國
立
交
通
大
學
生物科技系暨研究所
碩
士
論
文
建立一快速且便利的方式以增強轉殖基因在特定細胞中的
表現
Development of a rapid and convenient method to enhance the
transgenic expression in target cells
研 究 生:莊懷堯
指導教授:廖光文博士
建立一快速且便利的方式以增強轉殖基因在特定細胞中的表現
Development of a rapid and convenient method to enhance the transgenic expression in target cells 研 究 生:莊懷堯 Student:Huai-Yao Chuang 指導教授:廖光文 Advisor:Kuang-Wen Liao, Ph.D. 國 立 交 通 大 學 生物科技學系暨研究所 碩 士 論 文 A Thesis
Submitted to Institute of Biological Science and Technology College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in
Biological Science and Technology
August 2007
Hsinchu, Taiwan, Republic of China
建立一快速且便利的方式以增強轉殖基因在特定細胞中的表現 研 究 生:莊懷堯 指導教授:廖光文博士 國立交通大學生物科技學系暨研究所碩士班 摘要 基因治療對癌症病患提供了前所未有的治療策略及希望。不幸的是,無 論是基因傳遞或啟動子系統迄今仍未達到專一性之療效,除此之外,任一 系統的最佳化都極端困難。在本研究中我們希望介紹一個簡單的概念:亦 即部分專一的基因傳遞系統以及部分專一的啟動子相結合,將可對標靶細 胞達到更加專一性的表現。在第一部分,我們首先檢測與腫瘤相關的轉錄 因子在腫瘤或快速生長細胞中的活性。接著利用表現量較高的轉錄因子 (NF-κB,CREB 以及 HIF-1)之反應片段,取三倍體構築一轉錄因子相關之合
成啟動子(Transcription factor-based synthetic promoter,TSP) 。實 驗結果證實 TSP 在特定細胞中具有活性並有部分專一性。此外相對於 NF-κB 或 HIF-1 迷你啟動子,TSP 在抑制劑存在之下表現較佳的抵抗性。 在第二 部分,多功能胜肽 RGD-4C-HA 可專一性結合至 B16-F10 細胞表面之 integrin αvβ3 並且吸附至聚乙烯亞胺(Polyethyleneimine,PEI)。實驗結果顯示 RGD-4C-HA 能與聚乙烯亞胺形成複合物並且在
in vitro
實驗中引導專一性 的指向。最後,聚乙烯亞胺及胜肽複合物與 TSP 的結合能夠使轉殖基因專一性的表現在 B16-F10 細胞中。這種策略在
in vitro
實驗中已經證實為可 行,並且在in vivo
的專一性基因治療可能也具有潛力。Development of a rapid and convenient method to enhance the transgenic expression in target cells
Student:Huai-Yao Chuang Adviser: Dr. Kuang Wen Liao Institute of Biological Science and Technology
National Chiao Tung University
ABSTRACT
Gene therapy provides a novel strategy and a new hope for the patients with cancer. Unfortunately, the specificity of the delivery systems or the promoters did not achieve the specific efficacy so far and the perfection of either system will be extremely difficult. In this study we had introduce a simple concept that the combination of partial specific delivery and partial specific promoter activity may achieve more specific effect for specific expression in target cells. In the first part, the tumor related transcription factors were assayed in tumor or rapid-proliferating cells to determine their activities. The activities of NF-κB, CREB, and HIF-1 were higher and three copies of each response elements were used to construct a transcription factor-based synthetic promoter (TSP). The results showed that the expression of TSP was truly active and partial specific to cell types. In addition, it was more resistant than NF-κB or HIF-1 mini-promoters at the presence of inhibitors. In the second part, the multi-functional peptide RGD-4C-HA was designed to specifically target integrin αvβ3 on B16-F10 cells and absorbed to polyethyleneimine (PEI)
molecules. The results showed that RGD-4C-HA could associate with PEI to form complex and mediate specific targeting in vitro. Finally, the combination of PEI-peptide complex and TSP could enhance the specifically transgenic expression in B16-F10 cells. This strategy had been proven to work in vitro and might be also potential in specific gene therapy in vivo.
