梔子中Geniposide 於大鼠之代謝動力學及其與西藥之交互作用; Metabolic Pharmacokinetics of Geniposide in Gardenia Fruits and Interactions with Western Medicines in Rats
147
0
0
全文
(2) 目 錄 目錄… … … … … … … … … … … … … … … … … … … … … … … … … … … … … .I 附圖目錄… … … … … … … … … … … … … … … … … … … … … … … … … ...… III 附表目錄… … … … … … … … … … … … … … … … … … … … … … … … … … ....V 中文摘要… … … … … … … … … … … … … … … … … … … … … … … … … … ...IX 英文摘要… … … … … … … … … … … … … … … … … … … … … … … … … ...… XI 第一章 緒言… … … … … … … … … … … … … … … … … … … … … … … … … ...1 第二章 總論… … … … … … … … … … … … … … … … … … … … … … … … … ...4 第一節 梔子之文獻考察… … … … … … … … … … … … … … … … … … … .4 第二節 Geniposide 與 genipin 之結構、理化性質與藥理活性… … … .9 第三節 Methotrexate 之特性… … … … … … … … … … … … … … … … ...14 第四節 Phenytoin 之特性… … … … … … … … … … … … … ..… … … … ..20 第五節 Carbamazepine 之特性… … … … … … … … … … … … … … … ....24 第六節 多重耐藥性蛋白 (multidrug-resistance associated proteins) 之特 性… … … … … … … … … … … … … … … … … … … … … … … … ...29 第七節 有機陰離子運輸蛋白 (organic anionic transport proteins) 之特 性… … … … … … … … … … … … … … … … … … … … … … … … ...34 第三章 材料與方法… … … … … … … … … … … … … … … … … … … … … … .36. I.
(3) 第一節 實驗材料… … … … … … … … … … … … … … … … … … … … … ...36 第二節 實驗方法… … … … … … … … … … … … … … … … … … … … … ...45 一、Genipin 之 pKa 測定… … … … .… … … … … … … … … … … … … 45 二、Genipin 之安定性… … … … … … … … … … ..… … … … … … … … ..46 三、體循環前代謝-大鼠便腸道菌對 geniposide 之作用… … … … ..52 四、Genipin 於大鼠體內之代謝動力學… … … … … … … … ..… … … ..54 五、梔子水煎劑中 geniposide 之定量分析… … … … … … … … … … ..59 六、梔子水煎劑於大鼠體內之代謝動力學… … … … … … … … … … ...62 七、梔子水煎劑於大鼠體內對 methotrexate 動力學之影響… … … ..63 八、梔子水煎劑於大鼠體內對 phenytoin 動力學之影響… … … … ...65 九、梔子水煎劑於大鼠體內對 carbamazepine 動力學之影響… … ...69 第四章 結果與討論… … … … … … … … … … … … … … … … … … … … … … .72 第五章 結論與建議… … … … … … … … … … … … … … … … … … … … … … .89 參考文獻… … … … … … … … … … … … … … … … … … … … … … … … … … ...90. II.
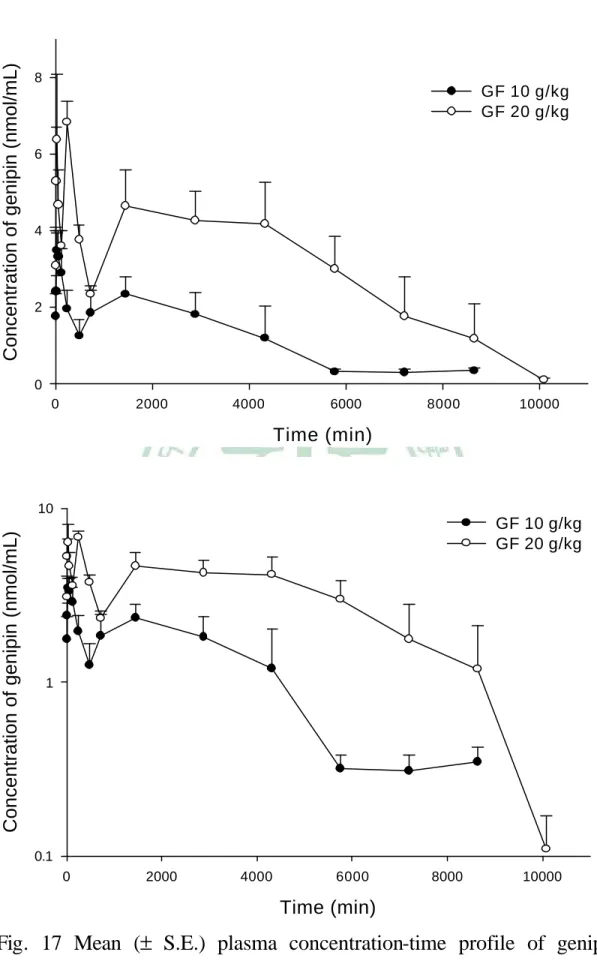
(4) 附 圖 Fig. 1 Fig. 2 Fig. 3 Fig. 4 Fig. 5 Fig. 6 Fig. 7 Fig. 8 Fig. 9 Fig. 10 Fig. 11 Fig. 12. Fig. 13. Fig. 14. Fig. 15 Fig. 16. Fig. 17. Fig. 18. Fig. 19 Fig. 20. 目 錄. The titration curve of genipin… … … … … … … … … … … … … … ....97 The first derivative (△ pH/△V) titration curve of genipin… … .........97 The second derivative (△ 2pH/△ V2) titration curve of genipin....… ..97 Stability of genipin in MeOH (n=3).… … … … … … … … … … … .....98 Stability of genipin in serum stored at -20℃ (n=3).… … … … … … .98 Effect of ascorbic acid on genipin stability in serum (n=3)..............99 Effect of thaw and sonication on genipin stability in serum (n=3)...99 Effect of incubation time with sulfatase at 37℃ on genipin emergence in serum (n=3).… … … … … ..… … … … … … … … ........100 Chromatograms of genipin in rat feces..… … … … … … … … … … .101 Mean (±S.D.) ratio-time profile of genipin after incubation of geniposide with rat feces (n=3).… … … … … … … … … … … … … ..102 Chromatograms of genipin in plasma… … … … … … … … … … … .103 Individual plasma concentration-time profiles of genipin ( ● ) and genipin sulfate (○) in nine rats after intravenous bolus of genipin (50 mg/kg).… … … … … … … … … … … … … … … … … … … … … … … 104 Mean (± S.E.) plasma concentration-time profiles of genipin (●) and genipin sulfate (○) after intravenous genipin (50 mg/kg) to nine rats (left) and the semi-log diagram (right)… ..… … … … … … … … ..… 105 Mean (± S.E.) plasma concentration-time profiles of genipin sulfate after oral administration of genipin (100, 200 mg/kg) to four rats (left) and the semi-log diagram (right).… … … … … … … … … … ...105 Chromatograms of geniposide (GP) in GF decoction… … ...… … ..106 Individual plasma concentration-time profiles of genipin sulfate in six rats after oral administrations of GF decoctions (10, 20 g/kg)..… … … … … … … … … … … … … … … … … … … … … .… … .107 Mean (± S.E.) plasma concentration-time profile of genipin sulfate after oral administrations of GF decoctions (10, 20 g/kg) to six rats and the semi-log diagram… … … … … … .… .… .… .........................108 Mean (± S.E.) serum concentration-time profile of MTX after oral MTX alone (●), and coadministration with 4 (○), 2 (▼) and 1 g/kg (▽) of GF decoction (upper) and the semi-log diagram (lower).… … … … … ..… … … … … … … … ...… … … … … … … .… ..109 Chromatograms of PHT in serum.… … … … … … … … … … … .… .110 Individual serum concentration-time profiles of PHT in six rats after III.
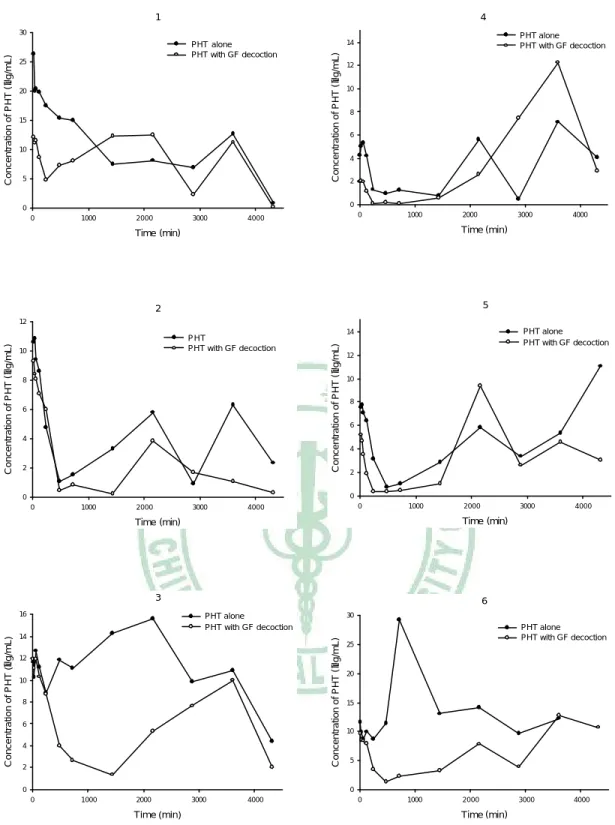
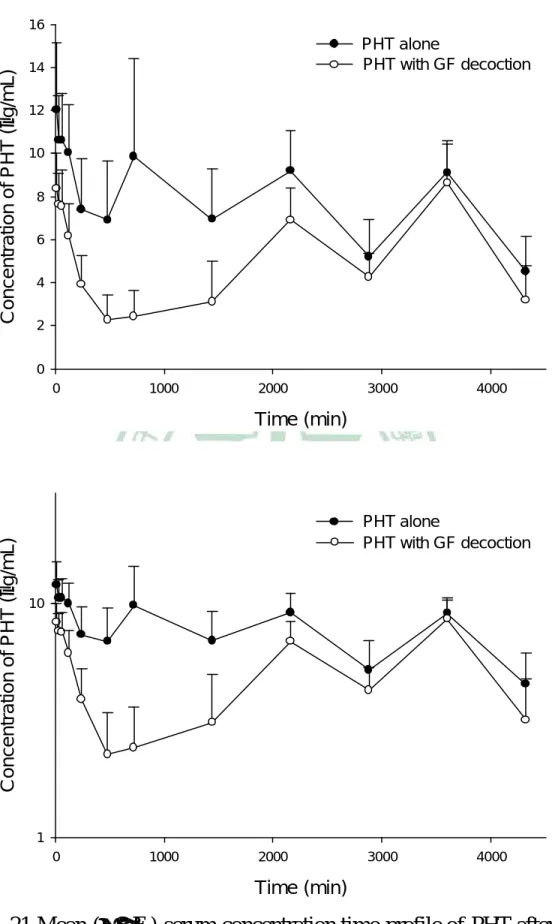
(5) Fig. 21. Fig. 22 Fig. 23. Fig. 24. oral PHT alone (●), and coadministration with 2 g/kg (○) of GF decoction.… … … … … ..… … … … ..… … … … … … … … .................111 Mean (± S.E.) serum concentration-time profile of PHT after oral PHT alone (●), and coadministration with 2 g/kg (○) of GF decoction (upper) and the semi-log diagram (lower).… … … … … … … … ..… 112 Chromatograms of CBZ in serum.… … … … … … … … … … ..........113 Individual serum concentration-time profiles of CBZ in six rats after oral CBZ alone (●), and coadministration with 4 g/kg (○) of GF decoction.… … … … … ..… … … … ..… … … … … … … … ................114 Mean (± S.E.) serum concentration-time profile of CBZ after oral CBZ alone (●), and coadministration with 4 g/kg (○) of GF decoction (upper) and the semi-log diagram (lower)… … … … .… … … … … ..115. IV.
