JOURNAL OF MOLECULAR RECOGNITION, VOL. 9,149-157 (1996)
Molecular Recognition with C-Clamp Porphyrins:
Synthesis, Structural, and Complexation Studies
Ying Liang, C. K. Chang*Shie-Ming Peng
Department of Chemistry, Michigan State University, East Lansing, MI 48824, USA
Department of Chemistry, National Taiwan University, Taipei, Taiwan
Porphyrin-based molecular clefts equipped with a strategically pmitioned carboxylic group have been synthesized. These ditopic porphyrins exhibit excellent binding affinity for many neutral substrates. X-ray structures of a porphyrin-water inclusion complex and a Zn(porphyrin)-methanol complex reveal how the carboxylic group interacts with substrates via H-bonding. Multipoint recognition is demonstrated in the differential binding of 1 f J - versus 1,2,4-triazole, suggesting the possible use of such receptors to separate heterocyclic bases.
Keywords: Porphyrin; Kemp’s triacid; €I-bonding; substrate binding; imidazole; purine; 1,2J-triazole, 1,2,4-triazole
Introduction
Molecular recognition plays an important role in biological systems. Highly specific interactions between proteins and ligands provide the basis for protein function. There have been extensive studies focusing on the design and synthesis of biomimetic host-guest systems that may provide insights to biological processes (Schneider and Durr, 1991). In the efforts at mimicking heme proteins, numerous model porphyrins have been synthesized for studies of mainly metal-centered interactions such as dioxygen binding (Momenteau and Reed, 1994). The porphyrin macrocycle, despite the crucial biological functions of its metal deriva- tives, usually does not have enough functionalized binding sites to interact with substrates. However, the porphyrin ring can be easily linked to other functional groups at the peripheral meso and Ppyrrole positions. Recent advances in the synthesis of functionalized prophyrins have made it possible to develop porphyrin-based receptors with a variety of sizes, shapes and functional surfaces. The relatively rigid and disk-shaped porphyrins are suitable to serve as a framework in building molecular clefts that function independently of the porphyrin. The presence of NH protons or metal ions at the porphyrin center can provide additional binding sites to aid the complexation of guest molecules. This flexibility in design and synthesis makes modified porphyrins useful not only in mimicking their biochemical functions, but also in developing new types of receptors for artificial molecular recognitions. The mecha- nism which allows the binding of specific ligands can be ascribed to hydrogen bonding, T T stacking and metal-
ligand coordination. Various porphyrin receptors have been designed for recognizing amino acids, nucleobase pairs, barbiturates and polyols (Bonm-Law and Sanders, 1995;
Imai et al., 1992; Kuroda and Ogoshi, 1994; Lindsey et al.,
1988; Mizutani et al., 1994; Slobodkin et al., 1992). Author to whom correspondence should be addressed
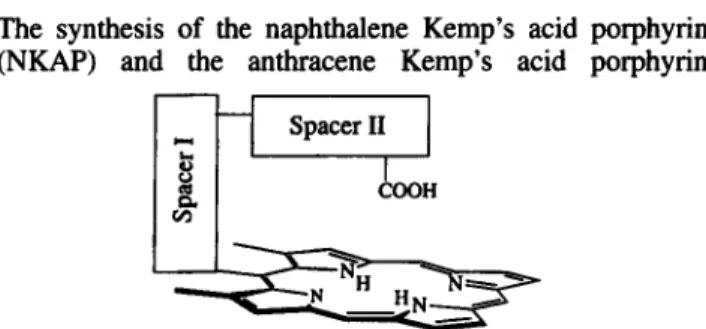
We wish to report here the study of a novel C-clamp shaped molecular receptor in which a porphyrin ring is supplemented by a carboxylic acid group pointing towards the porphyrin center. This acid functional group combined with the pyrrole NH or metal ion of the porphyrin furnishes a ditopic binding site capable of specific substrate recogni- tion. As shown in Fig. 1, the acid functionality is linked to the porphyrin ring via two perpendicular spacers. Spacer I is a rigid aromatic moiety attached to porphyrin meso-position and flanked by two methyl groups to limit its freedom of rotation. Spacer I1 is conveniently part of the Kemp’s triacid whose utility as a converging building block for molecular receptors has been well demonstrated by Rebek (Rebek,
1987, 1990). The combination of the Kemp’s triacid and naphthalene or an anthracene connector thus gives us a C- shaped receptor with a relatively rigid acid hovering over the porphyrin. The C-clamp porphyrins have been used to enhance
O2
binding to the Co(I1) porphyrin (Chang et af.,1995), and to form inclusion complexes with small neutral guests (Liang and Chang, 1995). We report here details of the synthesis, structures and molecular interactions with a number of substrates.
Synthesis
The synthesis of the naphthalene Kemp’s acid porphyrin (NKAP) and the anthracene Kemp’s acid porphyrin
Spacer I1
Figure 1. Essential components of the C-clamp porphyrin. CCC 0952-3499/96/020 149-09
150 Y. LIANG, C. K. CHANG AND S.-M. PENG
nucleophilic, but more exposed carboxylate was more reactive than the imide towards the highly hindered mesylate carbon. Hence, an alternative synthetic pathway was designed.