Acknowledgements
不知不覺兩年的時間就過去了,回想起碩士班的時光,真是如夢似幻, 從昨日無知的門外漢,到今天成為碩士班畢業生,歸根究柢,還是要感謝 許多人的支持與幫助,父母親生我育我,教化之功自是不在話下,然而在 學習的過程中,我的指導教授廖光文博士更是居功厥偉,承蒙他不嫌棄我 這個應化出身對生物一無所知的稚子,本著教育理念在各方面都不吝給予 指導,無論是學業或是待人接物,他告訴我如何能夠有效率的學習,是我 碩士班的學業成績,一直名列前茅,甚至得到從未得過的書卷獎,重拾我 對學習的信心,當研究遇到瓶頸時,也在在顯示其靈活的思維以及解決問 題的頭腦,讓我獲益良多,了解科學的態度,學到許多做人做事的道理, 如果沒有他,我不會學到這麼多。 再來我要感謝我實驗室的夥伴們,無論是面對實驗的難關或是情緒的低 潮,他們的陪伴給予我很多幫助,感謝靜宜學姐在各方面的指導,于鈴學 姐的辛苦交接,口試時鈺珊學姐的張羅準備,XX 王彥谷學長的 XX 講座,實 驗室最帥詹姆士與一點都不兇小護士的笑料(加油,你們是動物界的奇 蹟!),美工魔人 chenyu 的才藝,師弟 RT 的白爛與宅男同好,我兩個徒兒 小馬跟堅甫的跑腿打雜,子慧有禮貌的問好,以及其他的夥伴們,雖然相 處時間並不長,但是謝謝你們的幫助與陪伴,人生的路上有幸可以遇到你 們,希望將來仍能互相切磋與扶持,作一輩子的朋友。Contents
Abstract in Chinese
………i-iiAbstract
……….………iiiAcknowledgements
…...…..……….………ivContents
…...……...……….………v-ixList of tables
….…..…………..……….xList of figures
….………..………..……….xiAbbreviations
………..………xiiChapter 1 Introduction
1.1 Cancer gene therapy……..……….………...11.2 Delivery system for cancer gene therapy………..……….………3
1.2.1 Viral vectors………...3
1.2.2 Non-viral methods…….……….………….…..………....3
1.2.2.1 Polyethyleneimine (PEI)………...……….5
1.3 Promoters for cancer gene therapy……….…….…....6
1.3.1 Cancer specific promoters……….…7
1.3.2 Tumor related promoters………8
1.4 Activities of NF- B, HIF-1, and CREB in cancer progression and therapy……….…….9
1.4.2 Nuclear factor-kappaB (NF-κB).………...…....11
1.4.2.1 Biology of NF-κB……….12
1.4.2.2 NF-κB in cancer progression………..13
1.4.3 Hypoxia-inducible factors (HIFs)..………..…...15
1.4.3.1 Biology of HIFs……….………16
1.4.3.2 HIFs in cancer progression………..…………..16
1.4.4 cAMP response-element binding protein (CREB)……..16
1.4.4.1 Biology of CREB………..………17
1.4.4.2 CREB in cancer progression………….………18
1.4.5 Transcription factors interaction in cancer progression…18 1 . 5 S t r a t e g y . … … … . . . … … … 1 9
Chapter 2 Materials and Methods
2.1 Materials………..………..………....212.1.1 Primers ……….……….………...21
2.1.2 Cell lines ……….………..………....21
2.1.3 Plasmids ……….………..….……….22
2.1.4 Chemicals, enzymes, and reagents ………....23
2.1.5 Antibodies……….……….…...30 2.1.6 Kits………...……….………….…...…30 2.1.7 Buffers ……….31 2.1.8 Media ……….……….33 2.1.9 Equipment ……….……….34 2 . 2 Methods……….. ………....56
2.2.1 Construction of transcription factor-based synthetic promoter (TSP).56
2.2.1.1 Restriction enzyme digestion………….………..….…….57
2.2.1.2 DNA extraction………….……...……..…..……….…….58
2.2.1.3 Ligation……….……...………..……….…….58
2.2.2 Transformation of E. coli……….……..…………..58
2.2.2.1 Preparation of competent cells for heat shock……..…….58
2 . 2 . 2 . 2 Transformation of comp et ent cell by heat sho c k met h o d … … … . . … … … . … 5 9 2.2.3 Plasmid DNA extraction ………..………...59
2.2.3.1 Minipreparation method………….…..………..60
2.2.3.2 Midipreparation method…….……….……….…61
2.2.4 Cell culture ……….………...62
2.2.4.1 Procedures of subculture…….……….………..62
2.3 Transcription factors and TSP activity assay………..………..43
2.3.1 Transfection of mammalian cells……….………..44
2.3.1.1 Seeding cells……….………..…..44
2.3.1.2 Polyethyleneimine transfection……….……..…..44
2.3.1.3 LipofectamineTM 2000 transfection………..…..…..45
2 . 3 . 2 M e a s u r e m e n t o f r e p o r t e r g e n e e x p r e s s i o n b y f l o w cy t o me t r y … … … …… . . … … … . . .4 6 2.4 RGD-4C-HA binding assay………..46
2.5 The absorption of RGD-4C-HA to PEI assay……….……..47
2.5.1 Separating PEI-peptide complex with un-absorbed PEI and RGD-4C-HA by gel filtration column S-200.………..…..47
2.6 PEI-peptide complex transfection………..49 2.7 Statistical analysis………..49
Chapter 3 Results
3.1 Establishment of the transcription factor-based mini promoter (TSP)
s y s t e m … … … . . … … . . . . 5 0
3.1.1 Screening of the activities of several transcription factors in different
c e l l s … … … . . … … … . . . 5 0
3.1.2 Construction of the transcription factor-based synthetic promoter
(TSP)..………..……….……....51
3.1.3 Transcription factor-based mini promoter activity in different
cells………..………..……52
3.1.4 Inhibition effect of TSP in HeLa cells………....53 3.2 Design of RGD-4C-HA and the functional regions………..………..…....54 3.3 The binding affinity of RGD-4C-HA……….……….55 3.4 The absorption of RGD-4C-HA to PEI……….………..57 3 . 5 The enhancement of transgenic expression by PEI-peptide
complex………..………..……….58
3.6 The enhancement of transgenic expression by PEI-peptide complex
(HIF-1)……….………….…………58
3.7 Enhancement of transgenic expression by PEI-peptide complex combined
with TSP in B16-F10 cells………...……60
References
..……....……….……….………...78List of tables
Table 1. Primers used in this study.………..……….21
Table 2. Cell lines used in this study…..………...22
Table 3. Plasmids used in this study………..……...22
Table 4. Chemicals, enzymes, and reagents used in this study…..……….….23
Table 5. Antibodies used in this study……….………..…..30
Table 6. Kits used in this study……….….……….….30
Table 7. Buffers used in this study……….………..31
Table 8. Media used in this study………..………33
List of figures
Figure 1. The illustration of the strategy………….…..……….66 Figure 2. The activities of several transcription factors in different
cells……..………..……….67
Figure 3. Construction of transcription factor-based synthetic promoter
( T S P ) … … … . . . 6 8
Figure 4. Activities of the transcription factor-based synthetic promoter (TSP)
in different cells……….………..……...69
