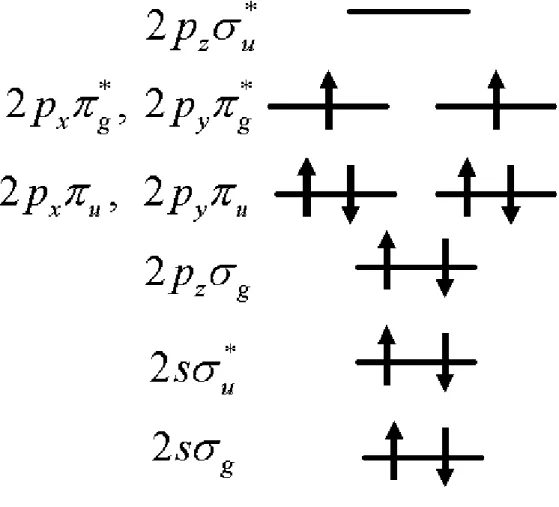國立交通大學
物理研究所
碩士論文
共軛高分子中的載子遷移率不對稱
研究生:皮旭庭
指導教授:孟心飛
中華民國九十五年六月
The Imbalance of Carrier Mobility in
Conjugated Polymers
研 究 生:皮旭庭 student: Pi,Shu-Ting
指導教授:孟心飛 Adviser: Hsin-Fei Meng
國立交通大學
物理研究所
碩士論文
A thesis submitted to
Institute of Physics College of Science
National Chiao Tung University
In partial fulfillment of the requirements for the
degree of master of science in physics
June 2006
Hsinchu, Taiwan, Republic of China
中華民國九十五年六月
共軛高分子中的載子遷移率不對稱
學生:皮旭庭 指導教授:孟心飛
國立交通大學物理研究所
摘要
在許多共軛高分子中,電子電洞的對稱不只是在能帶
結構上會被發現,在因晶格缺陷所造成的失序結構中
也同樣的被發現,但在實驗上卻被廣泛的看到電洞遷
移率遠大於電子遷移率的現象。我們提出在空氣中存
在了不可避免的氧吸附所造成的電子補捉來解釋此現
象。在本文中,我們可以定量的計算出關於吸附以及
電子電洞不對稱的許多性質,此外,也引入了一個用
來估計原子之間跳越積分的方法。
The Imbalance of Carrier Mobility
in Conjugated Polymers
Student: Pi,Shu-Ting Adviser: Hsin-Fei Meng
INSTITUTE OF PHYSICS
NATIONAL CHIAO TUNG UNIVERSITY
Abstract
Electron-hole symmetry is found to exist not only the band
structure but also the defect level caused by structure disorder.
The commonly observe
d higher hole mobility is explained by the
electron traps caused by oxygen molecule adsorption. We found
that defects will enhance oxygen adsorption and this is a key to
electron-hole symmetry breaking. In this paper, we calculate the
adsorption and imbala
nce properties quantitatively . Besides, a
method to estimate the hopping integral is also introduced.
謝 誌
一轉眼兩年就過去了,感覺還來不急留下些什麼,就
要跟這裡說聲再見了。交大的兩年對我人格的養成是
很重要的階段,除了學習到了科學研究的方法和基本
的觀念,滿足了我對物理的著迷,更讓我有了很多難
忘的回憶,和同學、老師、學長學弟妹之間的互動都
是我年輕的証明。臨別匆匆,這兩年改變我最多的莫
過於是指導我做論文的紀亙學長,從他身上我真正接
觸到了做研究是怎麼回事,我非常感謝他。另外我的
指導教授孟心飛老師也讓我獲益匪淺,他精準的眼
光,常常一語就到破了物理問題的核心,這點常讓我
覺得要跟他學的還很多。除了研究,我想更要感謝的
是我碩士班生活陪我走過酸甜苦辣的同學們:光胤、
德明、瑞仁、翔瑞、孟老師實驗室的學長們,以及天
天在所上搞笑的學弟妹們,要謝的人太多了。當然,
最後一定不能忘記的是我的家人,還有我家的貓咪,
如果沒有你們,我想這一切都不會存在,謝謝!
The imbalance of carrier mobility in
conjugated polymers
Shu-Ting Pi
Abstract
Electron-hole symmetry is found to exist not only the band structure but also the defect level caused by structure disorder. The commonly observed higher hole mobility is explained by the electron traps caused by oxygen molecule adsorption. We found that defects will enhance oxygen adsorption and this is a key to electron-hole symmetry breaking. In this paper, we calculate the adsorption and imbalance properties quantitatively . Besides, a method to estimate the hopping integral is also introduced.
Contents
1 Introduction 4
1.1 Introduction to Conjugated Polymer . . . 4
1.2 Transports in conjugate polymer . . . 4
1.3 Motivation . . . 6
1.4 More on this problem . . . 6
1.5 Oxygen molecule . . . 7 1.6 Thesis Sturcture . . . 7 2 Crystal Defects 12 2.1 The Hamiltonian . . . 12 2.2 Band Structure . . . 13 2.3 Summary . . . 15
3 Adsorption of oxygen molecules 21 3.1 Lennard-Jones Potenial . . . 21
3.2 Physical Meaning of Lennard -Jones Co¢ cients . . . 21
3.3 Estimate Lennard-Jones Potenial . . . 23
4 Hopping integral 28
4.1 Hamiltonian . . . 28
4.2 Estimate the Hopping Integral . . . 29
4.3 Hopping Integral in Energy Basis . . . 30
4.4 About Symmetry . . . 32 4.5 Summary . . . 32 5 Electron-Hole Imbanalce 36 5.1 Binding Energy . . . 36 5.2 Con…guration . . . 36 5.3 Matrix Element . . . 37 5.4 Imbalance . . . 39 5.5 Summary . . . 40
6 Conclusion and Outlook 44 6.1 Conclusion . . . 44
List of Figures
1.1 molecular sturcture of polyacetylene . . . 9
1.2 Pi band of polyacetylene. . . 10
1.3 oxygen molecule levels . . . 11
2.1 PPV surcuture . . . 12
2.2 PPV band structure in k-space . . . 16
2.3 PPV energy level with defect ( = 56) . . . 17
2.4 PPV density of state with defect ( = 5 6) . . . 18
2.5 PPV defect levels and defect order parameter . . . 19
2.6 defect level electron density on defect sites . . . 20
3.1 PRL 85 1710 . . . 24
3.2 PRB 67 165424 . . . 24
3.3 Lennard-Jones Potential . . . 26
3.4 adsorption energy and distance . . . 27
4.1 hopping tntegral with two atoms . . . 33
4.2 geometry of adsorption . . . 34
4.3 hopping integral between oxygen HOMO and PPV level . . . . 35
5.1 con…gurations . . . 41
5.2 imbalance energy in di¤erent order parameter . . . 42
1.