Rebek has reported the condensation of various amines with the acid chloride-anhydride (Rebek et al., 1985).
Therefore, we decided to choose this route and prepared an amine for coupling (Fig. 2). The mesylate reacted with excess sodium a i d e in DMF to give the azido-porphyrin in
99% yield. IR spectrum of this porphyrin showed a strong azide absorption band at 2096 cm-
'.
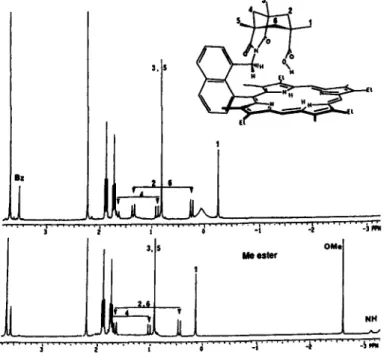
After reducing the zinc azido-porphyrin by LiAlH,, the amino porphyrin was coupled with the acid chloride-anhydride to successfully generate NKAP. The overall yield was 60% for NKAP. The anthracene homologue was prepared similarly with a slightly better yield (72%). 'H NMR of NKAP and AKAP showed that the capping cyclohexyl protons have sub- stantial upfield shift (Fig. 3). The gern-methyl group to the CO,H was found at-
0.26 ppm in NKAP and 0.37 ppm inFigure 2. General synthetic scheme. AKAP compared to 1.40 ppm in the starting Kemp's acid
chloride-anhydride, which is obviously due to the diamag- (AKAP) are outlined in Fig. 2. For NKAP, the starting ester netic ring current.
prophyrin (Chang and Kondylis, 1986) was converted into In the 'H NMR studies of NKAP, the typical NH proton its zinc complex before being reduced by LiAlH, in dry signals in the
-
3 to - 4 ppm region were not detected at THF, since direct reduction sometimes gave aluminum- room temperature; instead a broad peak around 0 ppm was inserted porphyrin which is nearly impossible to demetalate. observed (Fig. 3). This phenomenon suggests proton The resulting alcohol was treated with excess (10 equiv.) exchange among the NH protons of the porphyrin and the methanesulfonyl chloride at room temperature without carboxylic acid proton. When the acid group was methyl- heating to minimize decomposition. The excess reagent was ated by CH,N,, the NH protons emerged at-
3.2 ppm as a removed by vacuum pump. Reaction of this mesylate with doublet. Similar behavior for AKAP was observed. Here, the potassium imide-carboxylate salt did not give the intermolecular proton exchange is not probable since the C- NKAP, but instead an isomer. With the anthracene homo- shaped geometry obstructs close contact of the acid proton logue, the same reaction also occurred to yield an isomer. to another porphyrins NH protons. In order to probe this The first evidence of isomer formation came from the intramolecular exchange, variant temperature NMR (Sand- failure in transforming it into an ester by diazomethane, as strom, 1982;&,
1985) was employed to monitor the well as the inability to lose CO, in the mass spectrum, as dynamics of the proton exchange at 500 MHz in these two expected for carboxylic acid compounds. Clearly, the less systems. For NKAP, a sharp peak around-
4 ppm at&
r@o
LiAlH, 1 CHzOH 1. a*
-+gk&?q
323
2 CU~OMSr&
NKAP Spacer I = 1.8-naphthalene
4
($!
mZN3--
s [ G ] O 0
AKAP Spacer I = 1 .&anthracene
1
[jJ&
--*'
OM] ~~,.r
...
.
. . . . , . . . . 4 " ' -5 ir;'' ' " '
... ......I.. . . I . . .
...