Figure 5. The effect of inhibiting transcription factors to TSP in HeLa
cells………..…..….70
Figure 6. Design and illustration of the multi-functional peptide
R G D - 4 C - H A … … … . . … . . 7 1
Figure 7. The binding efficacy of RGD-4C-HA to different cells……….….72 Figure 8. PEI and RGD-4C-HA absorption assay………..……..74 Figure 9. The enhancement of transgenic expression by PEI-peptide complex
(pAAV-MCS-hrGFP)………75
Figure 10. The enhancement of transgenic expression by PEI-peptide complex
( p C R I I - h r G F P ) … … … . . … … … . . … 7 6
Figure 11. Enhancement of transgenic expression by PEI-peptide complex
Abbreviations
AAV Adeno-associated virus AFP α fetoprotein
ATF1 Activating transcription factor 1 CBP CREB binding protein
CCSST Clear-cell sarcomas of soft tissues
CEA Carcinoembryonic antigen CF Cystic fibrosis
CFTR Cystic fibrosis transmembrane regulator CREB cAMP response-element binding protein EWSR1 Ewing sarcoma gene
GCV Ganciclovir
HER-2/neu Human epidermal growth factor receptor 2/neu HIF-1 Hypoxia-inducible factor-1 HREs Hypoxia response elements
HSV-tk Herpes simplex virus thymidine kinase HTLV Human T-cell leukemia virus
hTR Human Telomerase RNA
hTRET Human telomerase reverse transcriptase IAPs Inhibitor of apoptosis proteins
IKK IκB kinase
IL-8 Interleukin-8 IPAS Inhibitory PAS domain protein kDa Kilo dalton
NF-κB Nuclear factor-kappaB PEI Polyethyleneimine TFBS Transcription factor binding sites TLC Thin layer chromatography
TSP Transcription factor-based synthetic promoter VEGF Vascular endothelial growth factor
Chapter 1 Introduction
Gene therapy was defined as transferring genetic material into cells in order to cure a disease or improve the clinical status of a patient. It has been a promising tool in diseases caused by genetic defects such as severe combined immunodeficiency, cystic fibrosis, hemophilia, Parkinson’s disease and Alzheimer’s disease [1]. For example, cystic fibrosis (CF) is a lethal autosomal recessive disease caused by a mutation in the cystic fibrosis transmembrane regulator (CFTR) gene for a chloride ion channel expressed in epithelial cells of lung. The gene therapy for CF is to deliver CFTR cDNA to the epithelial cells that line the lumen of the conducting airways of the lung by inhaled aerosol, liposomes, or viruses [2-5]. However, there are still some limitations to impede successful outcome of gene therapy. The targeting of multiple therapeutic genes into the gene-defective cells may be required in order to develop an effective therapeutic strategy. In cancer gene therapy, the specific regulation of gene expression in tumor cells should also be improved.
1.1 Cancer gene therapy
Development of cancer has been suggested to occur through a series of molecular events in which multiple genetic abnormalities accumulate within cells
and has been called ‘‘multistep carcinogenesis.’’ Mutations in oncogenes may result in excessive activity or expression of the oncogene product. In contrast, tumor suppressor genes may be mutated or deleted resulting in decreased activity or expression of the tumor suppressor gene product. Either case could result in abnormal growth regulation resulting in the cancer phenotype. As above, gene therapy is generally considered as a useful tool in the treatment of cancer. There are several ways to provide the benefits to the patients with cancer including the expression of tumor-suppressor genes in tumor cells, ablation of oncogene function by RNA interference and ribozymes, and expression of a suicide gene that converts a harmless prodrug into a potent toxin in tumor cells [6-8]. Tumor suppressor genes such as pRb and p53 play a critical role in the regulation of the cell cycle or promote apoptosis. Restoration of wild type p53 gene expression using a retroviral
p53 vector inhibited cell growth and induced apoptosis in human lung cancer cells
with mutated or deleted p53 genes [9, 10]. Besides, the products of the p16 tumor suppressor gene and a truncated Rb gene have been shown to suppress tumor growth in animal models [11, 12]. In ablation of oncogenes, adenovirus-mediated ribozyme targeting of HER-2/neu inhibited in vivo growth of breast cancer cells in a mouse model [13]. In similar way, intravenous tail injections of an Ad E1A construct in a mouse model inhibited the intratracheal growth of HER-2/neu overexpressing lung cancer cells [14]. In suicide gene therapy, the herpes simplex
virus thymidine kinase (HSV-tk) can specifically bind and phosphorylate nucleoside analogs such as acyclovir and ganciclovir (GCV), which blocks DNA synthesis and causes cell death. Human lung cancer cells have been shown to be selectively killed after transduction with retrovirus vectors carrying the HSV-tk gene and systemic administration of GCV [15]. As above, gene therapy provides a novel strategy and a new hope for the patients with cancer, although it is still not capable of completely eradicating malignant tumor cells in humans. The cancer gene therapy usually consists of two fields: the delivery system and the regulation system depend on promoter activities. In this study we focused on the non-viral delivery system and the promoter activities and they were discussed respectively in the following sections.