Introduction
1.1
Introduction to Conjugated Polymer
Recently, the research about new type semiconducor is a very hot topic. The rigmarole process and expensive price always let people unsatify in traditional inorangic semiconductors like Si,Ge...,etc.In this point of view,conjugated polymer semiconductors can provide a new and possible fuure.[6] In the fol-lowing, we will introduce some basic theoty about this new material.
1.2
Transports in conjugate polymer
Conjugated polymers are constitute by orangic elements which has a carbon backbone and some side chains. The backbone carbons are mainly bonded by sigma bonds and pi orbital are weakly overlap. In standare theory, because the sigma bonds are much tighter binding than pi bond, the sigma band sturcture is much far away from Fermi level than pi band structure. Therefore the pi band is the most important one and electrons are mainly transport in this band.[1]
(see …g.1.1 , molecular sturcture of polyacetylene)
In the above picture, we show how the pi orbital overlap and electrons just hop in these molecular orbital.
But there are more things we need to know about transport in conjugated polmer: quasi-particle, Peierls instability, missing n-type. In the following, we introduce quasi-particle and Peierls instability …rst.
As we know, the crystal is very soft in polymer, so there are strong electron-phnono interaction in this system. Electrons are actually transport in the form of polarons, soilitons or bipolarons which are the quasi-particle of electron–phnono interaction and these quasi-particles have larger e¤ective
mass than electrons.[?] No matter what results does polaron tell us, all we need to emphasize is that electron-phnono interaction is very important in conjugated polymers.[1]
In order to handle this electron-phnono interaction, a famous model called the "SSH Model" had already got many properties about the quasi-particle : silton. But there are something more important than quasi-particle which appears in this model, i.e. Peierls instability.[5]
Peierls, an famous British physicist, had pointed out that: in a 1D chain, if every atom contribute one electron, the band structure should be no band gap which can easily obtain by tight-binding mothod. But, when there exist strong electron-phonon interaction, there should be still band gap because of the crystal deformation and the chain will not be metallic state. This result makes conjugated polymer looks like semi-conductor.
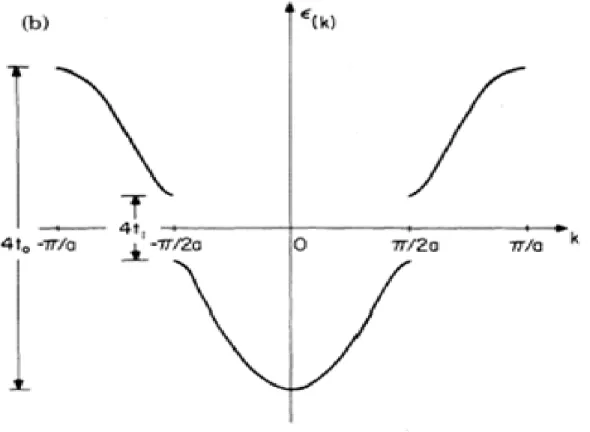
(see …g.1.2,pi band of polyacetylene)
In the above …gure, when electron-phnono interaction vanish, the parameter t1 will also vanish.
Beside electron-phonon interaction, the missing n-type also play important role in transport properities.
In most conjugated polymer, the polymer made FET are always p-type and the reason is unknown. Recently a paper, published in Nature, found that use of an appropriate hydroxyl-free gate dielectric— such as a divinyltetramethyls-iloxane-bis(benzocyclobutene) derivative (BCB)— can yield n-channel FET conduction in most conjugated polymers. The reason why n-type behaviour has previously been so elusive is the trapping of electrons at the semiconductor– dielectric interface by hydroxyl groups, present in the form of silanols in the case of the commonly used SiO2 dielectric. These …ndings should therefore open up new opportunities for organic complementary metal-oxide semicon-ductor (CMOS) circuits, in which both p-type and n-type behaviours are harnessed. [7]
Until now, some essentials of transport in polymer is known. In the next sec-tion, we will discuss a more intrinsic phenomenon: electron-hole imbalance.
Although we have introduced many applications about conjugated polymer, there are still many problems need to be solved. one of them is electron-hole mobility imbanalce. When making LED , it needs electrons and holes recom-bine in the emission layer. But in many polymer systems, the hole mobility is much higher than electron mobility and this makes the recombination in correct layer be di¢ cult.
This problem is not only a challenge in application area but also a inereasting puzzle in theoretic physics. Take PPV for example, PPV is also an P-type semiconductor but its Pi orbital band which is the transport orbital band has electron-hole symmetry structure. This means the electron and hole have the same e¤ective mass , so they should have similar transport proprtities. Some papers which caculate this problem by …rst-principle caculation even say that the electron and hole e¤ective mass di¤erence should be less than factor of 2.[11]
There are more things we need to be con…used : the oxygen! In many papers, the experimantal results show the electron-hole mibility imbalance will be enhanced if polymers are exposed to oxygen gas. [14] This fact hints us the oxygen molucules must play an important role in this problem. In next section, we will talk about oxygen more.