i . " i
MOLECULAR RECOGNITION WITH C-CLAMP PORPHYRINS 151
-
90°C was observed, which gradually broadened out. When temperature reached 20°C. a new signal centered at 0 ppm appeared as the averaged signal of the porphyrin NH protons and the acid proton. Over the entire temperature range accessible in CD2CI, (-
90 to 40 "C), the signal of the carboxylic acid proton was not obvious, possibly due to overlapping with other signals, and this acid proton was identified at 3.6 ppm by saturation spin transfer techniques (Dahlquist et al. 1975) under conditions of slow exchange. The coalescence temperature detected for NKAP was-
10°C and for AKAP 15°C. This agrees well with the expectation that in NKAP the acid proton is closer to the porphyrin center and less energy is needed for the coalescence to occur. Even though coalescence temperature may change due to changes in chemical shift for the two sites, this is not the major determining factor for NKAP and AKAP, since the difference in chemical shift of the two exchanging sites of these systems is quite small.Crystal Structure
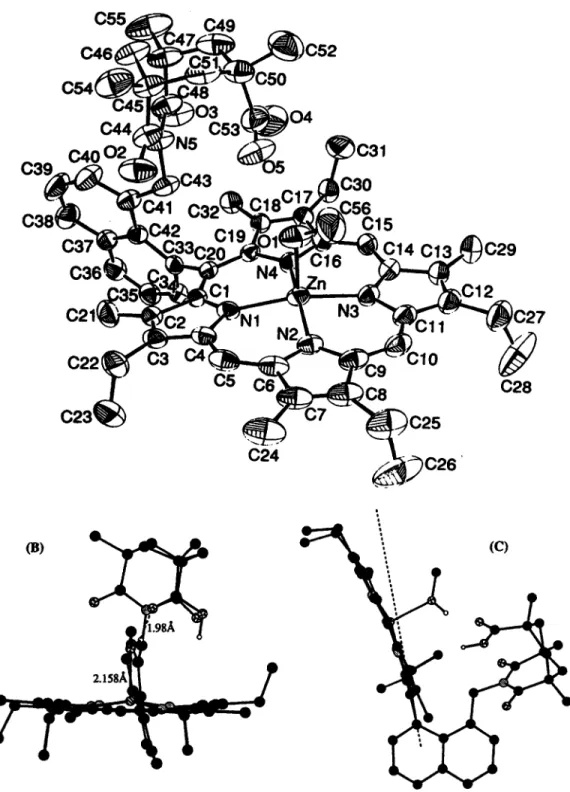
The crystal structure of NKAP-H20 inclusion complex has been published previously (Chang er al., 1995) and the structure of ZnNKAP-MeOH is shown in Fig. 4. The selected bond distances and angles of ZnNKAP are given in Table 1 .
The zinc complex of NKAP crystallized from methylene chloride solution layered with methanol has a methanol molecule as axial ligand with the 0-Zn distance of 2.1587(24)
A.
The methanolic proton is H-bonded to the carbonyl group of the Kemp's acid with a distance of 1.98(3)A.
In order to accommodate this H bond, the superstructure is twisted off center (Fig. 4b). The naph- thalene spacer and the porphyrin ring also distort severely from the ideal C20-C33 axis (Fig. 4c). The 'jaw-opening' observed in ZnNKAP is even greater than that in the free base NKAP. In both structures, the porphyrin skeleton remains essentially unchanged. The X-ray structures clearly indicate that the NKAP system is ideal for binding small, preferably monoatomic substrates. By inference supported by molecular modeling, the AKAP compounds with agreater porphyrin to acid distance should serve as good receptors for larger substrates.
Substrate Binding of Metal-free
Systems
The magnitude of molecular inclusion was followed by 'H NMR titration carried out in CDCl,. Upon stepwise addition of guest molecules into the porphyrin solution, all the protons of the Kemp's acid superstructure exhibited down- field shift due to less diamagnetic ring current. This downfield shift suggested that upon binding of the guest molecule, a 'jaw-opening' motion occurred at the por- phyrin-spacer I conjunction. On the other hand, along this motion, the naphthyl porton on C34 experienced an upfield shift due to the tilting of naphthalene, bringing this proton closer to the porphyrin. A similar shift was also observed for the corresponding proton on anthracene in AKAP. The
binding constants were derived from the chemical shift of naphthyl or anthryl methylene protons as well as by distinctive shifts of other protons during the titration process.
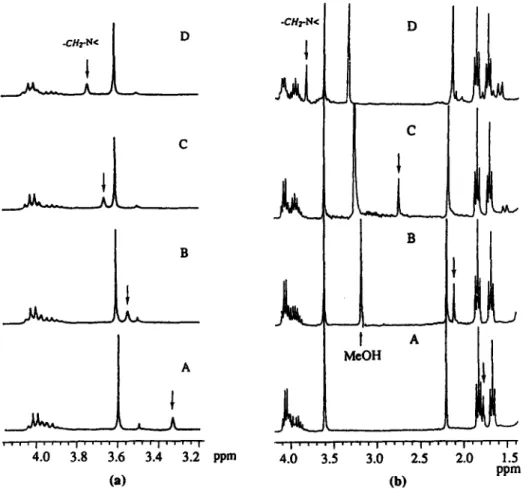
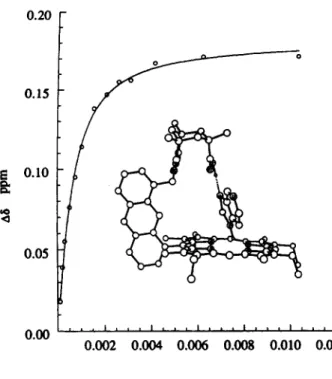
For studying the H,O binding to NKAP, it is crucial that all reagents and the NMR tube are as anhydrous as possible. The binding of water of NKAP is strong enough that the saturation point can be reached before the solubility of water in CDCl, becomes a problem (Staverman, 1941). Fig. 5(a) displays 'H NMR spectra showing the chemical shift of the naphthyl methylene proton at varying concentrations of water. For the AKAP binding of imidazole or purine, the interaction presumably relies on multipoint recognition as depicted by the computer model (Fig. 6). The titration data give a 1:l complex with binding constants of 2550 and 2600 M - for imidazole and purine, respectively. As purine
is a weaker base than imidazole (Table 2), the comparable binding strength of the two bases suggests that the intrinsic basicity is not the determining binding force. In our systems, T-T interactions between the guest and host
molecules need not to be considered. Therefore, the attractive forces come solely from the H-bonding network.