1.2 Delivery system for cancer gene therapy
The gene delivery systems can be divided into two categories: the viral vectors and non-viral vectors. Numerous viral or non-viral vectors for gene delivery to human body in vivo and in vitro have been developed. Whereas viral or non-viral vectors have certain advantages or limitations for themselves [16], they were both developing to reach available condition for the clinical treatment. In this study, we focused on the non-viral vectors as the gene delivery tool because of their safety, versatility and ease of preparation.
1.2.1 Viral vectors
Viruses are natural genetic material carriers that can efficiently introduce foreign DNA into specific cells. Viral vectors are available for clinical trials of gene therapy, such as almost 40% of all gene therapy clinical trials for cancer patients use adenoviral vector to deliver therapeutic genes [17]. Although recombinant adenoviral vectors have high titer advantage and are utilized for the clinical trials, their high immunogenecity for human impair their availabilities. Recently, adeno-associated virus (AAV) has been demonstrated that they are capable of inducing transgene expression in a broad range of tissues for a relatively long time without stimulation of a cell-mediated immune response [18]. However, the broad host tropism of AAV still remains a problem which impairs the availability in gene delivery. Moreover, the limited size of the genetic material through viral vectors is another limitation.
1.2.2 Non-viral methods
Investigators in non-viral vector development have introduced a variety of strategies to overcome barriers for gene delivery [19-21]. These include (a) polynucleotide degradation in the extracellular space, (b) internalization of the carrier, (c) intracellular trafficking from the endosome to the lysosome and the
escape of the polynucleotide from the endosome, (d) dissociation of polynucleotide from the carrier and (e) entry of the polynucleotide into the nucleus. Carrier molecules, such as polyethyleneimine (PEI), which can condense polynucleotides and provide protection against nucleases, is the major components of the delivery system. However, the positively charged DNA–cationic carrier complex tends to aggregate when injected into the blood and lacks tissue specificity. When entrapped in an acidic endosomal environment following endocytosis, components in the carrier that possess a proton sponge or endosomolytic activity will cause endosome rupture, thereby releasing the encapsulated polynucleotide. In this study, we used PEI as a gene delivery tool because of their ease of application.
1.2.2.1 Polyethyleneimine (PEI)
The cationic polymer polyethylenimine (PEI) has been widely used for non-viral transfection in vitro and in vivo and has an advantage over other polycations in that it combines strong DNA compaction capacity with an intrinsic endosomolytic activity. A large variety of different polymers and copolymers of linear, branched, and dendrimeric architecture, have been tested, in terms of their efficacy and suitability for in vitro transfection. It shows no morphology emerged as a general favorite [22]. The results from transfection experiments with PEI were impressive from the beginning. Depending on the linkage of the repeating
ethylenimine units, PEI occurs as branched or linear morphological isomers. Branched PEI derived vectors have been used to deliver oligonucleotides [23], plasmid DNA, and Epstein–Barr virus-based plasmid vectors [24] as well as RNA and intact ribozymes [25]. The efficacy of bPEI-derived vectors non-viral vectors and their cytotoxic effects depend to a remarkable extent on material characteristics like the molecular weight, the degree of branching, the cationic charge density and buffer capacity [26-28], polyplex properties, such as the DNA content, particle size and zeta potential and the experimental conditions like the polyplex concentration, the presence or absence of serum during transfection, the incubation time and the transfection model chosen for the gene delivery experiment. However, the use of PEI-derived gene delivery vehicles is still limited by a relatively low transfection efficiency and short duration of gene expression [29, 30]. The modification of PEI is a potential way to improve the therapeutic efficiency. Labeling of PEI/DNA complexes with receptor–ligand transferring could thereby enhance the gene expression in tumor cells, due to the efficient internalization of the transfecting complexes into the tumor cells via receptor-mediated endocytosis [31].
1.3 Promoters for cancer gene therapy
The specific expressions of therapeutic gene in different tumor cells are regulated by promoter sequences prior the transgenes. High specificity and efficacy
of transgene expression in cancer cells is not completely available until now. To address the purposes, the promoters of tumor associated antigen were used as tumor-specific promoters for gene therapy. Although the tumor-specific promoters are useful tools to accomplish specific expression in targeted tumor cells, low levels of gene expression is the chief defect of these tumor-specific promoters [32]. Several promoters were reported to specifically regulate certain expression of transgenes in different tumor cells [32-34] and these promoters may be classified as cancer specific promoters and tumor related promoters.