1.4
We have talked many things about the imbalance problem. But, in this sec-tion, we will talk about this more. In 1998, a paper which, published in Nature, says the electron-hole symmetry does exist in sloution by microwave experiments. [10] This result hints us the reasons which cause the imbalance shouldn’t come from the essence of polymer structure. It should something interact with polymer in the air. Besides, in some papers, we found that oxy-gen can enhance the imbalance and this mechanism is totally reversible.[14] As we know, no matter how prefect vaccum you can make, there are al-ways some oxygen molecules and they may form an unitentional doping in the bulk. According to the totally reversible process, we believe the oxygen doesn’t actually bond with polymer chain. It should just be adsorptive. Fur-thermore, many papers which discuss about oxygen adsorption shows defects can enhance it. [15]
Therefore, our main topic may be transformed to explain what happens when polymers are exposed to oxygen gas if defects exist.
Fortunately, in carbon nanotube researches, they have the same problem like us.[16] In carbon nanotube, they also found the e-h symmetry breaking and this result doesn’t match the symmetrical band structure. Further more, oxygen will enhance imbalance reversiblely, too. All the researches about this problem in CNT refer this phenomenon is caused by oxygen adsorption. As we know, our problem is also made by carbon and oxygen. Thus, why not using the same ideas in our system ?
1.5
Oxygen molecule
since we have thought oxygen is the key to imbalance, some introductions to oxygen molecule is needed. The molecular energy level of oxygen is like the fellowing …gure:[4]
(see …g 1.3 , the oxygen level)
In this picture, we found the HOMO of oxygen is two unoccupied state and this is because of Hund’s rule. This picture may tell us that adding an electron will not cost any energy. But in fact, the EA of oxygen molecule is around 1eV and IP is around 11 eV. This result tells us adding an electron to the HOMO needs about 10eV. This high energy di¤erence mainly comes from the exchange e¤ect and it will be important for us later.
Besides, the anti-bonding of HOMO is also important in the following. As a result, two conclusions for oxygen molecule are essential:
the exchange energy of oxygen HOMO is very high and this makes …ll an electron to oxygen hard.
anti-bonding of oxygen HOMO make coulomb interaction between oxy-gen HOMO and any single particle state become selective because of symmtery consideration.
1.6
Thesis Sturcture
Form last section , we know that the defects can induce oxygen adsorption and it may make the charge transfer form polymer to oxygen . Thus, we plan
to discuss our problem in four di¤erent parts. 1. Crystal Defects
In this part, we will analyse the energy and electron disturbation of defect states. What happens and what will be made when defects exist are our main topic in this chapter.
2. Adsorption of Oxygen Molecules
The adsorption of oxygen molecules is the main contributor that csuses e-h imbalance. We will give a simple model to estimate the adsorption energy and adsorption distance.
3. Hopping Integral
When oxygen molecules are close to the defects, the interaction between polymer chains and oxygen is turn on. To construct this perturbation in the Hamiltonian is our main goal in this part. Besides, some dis-cussions on molecular spatial symmetry are also included.
4. Electron-hole Imbalance
If we can get the perturbation term in the Hamiltonian, we can try to solve this Hamiltonian. We expect this slight perturbation could be the reason of electron-hole imbalance.
2.
Crystal Defects
2.1
The Hamiltonian
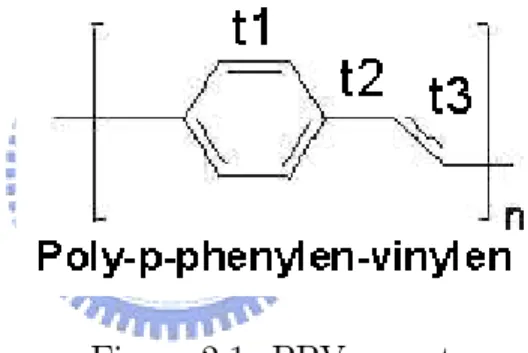
In this chaper, we will focus on physical defects in polymer system. The "physical defects" means the defects which is made from cyrstal disorder like malposition or non-prefect bonding ,etc. For simplicity, we choose PPV (100 unit cell with perodic boundary condiction) as our model system to illustrate the e¤ects when physical defects exist. The structure of PPV is :
Figure 2.1: PPV surcuture
In tight binding model, we can use three tight binding parameter t1,t2 and
t3 to constructure the Hamiltonian :
H = N X n=1 2 4 t1 0 @ C8(n 1)+1+ C8(n 1)+2+ C8(n 1)+1+ C8(n 1)+3 +C8(n 1)+2+ C8(n 1)+4+ C8(n 1)+3+ C8(n 1)+5 +C8(n 1)+4+ C8(n 1)+6+ C8(n 1)+5+ C8(n 1)+6 1 A 3 5 + N X n=1 h t2 C8(n 1)+6+ C8(n 1)+7 + t3 C8(n 1)+7+ C8(n 1)+8 i + 8N X "nCn+Cn+ h:c: (2.1)
where the last trem is on-site energy and we ofter set "n which is also the
Femi-energy be zero.
2.2
Band Structure
Form some experimental papers , we can get t1=-3.1eV,t2=-2.2eV and t3
=-3.0eV.[11] Thus, we can diagolal the above Hamiltonian and get the band stucture.
Furthermore, we can transform this equation to k-space by fourier transfor-mation. Thus, we have:
H =X k 8 < : t1[eika cos(3)(Ck1+C 2 k+ C 1+ k C 3 k+ C 4+ k C 6 k + C 5+ k C 6 k)] +t1[eika(Ck2+Ck4+ C 3+ k Ck5)] +t2[eika(Ck6+Ck7)] + t3[eika(Ck7+Ck8)] 9 = ;+ h:c: (2.2)
Where Ckn+is the creation operator of the state which has wave number k in n-th band.