The DSC (differential scanning calorimetry) analysis was also appplied to the solid crystals of NKAP-H,O. The DSC showed two endothermic peaks, with the first one attributa- ble to dehydration and the second peak coinciding with the melting of the compound. Continuous heating led to decomposition. The heat flow associated with the first peak
is 17.58 J g-', indicating that the water binding energy is about
-
15+ 1kJ
mol-l (Liang and Chang, 1995).Substrate Binding of Zinc Porphyrins
To estimate how much stabilization energy the C-clamp acid group can impart to metalloporphyrin-ligand binding, we examined the zinc porphyrin as a model. The same protocol of 'H NMR titration as mentioned previously was used for the C-clamp zinc porphyrin systems. A typical NMR titration of ZnNKAP-MeOH is shown in Fig. 5(b). For the control study using ZnOEP (octaethylporphyrin), whose guest-host interactions cannot be easily followed by 'H NMR, UV-vis spectroscopy was used to monitor ligand binding. The binding constants of these systems are listed in Table 2.
Methanol ligation to zinc porphyrin is well studied (Bonar-Law and Sanders, 1995). ZnNKAP binds MeOH more strongly than the unfunctionalized ZnOEP (AAG=3.1 kJ mol-l) due to the additional binding force derived from the H-bonding clearly shown in the crystal structure.
The most dramatic effect in molecular inclusion is seen in the case of triazoles. 1,2,4-Triazole, being a weaker base ( ~ K ~ 2 . 3 ) (Albert, 1968) than imidazole, is normally not a strong ligand for metalloporphyrins. But ZnAKAP maxi- mizes the binding by a three-point contact as depicted in the schematic structure (Fig. 7). The binding constant is 21
OW+
loo0 M - I , with a free energy gain of 9.9 kJ mol-I.'H- 1,2,3-triazole, capable of only a two-point interaction, displays a significant decrease in binding energy
(AAG 4.8 kJ mol- I). The binding of 1,2,4-triazole was
solution of the base to the zinc porphyrin dissolved in CDC13 due to insolubility of the base in pure CDC13. The 1,2,3-triazole study was measured in pure CDC13, as well as by the mixed solvent method. Both methods gave essen- tially the same binding constant within the margin of experimental error. Therefore, it is safe to assume that methanol cannot compete effectively with 1 ,2,4-triazole in binding with the host molecule, and the presence of small amounts
(4%)
of methanol does not distort the binding constant.Discussion
The X-ray structure of the NKAP-H,O complex allows a rare opportunity to examine the geometries of the acid-H,O interface. Ab initio calculations of benzoic acid mono- hydrate predict two possible conformations, syn and anti, with the syn being the more stable form (Nagy et al., 1994).
The
optimized geomeuic distances and angles are shown in Fig. 8. The salient feature is that the H-bond C 0 H . e - 0 is slightly shorter and less bent than the C = O - - . H bond. Our.
C24
C26
'Figure 4. (a) X-ray crystal structure of ZnNKAP-MeOH. (b) Side view of the X-ray structure of ZnNKAP- MeOH. (c) Side view from another angle showing the porphyrin outward bending.
MOLECULAR RECOGNITION WITH C-CLAMP PORPHYRINS 153
NKAP-H,O clearly displays a syn conformation with a slightly shorter and near-linear COH.
-
-0 and a more bent H bonding for the carbonyl group. The agreement between our structure and the theoretical model seems excellent given the other constraints that the water has to meet within this artificial molecular cleft.The molecular inclusion of C-clamp porphyrins is brought about by multiple H-bonding. It is always inter- esting to identify the contribution of each individual hydrogen bond within the overall free energy of complexa- tion. Selective removal of certain H-bonding participants from either the receptor or the substrate has been an important tactic in addressing this question. However, H- bond obliteration studies of this type must be treated with caution since other changes in the molecular properties, such as basicity and conformation, may contribute to the differences in binding energy. A comparison of ZnOEP vs ZnAKAP binding of IH- 1,2,3-triazole provides an estima- tion of the magnitude of H bonding strength of acid
C=O..
.
H. The energy difference here is 7.8kJ
mol-I. This AAG may be compared to the 3.1 kJ mol-' observed in MeOH binding to ZnNKAP vs ZnOEP. The difference clearly has something to do with the acidity of the proton (NH vs OH). Another useful comparison is the 3-point (1,2,4-triazole) vs 2-point ('H- 1,2,3-triazole) contact. TheBAG value of 4.8 kJ mol-l arises from more than just the
extra H-bond. The basicity difference of the two triazoles has significantly influenced the Zn-N bond (2.7 kJ mol-I)
whereas its effect on the NH-..O=C is possibly very small.
In conclusion, our functionalized porphyrin receptors, NKAP, AKAP and their zinc complexes, are capable of binding and recognizing various small neutral molecules. The shape and rigidity of superstructure porphyrin make possible some very effective complexation of substrates through multipoint recognition. The selectivity demon- strated by ZnAKAP suggests possible applications for the separation of heterocyclic bases. It is also possible to further modify the overhanging carboxyl group to effect more sophisticated substrate binding and recognition.
