polymer λmax,EL (nm) Vonb(V) Vonc(V) max brightness (cd/m2) P1 452 7.5 7.5 126 P2 408 7.5 7.5 29 P3 409 7.5 7.5 51 P4 423 8.5 8.5 104 P5 406 8.5 9.0 376 P6 431 6.5 7.0 306 P7 416 7.0 7.0 82 P8 417 6.5 7.0 462
aDevice structure: ITO/PEDOT:PSS/polymer/Ca/Al.bV onis the turn-on voltage of current.cV
onis the turn-on voltage of light.
Figure 8. Current-voltage and luminescence-voltage char-acteristics of ITO/PEDOT:PSS/P3/Ca/Al device.
Figure 9. Normalized EL spectra of ITO/PEDOT:PSS/Polymer/ Ca/Al devices.
Figure 10. Normalized EL spectra of ITO/PEDOT:PSS/P1/ Ca/Al device at different voltages.
that of the analogous dialkoxy-substituted polymer (PFPOC6) consisting of the same backbone without OXD pendants. Their photophysical and electrochemical prop-erties are not only affected by the OXD pendants but also affected by the terminal functional groups. Owing to the large steric hindrance of OXD pendants, the aggreagation of these polymers in solid state is reduced, which results in almost identical PL emissions in both solution and solid states. The incorporation of OXD pendants into polymer backbone leads to mitigating the interchain interaction and improving the electron af-finity and charge-injection properties effectively. Since only one emission peak is observed in both PL and EL spectra of these polymers, it is evidenced that effective energy transfer from the OXD pendants to the conju-gated polymer backbones has occurred, thus eliminating the light emission from the OXD pendants. By compar-ing P1 with P5, we recognize that asymmetrical OXD-substituted polymers possess better properties (higher quantum yield and lower aggregation in the solid state) than the symmetrical OXD-substituted polymer. The symmetrical OXD-substituted polymer (P1) has a longer PL emission wavelength than the asymmetrical OXD substituted polymers (P2-P8), which may be due to the improvement of the coplanarity between the polymer backbone and the symmetrical OXD pendants and/or the introduction of two electron-withdrawing OXD pendants.
Acknowledgment. We thank the financial support from the National Science Council of Taiwan (ROC) through NSC 92-2113-M-009-016.
References and Notes
(1) Burroughes, J. H.; Bradley, D. D. C.; Brown, A. R.; Marks, R. N.; Mackay, K.; Friend, R. H.; Burn, P. L.; Holmes, A. B.
Nature (London) 1990, 347, 539.
(2) (a) Kraft, A.; Grimsdale, A. C.; Holmes, A. B. Angew. Chem.,
Int. Ed. 1998, 37, 402. (b) Friend, R. H.; Gymer, R. W.;
Holmes, A. B.; Burroughes, J. H.; Marks, R. N.; Taliani, C.; Bradley, D. D. C.; dos Santos, D. A.; Bredas, J. L.; Lo¨gdlund, M.; Salaneck, W. R. Nature (London) 1999, 397, 121. (c) Johnson, M. J.; Sempel, A. Inf. Disp. 2000, 2, 12.
(3) (a) Gustafsson, G.; Cao, Y.; Treasy, G. M.; Klavetter, F.; Colaneri, N.; Heeger, A. J. Nature (London) 1992, 357, 477. (b) Greenham, N. C.; Friend, R. H. Solid State Phys. 1995,
49, 1.
(4) (a) Liu, M. S.; Jiang, X.; Liu, S.; Herguth, P.; Jen, A. K. Y.
Macromolecules 2002, 35, 3532. (b) Jenekhe, S. A.; Osaheni,
J A. Science 1994, 265, 765.
(5) Krebs, F. C.; Jorgensen, M. Macromolecules 2002, 35, 7200. (6) Chen, Z. K.; Meng, H.; Lai, Y. H.; Huang, W. Macromolecules
1999, 32, 4351.
(7) Marsella, M. J.; Fu, D. K.; Swager, T. M. Adv. Mater. 1995,
7, 145.
(8) Gillissen, S.; Jonforsen, M.; Kesters, E.; Johansson, T.; Theander, M.; Andersson, M. R.; Inganas, O.; Lutsen, L.; Vanderzande, D. Macromolecules 2001, 34, 7294.