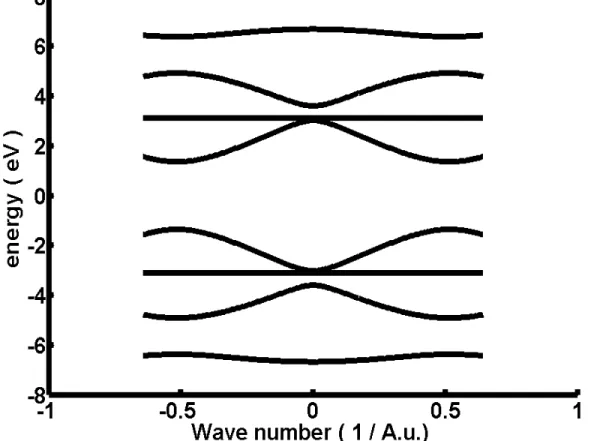
Thus, we can get the band structure in k-space by diagonalizing the above Hamiltonian:
(see …g 2.2, Band structure in k space)
When getting the band structure, we will discusse defects which are also the most interesting part in this chapter. As we know, there will be defect states if we change any tight binding parameter in any speci…ed unit cell. But, what kinds of defects is the most important one? The answer is certainly "defects which can create levels in the frobidden band" because electrons which are close to the Fermi level (Here EF=0 ) are the most active and dominating the system ones. From numerical tests, we found the only way to create levels in the frobidden band is to change t3 in any speci…ed cell and this conculsion also sati…es to experimental result.[12] In the following , we
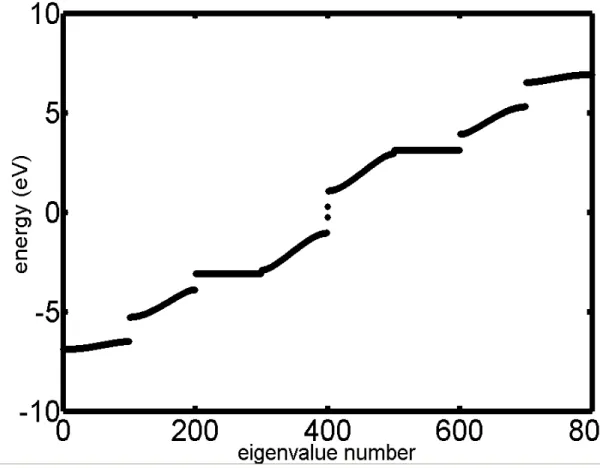
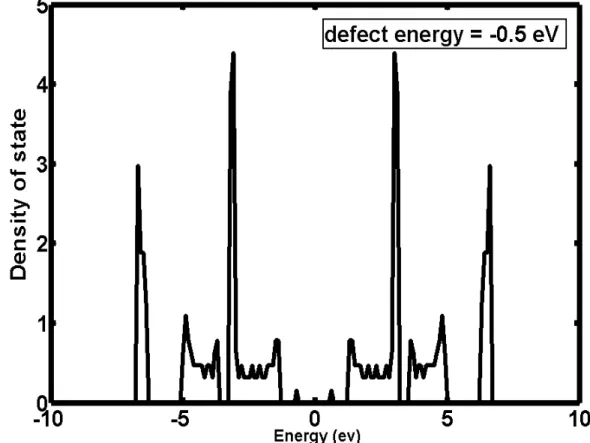
change t3 to -1.0eV for example, and plot the energy levels and density of states then. (take defect in 50th cell as our model)
(see …g 2.3, PPV energy level with defect) (see …g 2.4, PPV density of states with defect)
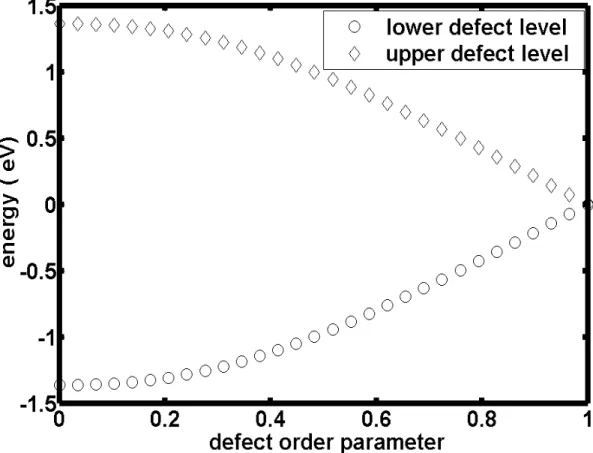
In the following pages, we choose the t3 bond in 50th cell as defect bond and this assumption will not a¤ect any results. Thus, we can observe how the defect levels change with the value of t3 by numerical caculation. In order to know how deep the defect is , we de…ne a parameter called the "defect order parameter" :
= (td t3 t3
) (2.3)
Where td is the new tight binding parameter of t3 in 50th cell.
Thus, when is zero there is no any defect. When is one there is a total broken chemical bond in t3-bond.
Also, we de…ne a name called the "defect sites". This means the polymer chain atoms which are nearest to the changed t3bond.
As the results shown in …g 2.5, we found the upper and lower defect states will be more and more close to the Fermi level and they will become degenerate states when td is equal to zero. Besides, we can also observe how the wave
function changes with td. From the following …gures, we found the defect
states will become more and more localized near the defect sites. When tdis
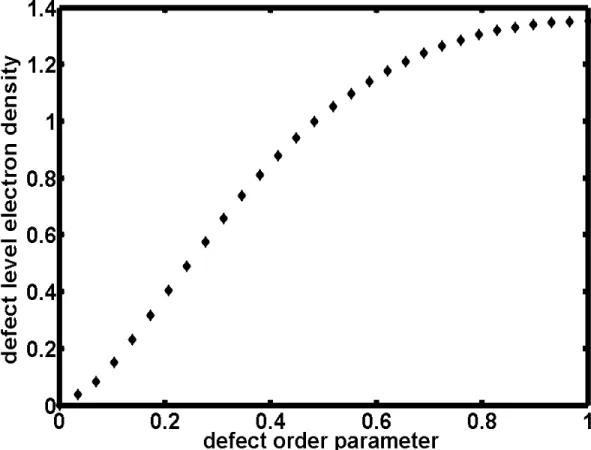
equal to zero, there will have maxium localization and 70% electrons in the lower defect will locate at the defect sites.