(6) 附 表. 目 錄. Table 1. The pH change of genipin solution (in water) titrated with 0.1N NaOH.............................................................................................116. Table 2. Intra-day and inter-day analytical precision and accuracy of genipin in plasma........................................................................................117. Table 3. Recovery (%) of genipin from plasma (n=3).................................117. Table 4. Plasma concentrations (nmol/mL) of genipin in nine rats after intravenous bolus of genipin (50 mg/kg).......................................118. Table 5. Plasma concentrations (nmol/mL) of genipin sulfate in nine rats after intravenous bolus of genipin (50 mg/kg)...............................118. Table 6. Individual pharmacokinetic parameters of genipin in plasma after intravenous bolus of genipin (50 mg/kg) to nine rats....................118. Table 7. Individual pharmacokinetic parameters of genipin sulfate in plasma after intravenous bolus of genipin (50 mg/kg) to nine rats............119. Table 8. Pharmacokinetic parameters of genipin parent from and genipin sulfate in plasma after intravenous bolus of genipin (50 mg/kg) … … … … … … … … … … … … … … … … … … … … … … … … … ....119. Table 9. Plasma concentrations (nmol/mL) of genipin sulfate in eight rats after oral administration of genipin (100 mg/kg)...........................120. Table 10. Plasma concentrations (nmol/mL) of genipin sulfate in four rats after oral administration of genipin (200 mg/kg)...........................120. Table 11. Individual pharmacokinetic parameters of genipin sulfate in plasma after oral administration of genipin (100 mg/kg) to eight rats … ....................................................................................................121. Table 12. Individual pharmacokinetic parameters of genipin sulfate in plasma after oral administration of genipin (200 mg/kg) to four rats … ....................................................................................................121. Table 13. Comparison of pharmacokinetic parameters of genipin sulfate in plasma after oral administrations of 100 and 200 mg/kg genipin … … … … … … … … … … … … … … … … … … … … … … … … … ....121. V.
(7) Table 14. Intra-day and inter-day analytical precision and accuracy of geniposide (n=3)............................................................................122. Table 15. Recovery (%) of geniposide from GF decoction (n=3).................122. Table 16. Plasma concentrations (nmol/mL) of genipin sulfate in six rats after oral administration of GF decoction (10 g/kg)..............................123. Table 17. Plasma concentrations (nmol/mL) of genipin sulfate in six rats after oral administration of GF decoction (20 g/kg)..............................123. Table 18. Individual pharmacokinetic parameters of genipin sulfate in plasma after oral administration of GF decoction (10 g/kg) to six rats … ....................................................................................................124. Table 19. Individual pharmacokinetic parameters of genipin sulfate in plasma after oral administration of GF decoction (20 g/kg) to six rats … ....................................................................................................124. Table 20. Comparison of pharmacokinetic parameters of genipin sulfate in plasma after oral administrations of 10 and 20g/kg GF decoction ........................................................................................................124. Table 21. Serum concentrations (µmol/mL) of MTX in six rats after oral administration of MTX (5 mg/kg) alone........................................125. Table 22. Serum concentrations (µmol/mL) of MTX in six rats after oral administration of MTX (5 mg/kg) with GF decoction (1 g/kg).....125. Table 23. Serum concentrations (µmol/mL) of MTX in six rats after oral administration of MTX (5 mg/kg) with GF decoction (2 g/kg).....126. Table 24. Serum concentrations (µmol/mL) of MTX in six rats after oral administration of MTX (5 mg/kg) with GF decoction (4 g/kg).....126. Table 25. Individual pharmacokinetic parameters of MTX in serum after oral administration of MTX (5 mg/kg) alone........................................127. Table 26. Individual pharmacokinetic parameters of MTX in serum after oral administration of MTX (5 mg/kg) with GF decoction (1 g/kg) … … .… … … … … … … … … … … … … … … … … … … … … … .......127. Table 27. Individual pharmacokinetic parameters of MTX in serum after oral administration of MTX (5 mg/kg) with GF decoction (2 g/kg) … … ................................................................................................127 VI.
(8) Table 28. Individual pharmacokinetic parameters of MTX in serum after oral administration of MTX (5 mg/kg) with GF decoction (4 g/kg) … … ................................................................................................128. Table 29. Comparison of pharmacokinetic parameters of MTX in serum after oral administration of MTX (5 mg/kg) alone and coadministration with GF decoctions (1, 2, 4 g/kg)..................................................128. Table 30. Intra-day and inter-day analytical precision and accuracy of PHT in serum (n=3)....................................................................................129. Table 31. Recovery (%) of PHT from serum (n=3).......................................129. Table 32. Serum concentrations (µg/mL) of PHT in five rats after oral administration of PHT (400 mg/kg) alone.....................................130. Table 33. Serum concentrations (µg/mL) of PHT in five rats after oral administration of PHT (400 mg/kg) with GF decoction (2 g/kg) ........................................................................................................130. Table 34. Individual pharmacokinetic parameters of PHT in serum after oral administration of PHT (400 mg/kg) alone.....................................131. Table 35. Individual pharmacokinetic parameters of PHT in serum after oral administration of PHT (400 mg/kg) with GF decoction (2 g/kg) … … ................................................................................................131. Table 36. Comparison of pharmacokinetic parameters of PHT in serum after oral administration of PHT (400 mg/kg) alone and coadministration with GF decoction (2 g/kg) in six rats… .......................................131. Table 37. Intra-day and inter-day analytical precision and accuracy of CBZ in serum (n=3)....................................................................................132. Table 38. Recovery (%) of CBZ from serum (n=3).......................................132. Table 39. Serum concentrations (µg/mL) of CBZ in five rats after oral administration of CBZ (400 mg/kg) alone.....................................133. Table 40. Serum concentrations (µg/mL) of CBZ in five rats after oral administration of CBZ (400 mg/kg) with GF decoction (4 g/kg) … … .… … … … … … … … … … … … … … … … … … … … … … … ...133. Table 41. Individual pharmacokinetic parameters of CBZ in serum after oral administration of CBZ (400 mg/kg) alone.....................................134 VII.
(9) Table 42. Individual pharmacokinetic parameters of CBZ in serum after oral administration of CBZ (400 mg/kg) with GF decoction (4 g/kg) … … ................................................................................................134. Table 43. Comparison of pharmacokinetic parameters of CBZ in serum after oral administration of CBZ (400 mg/kg) alone and coadministration with GF decoction (4 g/kg) in six rats… … .… ..............................134. VIII.
(10) 梔子中 Geniposide 於大鼠之代謝動力學 及其與西藥之交互作用. 研究生 中國醫藥大學. 賴姵妘 藥物化學研究所. 摘. 要. Geniposide 屬於 iridoid 類之配醣體,存在於茜草科 (Rubiaceae) 梔子 Gardenia jasminoides ELLIS. 之 乾燥成熟果實中,於體內可被腸道菌轉變為 genipin,再和氨基酸反應生成梔子藍色素。本研究探討靜脈注射、口服 genipin 與口服梔子水煎劑於大鼠體內之代謝動力學及其對 MRP2 受質之 西藥 methotrexate (MTX)、phenytoin (PHT) 及 carbamazepine (CBZ) 動力 學之影響。 梔子水煎劑利用高效液相層析法進行分析。大鼠靜脈注射、口服 genipin 與口服梔子水煎劑後,於各時間點以心臟穿刺採血,血漿中 genipin 及其 sulfate 與 glucuronide 之定量,係以 sulfatase、glucuronidase 分別水解, 水解前、後利用高效液相層析法定量,以 WinNonlin 非室體模式計算動力 學參數。結果顯示,靜脈注射 genipin 後,立即代謝成 sulfate 結合態代謝. IX.
(11) 物,其原形分子只短暫存在於血清中;口服給予 genipin 後,主要以 genipin sulfate 存 在 循 環 中, 且口服 200 mg/kg genipin 的大鼠死亡率竟高達 77.8%。口服梔子水煎劑主要以 genipin sulfate 循環於體內,其存在體內的 時 間 達 六 天 之 久 ,且 有 明 顯 的 腸 肝 循 環 現 象 ,而 genipin 與 genipin glucuronide 則無法偵測到。 MTX 為抗癌藥物及免疫抑制劑,治療指數狹窄,為 MRPs 1, 2, 3, 4 之 受質。本研究探討梔子水煎劑於大鼠體內對 MTX 動力學之影響。利用螢光 偏極免疫分析法定量 MTX 之血中濃度,以 WinNonlin 非室體模式計算動 力學參數。結果顯示,併服 2 g/kg 梔子水煎劑,對 MTX 之血藥面積及滯 留時間顯著增加了 477.1% 及 397.7%,甚至導致部分大鼠死亡 (4/6)。因 此,建議為確保療效及用藥安全,使用 MTX 之病人盡量避免併服梔子水煎 劑及其相關方劑。 PHT 及 CBZ 皆為臨床常用之抗癲癇藥物,其治療指數狹窄,為 MRP 2 之受質,本研究探討梔子水煎劑於大鼠體內對 PHT 及 CBZ 動力學之影 響。利用高效液相層析法定量血清中 PHT 及 CBZ。結果顯示,併服梔子 水煎劑,造成 PHT 給藥後 24 小時內之早期暴露及對 CBZ 之全期暴露。 為了保障藥效,服用 PHT 及 CBZ 之病患,應避免與梔子水煎劑併服。. X.
(12) Metabolic Pharmacokinetics of Geniposide in Gardenia Fruits and Interactions with Western Medicines in Rats. Pei-Yun Lai Institute of Pharmaceutical Chemistry, China Medical University, Taichung, Taiwan. ABSTRACT. Geniposide is an iridoid glycoside in the fruits of Gardenia jasminoides ELLIS. It has been reported that geniposide is transformed to genipin by intestinal bacteria in animals. We attempted to investigate the pharmacokinetics of geniposide in rat after oral administration of traditional decoction of Gardenia fruits (GF). The concentration of geniposide in the GF decoction was determined by HPLC method. In addition, the pharmacokinetics of genipin was investigated after intravenous and oral administrations. Based on the pharmacokinetic study, we proposed possible pharmacokintic interactions between GF decoction with methotrexate (MTX), phenytoin (PHT) and carbamazepine (CBZ), which are substrates of MRP2. Blood samples were withdrawn via cardiopuncture at specific time points after drug administration. Plasma samples were assayed by HPLC method prior to and after enzymatic hydrolysis with sulfatase and glucuronidase, respectively. Pharmacokinetic parameters were calculated using noncompartment model of WinNonlin. Our results indicated that after oral administration of GF decoction, XI.
(13) no free form of genipin was detected, and genipin sulfate was found predominately circulating in the bloodstream until the 6th day after dosing. Likewise, after oral and intravenous dosing of genipin, genipin sulfate was found predominant in the bloodstream, whereas glucuronides were negligible. Therefore, the biological activities of the genipin sulfate awaits further studies. To our surprise, oral administration of 200 mg/kg genipin resulted in a high mortality of 77.8% in rats. MTX, an immunosuppressant with narrow therapeutic window, is a substrate of MRP 1, 2, 3, 4. In this study, rats were orally given MTX alone and coadministered with 4, 2 and 1 g/kg of GF decoctions, respectively. The serum MTX levels were assayed by FPIA method. The coadministration of 2 g/kg of GF decoction markedly increased the AUC and MRT of MTX by 477.1% and 397.7%, respectively, and resulted in death of rats (4/6). Therefore, concurrent use of GF may enhance the efficacy and toxicity of MTX. PHT and CBZ are anti-epileptic drugs with narrow therapeutic window. This study investigated the effects of GF on the pharmacokinetics of PHT and CBZ. The blood concentrations of PHT and CBZ were assayed by HPLC methods. The AUC0-1440 of PHT and AUC0-t of CBZ were significantly decreased after coadministration of GF, respectively. For ensuring the efficacy of PHT and CBZ, the coadministration with GF decoction should be avoided.. XII.