1.3.1 Cancer specific promoters
Cancer specific promoters are specific for the malignant process, such as telomerase related promoters. The activation of the telomerases activity is always considered as a critical step in cancer progression. The activities of telomerases exist in approximately 90% of human cancer cells, but are much lower or undetectable in normal somatic tissues [35-37]. Telomerase consists of an RNA component [human Telomerase RNA(hTR)] and a reverse transcriptase component [human telomerase reverse transcriptase (hTERT)] in human [38]. The hTR is not translated and remains as RNA and the hTERT functions as adding single-stranded telomere repeats into chromosome. Researchers had measured the expression levels of hTR and hTERT in a panel of 10 cell lines to demonstrate that the promoters of
hTR and hTERT are tumor-specific in tumor cells but not normal cells [39]. Moreover, there are cancer specific promoters oncofetally related with tissue specificity. Certain types of tumor often have genes overexpression of oncofetal origin that are silent in normal tissue. The most well-characterized promoters of these tumor-specific genes are the carcinoembryonic antigen (CEA) [40, 41] and α fetoprotein (AFP) [42, 43]. They are expressed in adenocarcinomas and hepatocellular carcinomas, respectively. These promoters have a potential in targeting a wide range of different tumor types and have been developed in cancer gene therapy.
1.3.2 Tumor related promoters
Tumor related promoters including tumor microenvironment-related promoters and tumor vasculature-related promoters. The former is responding to the tumor microenvironment and physiology such as hypoxia and glucose regulation. Many genes are transcriptionally upregulated in response to hypoxia which are mediated by the inducible transcription complex, hypoxia-inducible factor-1 (HIF-1). HIF-1 binds to hypoxia response elements (HREs) within these genes and activates the downstream gene expression. Therefore, HREs may be used to drive transgene expression specifically within tumor hypoxia areas. It is extremely important to target this population of cells since they are highly resistant
to other forms of treatment, such as radiotherapy and chemotherapy [44]. In addition to oxygen starvation, tumors can also be deprived of glucose that leads to the increased expression of genes involved in glucose metabolism. The promoters of these genes are also used to drive transgene expression specifically within a tumor [45, 46].
Another tumor related promoters are tumor vasculature-related promoters which are more active in the tumor vasculature than normal one. It has been reported that genes are upregulated in proliferating endothelium cells of tumor blood vessels [47]. The endothelial-specific kinase inserts domain receptor (KDR/flk-1) and E-selectin promoter have been indicated to enhance transgene expression in tumor endothelium [48]. Recently, it was demonstrated that the KDR/flk-1 promoter is not only endothelial cell-specific, but also actives in human ovarian cancer cell lines[49].
The use of cancer specific or tumor related promoters is promised to improve the safety of cancer gene therapy. However, the activities of these promoters are much weaker than the current benchmark CMV promoter [32]. The herapeutic efficacy might be limited when employing these kind of weak promoters.
1.4 Activities of NF-
κB, HIF-1, and CREB in cancer
1.4.1 Transcription factor binding sites for expression
Eukaryotic transcriptional regulatory factors are conducted synergistically by multiple transcriptional regulatory factors [50]. These factors can bind to the promoter regions called transcription factor binding sites (TFBSs). TFBSs are usually short (about 5-15 base-pairs) and they are frequently degenerate sequence motifs [51]. The sequence degeneracy of TFBSs has been selected through evolution and is beneficial, because it confers different levels of activity upon different promoters, causing certain genes in specific cells to be transcribed at higher levels than other cells [51]. Although the sequences of TFBSs are degenerated, they still have consensus sequences, such as NF-κB element consensus sequence 5’-GGGPuNNPyPyCC-’3, which can be recognized by specific transcription factors. The orientations and functions of TFBSs are not absolutely correlated. The positions within a promoter can be varied in yeast, and in higher eukaryotes they can be placed upstream, downstream, or in the introns of the genes which they regulate. In addition, they can be placed close to or far away from regulated genes [51]. When one transcription factor interacts with other transcription factors and results in high levels of a transcriptional activation, it is called “synergism or synergistic effect”. This phenomenon usually forms a ternary protein-protein-DNA complex which leads to altered DNA conformation and allowed other factors to bind on [52-54]. Interactions between two factors may be
direct or mediated by co-activators [40, 52]. For example, the coordination of c-Rel and ATF-1/CREB2 is mediated by p300/CREB-BP [53]. In some cases, two factors binding to DNA independently can still activate transcription synergistically [55-57]. A number of factors are known to bend the DNA structure and thus permit binding of other factors [58, 59]. For example, Fos and Jun can induce a corresponding alteration in the conformation of the DNA helix [59]. Furthermore, a variety of elements can contribute to promoter activity, but none is essential for all promoters [60]. Some transcription factors are specific in tissues and contributing to cell development [61]. Transcription factors play a major role in tumor progression. For example, NF-κB promotes cell cycle progression, regulates apoptosis, and facilitates cell adhesion [62]. Recently, many strategies have been used to enhance the potency of promoters needed to retain the tumor specificity in order to maintain potential therapeutic benefits. It is noticed that the transcription factors can recognize DNA sequence specifically and can be utilized in the promoter specificity. In next section, the roles and applications of NF-kB, HIF-1, and CREB in cancer progression and therapy will be discussed.
1.4.2 Nuclear factor-kappaB (NF-κB)
Nuclear factor-kappaB (NF-κB) is a common transcriptional factor that regulates many gene expressions. Many diseases are related to NF-κB, such as
cardiovascular diseases [63], muscular dystrophy [64], inflammatory diseases [65], and cancers [66]. In this section the relationship of NF-κB and cancers are discussed.