(9) (a) Cui, Y.; Zhang, X.; Jenekhe, S. A. Macromolecules 1999,
32, 3824. (b) Jandke, M.; Strohriegl, P.; Berleb, S.; Werner,
E.; Brutting, W. Macromolecules 1998, 31, 6434.
(10) (a) Jenekhe, S. A.; Zhang, X.; Chen, X. L.; Choong, V. E.; Gao, Y.; Hsieh, B. R. Chem. Mater. 1997, 9, 409. (b) Jen, A. K.; Wu, X. M.; Ma, H. Chem. Mater. 1998, 10, 3824.
(11) (a) Zhang, X.; Jenekhe, S. A. Mater. Res. Soc. Symp. Proc.
1998, 488, 539. (b) Wang, Y. Z.; Gebler, D. D.; Fu, D. K.;
Swager, T. M.; MacDiarmid, A. G.; Epstein, A. J. Synth. Met.
1997, 85, 1179.
(12) (a) Wang, C.; Jung, G. Y.; Hua, Y.; Pearson, C.; Bryce, M. R.; Petty, M. C.; Batsanov, A. S.; Goeta, A. E.; Howard, J. A. K.
Chem. Mater. 2001, 13, 1167. (b) Zhan, X.; Liu, Y.; Wu, X.;
Wang, S.; Zhu, D. Macromolecules 2002, 35, 2529. (c) Lee, Y. Z.; Chen, X.; Chen, S. A.; Wei, P. K.; Fann, W. S. J. Am.
Chem. Soc. 2001, 123, 2296. (c) Peng, Z.; Zhang, J. Chem. Mater. 1999, 11, 1138. (d) Chung, S. J.; Kwon, K. Y.; Lee, S.
W.; Jin, J. L.; Lee, C. H.; Lee. C. E.; Park, Y. Adv. Mater.
1998, 10, 1112. (e) Kim, J. H.; Park, J. H.; Lee. H. Chem. Mater. 2003, 15, 3414.
(13) Adachi, C.; Tsutsui, T.; Saito, S.Appl. Phys. Lett. 1990, 56, 799.
(14) (a) Ruiz, J. P.; Dharia, J. R.; Reynolds, J. R.; Buckley, L. J.
Macromolecules 1992, 25, 849. (b) Chen, Z. K.; Meng, H. Lai,
Y. H.; Huang, W. Macromolecules 1999, 32, 4351. (c) Meng, H.; Yu, W. L.; Hunang, W. Macromolecules 1999, 32, 8841. (d) Pu, Y. J.; Soma, M.; Kido, J.; Nishide, H. Chem. Mater.
2001, 13, 3817. (e) Chen, Z. K.; Lee, N. H. S.; Huang, W.;
Wu, Y. S.; Cao, Y. Macromolecules 2003, 36, 1009. (f) Wu, F. I.; Reddy, S.; Shu, C. F.; Liu, M. S.; Jen, A. K. Y. Chem. Mater.
2003, 15, 269.
(15) (a) Moratti, S. C.; Cervimi, R.; Holmes, A. B.; baigent, D. R.; Friedn, R. H.; Greenham, N. C.; Gruner, J.; Hamer, P. J.
Synth. Met. 1995, 71, 2117. (b) Yang, J. S.; Swager, T. M. J. Am. Chem. Soc. 1998, 120, 5321.
(16) Bo, Z.; Zhang, C.; Severin, N.; Jurgen, P. R.; Schluter, A. D.
Macromolecules 2000, 33, 2688.
(17) Liu, B.; Yu, W. L.; Lai, Y. H.; Huang, W. Chem. Mater. 2001,
13, 1984.
(18) Song, H. H.; Lin, H. C. Liq. Cryst. 2004, 31, 831.
(19) Xu, B.; Pan, Y.; Zhang, J.; Peng, Z. Synth. Met. 2000, 114, 337.
(20) Liu, M. S.; Jiang, X.; herguth, P.; Jen, A. K. Y. Chem. Mater.
2001, 13, 3820. (b) Wilson, J. N.; Windscheif, P. M.; Evans,
U.; Myrick, M. L.; Bunz, U. H. F. Macromolecules 2002, 35, 8681.
(21) Egbe, D. A. M.; Cornelia, B.; Nowotny, J.; Gunther, W.; Klemm, E. Macromolecules 2003, 36, 5459.