Experimental Section
Materials and measurements
Reagents and solvents for synthesis were used as received unless otherwise stated. CH,Cl, and DMF were distilled from CaH,. THF and toluene were freshly distilled from
Tablel. Selected bond distances
(A)
and angles (deg) and their estimated standard deviations for ZnNKAP Zn-N1 2.0482(24) N 1 -Zn-N2 91.36(10) Zn-N2 2.063(3) Nl-Zn-N4 87.45(9) Zn-N3 2.063(3) N2-Zn-N3 87.44(10) Zn-N4 2.0578(24) N3-Zn-N4 90.63(9) Zn-0 2.1587(24) C2O-C33-C42 126.0(3) 0(5)-H0(1) 1.98(3) c42-c41-c43 120.8(3)Distance (A) Angles (degl
0(1)-H0(1) 0.76(3) 0(1)-H0(1)-0(5) 159(4)
LiAlH,. UV-visible spectra were measured on a Cary 219
or a Shimadzu 160 spectrophotometer. 'HNMR spectra were recorded on Varian Gemini300 in '100%' CDCl, (minimum 99.8 atom % D, Cambridge Isotope Labora- tories) with the residue CHC& as the internal standard set at 7.24 ppm. Variant temperature NMR measurements were conducted on a Varian 500 MHz NMR spectrometer. Mass spectra were measured from a benchtop VG Trio-1 mass spectrometer or FAB (fast atom bombardment) on a JEOL HX-110 HF double focusing spectrometer in the positive ion detection mode. Infrared spectra were obtained from sample film on an NaCl plate and recorded on a Nicolet IW 42 spectrometer.
5-[8-(Hydroxymethyl)-l-naphthyl]-2,8,13,17-tetraethyl-3,7,12,
18-tetramethylporphyrin (1). To 5-(8-methoxycarbonyl- 1
-
naphthy1)2,8,13,17
-
tetraethyl-
3,7.12.18-
tetramethylpor-
phyrin (Chang and Kondylis, 1986) (2.1 g, 3.17 mmol) dissolved in methylene chloride (400ml) was added a saturated methanolic solution of zinc acetate containing sodium acetate. The mixture was heated to reflux for 10 minutes before washing with water (50ml), dried over Na,SO, and evaporated to dryness to give the zinc porphyrin. To this material dissolved in freshly distilled THF (300ml), a suspension of lithium aluminum hydride (LiAlH,, 0.5 g) in THF was added slowly. The reaction solution was stirred at room temperature for another hour. Completion of the reaction was monitored on TLC by the disappearance of the fast moving starting material. After the reduction was complete, the mixture was quenched by addition of ice water and the product was isolated by extraction with CH,C12 from water. The crude product was purified by column chromatography over silica gel using 2% MeOWCH,Cl,. The purified zinc porphyrin was redissolved in methylene chloride (200 ml), and demeta- lated by washing with 10% HCI (200ml). The organic solution was then washed with saturated NaHCO, and water and dried over anhydrous Na,SO,. Evaporation afforded 1.63g (81%) purple crystal of 1. IH NMR (300 MHz, CDCl,) Sppm -3.09 (2H, d, NH), 0.20 (lH, t, OH), 1.71 (6H, t, Et), 1.88 (6H, t Et), 2.13 (6H, s, Me), 3.08 (2H, d, CH,O-), 3.65 (6H, s, Me), 3.96 (4H, m, Et), 4.08 (4H, q, Et), 9.97 (lH, s, meso), 10.17 (2H, s, meso), naphthyl: 7.61 (lH, s), 7.63 (lH, d), 7.78 (lH, t), 8.01 (lH, d); 8.17 (lH, t), 8.35 (lH, d); MS, m/e (relative intensity) 634 (M+, 86.89); UV-vis A,, nm (EM) 625 (2100), 572 (6400), 538 (6500), 503 (15000), 405 (19oooO).5
-
[8-
(Methanesulfonylmethyl)-
1-
naphthyl]-
2,8,13,17-
tetra-
e t h y l - 3 , 7 , 1 2 , 1 8 - t y I p o ~ h ~ n (2). The above alcohol 1 (100 mg, 0.16 mmol) was dissolved in 5 mldry
methylene chloride plus 3 drops of trimethylamine. To this mixture stirred under argon in an ice bath, methanesulfonyl chloride (2 ml) was slowly added. The system was continuously stirred for 14 hours at room temperature. The solvent and excess methanesulfonyl chloride was pumped away by a vacuum pump (room temperature until almostdry,
then at60 "C for 2 hours). This product 2 was used for the next step without further purification.
Naphthalene Kemp's acid isomer porphyrin (3). To the above porphyrin mesylate (15 mg, 0.024 mmol), the Kemp's imide
C
I
I
B
I *
D
c CI
t
A MeOHL
1
d.