(14) 第 一 章. 緒. 言. 梔子始載於《神農本草經》,列為中品。《中國藥典》記載梔子來源 於茜草科 (Rubiaceae) 植物梔子 Gardenia jasminoides ELLIS. 的乾燥成熟果 實。具有瀉火除煩,清熱利尿,涼血解毒的功效。亦具利膽保肝、降壓、 鎮靜、鎮痙、降溫、鎮痛、促進胰腺分泌作用、抑菌作用、抗炎及治療軟 組織損傷的作用等藥理活性 (徐等,1996; Chang & But, 1987; Tseng et al., 1995)。 Geniposide 屬於 iridoid 類之配醣體,為梔子所含之主要成分之ㄧ,水溶性 頗佳,故易溶於傳統水煎劑中,具利膽保肝 (徐珞珊等,1996)、抑制胃液分 泌及腸胃道蠕動、鎮靜、抑制氣喘、抗凝血 (Suzuki et al., 2001)、抗發炎等藥 理活性;Geniposide 於體內可被動物之腸道菌轉變為 genipin (Akao et al., 1994; Ueno et al,. 2001),再和氨基酸反應生成梔子藍色素 (Fujikawa et al., 1987; Hendry & Houghton, 1996; Park et al., 2002; Lee et al., 2003)。Genipin 為 geniposide. 之非醣體,具利膽保肝 (徐珞珊等,1996)、抑制膽固醇結石的形成 (朱振家等, 2001)、抑制胃腸運動、鎮靜、促進胰腺分泌作用、抗發炎作用 (朱江等,2005; Koo et al., 2004; 2006) 等藥理活性,亦可作為生物材料交聯劑及其與胺基酸結. 合生成之梔子藍色素 (Fujikawa et al., 1987; Hendry & Houghton, 1996; Park et al., 2002; Lee et al., 2003) 可當食用色素。文獻中已有研究報導梔子水煎劑口服給. 予小鼠,其 geniposide、genipin 於體內之代謝動力學 (Ueno et al., 2001),與. 1.
(15) 靜脈注射給予 geniposide 後,探討其於大鼠體內代謝動力學之研究 (Tseng & Tsai, 2004);但 genipin 之結構具 hemiacetal 官能基,易開環,化性較不. 安定,因此本研究以 HPLC 方法先行探討 genipin 於甲醇及血清中之安定 性;再以大鼠為模式,探討靜脈注射、口服 genipin 及口服梔子水煎劑之代 謝動力學。 在台灣的醫療環境中,除現代醫學外,傳統中醫藥仍被國人所接受而 大量使用,但民眾普遍認為中藥藥性溫和、安全,西藥藥效迅速,兩者並 用,雙重保險,因此常在非醫囑的情況下自行併用中藥。因此,面對此複 雜的用藥模式下,其中實是潛藏著許多未知的風險有待探究。 Methotrexate (MTX) 為葉酸衍生物,於低劑量使用時為一免疫抑制 劑,高劑量則用於治療癌症,但其治療指數狹窄,是癌症治療中唯一需監測 血 中 濃 度 的 藥 物 。 近 年 研 究 顯 示 , MTX 為 多 重 耐 藥 性 蛋 白 (multidrug-resistance associated proteins; MRPs) 及 有 機 陰 離 子 運 送 蛋 白 (organic anionic transporters; OATs) 之受質 (Ng et al., 2003; Mizuno et al., 2003; Lauretta MS et al., 2004; Elaine et al., 2005),又有報導指出,配醣體於吸收後會形. 成 sulfates 及 glucuronides 等結合態代謝物,研究報告指出此等代謝物為 MRPs 與 OATs 之受質 (Ballatori et al., 2005; Sweet et al., 2005)。MTX 與梔子配 醣體代謝後之結合態代謝物之間是否會相互競爭 MRPs、 OATs 而影響 MTX 於體內之運送與排除過程,值得探討。因此本研究以大鼠模式探討併. 2.
(16) 服梔子水煎劑對 MTX 動力學之影響。 癲癇是由大腦神經細胞異常放電引起的突然性、反覆性和短暫性的大 腦功能失調,可以表現為運動、感覺、意識、精神等多方面的功能障礙 (NCCHC, 2002)。台灣在流行病學上的探討發現約有 0.3%~1% 的人口患有癲. 癇,無論在發達國家還是在發展中國家,癲癇都是一項重要的公共衛生問 題,國際衛生組織估計全世界大約有五千萬癲癇患者。抗癲癇藥物必須長 期、不間斷、定時、定量服用,癲癇患者常因控制狀況不佳而需併用許多 不同的抗癲癇藥物治療,然而,除了標準的治療藥物外,癲癇病患併用中 草藥之安全性亦陸續被報導 (蔡,2000; 陳,2000),顯示抗癲癇藥與中藥之交 互作用已成為ㄧ令人關注的用藥安全問題。梔子被報導可用於鎮靜、鎮痙, 另外,Phenytoin (PHT) 及 Carbamazepine (CBZ) 為臨床常用的抗癲癇藥 物,其治療範圍狹窄,主要經由體內肝酵素系統代謝,亦會影響肝酵素系 統之作用 (Brodie, 1992; Neels et al, 2004),且連續七年皆為行政院衛生署藥物不 良反應通報系統中「最常通報之可疑藥品前 10 名」之ㄧ,因此已有不少抗 癲癇藥物與其他藥物併用產生交互作用之臨床報告 (Mattson et al., 1986; Brodie et al., 1996; Brodie & Dichter, 1996;Haag, 2004),但抗癲癇藥物與中草藥併用可能. 之交互作用研究目前鮮見。因此,本研究將以大鼠為模式,分別探討併服 梔子水煎劑對 PHT 及 CBZ 動力學的影響,探討其併用之交互作用及可能 潛在的問題,希望能提供藥物臨床應用及發展用藥安全之參考。. 3.
(17) 第 二 章 第一節. 梔子之文獻考察. 總. 論. (徐等,1996). [別名] 黃梔子,山枝子,大紅梔 (江蘇),白蟬 (廣東) [基源] 茜草科 (Rubiaceae) 梔子 Gardenia jasminoides ELLIS. 之乾燥成熟果 實。 [形態特徵] 梔子為常綠灌木,高 0.5~2 公尺,枝圓柱形,灰色。葉對生或 3 葉輪生,革質,長橢圓形或長圓狀披針形,長 5~14 cm,寬 2~7 cm, 全緣,側脈纖細,明顯;葉柄短,托葉鞘狀膜質。花單生於枝頂,芳香, 花梗短 (圖一);萼管倒圓錐狀,有稜,裂片線形;花冠未開放時旋卷, 開放後呈高腳杯狀,五至多裂,初為白色,後變為乳黃色;雄蕊與花冠 裂片同數,著生於花冠喉部;子房內含。果黃色,卵形或長橢圓形,有 翅狀縱稜 5~8 條 (圖二)。種子多數。花期 5~7 月,果期 8~11 月。. 圖一. 圖二 4. 圖三.
(18) [生態環境] 喜溫暖濕潤的氣候,以排水良好、肥沃疏鬆而較濕潤的砂質壤土或 粘質壤土為佳。 [產地] 主產於湖南、湖北、江西、浙江、福建、四川、河南、江蘇、安徽、 廣東、廣西、雲南等地。以湖南產量大,浙江質量佳。 [採集加工] 秋季果實成熟時採收,除去果梗及雜質,略蒸或置沸水中略燙,或 蒸至上氣,取出,曬乾。 [藥材性狀] 果實長橢圓形或卵圓形,長 2~4.5 cm,直徑 0.8~2 cm,表面棕紅 色或黃棕色,微有光澤,有翅狀縱稜 5~8 條,兩翅稜間有縱脈 1 條, 頂端有宿萼,具 5~8 個長形裂片,多碎斷,基部有果梗痕;果皮薄, 內表面鮮黃色,有光澤,具 2~3 條隆起的假隔膜,折斷面鮮黃色。種 子多數,扁長圓形,具成球狀團塊,表面紅棕,有細點狀突起 (圖三)。 [化學成分] 主 要 成 分 為 gardenoside 、 geniposide 、 genipingentiobioside 、 shanzhiside、gardoside、scandoside methyl ester、crocin、gardenin 及 ursolic acid 等。. 5.
(19) COOCH3. COOH. H. HO O. H OH. HOH2C. O HO. O-glc. O. HO. H. gardenoside. H. COOCH3. gardoside. geniposide. R=glc. R1=H. genipingentiobioside. R=gen. R1=H. scandoside methyl ester. R=glc. R1=β-OH. O H CH2OH OR. O-glc. O. O-glc. shanzhiside. H. R1. COOH H. H. CH3. H3C CH3. CH3. H3 C. O. H. O. O. CH3. O. OH CH3. O. H. O. O CH3. CH3. O. CH3 HO. OH. CH3. O. H3C. gardenin. H CH3. ursolic acid OH OH HO O OH O OH. OH. CH3. HO. O. CH3. O. O. O O. O. CH3. HO O HO O OH HO. crocin. OH. 6. CH3. OH OH.
(20) [藥理作用] 1. 利膽保肝:梔子煎劑灌服,對結紮總膽管兔有抑制血中膽紅素升高的作 用。Crocin 及 crocetin sodium 能增加小鼠、兔的膽汁分泌量,也能抑 制結紮總膽管之兔血中膽紅素的出現。 2. 降壓:煎劑灌胃或乙醇提取物靜脈注射,對貓、兔、大鼠均有降壓作用, 此作用可被阿托平阻抑;對麻醉貓、兔及不麻醉兔均可使心率減慢。梔 子的降血壓作用部位在中樞,主要是加強了延腦副交感中樞緊張度所致。 3. 鎮靜、鎮痙、降溫、鎮痛:Ursolic acid 腹腔注射,可降低小鼠的自發活 動及對抗 pentylenetetrazol 的驚厥作用,對閥下劑量的戊巴比妥鈉有協 同睡眠作用,對大鼠有明顯降溫作用。 4. 促進胰腺分泌作用:用膽胰插管研究顯示梔子有明顯的利胰、利膽及降 胰酶效應,治療胰腺炎;使膜脂的過氧化產物減少作用。 5. 抑菌作用:梔子煎劑對白喉桿菌、金黃色葡萄球菌、傷寒桿菌有抑制作 用,對多種皮膚真菌也有不同程度的抑制作用。 6. 抗炎及治療軟組織損傷的作用:梔子以乙醇迴流提取液,對小鼠、家兔 軟組織損傷具抗炎作用;對小鼠急性耳腫脹有明顯抑制作用;對大鼠亞 急性足蹠腫脹有抑制作用。 7. 其他:梔子乙醇提取物灌服小鼠或大鼠均引起肝毒性,肝臟呈綠色, CYP450 含量和 p-nitroanisole-O-demethylase 活性均降低。. 7.