1.4.2.1 Biology of NF-κB
NF-κB was first found in B-lymphocytes [67] but NF-κB didn’t only restrict to B-lymphocytes. For example, the stimulation of NF-κB by lipopolysaccharide [68] or phorbol ester was observed in a T cell line [53] and a non-lymphoid cell line [69] [70]. NF-κB belongs to the Rel family transcriptional factors, including Rel-A (also known as p65), Rel-B, c-rel, p50/p105 and p52/p100 [71]. The mature DNA-binding forms of p105 and p100 are shortened forms called p50 and p52, respectively. Unlike most transcriptional factors, proteins of this family reside in the cytoplasm and must translocate into the nucleus to work [72]. All NF-κΒ proteins contain a highly conserved Rel-homology domain (RHD) that is responsible for DNA binding, dimerization, nuclear translocation and interaction with the IκΒ proteins. The IκΒ proteins, including IκΒα, β and ε, bind to NF-κΒ via ankyrin repeats and block its nuclear import and transcriptional activity [71]. Generally, NF-κB dimerization is the classical p50-p65 heterodimer which binds on the 5’-GGGANNYYCCC-3’ consensus sequence [40] to regulate gene expression. NF-κB can regulate many gene expressions, such as cytokines/chemokines, cell
adhesion molecules, acute phase proteins, and cell-surface receptors, regulators of apoptosis and transcription factors.
1.4.2.2 NF-κB in cancer progression
The NF-κB family might act as tumorigenic transcription factors was first put forward upon the cloning of the p50/p105 subunit [73, 74] and analyzed its sequence. Sequence analysis revealed remarkable homology for over 300 amino acids at the amino-terminal end to the oncogene, v-rel. The v-rel is a potent transforming oncogene from the avian reticuloendotheliosis virus [75]. In many cancers, aberrant activation and nuclear localization of NF-κB is actually quite frequent but most often results from defects in the pathways regulating NF-κB [76, 77]. IκB kinase (IKK) can inhibit IκB resulting in enhancing NF-κB activation [76, 77]. Some oncogenesis are correlated with the levels of IkBα and IkBβ proteins and coincided with the activation of IKK that govern the destruction of IkB factors [78]. Other ways, the loss of negative feedback mechanisms, which inhibit the NF-κB response, can result in its aberrant activity. An example of this is the CYLD tumor suppressor gene, which is associated with a predisposition to familial cylindromatosis (tumors of skin appendages). Losses of CYLD can lead to NF-κB activation [79]. In addition, the microenvironment of a solid tumor frequently contains high levels of inflammatory cytokines and/or hypoxic conditions, which
both stimulate nuclear translocation of NF-κB [76, 77]. The constitutive activation of NF-κB also appears to have a role in cell proliferation. NF-κB prevent Hodgkin's lymphoma cells from undergoing apoptosis under stress conditions [80]. It was further shown that growth factors such as epithelial growth factor [81] and platelet-derived growth factor induce proliferation of tumor cells through activation of NF-κB [82]. NF-κB signaling was also shown to promote pheochromocytoma 12 (PC12) cells survivals by nerve growth factor ligand, TrkA [83]. Recently, research has indicated that NF-κB possesses the prosurvival and antiapoptotic functions [84]. Several gene products that negatively regulate apoptosis in tumor cells, including inhibitor of apoptosis proteins (IAPs) 1 and 2, X-linked IAP, cellular Fas-associated death domain-like interleukin-1β converting enzyme (FLICE)-like inhibitory protein (cFLIP), were shown to be controlled by NF-κB activation [84]. The production of angiogeneic factors, such as vascular endothelial growth factor (VEGF) and Interleukin-8 (IL-8) has been shown to be regulating by NF-κB activation. NF-κB expression was associated with VEGF expression and microvessel density in human colorectal cancer [85]. IL-8 also activate by NF-κB. Bombesin (BBS)-like peptide treated PC-3 cell stimulated an NF-κB-dependent migration of human umbilical vascular endothelial cells in vitro by activating VEGF and IL-8[86]. These findings suggest that increased expression of NF-κB contributes to tumor angiogenesis in cancer.
1.4.3 Hypoxia-inducible-factors (HIFs)
Cancer cells always have a higher growth rate whereas their expansion relies on nutrient supply. Oxygen limitation is central in controlling neovascularization, glucose metabolism, survival and tumour spread. Hypoxia occurs when available oxygen falls below 5%, triggering a complex cellular and systemic adaptation mediated primarily through transcription by hypoxia-inducible factors (HIFs). HIF-1α was first identified as a crucial regulator of erythropoietin expression inresponse to low oxygen [87]. HIF-2α and HIF-3α have also been described, with HIF-3α, also known as IPAS (inhibitory PAS domain protein), functioning as an inhibitor of transcription [88, 89].
1.4.3.1 Biology of HIFs
HIF was shown in vitro, in a variety of cell culture systems, to be activated at a cut-off point of about 5% oxygen (40 mmHg), and to progressively increase its activity with a decrease in oxygen gradient down to 0.2–0.1% oxygen (1.6–0.8 mmHg), close to anoxia. HIF belongs to the large family of basic-helix–loop–helix (bHLH) proteins and is a heterodimer of a constitutively expressed and stable HIF-1β subunit, and one of three oxygen-regulated HIF-α subunits (HIF-1α, HIF-2α or HIF-3α). HIF-1α and HIF-2α, complexed with the b-subunits ARNT and (more rarely) ARNT2, bind DNA at hypoxia response elements (HREs) [90, 91].
HIF subunits are continuously transcribed and translated, and their stability is regulated by oxygen availability. HIF activation is a multi-step process involving HIF-α stabilization, nuclear translocation, heterodimerization, transcriptional activation and interaction with other proteins [92, 93].