4.0 3.5 3.0 2.5 2.0 1.5 PPm (b)Figure 5. (a) 'H NMR titration of water to NKAP. [NKAPl=3 rnM, [H,01 in A: 0.000 M; B: 0.022 M;
C:0.037 M; D:0.047 M. (b) 'H NMR titration of MeOH to ZnNKAf? IZnNKAPl=0.014 M, [MeOHl in
A:0.000 M; B0.013 M; C:0.066 M; D:0.500 M.
potassium carboxylate (30 mg, 0.091 mmol) and dry tolu- ene (20ml) were added. The mixture was refluxed under argon for 12 hours. The excess potassium salt was removed by filtration. After removal of the solvent under vacuum, the product was dissolved in methylene chloride and purified on TLC plate using 1% MeOH/CH,Cl,: yield, 2.87 mg, 14%. 'HNMR (300 MHz, CDCl,) 6 ppm -3.06(2H, d, NH), 1.71 (6H, t, Et), 1.86 (6H, t, Et), 2.12 (6H. s, Me), 3.61 (6H, s, Me), 3.86 (2H, q, Et), 3.96 (2H, s, -CH,N<), 4.04 (6H, m, Et), 9.91 (lH, s, meso), 10.11 (2H, s, meso), naphthyl: 7.42 (lH, d), 7.64 (lH, t), 7.71 (lH, t), 7.83 (lH, d), 8.21 (lH, d), 8.31 (lH, d); chair: -0.60 (3H, s Me), 0.31 (lH,
d), 0.64 (6H, s, Me), 0.78 (lH, d), 1.40 (lH, d), 1.63 (2H, d), 6.36 (lH, NH); FABMS, m/e (relative intensity) 856 M+ 1, 100); UV-vis A,, nm (relative intensity) 627 (l.O), 574 (8.3), 539 (10.2), 504 (15.5), 409 (219.7).
5
-
[8-
(Azidomethyl)-
1-
naphthyl]-
2,8,13,17-
tetraethyl -3,7,12,18-tetramethylporphyrin (4). The porphyrin mesylate 2 was dissolved in dry DMF (10 ml) and sodium azide (300 mg) was added. The mixture was heated to 90 "C for 5 hours under argon. After the reaction was done, water was added to the solution and the porphyrin was extracted to the organic layer by methylene chloride. The crude product was
Table 2. Association constants of porphyrins with substrates
Receptor NKAP AKAP AKAP ZnOEP ZnNKAP ZnOEP ZnAKAP ZnOEP ZnAKAP Substrate H,O lrnidazole purine MeOH MeOH 1,2,3-triazole 1,2,3-triazole 1,2,4-triazole 1.2.4-triazole - K ( M - ' ) A A G W mol-'l PK. l(20") 6627 7.0 2550+90 2.4 2600 f 300 - 2 7 2 1 2523 3.1 (vs ZnOEP) - 2 1.2 130220 1.2 3000+70 7.8 (vs ZnOEP) 2.3 390k40 2.7 (vs 1,2,3-triazole) 2.3 21 0002 1000a 9.9 (vs ZnOEP) 4.8 (vs 1,2,3-triazole)
a The error margin was improved from the early report (Liang and Chang,
MOLECULAR RECOGNITION WITH C-CLAMP PORPHYRINS I55
purified by column chromatography over silica gel using methylene chloride as solvent and crystalized from CH,ClJ CH,OH to give 103 mg (99%) of 4. 'H NMR (300 MHz, Oe2O
F
0.15 t p ' 10 a 0.05i'
0.00 0.002 0.004 0.006 0.008 0.010 0.012 [Purine] MFigure 6. Schematic structure and curve fitting of AKAP-purine binding.
CDCl,)
S
ppm-
3.08 (2H, d, NH), 1.73 (6H. t, Et), 1.89 (6H. t, Et), 2.12 (6H. s. Me). 2.95 (2H, d, CH,N,), 3.66 (6H,s. Me). 3.98 (4H. q, Et), 4.10 (4H. q, Et). 9.99 (1H. s, meso), 10.19 (2H. s, meso), naphthyl: 7.49 (lH, d), 7.64 (lH, t),
7.80 (lH, t), 8.03 (lH, dd); 8.22 (lH, d), 8.36 (lH, dd); FAB-MS, m/e (relative intensity) 660 (M+ 1, 3.17); UV-vis A,, nm
(EM)
625 (2100), 572 (6400), 538 (6500), 503 (15OOO), 405 (19oooO); IR: 2096 cm-' (strong).5- [8-(Aminomethyl)- 1 -naphthyl]-2,8.13,17-tetraethyl-3,7,12,
18-tetramethylporphyrin (5). Zinc was inserted into 50 mg of porphyrin 4 using the procedure described previously for the ester porphyrin 1. To the dried zinc porphyrin dissolved in freshly distilled THF (15 ml), cooled in an ice bath and stirred under argon, a suspension of LiAlH, (20 mg) in THF was added slowly. The mixture was stirred for another 10 minutes. The reduction was monitored by TLC as the product has a relatively smaller Rf value compared to that of the starting material. After the reaction was done, ice water was added cautiously to the reaction flask. The THF was evaporated, and the porphyrin was extracted into methylene chloride and evaporated to dryness. Column chromatog- raphy (silica, CH,Cl,) provided the pure zinc amino-porphyrin. The zinc complex was then partitioned in CH,Cl, and 10% HCl, the CH,Cl, layer of porphyrin was washed with saturated NaHC03 and water, and evaporated to give 29 mg (61%) of 5.