(21) [毒理學] 梔子醇滲漉濃縮液 (300%) 一次給藥,腹腔注射和灌服小鼠的 LD50 分別為 17.1 g/kg 和 107.4 g/kg。梔子水提物對小鼠的急性毒性很低,腹 腔注射給藥的 LD50 為 5 g/kg。梔子乙醇提取物小鼠灌服的 LD50 為 107.4 g/kg,腹腔注射為 27.5 g/kg,連續 4 天,使小鼠 hexobarbitone 睡 眠時間延長,肝臟呈灰綠色,使大鼠. CYP450 含 量 和. p-nitroanisole-O-demethylase 活性降低,肝呈灰綠色。梔子骨髓細胞微核 子試驗、Ames 試驗和睪丸染色體畸變試驗表明,梔子無致突變,致畸 效應。 [性味功能] 苦,寒。入心、肺、三焦經。瀉火除煩,涼血解毒,清熱利濕。 [主治] 熱病心煩不寧;溼熱黃疸;血熱妄行之吐血、衄血、尿血;熱毒瘡 盪,扭挫傷,瘀血腫痛 (Chang & But, 1987; Tseng et al., 1995)。 [臨床應用] 治療急性黃疸型肝炎、扭挫傷、局部止血劑。 [用法用量] 6~9 g,水煎服;外用適量。. 8.
(22) 第二節 Geniposide 與 genipin 之結構、理化性質與藥理活性 1. Geniposide (Budavari, 2001) 【化學名】 Methyl 9-(hydroxymethyl)-2-[3,4,5-trihydroxy-6-(hydroxymethyl) oxan-2-yl]oxy-3-oxabicyclo[4.3.0]nona-4,8-diene-5-carboxylate 【結構式】 OH HO. OH OH. HO O. O H. H. O. H MeO. O. 【分子式】C17H24O10 【分子量】388.37 【物化性質】 白色結晶粉末,可溶於甲醇。UV max (MeOH):238 nm。 【藥理活性】 (1) 利膽保肝:促使大鼠膽汁分泌增加;灌服小鼠致腹瀉,ED50 為 0.3 g/kg;促進膽汁分泌作用,而對血液中血細胞數、血色素、血糖、GOT、 GPT 等均無顯著影響。Geniposide 灌服,對異硫氰酸 α-萘酯引起的急 性黃疸,可使 GPT、血清膽紅素明顯降低 (徐等,1996)。 9.
(23) (2) 抑制胃液分泌及胃腸運動:十二指腸給予 geniposide,能使幽門 結紮大鼠胃液分泌減少,總酸度下降,pH 值升高。小鼠和豚鼠離體迴 腸試驗顯示 geniposide 有較弱的抗乙醯膽鹼和抗組織胺作用。靜脈注 射,抑制大鼠胃腸運動,並能抑制 pilocarpine 引起的運動亢進。 (3) 促進胰腺分泌作用:geniposide 有最顯著的降低胰澱粉酶作用, 而其酶解產物 genipin 的增加胰膽流量作用最強,持續時間較短。 (4) 鎮靜、鎮驚、降溫、鎮痛:皮下注射,可抑制醋酸引起的扭體 反應。 (5) 抑制氣喘:於大鼠實驗顯示抑制 5-lipoxygenase 可治療氣喘 (Nishizawa et al., 1988)。. (6) 抑制 CYP3A 和增加老鼠肝臟 glutathione 的量 (Kang et al., 1997)。. (7) 抑制血小板凝集 (Suzuki et al., 2001)。 (8) 保護癌症病患於高劑量放射治療時的傷害 (Hsu et al., 1997)。. 10.
(24) 2. Genipin (Budavari, 2001) 【化學名】 Methyl 2-hydroxy-9-(hydroxymethyl)-3-oxabicyclo[4.3.0]nona-4,8diene-5-carboxylate 【結構式】 OH OH H O. H MeO. O. 【分子式】C11H14O5 【分子量】226.23 【物化性質】 白色結晶粉末,可溶於酒精,甲醇,丙酮,微溶於水。UV max (MeOH):240 nm。 【藥理活性】 (1) 利膽保肝:genipin 靜脈注射或十二指腸給藥,均可使大鼠膽汁 分泌增加。有促進膽汁分泌作用,而對血液中血細胞數、血色素、血糖、 GOT、GPT 等均無顯著影響 (徐等,1996)。對地鼠膽固醇結石的形成具 有抑制作用 (朱等,2001),促使大鼠膽汁分泌增加 (Masatoshi H, 1974; Wang, 1991; 彭等,2003)。. (2) 抑制胃腸運動:genipin 靜脈注射可抑制大鼠胃腸運動,並能抑 制 pilocarpine 引起的運動亢進。 11.
(25) (3) 鎮靜、鎮驚、降溫、鎮痛:genipin 皮下注射,可抑制醋酸引起 的扭體反應。 (4) 生物材料交聯劑:可交聯修飾動物組織、膠原蛋白、明膠與幾 丁聚醣等形成穩定的交聯製品 (Sung et al., 1998; 黃&願,2003; Huang & Gu, 2003);細胞毒性小,對小鼠 LD50 (i.v.) 為 382 mg/kg,生物相容性好,不. 發生鈣化現象和降解,可成為植入式藥物緩釋系統的良好載體。 (5) 食用色素 (Fujikawa et al., 1987; Hendry & Houghton, 1996):和氨基酸 反應生成梔子藍色素 (Fujikawa et al., 1987; Hendry & Houghton, 1996; Park et al., 2002; Lee et al., 2003),但這種藍色素在酸性環境中不能穩定存在,因而在. 應用上存在一定的侷限性;可用作測定氨基酸含量的顯色劑 (Lee et al., 2003)。. (6) 其 他 : 對 Al z he i me r ’ sa my l oi d β pr ot e i n毒性有保護作用 (Yamazaki et al., 2001) ;具抗發炎作用 (朱等,2005;Koo et al., 2004; 2006)。. Genipin 口服、腹腔注射、靜脈注射對小鼠的 LD50 分別為 237、190 與 153 mg/kg。. 12.
(26) 3. Geniposide 與 genipin 之吸收、代謝 Geniposide 屬於 iridoid 類配醣體,geniposide 在體內可被動物體內之 腸道菌轉變為 genipin (Akao et al., 1994; Ueno et al,. 2001),再和氨基酸反應生成 梔子藍色素 (Fujikawa et al., 1987; Hendry & Houghton, 1996; Park et al., 2002; Lee et al., 2003),如圖四所示。. Amino acids COOCH3. COOCH3. O. O. CH2 O Glucose OH. Blue pigment. CH2 OH OH. Geniposide. Genipin Hydrolysis (glucosidase). 圖四. Mechanism of blue pigment formation from gardenia fruit (Fujikawa et al, 1987).. 13.
(27) 第三節 Methotrexate (MTX) 之特性. (Budavari, 2001). 【化學名】N-[4-[[(2,4-Diamino-6-pteridinyl) methyl] methylamino] benzoyl]-L-glutamic acid 【結構式】 H2N. N. N CH3. N. N N. O H N. NH2. OH O. O. OH. 【分子式】C20H22N8O5 【分子量】454.45 【物化性質】 水合物,從稀鹽酸析出黃色晶體,185-204℃ 分解;幾不溶於水、乙 醇、氯仿、乙醚,易溶於鹼金屬的氫氧化物及其碳酸鹽的稀溶液中,微溶 於 6N 鹽酸;UV max (0.1 N HCl):244,307 nm;UV max (0.1 N NaOH): 257,302,370 nm;對大鼠之 LD50:14 mg/kg (iv)。 【藥理活性】 MTX 是葉酸衍生物,為抗代謝藥物 (Lauretta et al., 2004; Kuo et al., 2004)。MTX 和它的代謝物聚穀氨酸酯 (polyglutamate) 與葉酸 (folic acid). 的化學構造相似,會一起競爭二氫葉酸還原酶 (DHFR) 的葉酸結合位 14.
(28) 置,與 DHFR 緊密的結合造成細胞內四氫葉酸 (FH4) 的合成受阻和還原 形葉酸的缺少,導致胸腺嘧啶核苷 (thymidine) 和嘌呤核苷酸 (purine nucleotides) 的合成降低。這些作用最後導致細胞凋亡。它的 polyglutamate 也會在細胞內抑制其它的酵素 ,特別是 amino-imidazole carboxamide ribonucleotide (AICR) transformylase、thymidylate synthase (Kuo et al., 2004; Rubino, 2001; Johnstone et al., 2000)。. [藥物動力學] MTX 主要經腎臟排除 (80-90 %),大多以原形藥排出體外,少部分由 膽汁排除 (10 %),在低劑量時 (低於 30 mg/m2),半生期約為 3 至 10 小 時;高劑量時,其半生期則達 8 至 15 小時。 劑量小於 30 mg/m2 時口服吸收良好,大於 80 mg/m2 時只有約 20%的人吸收較好,食物與牛奶會降低其生體可用率。高度集中分佈於腎 臟、膽囊、脾臟及肝臟,尤其會在肝臟停留長時間,會經過胎盤,隨著乳 汁擴散,不容易通過血腦障壁,血漿蛋白結合率約為 50%。 MTX 具有潛在的嚴重毒性,其發生毒性之情況與嚴重程度可能與劑 量或用藥次數有關,但在高、低劑量都曾發現過毒性作用。MTX 在體內 會因酵素作用而產生不同活性之代謝物,分述如下 (Kuo et al., 2004; Rubino, 2001; Johnstone et al., 2000):. 1. 7-Hydroxy-methotrexate 由肝臟之 aldehyde oxidase 在 pterine ring 上的 C-7 進行氧化所 15.
(29) 得,其活性約為 MTX 之 1/100~1/200。 2. 2, 4-Diamino-N10-methylpteroic acid (DAMPA) MTX 在體內由 carboxypeptidase 代謝成 DAMPA,其代謝量大約 是 MTX 注射量的 5%,其活性約為 MTX 的 1/200。 3. MTX polyglutamates (MTX-(Glu)n) MTX 受 polyglutamate synthase 的 作 用 而 轉化 成鍵結 2-7 個 glutamate 的代謝物。有研究指出不同 glutamyl chain length 的代謝物 於細胞內有不同程度的滯留,而 MTX-(Glu)n 之滯留會增強此藥之細 胞毒性。較長鏈之代謝物對 DHFR 有較高的親和力,因而對 DNA 合 成及細胞生長有較久之抑制作用,如圖五所示。. 圖五. Structures of MTX and its major metabolites (Rubino, 2001).. 16.
(30) [臨床應用]. 圖六. MTX 對 二 氫 葉 酸 還 原 酶 (DHFR) 有 高 度 親 和 力 , 與 葉 酸 競 爭 DHFR,使葉酸不能轉變為四氫葉酸,且去氧尿苷酸不能轉變為去氧嘧啶 核苷酸,阻止 DNA 合成,亦干擾 RNA、蛋白質合成,如圖六所示 (Ranganathan, 2003)。MTX 屬細胞周期特異性藥,主要作用於 G1 及 G1/S. 轉換期細胞。臨床上高劑量 (12 g/m2) 可用於治療成骨肉瘤 (Postovsky& Arush, 2005; Crews et al., 2004)、腦膜性白血病 (Aquerreta et al., 2004)、急性白血. 病 (Fernandez et al., 2004 ),低劑量用於治療牛皮癬 (Chladek et al., 2002;Chladek et al., 2005)、風濕性關節炎 (Godfrey et al., 1998; El Desoky, 2001; Zhou et al., 2004; Ranganathan & McLeod, 2005;)、眼色素層炎 (Puchta et al., 2005)、自體免疫疾病 (Grim et al., 2003),腸道發炎反應 (Egan et al., 1999)。. 17.