(Note: In …g.2.6, we plot how many electrons which belong to lower defect state are located in the defect sites)
( See …g 2.5, defect level and defect order parameter) ( See …g 2.6, electron density and defect order parameter )
2.3
Summary
In this chapter, we can conclude many things:
1. When defect is deeper, the defect levels will be closer to Fermi level. 2. When defect is deeper, the defect states will be more localized in defect
sites.
3. Whether there exist defects or not, there will always have electron-hole symmetry. This means that defects can’t be reasons for imbalance and this result also match experimental result.[13]
3.
Adsorption of oxygen molecules
3.1
Lennard-Jones Potenial
From the discussion in chapter zero, we know that oxygen molecules need to approach the defect sites when getting into the bulk or they will have no chance to interact. Therefore, giving an appropriate estimation to the adsorption energy and adsorption distance will help us to understand the process of oxygen adsorption. To solve this problem, we need to write down the potential between oxygen and carbon …rst.
In gereral, the potential between two molecules can be express as a Lennard-Jones potential:
V =
r12 r6 (3.1)
where the power 12 term is a repulsion force which comes from the core electrons of two molecules and the power 6 term comes from the dipole-dipole interaction( i.e. Van der waal’s force) which is an attractive force. But, the question is how to estimate these two coe¢ cients of r six and r twelve term?
3.2
Physical Meaning of Lennard -Jones Co¢ cients
Before any estimation, we should talk about the physical meaning of these two co¢ cients. First, we know the r twelve term mainly comes from core electron replusion between two molecules. So, we believe the co¢ cient should be only a function of what the two molucules are. In other words, it should be independent to the band structure or any other condensed matter e¤ects.
As we know the attractive potential of two molecules mainly comes from dipole-dipole interaction. Th dipole-dipole interaction is due to the electrons in one atom seeing the positive atomic nucleus in another atom, and there exist two dipole to interact each other therefore. In general, this interaction can be express as:[22]
Vdip dip =
e2
r3 f(~p1 ~p2) 3(~p1 z)(~^ p2 z)^ g (3.2)
where ~p1 and ~p2 are dipole moments of atom 1 and atom 2 and ^z is the unit
vector which is parallel to the connection axis. Thus, we can get the second order perturbation:
E(2)(r) = e 4 r6 X k6=0 j < k(0)jVdip dipj0(0) >j2 E0(0) Ek(0) (3.3)
Here, the energy Ek(0) is corrsponding di¤erent single particle states of the isolated Hamiltonian (i.e. Hpolymer+Hoxygenwithout any interaction between
them.) . In other words , the eigenstate are all costituted by j"polymer >
jEoxygen > ,where the j"polymer > and jEoxygen > are eigenstate of isolated
polymer chain and isolated oxygen molecule. So, we should calculate eq.3.3 with all the electron …lled in system. In order to simplify our calculation, we just extract the most important term in eq.3.3 and approximation will not a¤ect our results if defect states energy are very close. Thus, we have:
E(2)(r) ' e
4
r6
j < k(0)jVdip dipj0(0)>
(Elower def ect Eupper def ect) + (E E )
(3.4) ' e 4 r6 j < d+; jVdip dipj ; d > j2 ("d+ "d ) + (E E ) (3.5)
where, j ; d >= j > jd > and E E is rough 4:3 eV.[20] Therefore, the attractive term in Lennard-Jones potential:
r6 ' e4 r6 j < d+; jVdip dipj ; d > j2 ("d+ "d ) + (E E ) ) = e4 j < d+; jVdip dipj ; d > j 2 ("d+ "d ) + (E E ) (3.6) From the above equation, is certainly di¤erent accroding to di¤erent band structures . For simlicity, we make an assumation further .That is the numer-ator can be seen as a constant , because the matrix element in denominnumer-ator should be only depentent on what the two atoms are. From the previous discussion, all that we need to do is to search if there are any papers which have already obtained and the numerator of bwtween carbon and oxy-gen in the Lennard-Jones and put them in our case. Fortunately, in carbon nanatube research, there already had many papers which discussed the in-teraction between carbon and oxygen. In the next section, we will calculate it.
3.3
Estimate Lennard-Jones Potenial
Form some caculations, we know that if we get the minial energy and the its corrsponding distance we can get a and b.
a = Vmin rmin12 (3.7)
b = 2Vmin r6min
Here, we de…ne a new parameter = e4 j < d+jVdip dipjd > j2 which is
considered as a constant in our model.
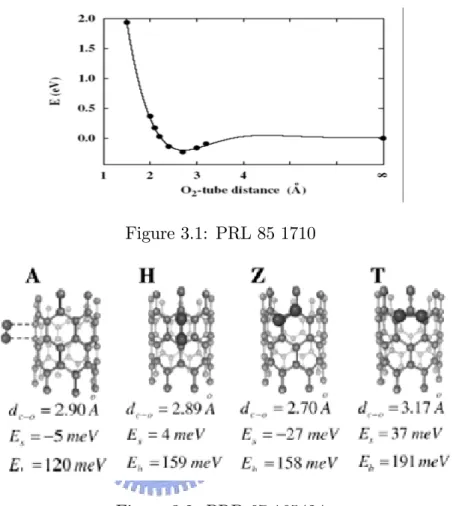
Let’s review two important papers about carbon nanatubes. In the …rst
pa-per, we can get =37500, =193.7 and pi band gap is 1.0eV (i.e. =193.7*(1.0+4.3) ).