'H
NMR (300 MHz, CDCl,) 6ppm
-
3.09(2H, br, NH), 0.89 (2H, t, NH,), 1.70 (6H, t, Et), 1.88 (6H, t, Et), 2.12 (6H, s, Me), 2.39 (2H, s, CH,N), 3.65 (6H, s, Me), 3.88 (2H. q, Et), 3.97 (2H. q Et), 4.08 (4H, q. Et), 9.97 (1H. s, meso), 10.16 (2H, s, meso), naphthyl: 7.52#
I #
Figure 7. (a) ZnAKAP-1,2,4-triazole. (b) ZnAKAP-1,2,3-triazole.
H
(lH, d), 7.61 (lH, t), 7.77 (lH, t), 8.00 (lH, dd); 8.13 (lH, dd), 8.32 (lH, dd); MS, d e (relative intensity) 633 (M+, 2.56); UV-vis A,, nm (EM) 625 (2100), 572 (6400), 538 (6500). 503 (15000), 405 (19oooO).
5
-
(8-
[endo-
7-
(Hydroxycarbonyl)-
1,5,7-
trimethyl-
2,4-
dioxo- 3-
azabicyclo[3,3,l]non -3-
yl] -methyl-
1-
naphthyl}-
2,8,13,17-
tetraethyl-3,7,12,18-tetramethylporphyrin
(NKAP).
The Kemp's imide-acid chloride was prepared as reported previously (Rebek et al., 1985). 5-[8-(Aminomethyl)-l-naphthyl]-2,8,13,17-tetraethyl-3,7,12,18-tetramethyl-por-
phyrin 5 (57 mg, 0.09 mmol) was dissolved in freshly distilled toluene (5 ml) and stirred under argon. To this solution, a catalytic amount of dimethylaminopyridine (DMAP) and 2,6-di-t-butylpyridine (46 mg) and Kemp's imide-acid chloride (28 mg, 1.0 mmol) were added. The mixture was refluxed under argon for 19 hours. The solvent was evaporated and methylene chloride was added. Pur- ification from preparation TLC plate (silica gel, l% CH,OH/CH,Cl,) and recrystallization from CH,C&/CH,OH gave 77mg (75%) of NKAP. mp: 298 "C dec; 'H NMR (300MHz, CDCl,) Gppm 1.73 (6H, t, Et), 1.88 (6H, t, Et), 2.21 (6H, s, Me), 3.28 (2H, s, CH,N), 3.62 (6H, s, Me), 3.824.08 (8H. m, Et), 9.93 (lH, s, meso), 10.19 (2H. s, meso), naphthyl: 6.67 (lH, d), 7.45 (lH, t), 7.69 (lH, t), 7.78 (lH, dd); 8.04 (IH, d), 8.27 (lH, dd), kemp: -0.26 (3H, s, Me), 0.23 (2H, d, CH,), 0.80 (6H, s, Me) 0.88 (lH, d, CH,), 1.30 (2H, t, CH,), 1.63 (lH, d, CH,); MS, d e (relative intensity) 855 (M+, 7.86); UV-vis A,,, nm (EM) 622 (2000), 570 (6300), 540 (6000), 507 (12000), 406 (14OOOO).
Determination of Binding Constants
A. NMR method. The receptor molecules NKAP, AKAP, ZnNKAP and ZnAKAP (2
-
5 x lo-, M in 0.8 ml CDCl,)were titrated with a solution of the substrate (2-5 M)
dissolved in CDCl, or methanol-d,. The downfield shifts of the naphthyl and anthryl methylene protons linked to the imide were monitored as a function of substrate concentra- tion. Addition was continued until up to 10-15 equivalents of the substrate had been added. The resultant titration curve was analysed by nonlinear regression methods (Wilcox and Cowart, 1986).
B. UV-Vis method. A similar protocol to A was followed. The increase in absorbance of the Soret peak of the
Refert
Albert, A. (1968). Heterocyclic Chemistry. 2nd edition, Oxford University Press, New York, pp. 441.
Bonar-Law, R. R and Sanders, J. K. M. (1995). Polyol recognition by a steroid-capped porphyrin. Enhancement and rnodula- tion of misfit guest binding by added water or methanol. J. Am. Chem. SOC. 117,259-271.
Chang, C. K. and Kondylis, M. R (1986). Intramolecular hydro- gen-bonding affecting dioxygen binding to cobalt(l1) porphyrins. J. Chem. SOC. Chem. Commun. 316-318.