(31) [副作用]. 圖七. MTX 經 由 reduced folate carrier (RFC) 帶 進 細 胞 後 , 部 分 會 被 multidrug resistance proteins (MRP) 所排出,部分與 folylpoly-γ-glutamate synthetase (FPGS) 反應形成 polyglutamylated MTX (MTX-(Glu)n) 而抑制 DHFR,導致阻止葉酸之形成,如圖七所示 (Borst et al., 2000)。MTX-(Glu)n 因 分子量大,對 MRP 的親和力降低,無法排出細胞外,因此會長時間存在 於細胞,對組織便產生毒性。MTX 之毒性如下: 1. 肝毒性:MTX 具有潛在之急性及慢性肝臟毒性 (纖維變性及硬化)、 肝功能損害 (West, 1997; Kremer et al., 1986);慢性毒性可能導致死亡。長 期使用後或總劑量達 1.5 g 後可能發生慢性毒性。 2. 腎毒性:高劑量 MTX 會引起血尿、蛋白尿、尿毒症,腎臟損害而導 致急性腎衰竭,其腎毒性主要因 MTX 及 7-hydroxymethotrexate 在腎 小管中沈澱而引起。 3. 肺毒性:肺部病變 (Carson et al., 1987; Ameen et al., 2001) 如發燒、咳嗽、 呼吸困難、血氧過少以及浸潤現象。 18.
(32) 4. 骨髓抑制 (Wheeler et al., 1995):導致貧血、白血球減少症及或血小板減 少症 (Postovsky t al., 2005; Crews et al., 2004; Aquerreta et al.,2004; Megia et al., 2004; Gutierrez-Urena et al., 1996)。高劑量與某些非類固醇抗炎藥物併用. 時,發生嚴重骨髓抑制及胃腸道毒性 (Egan et al., 1999)。 5.. 胃腸道反應:噁心 (Jones & Patel, 2000)、嘔吐、腹痛 (Buchbinder et al.,1993; McKendry & Dale, 1993)。. [抗藥性] 產生抗藥性原因如下: 1. DHFR 對 MTX 的親和力改變。 2. 癌細胞合成更多的 DHFR。 3. MTX polyglutamate 的形成降低。 4. RFC 的表現減少,降低 MTX 進入細胞 (Jansen & Pieters, 1998)。 [藥物交互作用] MTX 與蛋白質廣泛結合且其結合可能被某些藥物所取代,如水楊 酸、磺胺藥、利尿劑、降血糖藥、三苯內醯 (diphenylhydantoins)、四環黴 素、氯黴素、對胺苯甲酸 (p-aminobenzoic acid) 以及酸性抗原藥物等。因 此,與這類藥物同時使用時,可能引起毒性增加。. 19.
(33) 第四節 Phenytoin (PHT) 之特性 【化學名】5,5-Diphenyl-2,4-imidazolidinedione (Budavari, 2001) 【結構式】 H N. O. O NH. 【分子式】C15H12N2O2 【分子量】252.27 【物化性質】 白色粉末,mp 295-298℃,pKa 8.3。幾乎不溶於水,1 g 溶於約 60 mL 乙醇、30 mL 丙酮,溶於鹼金屬氫氧化物。 鈉鹽,C15H11N2NaO2,sodium 5,5-diphenyl hydantoinate,白色粉末; 肥皂樣苦味,稍吸濕。甚至弱酸 (包括從空氣中吸收的 CO2) 亦易於使 之解離而再成為 phenytoin。1 g 溶於 10.5 mL 乙醇;~66 mL 水 (水溶 液混濁,若將 pH 調至其飽和溶液 pH 值 11.7 以上時可不混濁)。不溶 於乙醚、氯仿。 【藥物動力學】 Phenytoin 口服吸收良好,血中蛋白質結合率約 80-90%,故血中白. 20.
(34) 蛋白低下及尿毒症病患需注意給藥劑量。分布體積約 0.6 L/kg,約 90% 由肝臟 CYP2C9 和 CYP2C19 代謝,其代謝產物主要為 glucuronide, 少於 2% 以原形由尿液排除。血中濃度低時,其代謝速率受血中濃度影 響,而血中濃度達 10 mg/L 時,代謝呈現飽和狀態,如再給予更多 phenytoin,則血中濃度快速升高,造成中毒。Phenytoin 易誘導代謝酵素, 增加其他藥物之代謝。其半衰期因人而異,差異甚大,約 12 至 36 小 時 (Neels et al, 2004)。 文獻報導顯示,PHT 於 P-gp、 MRP1/MRP2 被抑制之大鼠腦內濃 度 明 顯 增 加 , 表 示 PHT 為 腦 部 multidrug transporters , P-gp 或 MRP1/MRP2 之受質 (Potschka et al., 2001)。 【臨床應用】 PHT 屬於第一線的抗癲癇藥物,一般用量為 200-500 mg/d;治療濃 度為 10-20 mg/L (Neels et al, 2004)。一般說來,其治療癲癇重積有兩個時 機,一是接續在 benzodiazepines (BZDs) 快速的終止癲癇發作之後,利 用其長效的抗癲癇作用,維持癲癇不再發作;一是當 BZDs 藥物治療失 敗時,PHT 作為替代藥物 (Lowenstein & Alldredge, 1998);PHT 的脂溶性比 BZDs 低,進入腦內的速度較緩慢,需要花費比 BZDs 較長的時間去控 制癲癇 (Browne, 1990)。儘管有許多副作用及藥物不良反應,PHT 仍被使 用為抗癲癇藥物,其治療急性癲癇發作和癲癇重積的效果卓越,另外可. 21.
(35) 用於治療慢性癲癇,特別是局部發作和繼發性全身性發作。 【副作用】 PHT 治療最理想的血中濃度為 8-10 mg/L,超過 20 mg/L 常伴隨神 經毒性,包括疲倦、肌肉顫抖、認知困難、眼球震顫、複視、運動失調, 超過 30 mg/L 易困倦、昏睡,當血中濃度大於 50 mg/L 時則會昏迷 (Moyer, 1999; McNamara , 2001; Haag, 2004)。. 另外,PHT 具有多毛副作用,與劑量有關;長期投予 PHT 可能造 成的嚴重不良反應,包括史帝文生-強生氏症候群 (Stevens Johnson syndrome) 以及肝臟毒性,目前臨床上的處置除了停藥外,建議採取症 狀緩解、支持療法以及類固醇的治療。對於低白蛋白血症患者須嚴密加 強 PHT 之血中濃度監測 (Haag, 2004)。小鼠 LD50 92 mg/kg (i.v.);110 mg/kg (s.c.)。 【藥物交互作用】 抑制. PHT 代 謝 之 藥 物 包 括. allopurinol 、 amiodarone 、. chloramphenicol、cimetidine、cotrimoxazole、disulfiram、fluconazole、 isoniazid、itraconazole、omeprazole、oral anticoagulants、warfarin 和某 些 sulfonamides (Brodie et al., 1996; Haag, 2004),因此臨床使用上必須監測血 中濃度,以免產生毒性及副作用;而 rifampicin 和 ritonavir 會促進 PHT 被代謝,減少其血中濃度。. 22.
(36) PHT 會誘導脂溶性藥物之氧化代謝,包括 carbamazepine、valproic acid、ethosuximide、anticoagulant agents、corticosteroids、benzodiazepines、 cyclosporin 、 tacrolimus 及. oral contraceptives (ethinyl estradiol 、. progestagens)。. 23.
(37) 第五節 Carbamazepine (CBZ) 之特性 【化學名】5H-Dibenz [b,f]-azepine-5-carboxamide (Budavari S, 2001) 【結構式】. N O. NH2. 【分子式】C15H12N2O 【分子量】236.27 【物化性質】 mp 190-193℃,無水乙醇/苯結晶,pKa 7.0。溶於乙醇、丙酮、丙二 醇,幾不溶於水。 【藥物動力學】 CBZ 之水溶性不佳,影響其吸收。血中蛋白質結合率約 70-80%, 主要由肝臟代謝,是 CYP3A4、CYP2C8 及 MRP 2 (Potschka, 2003) 之受 質,會誘導 CYP3A4 及 CYP2C9 (Neels et al., 2004) ,也會誘導腸的 MDR1mRNA,MRP2mRNA 及 MRP 2 (Giessmann et al., 2004),又有報導指 出與 CYP3A4 及 P-gp 相關連性 (Kim et al., 1999; Schuetz et al., 1996; Fromm et al., 2000; Rae et al., 2001)。但另有研究顯示,CBZ 與 P-gp 之相關性仍無. 定論 (Owen et al., 2001; Giessmann et al., 2004)。. 24.
(38) CBZ 約 2% 以原形由尿中排除,代謝產物為 CBZ-10,11-epoxide (屬活性代謝產物) 及 CBZ 的 N-或 O-glucuronides (Maggs, 1996),如圖 八、九所示,口服生可用率於大鼠及人皆大於 70% (Faigle & Feldman , 1995; Shinoda et al., 1995)。單一給藥時半衰期約 30 小時,當多次給藥且與其它. 會誘導肝代謝酵素的藥物 (如:phenytoin、phenobarbital、valproic acid 和 lamotrigine) 併用時,半衰期可能縮短為 15 小時 (McNamara et al., 2001)。CBZ-10,11-epoxide 排除半衰期為 6 小時,血中蛋白質結合率為. 50~60%。. N CONH2. Carbamazepine P-450. O. HO. OH. epossidoidrolase N. CONH2. CONH2. Carbamazepine epoxide. 圖八. O- and N-glucuronide. N. Carbamazepine diol. Carbamazepine is metabolized in the liver by oxidation to an active metabolite, carbamazepine 10,11-epoxide, which undergoes further metabolism. Carbamazepine is a potent enzyme inducer and can induce its own metabolism; this appears to be mediated via its effects on the CYP3A4 isozyme.. 25.
(39) 圖九. Proposed scheme for the formation of reactive metabolites from carbamazepine in humans. (Pearce et al., 2002). 【臨床應用】 一般用量:初始劑量為 100~400 mg/d,可逐漸增加劑量至 400~1200 mg/d,最大劑量為 1800 mg/d;治療濃度:4~12 mg/L (CBZ),0.2~6 mg/L (CBZ-10,11-epoxide)。CBZ 廣泛使用於治療多表現型的局部癲癇、大發 作與其他混合型癲癇及各種神經性疼痛、精神疾病之治療 (Brodie & Dichter, 1996;McLean & McDonald, 1986)。. 26.
(40) 【副作用】 CBZ 可避免動作電位反覆去極化,阻斷鈉離子通道。1960 年首先 被使用於治療三叉神經痛及預防躁鬱症,但需由低劑量開始以免產生 CNS 副作用 (複視、頭痛、頭昏、噁心),其他副作用包括:紅疹 (Ju & Uetrecht, 1999),眼球震顫,嘔吐,再生不良性貧血,顆粒白血球缺乏症,. 尿滯留 (Brodie & Dichter, 1996)。 CBZ 公認的治療血中濃度為 4~12 mg/L,但有些病患 CBZ 血中濃 度在此範圍內仍呈現中毒症狀,其原因為 CBZ-10,11-epoxide 之血中濃 度過高 (Shen et al., 2001);CBZ 口服給藥後 4~8 小時達最高血中濃度, 但給予大劑量時,可能延後至 24 小時;血中濃度高於 9 mg/L 時常出 現 CNS 副作用;治療前須測定血球數並作肝功能檢查,治療第 1 個月 內每週計數血球,之後每月測 1 次,肝功能亦須定期檢查;如發現皮膚 有過敏反應或肝功能有惡化跡象,則應停止服用,有時會發生非進行性 或症狀起伏不明顯的血小板減少,一般不須停藥,但如果發展為進行性 白血球減少或出現臨床徵兆如發燒及喉嚨痛則須停藥 (Richens et al., 1994)。. 【藥物交互作用】 CBZ 由 肝 臟 酵 素. CYP3A4 、 CYP2C8 代 謝. (小部分係由. CYP1A2 、 CYP2C19) , 氧 化 形 成 具 抗 癲 癇 活 性 之 中 間 產 物 CBZ-10,11-epoxide , 之 後 再 水 解 為 不 具 活 性 之 27.