In the second one, we can caculate is 30000~40000 and is around 300. All the adsorption distance are around 2.5~3 A and adsorption energy is about 150 meV. We believe that in our system the order of and is close to these results.
Although the ‡uctuation of is not small, but it does less a¤ect the results of distance and energy because of its corrsponding to a high order r term.
Figure 3.1: PRL 85 1710
Figure 3.2: PRB 67 165424
All results in these two papers tell us: is about 200~300 and is about 20000~40000 . This means our assumation (i.e. and are almost constant) is quite reasonable. For simplicity, we just take the and in the …rst paper as our system parameter to help us estimate the adsorption properities qualitatively. Thus, we have:
V = 37500 r12 193:7 (4:3 + 1:0) r6 [4:3 + (" d+ "d )] (3.8)
with di¤erent defect order parameter.
( See …g 3.4 , adsorption energy and defect order parameter ) ( See …g 3.4 , adsorption distance and defect order parameter )
3.4
Summary
To sum up this chapter, we can conclude:
1. Deeper defect induces higher adsorption energy and shorter adsorption distance.
2. Defects can enhance adsorption actually.
3. In other words, deeper defect induces strong interaction between oxygen and polymer chain.
4. Futhermore, we can assert the oxygen must be adsorptive near defect sites because the attractive trem is mainly contributed by the defect levels.
4.
Hopping integral
4.1
Hamiltonian
Accroding to the analysis in last chapter, we know that defects can induce oxygen adsorption near the defect sites.When oxygen molecule is close to polymer chain, the interaction between oxygen molecule and polymer chain will turn on. Thus, the total Hamiltonian can be wrote as:
H = Hpolymer+ Hoxygen+ Vint (4.1)
where ,Vint is the perturbation term.
Hpolymer = eq 2:1 (4.2) Hoxygen= ["(n 1 + n 2) + U 2(n 1 1)(n 2 1)] J 2 ! S 1 ! S 2 (4.3) n 1 = C + 1C 1and n 2 = C +
2C 2is charge number operator of oxygen z, x:
! S
1
! S
2 is spin copuling operator and that why the ground state of oxygen
is triplet. Here, we choose the axis which connects two oxygen atom as y axis. (Note: the highest occupied molecular orbitals [HOMO] are degenerate ). "o is the onsite energy of oxygen HOMO state and U is exchange energy
of this orbital. When charges transfer to oxygen and make it be negative, the total energy of oxygen moleule will rise because of its coulomb exchange e¤ects. So, U should be a positive number.
On the other hand, the interaction Vintcan be represented as a perturbation
term in the Hamiltonian like this:
(reason for this term is in …g 4.2 )
where Cd+1 and Cd+2 are creation operators of the two defect site atoms and Co+1 and Co+2 are creation operators of the two oxygen atoms.
The relation between Cd+1 , Cd+2 , C+
1 and C + 2 is: C+ 1 = 1 p 2(C + o1Z C + o2Z) (4.5) C+ 2 = 1 p 2(C + o2X C + o2X) (4.6) Here, Co+nX and C +
onZ are the ceration operators of 2Px and 2PZ moleculear
orbital of nth oxygen atom.
Our main problem is what the value of U , " and t is?
From some experimential results, the EA (electron a¢ nity) and IP (ioniza-tion potential) of oxygen molecule is -0.45eV and -12eV in gas phase.(here, the minus means we set vacuum is 0 ) As we know the HOMO of oxygen are degenerate states. Thus, we can de…ne EA and IP by:
(2" J
2) + IP = " and (3" + U ) + EA = 2" J 2 from experimental result: J =1eV. So, U = IP EA J = 10:6eV
In solid state system, EA and IP should be closer to the Fermi level but U is still not too small , because EA of oxygen in solid may be higher than gea phase value sign…catly due to solid state polarization e¤ects and structural relaxation.[2] Even though we don’t know the exact value of EA and IP in solid state, we can still handle our problem. In next chapter, we will show that only EA is important and it should range between -0.45 and -3.5 eV.
4.2
Estimate the Hopping Integral
Next, we will estimate t. In general, we can replacec t by a single particle matrix element. Here, we assume t is:
t'< 1s(a)j 1
4 rj1s(b) > (4.7) where j1s(a) > and j1s(b) > are the e¤ective 1S orbital of caroon and oxygen. So, if we can found the e¤ective Bohr radius of these two atoms , we can get the matrix element.
This matrix element with di¤erent Bohr radius is:
< 1s(a)j 1 4 rj1s(b) >= 4(a1 1a2) 3 2a2 1a22 h e a1r (ra2 1 ra22) + 2a1a22(e r a2 e r a1) i 4 r(a2 1 a22) (4.8)
where a1 and a2 are the Bohr radius of carbon and oxygen.
In CRC Handbook, we can …nd that the e¤ective Bohr radius of carbon and oxygen is 0.77 A and 0.65 A . The dielectric constant in polymer is 3. Thus, we can get the following …gure:
(see …g 4.1 , Hopping integral and distance)
When distance is around 1.2~1.3 A , the integral is around 2.5~3.5eV. This result is quiet reasonable.