Chang, C. K., Liang, Y., Aviles, G. A. and Peng, S.-M. (1995). Conformational control of intramolecular hydrogen bond- ing in heme models: maximal Co"-0, binding in a C-clamp porphyrin. J. Am. Chem. SOC. 117,4191-4192.
molecular complex and the decrease of the receptor Soret peak were monitored upon the addition of substrate molecules.
C. DSC method. DSC measurements were canied out for the NKAP-H,O crystal under N, with heating rate at 5 "Chin.
Crystal Structure Determination
Data collection was performed on a Nonius diffractometer at room temperature using graphite-monochromated Cu Kcu radiation, The crystals used for analysis were of approx- imate dimensions 0.30 x 0.35 x 0.40 mm for NKAP and 0.40 x 0.40 x 0.70 mm for ZnNKAP. The unit cell parame- ters were determined by a least-squares fit of 25 machine-centered reflections having 28 values in the ranges of 39.6-44.4" for NKAP and 20.10-27.12" for ZnNKAP. The intensity data were reduced and corrected for Lorentz and polarization factors using the applied programs. Semi- empirical absorption corrections were applied. The crystal structures were solved by direct methods using the NRCVAX program package. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms of the car- boxylic group and methanol were located from the difference map and refined positionally. All other hydrogen atoms were calculated with fixed isotropic thermal parame- ters. The highest difference Fourier peak was 0.31 and 0.35 e
A-3
for NKAP and ZnNKAP, respectively. For NKAP, the final R is 0.047 and R, is 0.044; and for ZnNKAP, R and R, are 0.040 and 0.042, respectively. The crystal structure data are available as Supplementary Material which includes crystallographic data, all refined and calculated atomic coordinates and Beq values, and bond distances and angles for ZnNKAP.Supplementary material. Crystal structure data (5 pages) available from the author.
Acknowledgements
This work was supported in part by the NIH. We thank Dr G. M. Avil6s for her efforts in the initial synthesis of Kemp's isomer porphyrin.
mces
Dahlquist, F. W., Longmuir, K. J. and Du Vernet, R. 6. (1975). Direct observation of chemical exchange by a selective pulse NMR technique. J. Magn. Reson. 17, 406-410.
Imai, H., Nakagawa, S. and Kyuno, E. (1992). Recognition of axial ligands by a zinc porphyrin host on the basis of nonpolar interligand interaction. J. Am. Chem. SOC. 114,
6719-6723.
Kuroda, Y. and Ogoshi, H. (1994). Molecular recognition of modified porphyrins. Synlett. 319-324.
Liang, Y. and Chang, C. K. (1995). Inclusion complex and substrate recognition by C-clamp porphyrins containing Kemp's triacid. Tetrahedron Lett. 36, 3817-3820.
MOLECULAR RECOGNITION WITH C-CLAMP F'ORF'HYRINS I57 J., Jr. (1988). Molecular recognition: multipoint contacts
with new sizes and shapes. J. Am. Chem. Soc. 110, 6576-6577.
Mizutani, T., Ema, T., Tomita, T., Kuroda, Y. and Ogoshi, H. (1994). Design and synthesis of a trifunctional chiral por- phyrin with C2 symmetry as a chiral recognition host for amino acid esters. J. Am. Chem. SOC. 116,4240-4250. Momenteau M. and Reed C. A. (1994). Synthetic heme dioxygen
complexes. Chem. Rev. 94,659-698.
Nagy, P., Durant, G. J. and Smith D. A. (1994). Competing intra- and intermolecular hydrogen bonds for organic solutes in aqueous solution. In: Modeling the Hydrogen Bond. ed. D. A. Smith. American Chemical Society, Washington DC, pp. 60-79.
Oki, M. (1985). Methods in Stereochemical Analysis, Vol. 4:
Applications of Dynamic NMR Spectroscopy to Organic Chemistry. VCH Publishers, Deerfield Beach pp. 1-12. Rebek, J., Jr. (1987). Model studies in molecular recognition.
Science 235,1478-1 484.
Rebek, J., Jr. (1990). Molecular recognition with model systems.
Angew. Chem. Inr. Ed. Engl. 29,245-255.
Rebek, J., Jr., Marshall, L., Wolak, R., Parris, K., Kolloran, M., Askew, B., Nemeth, D. and Islam, N. (1985). Convergent functional groups: synthetic and structural studies. J. Am.
Chem. SOC. 107,7476-7481.
Sandstrom, J. (1982). Dynamic NMR Spectroscopy. Academic Press, New York, pp 97.
Schneider, H. J. and Durr, H. (Eds) (1991). Host-Guest Molecular Interactions: From Chemistry to Biology. Wiley, Chichester. Slobodkin, G., Fan, E. and Hamilton, A. D. (1992). Molecular
recognition: porphyrin-containing receptors as analogs of barbiturate-induced cytochrome p. 450. New J. Chem. 16,
643-645.
Staverman, A. J. (1941). The miscibility of water and alkylha- tides. Recl. Trav. Chim., 60,836-841.
Wilcox, C. S. and Cowart, M. D. (1986). New approaches to synthetic receptors. Synthesis and host properties of a water soluble macrocyclic analog of Trogers base. Terra-