1.4.3.2 HIFs in cancer progression
HIF can induce a vast array of gene products controlling energy metabolism, neovascularization, survival, pHi and cell migration, and has become recognized as a strong promoter of tumor growth [94]. The chemokine receptor CXCR4, a major metastatic mediator, is upregulated by HIF [95]. In addition, metalloproteinases (MMPs) 2 and 9 are regulated by hypoxia [96]. Another key mediator of metastasis is lysyl oxidase which is also a HIF target strongly associated with hypoxia. Inhibition of the lysyl oxidase blocks in vitro migration and in vivo metastasis from subcutaneous xenografts or after tail vein injection [97].HIF-1α is also associated with VEGF-C expression in invasive ductal carcinomas.
1.4.4 cAMP response-element binding protein (CREB)
cAMP response-element binding protein (CREB) has been found to mediate transcriptional responses to a variety of growth factor and stress signals. CREB regulate many gene expressions. Genome-wide studies put the number of putative
CREB target genes at about 5000, or nearly one-quarter of the human genome. CREB or related factorswhoseaberrant expression is often associated with certain cancers [98]. In this section, the relationship between CREB and cancer will be discussed.
1.4.4.1 Biology of CREB
CREB is a member of the CREB/ATF-1 (activating transcription factor 1)/CREM (CRE modulator) transcription factor family that mediates cyclic AMP (cAMP), growth factor-dependent, and calcium-dependentgene expression through the cAMP response element [99]. CREB is a 43-kDa basic/leucine zipper (bZIP) transcription factor that is expressed at the RNA levelin most tissues. CREB binds to the consensus octanucleotide CRE element (5’-TGANNTCA-3’) as a homodimer and heterodimers in conjunction with other members of the CREB/ATF superfamily of transcription factors [100]. In resting cells, CREB exists in the unphosphorylated state that is transcriptionally inactive but can still bind to DNA.Upon cell activation, CREB becomes phosphorylated, which induces its transcriptional activity by promoting its interaction with the 256-kDa co-activator protein CREB binding protein (CBP). CBP serves as a molecular bridge that allows CREB to recruit and stabilizethe RNA polymerase II complex at the TATA box, leading to switch certain genes on or off.
1.4.4.2 CREB in cancer progression
A potential role for the CREB family in cellular transformation was first appreciated in clear-cell sarcomas of soft tissues (CCSST) [101]. CCSST is an unusual malignancy of adolescents and young adults that typically arises in the deep soft tissues of the lower extremities close to tendon, fascia, and aponeuroses [102]. CCSST is typified by a chromosomal t(12;22)(q13;q12) translocation resulting in a fusion between the Ewing sarcoma gene (EWSR1) and activating transcription factor 1 (ATF1) [63]. The EWS–ATF1 can enhance expression of numerous CREB target genes by functioning as a strong activator. Indeed, disrupting EWS–ATF1 activity appears sufficient to block cell proliferation and promote cell apoptosis [63, 103]. Virally encoded oncoproteins such as hepatitis B virus and human T-cell leukemia virus (HTLV-1) tax also influence CREB activity in their efforts to promote cellular transformation [104, 105]. Based on this evidence, CREB will appear to cooperate with other factors, either in the context of a fusion protein or as part of a complex with an oncoprotein, to induce transformation. But whether CREB alone is capable of promoting tumorigenesis remained unclear [98].
The activity of many inducible transcription factors, such as NF-κB, is regulated through their association with cellular co-activators [106] . Interaction with the co-activator CREB binding protein (CBP) appears to be necessary to optimize the transcriptional activity of NF-κB. The interaction of the p65 (Rel A) subunit of NF-κB with CBP involves the KIX region of CBP, which is the same region responsible for binding the transcriptionally active serine-133-phosphorylated form of CREB [107, 108]. In human germline (GL) Iγ1promoter, NF-κB interacts with CREB to enhance gene expression. The Human Iγ1 promoter has NF-κB binding sites and CREB sites; they are communicating with each other via direct or indirect interactions. When using EMSA to observe NF-κB and CREB, it was found that the co-activator p300 interacts with CREB and NF-κB [109].
1.5 Strategy
Specific expression of the therapeutic gene in target cells depends on the specific delivery or the specific promoter activity. Either one of the two systems can be improved to become completely specific therapy without side effects. However, the both systems do not achieve the specific efficacy so far and the perfection of either system is extremely difficult. In this study we introduced a simple concept that the combination of partial specific delivery and partial specific promoter activity may achieve more specificity for target cells (Figure 1). Besides,
this strategy can be done in a rapid and convenient fashion. The first part in our study is to rapidly create a novel promoter based on the activities of transcription factors. The transcription factors which are important in cancer progression will be roughly assayed in several tumor or rapid-proliferating cells. The response elements with higher activities in tumor cells will be processed to create a novel mini-promoter. This transcription factor-based synthetic promoter (TSP) which consists of several kinds of response elements might be flexible and partial specific in tumor cells. The second part is to enhance the delivery efficiency of PEI by a convenient method of peptide absorption. The multi-functional peptide RGD-4C-HA possesses the ability of specific targeting and can absorb to PEI. RGD-4C-HA contains RGD-4C sequence which was proved to specifically bind to integrin αvβ3 [110-112]. In addition RGD-4C-HA contains a negatively charged tail
which can absorb to the positively charged PEI by electrostatic forces. This modification of PEI is rapid and convenient in laboratory compared to the complicated chemical coupling or modification of the functional groups. RGD-4C-HA should improve the delivery efficiency and specificity of PEI for integrin αvβ3 expessing cells such as B16-F10 cells. The partial specific promoter
and the partial specific delivery system can be developed in a rapid and convenient method as described above. Finally, the combination of the two systems should achieve more specificity than either system alone.