4.3
Hopping Integral in Energy Basis
In order to specify our problem , we need to build a geometry relation of oxygen and defect sites. In many papers which discuss about adsorption of oxygen and carbon think the bond of oxygen is parallel to the polymer plane.[17] [18] Besides, there are also little paper think the bond of oxygen is normal to the bond of carbon.[19] In our problem, we believe the actual picture should be like the …rst status. (i.e. the bond of oxygen molecule is parallel to the polymer plane). Thus, we will assume the adsorption is like
(see …g 4.2 , Geometry of asdorption)
As a result of this geometry, the main contributor of Carbon-Oxygen overlap is certainly z orbital because this wave function is combined by 2Pz orbital
and its space distribution overlap with carbon highly. Therefore, in order to reduce our symbols, we will replace z by just :
Furthermore, we can also calculate single particle states hopping. As we know, when we diagonal the Hamiltonian of polymer chain, we can get all the single particle levels and there are only site jd1 >and jd2 >exist hopping
intergal with oxygen atoms. So, for every single particle state, we have:
j"n>= 8N X n=1 anjn >) t"n =< "njVintj >= 8N X n=1 an < njVintj > Because, j >= p1 2(jO1 > jO2 >) . Thus, t"n = 1 p 2 P8N n=1an(< njVintjO1 > < njVintjO2 >) = pt 2 P8N n=1an + < nj(Cd+ 1Co1 + C + d2Co2) + h:c:jO1 > < nj(Cd+ 1Co1 + C + d2Co2) + h:c:jO2 > = pt 2 P8N n=1an 8 > > < > > : + < njCd+ 1Co1jO1 > + < njC + o1Cd1jO1 > + < njCd+ 2Co2jO1 > + < njC + o2Cd2jO1 > < njCd+ 1Co1jO2 > < njC + o1Cd1jO2 > < njCd+ 2Co2jO2 > < njC + o2Cd2jO2 > 9 > > = > > ; = pt 2(ad1 ad2)
where jn > is polymer site basis, jOn>is oxygen site basis, j"n >is polymer
energy site basis and j > is oxygen molecule energy basis.
So, we can caculate all the single particle energy hopping integral with oxygen molecule.
4.4
About Symmetry
As a result in last section, we found the hopping integrals of single particle energy levels are maxium at upper defect level and almost zero at lower defect level. It is totally because the upper defect is highly localized and anti-bonding at the defect sites just like the oxygen is anti-bonding, too. If we assume the oxygen molecule is normal to the polymer plane, the maxium will occur in the lower defect level.
In our system, we wish to construct an electron trap to explain electron-hole imbalance. So, let the upper defect level have larger hopping integral is better and we will show this in the next chapter.
4.5
Summary
In this chapter, we have already gotten some important things:
1. We build a standard process to estimate the hopping integral by using a semi-empirical method and get reasonable results.
2. We calculate the hopping integral between di¤erent polymer single par-ticle energy levels and oxygen energy level.
3. Whether the Oxygen molecule is normal or parallel to the polymer plane , symmetry is always an important property which is deeply a¤ect the hopping integral. In our system, we prefer the parallel case and it will be explained in the next chapter.
5.
Electron-Hole Imbanalce
5.1
Binding Energy
From the previous chapters, we know that when oxygen molecule is close to the defect sites, there will be a perturbation acting on polymer system. In order to quantify the degree of imbalance. we de…ne an energy called the "binding energy".
When there is a hole injection into the polymer system, it will stay in the lower defect state. Similarily, when there is a electron injection into the polymer system, it will stay in the upper defect state. Our de…nation of binding energy is:
Binding energy = the energy that move a carrier from ground state to continious state:
In polymer device physics, the mobility is roughly proporation to e kT. In
room tempture, kT 25meV. So, if the electron binding energy can be larger than hole binding energy 0.1 eV at least , there will be large mobility di¤erence between them.
By this de…nition, we found that an electron injection or a hole injection will have the same binding whether there is a defect or not.
Can the perturbation induce any symmetry breaking of binding energy ?
5.2
Con…guration
1. O+2 is found rarely but O2 is often found in many physical system. This means oxygen molecule can be an electron trap.
2. Because defect states are highly localized in space. Thus, defect energy levels doesn’s like to be totally occupied by carriers because it might casuse strong coulomb replusive energy.[23]
Now, let’s consider two di¤ferent system.
1. electron case: there is an electron injection. 2. hole case: there is a hole injection.
Thus, we can …gure out the most important condigurations:
(see …g 5.1 , con…guration, the upper is electron case and the lower is hole case )
As a result , we can constructure the following matrix:
0 @ < gjHjg > < gjHjex > < gjHjct > < exjHjg > < exjHjex > < exjHjct > < ctjHjg > < ctjHjex > < ctjHjct > 1 A , electron case . < gjHjg > < gjHjex >
< exjHjg > < exjHjex > , hole case
5.3
Matrix Element
From the above analysis, we can …nd that the charge transfer state is only appare in electron injection case. The diagonal terms are certainly that sum all the occupied single particle state energy. For simplicity, we choose the ground state of netural system as zero point. Then , we have:
electron case:
< gjHjg >= "d+
< exjHjex >= "4N +2
< ctjHjct >= " + U
= (IPo IPc) + [(EAo IPc) (IPo IPc)]
= EAo IPc
hole case:
< gjHjg >= "d+
< exjHjex >= "4N 1
Here, we shift EA and IP of oxygen because we have taken the IP=-5eV of carbon in polymer (i.e. the Fermi level) be zero.
Next, we calculate o¤-diagonal terms. No matter what the bra and ket are, the main problem we need to solve is :
< jHj' >=< jHpolymerj' > + < jHoxygenj' > + < jVintj' >
In our calculation, we always take the energy basis of Hpolymer Hoxygen
, so the …rst two terms are just that sum all the electrons in polymer and oxygen single particle state. Thus, our main problem is how to solve the last term. As we know, the interaction term is written in site basis and the wave-function is written in energy basis. So, to calculate the last term is not easy. In physiscal system, the most active charge is the one which is in the highest occupied level and this charge is also the most possible one to transfer from polymer to oxygen. To simplify our calculation, we can just take the single particle level’s hopping integrals which belongs to the highest occupied level as those matrix elements if there is any charge transfers to oxygen from bra
state to the ket state. Otherwise, take the matrix element be zero. Thus, we have: o¤-diagonal term electron case: < gjHjex >= 0 < gjHjct >= td+ < exjHjct >' 0 hole case: < gjHjex >= 0
5.4
Imbalance
Now, we have already get all the informations of our problem. Similarily, we de…ne "imbalance energy" :
imbalance energy = the binding energy di¤erence of electron case and hole case.