(41) trans-10,11-dihydro-10,11-dihydroxycarbamazepine。CBZ 不僅會誘導自身 代謝,也會促使其他脂溶性藥物受在肝臟氧化代謝之速率增加 (Brodie, 1992),導致口服的 anticoagulants、disopyramide 和 quinidine 等藥療效. 減低,亦使 cyclosporin、tacrolimus、protease inhibitors 和 benzodiazepines 之血中濃度降低,使用口服避孕藥之女性平均每天需增加 35~50 μg estrogen 之用量 (Brodie & Dichter, 1996;Mattson et al., 1986)。CBZ 也會增加 valproic acid 和 ethosuximide 的代謝。Danazol、isoniazid、cimetidine、 fluoxetine、fluvoxamine、erythromycin、dextropropoxyphene、diltiazem、 verapamil、nefazodone、protease inhibitors 和 lithium 等會抑制 CBZ 之 代謝,增加其血中濃度 (Haag, 2004; Brodie & Dichter, 1996)。. 28.
(42) 第六節 多重耐藥性蛋白 (multidrug-resistance associated proteins; MRPs) 之特性 【背景】 MRP 1 於 1992 年首次被發現 (Cole et al., 1992),MRPs 是一群藥物 的運輸蛋白,在 ATP-binding cassette (ABC) transport protein 中屬於 C 的家族 (ABCC),在體內可以執行許多功能,如調節藥物在體內的排除, 也會使癌細胞對藥物產生抗藥性。MRPs 家族已發現有九種亞型:MRPs 1-9 (Ng et al., 2003; Mizuno et al., 2003; Chu et al., 2004; Lauretta et al., 2004; Elaine et al., 2005; Ballatori et al., 2005; Tian et al., 2005)。. 【結構與功能】 MRPs 1, 2, 3, 6, 7 具有 17 個 transmembrane helices,以及 2 個 nucleotide binding domains,N 端在細胞外,C 端在細胞內,MRPs 4, 5, 8 具 有 12 個 transmembrane helices , 以 及 2 個 nucleotide binding domains,N、C 端皆在細胞內,如圖十所示 (Lauretta et al., 2004)。 MRPs 1, 3, 4, 5, 6 存在於器官細胞膜上之 basolateral 側,MRPs 2, 4 則存在於 apical 側,MRPs 7-9 尚不清楚其分布位置 (Gary & Martin, 2003)。存在於 basolateral 側之 MRPs 對藥物是造成再回收的作用;存. 在於 apical 側之 MRPs 對藥物是造成外排的作用。藥物之結合態代謝 物,如 glucuronides、sulfates 或 glutathiones conjugates 等皆為 MRPs 29.
(43) 之受質。其中 glucuronides conjugate 為 MRPs 1, 2, 3, 4, 7 之受質, sulfates conjugate 為 MRPs 1-3 之受質,glutathiones conjugate 則為 MRPs 1-6 之受質 (Ballatori et al., 2005)。. 圖十. Proposed structural topology for the MRP proteins.. 【組織分布】 主要位於肝細胞、腎小管、胰臟、小腸、結腸、肺臟、膽囊、腦細 胞、唾腺、乳房上皮細胞、睾丸及骨骼肌之細胞等。 MRPs 於肝、腎 (Bleyer, 1978) 及腦 (Kusuhara & Sugiyama, 2005) 之 分布如圖十一~十三;組織分布、受質及抑制劑分列於下表一、二 (Tian et al., 2005)。. 30.
(44) 圖十一. 圖十二. 圖十三. Schematic diagram of drug transporters expressed in the liver in rat and human.. Schematic diagram of drug transporters expressed in the kidney in rat and human.. Schematic diagram of the efflux transport mechanisms at the BBB and BCSFB.. 31.
(45) 表一 Tissue Distribution, Expression levels of the MRP Family (Tian et al., 2005) Name MRP1. Symbol ABCC1. Tissue location All major tissues. Expression levels Differ in various organs and cell lines. MRP2. MRP3. ABCC2,. Liver, kidney, intestine,. cMOAT. brain. ABCC3. Small intestine, pancreas,. Low level in liver, brain,. colon, placenta, adrenal. kidney, and prostate. gland MRP4. ABCC4. Kidneys. Low levels in other tissues. MRP5. ABCC5. Most tissues. Low levels. MRP6. ABCC6. Liver , kidney. Low levels in other tissues. MRP7. ABCC10. Most tissues. Very low levels. MRP8. ABCC11. Normal breast, testis. Low levels in liver, brain, and placenta. MRP9. ABCC12. Breast cancer, normal breast, testis, brain, skeletal muscle, ovary. 32. Low levels.
(46) 表二. Substrates, and Inhibitors of the MRP Family (Tian et al., 2005). Name. Major drug substrates. Inhibitors. MRP1. Doxorubicin, vincristine, etoposide, MTX, camptothecin, CPT-11, SN-38, cyclophosphamide, conjugates. Probenecid, sulfinpyrazone, indomethacin, verapamil, quercetin, genistein, cyclosporine, PAK-104P, steroid analogs, MK571, ONO-1078, sulphonylurea, glibenclamide. MRP2. Conjugates, cisplatin, etoposide, vinca alkaloids, anthracyclines. camptothecins, MTX. MK571, furosemide. MRP3. Etoposide, teniposide, dinitrophenyl S-glutathione, acetaminophen glucuronide, vincristine, MTX. Etoposide, MTX. MRP4. MTX, 6-thioguanine, PMEA, 6-mercaptopurine, topotecan. MK571, celecoxib, rofecoxib, diclofenac. MRP5. 6-Mercaptopurine, 6-thioguanine, PMEA, heavy metals, S-(2,4dinitrophenyl)glutathione. Probenecid, sulfinpyrazone, benzbromarone, MK571. MRP6. LTC4, N-ethylmaleimide S-glutathione, dinitrophenol glutathione, etoposide, doxorubicin, cisplatin, daunorubicin. Indomethacin, probenecid, benzbromarone. MRP7. ?. ?. MRP8. 5-FU, ddC, PMEA, MTX, bile acids. ?. MRP9. ?. ?. ?=Undetermined.. 33.
(47) 第七節 有機陰離子運送蛋白 (organic anionic transporters; OATs)之 特性 OATs 家族中目前已發現五個成員:OATs 1-5,許多動物身上都有 OATs 家族,其中大鼠 OATs 家族之分布與人類相當近似。 OATs 之受質是有機陰離子,正常環境下 OATs 必須負起體內離子的 平衡。因此 OATs 特別重要的任務是排除體內第二相代謝所產生的物質, 如 glucuronides、sulfates 及 glutathiones 等 conjugates,將之排至尿中。 OATs 家族與 MRPs 家族之受質有許多重複 (Terlouw et al., 2003; Sweet; 2005);其於腦及肝之分布於圖十三、十四所示 (Tirona & Kim, 2002; Kusuhara & Sugiyama, 2005),其在各種動物體內主要的分布及其受質藥物分列於表三及. 表四 (Terlouw et al., 2003; Sweet; 2005)。. 圖十四. Schematic representation of drug transporters in liver.. 34.
(48) 表三 OATs 在各種動物體內主要的分布 (Terlouw et al., 2003; Sweet; 2005): Transporter Species Name Tissue Membrane type distribution localization OAT1 Mouse mOAT1 K, B BLM Rat rOAT1 K, B BLM Human hOAT1 K, B BLM Rabbit rbOAT1 K Pig pOAT1 K OAT2 Rat rOAT2 K, L APM, BLM Human hOAT2 K, L BLM Mouse mOAT2 K OAT3 Rat rOAT3 K, L, B BLM Human hOAT3 K, B BLM Mouse mOAT3 K OAT4 Human hOAT4 K, P APM, BLM OAT5 Mouse mOAT5 K Human hOAT5 L m: mouse, r: rat, h: human, f: flounder, Ce: C-elegans, rb: rabbit, p: pig, K: kidney, L: liver, B: brain, P: placenta, BLM: basolateral membrane, APM: apical membrane. 1. 表四 OATs的受質藥物 (Terlouw et al., 2003; Sweet; 2005): Nonsteroidal anti-inflammatory drug Diclofenac, Ibuprofen, Indomethacin, Ketoprofen, Naproxen, Phenacetin, Piroxicam, Salicylate Uremic toxins Hippuric acid, Indoxyl sulfate Antivirals Acyclovir, Adefovir, Azidothymidine, Cidofovir, Ganciclovir Antibiotics Cephalosporins, Penems, Penicillins Chemotherapeutics Methotrexate Heavy metals Cadmium, Mercury Mycotoxins Ochratoxin A Chlorinated phenoxyacetates 2,4-Dichlorophenoxyacetic acid Neurotransmitter metabolites 3,4-Dihydroxymandelic acid 3,4-Dihydroxyphenylacetic acid Chlorinated haloalkenes 1,2-Dichlorovinyl-L-cysteine Homovanillic acid Hydroxyindoleacetic acid. 35.
(49) 第 三 章. 材 料 與 方 法. 第一節 實驗材料 一、試藥 1. Acetic acid, glacial. J.T. Baker, Inc. (Phillipsburg, NJ, U.S.A.). 2. Acetonitrile (LC Grade). J.T. Baker, Inc. (Phillipsburg, NJ, U.S.A.). 3. L (+)-Ascorbic acid. Riedel-deHaën AG (Seelze, Germany). 4. Carbamazepine 5. Caffeic acid 6. 5, 7-Dimethoxycoumarin 7. Dimethylsulfoxide (DMSO) 8. Genipin 9. Geniposide. Sigma-Aldrich Chemical Co. (St. Louis, MO, U.S.A.) Sigma Chemical Co. (St. Louis, MO, U.S.A.) Aldrich Chemical Company (Milwaukee, WI, U.S.A.) Merck-Schuchardt (Hohenbrunn, Germany) Challenae Bioproducts Co. Ltd (San Jose, CA, USA) Wako Pure Chemical Industries, Ltd. (Osaka, Japan). 10. Ethyl acetate (LC Grade). J.T. Baker, Inc. (Phillipsburg, NJ, U.S.A.). 11. Ethyl alcohol. 台灣菸酒公賣局 (Taiwan). 12. Ethyl ether 13. Heparin 14. Hydrochloric acid 15. Methotrexate (25 mg/mL). Shi ma ky u’ sPur eChe mi c a l s( Os a ka , Japan) Novo Nordisk (Bagsvaerd, Denmark) Wako Pure Chemical Industries, Ltd. (Osaka, Japan) Wyeth Pharma Gmbh (Wolfratshausen, Germany). 36.
(50) 16. Methyl alcohol (LC Grade) 17. Methyl paraben 18. Mili-Qplus water (Milli-Q® ) 19. Ortho-phosphoric acid (85%) 20. Phenytoin 21. Polyethylenglycol 400 (PEG 400) 22. Sesamol 23. Sodium acetate, anhydrous. J.T. Baker, Inc. (Phillipsburg, NJ, U.S.A.) Sigma-Aldrich Chemical Co. (St. Louis, MO, U.S.A.) Millipore corporation (Billerica, MA, U.S.A.) Riedel-deHaën AG (Seelze, Germany) Sigma Chemical Co. (St. Louis, MO, U.S.A.) Merck-Schuchardt (Hohenbrunn, Germany) Sigma Chemical Co. (Saint Louis, MO, U.S.A.) Sigma Chemical Co. (Saint Louis, MO, U.S.A.). 24. Sodium hydroxide. Merck-KgaA (Darmstadt, Germany). 25. Sulfatase (type H-1, from Helix pomatia) 26. TDx® TDxFLx® Methotrexate monoclonal whole blood reagent pack. Sigma Chemical Co. (St. Louis, MO, U.S.A.) Abbott Laboratories (Abbott Park, Il, U.S.A.). 37.
(51) 二、器材及儀器設備 1. 酸鹼測定儀. Wissenschaftlich-Technische Werkstätten GmbH & Co. KG (Weilheim, Germany). Microprocessor pH-mV meter 2. 高速離心機. Hermle Labotechnik GmbH (Wehingen, Germany). Z 200 M/H 3. 渦旋振盪器. Scientific Industries Inc. (Bohemia, NY, U.S.A.). Vortex Genie G-560 4. 超音波振盪器. Branson Ultrasonics Co. (Taiwan). Bransonic 8120 5. 控溫往復式振盪水槽. Yih Der Instruments Co., Ltd. (Taiwan). BT-350 6. 氮氣濃縮裝置. Organomation Associates Inc. (Berlin, MA, U.S.A.). N-EVAP 112 R-MT 7. 分析天平. Mettler Toledo (Switzerland). AB 104 8. 微量移液管. Gilson S.A.S. (Entrepreneurs, Villiers Le Bel, France). Pipette 2-20 L, 10-100 L, 20-200 L, 100-1000 L 9. 水壓抽氣機. Tokyo Rikakikai Co. Ltd. (Tokyo, Japan). Eyela Aspirator A-2S 10. 電熱板. Shin Kwang Machinery Industry (Taiwan). HP-20 11. 高效液相層析儀-紫外光檢出器 (High Performance Liquid. 38.
(52) Chromatography-UV)包括: 幫浦 LC-10ATVP 層析管 Cosmosil 5C18-AR-Ⅱ (150×4.6 mm i.d, 5 m) Apollo C18 5(250×4.6 mm). Shimadzu (Kyoto, Japan) Nacalai Tesque (Kyoto, Japan) Alltech P.J. Cobert Associates, Inc.. 管柱前濾膜 Xpertec® 0.5 m (UPCH A-102X). P.J. Cobert Associates, Inc. (St. Louis, Missouri, USA). 紫外光偵測器 UV-VIS detector SPD-10AVP 自動進樣器 SIL-10AF 除氣裝置 ERC-3415α 樣品冷卻槽. Shimadzu (Kyoto, Japan) Shimadzu (Kyoto, Japan) Shimadzu (Kyoto, Japan) Shimadzu (Kyoto, Japan). 12. 螢光偏極免疫分析儀. Abbott Laboratories (Abbott Park, Illinois, U.S.A.). TDxFLx Analyzer 13. 拋棄式注射針及針筒. Terumo Medical Corporation (Elkton, MD, U.S.A.). 1.0 mL syringe (0.45×13 mm) 3.0 mL syringe (0.55×25 mm) 5.0 mL syringe (0.65×32 mm) 14. 胃管. 晶龍科技儀器有限公司. (0.9×L 70 mm, 1.5×L 120 mm). (Taiwan). 15. 微量吸管尖. Axygen Scientific, Inc. (Union City, CA, U. S. A.). Tips (200 L, 1000 L) 16. 微量離心管. Axygen Scientific, Inc. (Union City, CA, U. S. A.). Microtubes (1.7 mL) 17. 濾膜. Millipore carrigtwohill, Co. 39.
(53) Millex® (0.45 m, 13 mm; 0.22 m, 13 mm). (Cork, Ireland). 18. 玻璃試管 (12×75 mm). Kimble glass Inc. (U.S.A.). 19. 血清塞. 弘光企業有限公司 (Taiwan). 20. 混合氣體. 吉源行有限公司 (Taiwan). 40.
(54) 三、藥材 梔子購自台中市欣隆藥行。 四、動物 Sprague-Dawley (SD) 大鼠,購自國家實驗動物中心,購入後飼養於 中國醫藥大學動物中心,環境溫度控制在攝氏 221℃,12 小時光照與. 黑暗循環之環境中,相對溼度維持 5010%。動物試驗遵循中華民國實 驗動物學會 2002 年所編纂之「實驗動物飼養與使用操作手冊」 (A guide book for the care and use of laboratory animals)。 五、溶液製備 (一) Geniposide 及 genipin 標準溶液 精確稱取 geniposide 及 genipin 各 1.0 mg,分別加入甲醇定容至 1.0 mL,即得 1.0 mg/mL 之儲備溶液,再以甲醇稀釋成各種所需濃度 之標準溶液;genipin 需於使用前新鮮配製。 (二) 內標準溶液 1. 精確稱取 sesamol 5.0 mg,加入甲醇定容至 5.0 mL,即得 1.0 mg/mL 之儲備溶液,再以甲醇稀釋成各種所需濃度之內標準溶液。 2. 精確稱取 caffeic acid (CA) 5.0 mg,加入甲醇定容至 5.0 mL,即得 1.0 mg/mL 之儲備溶液,再以甲醇稀釋成各種所需濃度之內標準溶液。. 41.
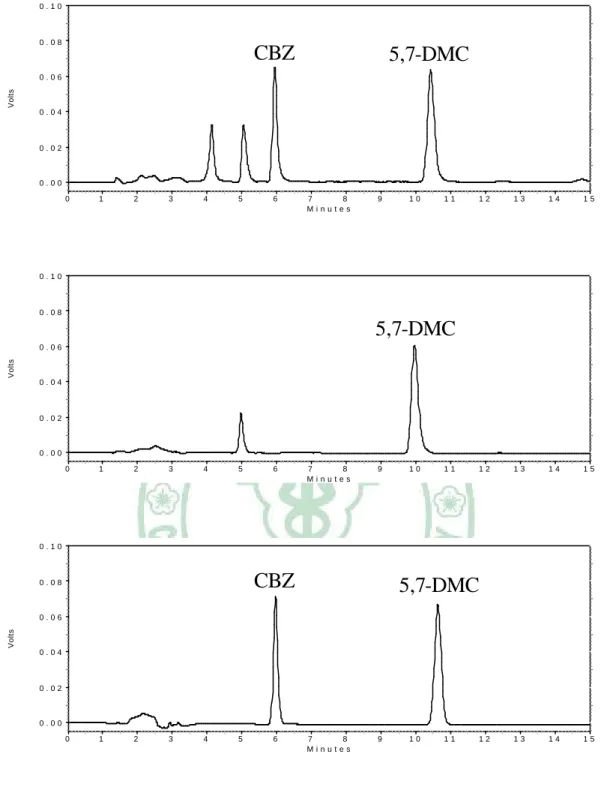
(55) 3. 精確稱取 methylparaben (MP) 10.0 mg,加入乙酸乙酯定容至 10.0 mL,即得 1.0 mg/mL 之儲備溶液,再以乙酸乙酯稀釋成各種所需濃 度之內標準溶液。 4. 精確稱取 MP 10.0 mg,加入甲醇定容至 10.0 mL,即得 1.0 mg/mL 之 儲備溶液,再以甲醇稀釋成各種所需濃度之內標準溶液。 5. 精確稱取 5, 7-dimethylcoumarin (5,7-DMC) 10.0 mg,加入乙酸乙酯定 容至 10.0 mL,即得 1.0 mg/mL 之儲備溶液,再以乙酸乙酯稀釋成各 種所需濃度之內標準溶液。 6. 精確稱取 5,7-DMC 10.0 mg,加入甲醇定容至 10.0 mL,即得 1.0 mg/mL 之儲備溶液,再以甲醇稀釋成各種所需濃度之內標準溶液。 (三) 人工腸液之製備 取磷酸二氫鉀 (KH2PO4) 6.8 g 溶於水 250 mL 中,再加入 0.2 N 氫氧化鈉溶液 190 mL 及水 400 mL ,以 0.2 N 氫氧化鈉溶液調節 pH 至 7.5 0.1 後,加水至 1 L。 (四) 緩衝液 (pH 5.0) 稱取無水醋酸鈉 0.82 g,加水溶解後,稀釋至 100 mL 即為 0.1 N 醋酸鈉溶液。量取醋酸 (d = 1.049) 0.6 mL,加水稀釋至 100 mL 即為 0.1 N 醋酸溶液。取 0.1 N 醋酸鈉溶液 (sodium acetate) 68.0 mL,加 入 0.1 N 醋酸溶液至 100 mL,再加 1.0 N 氫氧化鈉將 pH 值調至. 42.
(56) 5.00.1。 (五) 0.1 N 氫氧化鈉溶液 取氫氧化鈉 (sodium hydroxide) 0.4 g,加水溶解後,稀釋至 1 00 mL。 (六) 1.2 N 鹽酸溶液之製備 取濃鹽酸 11.4 mL,加水稀釋至 100 mL。 (七) Sulfatase 溶液 取 Sulfatase (15300 units/g, type H-1) 3.3 g,以 pH 5.0 緩衝溶液 溶解使成 50 mL,儲備於 -20℃ 備用。 (八) 抗壞血酸溶液 稱取抗壞血酸 100 mg,加水至 1 mL 即得 100 mg/mL 之抗壞血 酸溶液,使用前新鮮製備。 (九) Methotrexate (MTX) 溶液 精確量取 MTX 2.0 mL (25 mg/mL),加水定容至 20.0 mL,即得 2.5 mg/mL 之 MTX 溶液,使用前新鮮製備。 (十) Phenytoin (PHT) 溶液 精確稱取 PHT 400.0 mg,加少量水混合均勻,加 0.1N NaOH 120 L,再加水定容至 10.0 mL 即得 40.0 mg/mL 之 PHT 溶液,使用前 新鮮製備。. 43.
(57) (十一) Carbamazepine (CBZ) 溶液 精確稱取 CBZ 400.0 mg,加 DMSO 1 mL,PEG 400 5 mL,混合 均勻,再加熱水定容至 10.0 mL,即得 40.0 mg/mL 之 CBZ 溶液, 使用前新鮮配製。. 44.
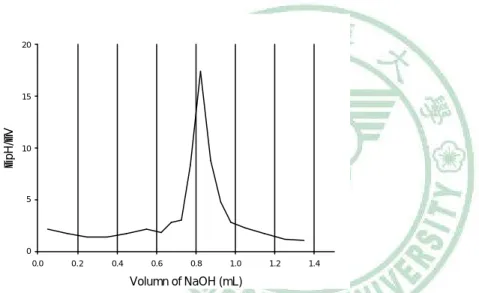
(58) 第二節 實驗方法 一、 Genipin 之 pKa 測定 稱取 genipin 22.6 mg,加水至 100 mL,置於攪拌器上,放入 磁石及 pH meter 之電極,以 0.1 N NaOH 滴定,記錄 NaOH 體積 及 pH 值之變化,繪製滴定曲線圖,並求其滴定中點。當達滴定中 點 (50% 中和) 時,解離態與不解離態的濃度正好相等,此時 pH = pKa + log ( [A-] / [HA] ) = pKa + log 1 = pKa. 45.