In our system: = 1 2 ["d+ (EAo IPc)] + q "d+ (EAo IPc) 2 t2 d+
Like those analyses in the previous chapters, we know we will get a adsorption distance, hopping inegral and coupling energy between ground state and charge transfer state in the electron case when we get a defect order parameter . Thus, we can analysis the realation between defect order parameter and imbanalce energy.
Beside, we can also let td+ slightly change if our estimate on it is not
accurate enough.
(see …g 5.3 , imbalance energy and td+ in a …xed defect order
parameter) Finally, we will discuss some things about EA.
In gas phase , EA is -1eV. In most polymer system, the conduction band is around 1.5~2eV higher than Fermi-level (EF is -5eV to vacuum). It seem strange if EA is lower than conduction band . Because, when defect exist and the upper defect state might be higher than EA (i.e. acceptor level), the charge transfer state will be ground state in electron case. And this fact is unreasonable because the oxygen molecuse is just adsorptive not bonding. Therefore, we believe the EA of oxygen in polymer is -1 ~ -3.5 eV.
5.5
Summary
In this chapter, we get many important things:
1. Deeper defect will induce stronger e-h imbalance.
2. In most case, the imbalance energy can be from 0.1eV to 0.8eV easily. In otherwise, the electron-hole mobility di¤erence can be form 102to 1014. This means that althouhg oxygen molecule adsorption is an
slight perturbation to the system, it can induce an extreme symmetry breaking in polymer transport properties.
6.
Conclusion and Outlook
6.1
Conclusion
In the previous chapter, we have gotten many important physical concepts.When defect exist , it will create some energy levels in the frobidden band and those electrons which locate at defect state will deeply a¤ect the electronic proper-ties. In polymer system, defects are more interesting because it will enhance oxygen adsorption. As we know, no matter how prefect vacuum you can make, there always exist a few oxygen molecules in the air. In our cacula-tion, even thouhg there are only one oxygen molecule and one defect, there are still signi…cant symmetry breaking. So, we can assert that electron-hole transport imbalance is almost an unavoidable result and oxygen is the key. Altogether, our central concepts is:
"Defect will enchance oxygen adsorption and it will cause charge transfer to oxygen. Thus, an electron trap is formed. Transport symmetry is breaking."
6.2
Outlook
In this thesis, we have discussed many things about polymer physics. But, there are still many problems we need to solve. For example, in the imbalance problem, there are many other reasons for this phenomenon[21] and oxygen adsorption may not be the only thing that can cause this result. So, we could expect there will be more experiments to tell us more about oxygen and polymer behavior.
Polymer physics is much di¤erent from inorganic semiconductor physics and it is also an interseting area in theoretic physics. We believe there will be more and more exciting discoveries in the future.
Bibliography
[1] H.G. Kiess, etc al., Conjugated Conducting Polymers , 1st edition, Ch1 & 2 (1992)
[2] I.H. Campbell etc al., Physics of orangic electronic devices in solid state physics, 55 (2001)
[3] Richard P. Feynman, Statistical Mechanics , Ch8 (1997)
[4] D.O. Hayward, Quantum Mechanics for Chemists, 1st ed, Ch8 (2002) [5] W.P. Su, J.R. Schrie¤er, A.J. Heeger, Phys. Rev. Lett. 42 1698 (1979) [6] For a review, see, R. Friend, etc al. , Nature 397 127
[7] Lay-Lay Chua, etc al. , Nature 434 194 (2005)
[8] P. W. M. Blom and M. C. J. M. Vissenberg, Mater. Sci. Eng. 27,53 (2000).
[9] L. Bozano, S. Carter, J. Scott, G. Malliraras, and P. Brock, Appl. Phys. Lett. 74, 1132 (1999).
[10] R. J. O. M. Hoofman, M. P. de Haas, L. D.A. Siebbeles, and J. M. Warman, Nature (London) 392, 54 (1998).
[11] P. Gomes da Costa, R. Dandrea, and E. M. Conwell, Phys. Rev. B47, 1800 (1993).
[12] H. F. Meng and T. M. Hong, Physica B 304, 119 (2001).
[13] P. Stallinga, H. Gomes, H. Rost, A. Holmes, M. Harrison, and R.Friend, Synth. Met. 111, 535 (2000).
[14] M.S.A Abdou, F.P. Or…no, T. Son, and S. Holdcroft, J. Am. Chem. Soc.119,4518 (1997)
[15] Greg Mills, Mark S. Gordon, Horia Metiu, J. Chem. Phys.118,4198 (2003)
[16] Seung Mi Lee, Young Hee Lee, Yong Groo Hwang, J.R. Hahn, H. Kang, Phys. Rev. Lett. 82, 217 (1999)
[17] Seung-Hoon Jhi, Steven G. Louie, Marvin L. Cohen, Phys. Rev. Lett. 85, 1710 (2000)
[18] S. Dag, O. Gulseren, T. Yildirim, S. Ciraci, 67, Phys. Rev. B. 165424 (2003)
[19] D.A dos Santos, J.L. Bredas, Synthetic Mental , 101, 486 (1999) [20] John C. Slater, Quantum theory of atomic structure , (1960) [21] Hsin-Fei Meng, Yi-Shiou Chen, Phys. Rev. B ,70 ,115208 (2004) [22] J.J Sakurai, Morden Quantum Mechanics ,Revised Edition,312 (1994) [23] Ashcroft and Mermi, Solid State Physics, 581 (1972)