(59) 二、 Genipin 之安定性 (一) 甲醇中 genipin 之安定性 精確稱取 genipin 1.0 mg,加入甲醇定容至 1.0 mL,經系列 稀釋配製成 5.0 g/mL 之 genipin 標準品溶液,加入等體積含內 標 (10.0 g/mL sesamol) 之甲醇後,分別於 0、16、22、40、70、 164 小時,取 20 L 以 HPLC 分析。 (二) 血清中 genipin 之安定性 1. 儲備於 -20℃ 對血清中 genipin 安定性之影響 取空白血清 9 mL,加入 genipin 標準品溶液 1 mL,使其濃 度為 10.0 g/mL,分裝至微量離心管,分成三組,每組三管,每 管 200 L,第一組加入含內標 (1.0 g/mL MP) 之乙酸乙酯 200 L 萃取之,於渦旋振盪器上混合,經 9860 × g 離心 15 分鐘, 取出乙酸乙酯層,以氮氣吹乾後,貯於 -20℃,俟後進行 HPLC 分析。其餘的血清檢品則先貯於 -20℃,分別於 1 及 3 天後取 出,處理步驟如第一組。 2. 抗壞血酸對血清中 genipin 安定性之影響 取空白血清,加入 genipin 標準品溶液,使其濃度為 5.0 g/mL,分裝至微量離心管,分成三組,每組三管,每管 200 L, 第一組加入 pH 5.0 之緩衝溶液 100 L、100 mg/mL 抗壞血酸溶. 46.
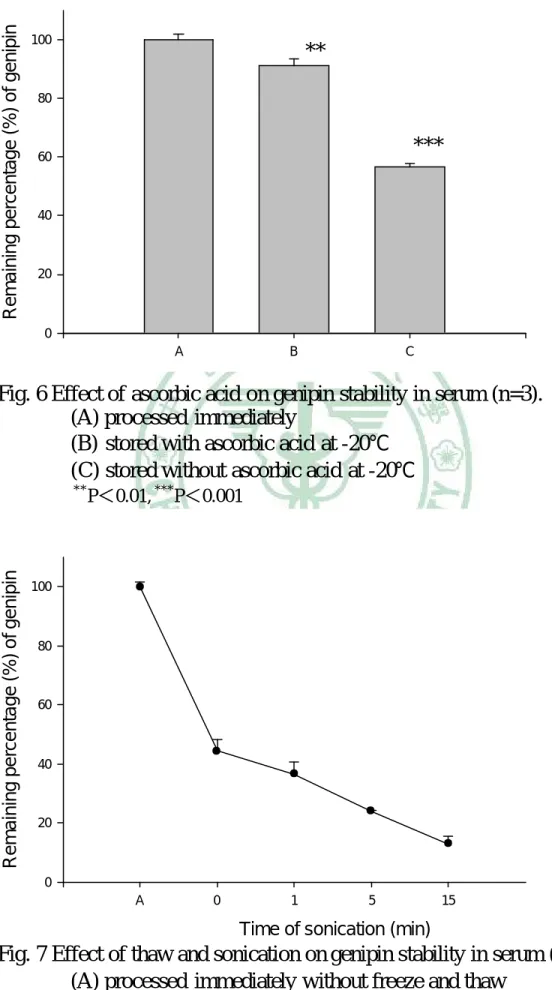
(60) 液 100 L,再加含內標 (1.0 g/mL MP) 之乙酸乙酯 500 L 萃 取之,於渦旋振盪器上混合,經 9860 × g 離心 15 分鐘,取出 乙酸乙酯層,以氮氣吹乾後,貯於 -20℃,俟後進行 HPLC 分 析。第二組加入 100 mg/mL 抗壞血酸溶液 50 L,第三組加入 水 50 L,皆貯於 -20℃,於第二天取出,分別加入 pH 5.0 之 緩衝溶液 100 L 及 100 mg/mL 抗壞血酸溶液 50 L 後,其餘 處理步驟如第一組。 3. 超音波震盪解凍對血清中 genipin 安定性之影響 取空白血清,加入 genipin 標準品溶液,使其濃度為 5.0 g/mL,分裝至微量離心管,分成五組,每組三管,每管 200 L, 第一組加入 pH 5.0 之緩衝溶液 100 L、100 mg/mL 抗壞血酸溶 液 50 L,再加含內標 (1.0 g/mL MP) 之乙酸乙酯 350 L 萃 取之,於渦旋振盪器上混合,經 9860 × g 離心 15 分鐘,取出 乙酸乙酯層,以氮氣吹乾後,貯於 -20℃,俟後進行 HPLC 分 析。其餘四組則先貯於 -20℃,於第二天取出,分別經超音波震 盪器震盪 0、1、5、15 分鐘後,其餘處理步驟如第一組。 (三) 血液檢品之前處理及安定性 1. 抗壞血酸對血清中 genipin 於 37℃ 下安定性的影響 取大鼠靜脈注射 genipin 後 3 分鐘之血清檢品,置於冰溶. 47.
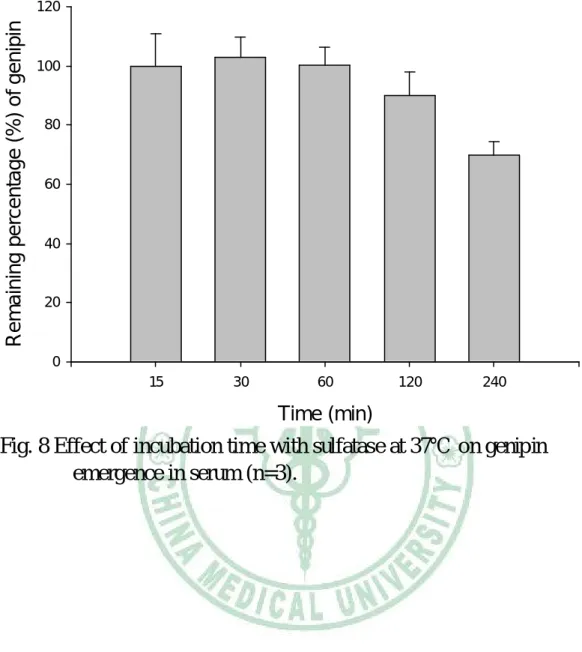
(61) 上,分裝至外覆鋁箔紙之試管,分成四組,每組三管,每管 200 L,加入 1,000 unit/mL sulfatase 溶液 100 L,第一組加入 200 mg/mL 抗壞血酸溶液 50 L,第二組加入 100 mg/mL 抗壞血酸 溶液 50 L,第三組加入 50 mg/mL 抗壞血酸溶液 50 L,第四 組則加入水 50 L,並栓上血清塞,以注射針筒抽去管內之空 氣。置於 37℃ 水浴中 1 小時之後,加入含內標 (1.0 g/mL MP) 之乙酸乙酯 350 L 萃取之,於渦旋振盪器上混合後,置於微量 離心管,經 9860 × g 離心 15 分鐘後,取出乙酸乙酯層,以氮 氣吹乾,加甲醇溶解 50 L,取 20 L 以 HPLC 分析。 2. 抽氣及避光對血清中 genipin 於 37℃ 下安定性的影響 取大鼠靜脈注射 genipin 後 3 分鐘之血清檢品,置於冰溶 上,分成三組,每組三管,每管 200 L,第一組置於外覆鋁箔 紙之試管,第二組置於透明玻璃試管,第三組置於微量離心管, 各加入 1,000 unit/mL sulfatase 溶液 100 L,100 mg/mL 抗壞血 酸溶液 50 L,前兩組栓上血清塞,並以注射針筒抽去管內之空 氣。置於 37℃ 水浴中 1 小時後,加入含內標 (1.0 g/mL MP) 之乙酸乙酯 350 L 萃取之,於渦旋振盪器上混合,置於微量離 心管,經 9860 × g 離心 15 分鐘後,取出乙酸乙酯層,以氮氣 吹乾,加甲醇 50 L 溶解,取 20 L 以 HPLC 分析。. 48.
(62) 3. 血清中 genipin 結合態代謝物酶解之最佳時間 取大鼠靜脈注射 genipin 後 3 分鐘之血清檢品,置於冰溶 上,分裝至微量離心管,每管 200 L,加入 sulfatase 溶液 100 L,100 mg/mL 抗壞血酸溶液 50 L。置於 37℃ 之往復式振盪 水槽,以 100 rpm 震搖,分別於 0、15、30、60、120、240、 360 分鐘後,取出檢品,每組三管,各加含內標 (1.0 g/mL MP) 之乙酸乙酯 350 L 萃取之,於渦旋振盪器上混合,置於微量離 心管,經 9860 × g 離心 15 分鐘後,取出乙酸乙酯層,以氮氣 吹乾,加甲醇 50 L 溶解,取 20 L 以 HPLC 分析。 4. 凍存對血清中 genipin sulfate 安定性之影響 取大鼠靜脈注射 genipin 後 3 分鐘之血清檢品,置於冰溶 上,分裝至微量離心管,分成兩組,每組三管,每管 200 L, 第一組加入 1,000 unit/mL sulfatase 溶液 100 L、100 mg/mL 抗 壞血酸溶液 50 L,於渦旋振盪器上混合,置於 37℃ 之往復式 振盪水槽,以 100 rpm 震搖,30 分鐘後取出,再加入含內標 (1.0 g/mL MP) 之乙酸乙酯 350 L 萃取之,於渦旋振盪器上混合, 經 9860 × g 離心 15 分鐘,取出乙酸乙酯層,以氮氣吹乾後, 貯於 -20℃,俟後進行 HPLC 分析。第二組則先貯於 -20℃,於 第 3 天取出,處理步驟處理如第一組。. 49.
(63) (四) 血清檢品前處理方法之比較 1. 乙酸乙酯萃取之法 取空白血清 900 L,加入 100.0 g/mL 之 genipin 標準品 溶液 100 L 使成濃度為 10.0 g/mL 之血清標準溶液。取 100 L 置於微量離心管,加入含內標 (4.0 g/mL MP) 之乙酸乙酯 100 L 萃取之,於渦旋振盪器上混合,置於微量離心管,經 9860 × g 離心 15 分鐘後,取出乙酸乙酯層,以氮氣吹乾,加甲醇 50 L 溶解,取 20 L 以 HPLC 分析。 2. 加甲醇去蛋白法 取 genipin 血清標準溶液 100 L 置於微量離心管,加入含 內標 (1.0 g/mL MP) 之甲醇 400 L,於渦旋振盪器上混合,經 9860 × g 離心 15 分鐘後,以氮氣吹乾,加甲醇 50 L 溶解, 取 20 L 以 HPLC 分析。 (五) 高效液相層析儀 (HPLC) 分析條件 分析管柱:Cosmosil® 100 RP-18 e (5 m, 150×4.6 mm) 移 動 相:0.1 % phosphoric acid:acetonitrile = 84:16 沖提時間:25 min 流. 速:1.0 mL/min. 檢測波長:240 nm. 50.
(64) (六) 數據分析 比較所測得 genipin 與內標之波峰面積比。. 51.
數據

+7

Outline
相關文件
Practice with your teacher - Show and tell Hi, Mike.. How
Do you want bacon and eggs?.
It’s (between/next to) the church and the
Listen - Check the right picture striped hat polka dotted hat.. Which hat do
Play - Let’s make a big family How many people are in your family1. Write it
Sam: I scraped my knee and bumped my head.. Smith: What happened
straight brown hair dark brown eyes What does he look like!. He has short
While Korean kids are learning how to ski and snowboard in the snow, Australian kids are learning how to surf and water-ski at the beach3. Some children never play in the snow