Chapter 2 Materials & Methods
2.1 Materials
2.1.1 Primers
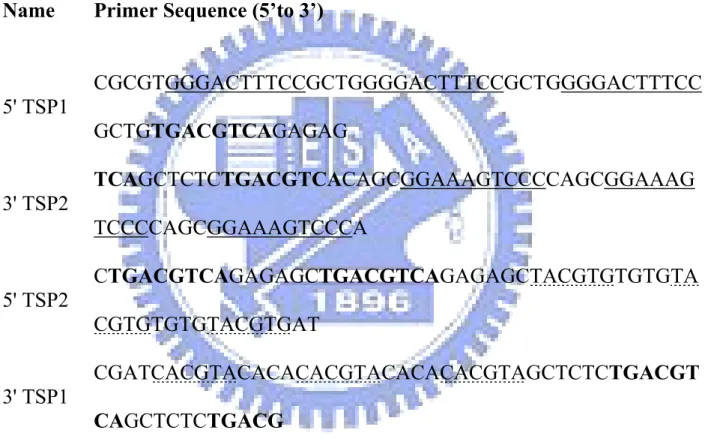
Table 1: Primers used in this study Name Primer Sequence (5’to 3’)
5' TSP1 CGCGTGGGACTTTCCGCTGGGGACTTTCCGCTGGGGACTTTCC GCTGTGACGTCAGAGAG 3' TSP2 TCAGCTCTCTGACGTCACAGCGGAAAGTCCCCAGCGGAAAG TCCCCAGCGGAAAGTCCCA 5' TSP2 CTGACGTCAGAGAGCTGACGTCAGAGAGCTACGTGTGTGTA CGTGTGTGTACGTGAT 3' TSP1 CGATCACGTACACACACGTACACACACGTAGCTCTCTGACGT CAGCTCTCTGACG
The primers were purchased from commercial (MDBio, Taiwan, ROC, ROC). The binding sites of NF-kB (underlined), CREB (bold), and HIF-1 (dotted) were labeled.
Table 2: Cell lines used in this study
Cell line Description ATCC #
B16-F10 mouse melanoma cells CRL-6475 Balb/3T3 mouse embryo fibroblast cells CCL-163 HeLa human cervical carcinoma cells CCL-2
2.1.3 Plasmids
Table 3: Plasmids used in this study
Plasmid Description Source
pAAV-MCS With multiple cloning site
Stratagene, Cedar Creek, TX
pAAV-MCS-hrGFP
With humanized renilla green fluorescent protein
From Dr. Liao’s Lab
pAP-1-hrGFP
Containing 7 copies of AP-1 binding site
Stratagene, Cedar Creek, TX
pARE-hrGFP AmpR assay plasmid From Dr. Liao’s Lab
pAsRed2-N1 With red fluorescent protein
Becton Dickinson, Moutain View, CA
pCRII-hrGFP
Containing 7 copies of HIF-1 binding site
From Dr. Liao’s Lab
pCRE-hrGFP
Containing 4 copies of CREB binding site
Stratagene, Cedar Creek, TX
pD5-hrGFP With synthetic promoter From Dr. Liao’s Lab
pNF-κB-hrGFP Containing 5 copies of NF-κB binding site
Stratagene, Cedar Creek, TX
pNFAT-hrGFP
Containing 4 copies of NFAT binding site Stratagene, Cedar Creek, TX MZF-1-hrGFP Containing 3 copies of MZF-1 binding site
From Dr. Liao’s Lab
2.1.4 Chemicals, enzymes, and reagents
Table 4: Chemicals, enzymes, and reagents used in this study
Chemical Company
100 bp DNA ladder
Protech, Taiwan, ROC
ROC
Acetic acid
Showa, Tokyo, Japan
Adenosine triphosphate (ATP)
Epicentre, Madison, WI Agar Amresco, Solon, Ohio Agarose MDBio, Taiwan, ROC
Albumin bovine Fraction V (BSA)
MP Biomedicals, Irvine, CA
Ampicillin
Amresco, Solon, Ohio
ApaI (restriction enzyme) Promega, USA
BamHI (restriction enzyme)
Fermentas,
Burlington, Canada
Bsu15I (ClaI)
Fermentas,
Burlington, Canada Calcium chloride, dyhidrate J.T.Baker,
Phillipsburg, NJ
Coomssie Brilliant blue
Amresco, Solon, Ohio
Deoxy-nucleotide triphosphates (dNTP) Promega, USA
Dimethyl sulfoxide (DMSO)
MP Biomedicals, Irvine, CA
Disodium hydrogen phosphate anhydrous (Na2HPO4)
Scharlau, Barcelona, Spain
Dulbecco’s modified Eagle’s medium (DMEM)
Sigma, St. Louis, MO
Ethanol
Sigma, St. Louis, MO
Ethidium bromide (EtBr)
Amresco, Solon, Ohio
Ethylenediaminetetraacetic acid (EDTA) Tedia, Fairfield, OH
Fetal bovine serum
Biological
Industries, Kibbutz Beit Haemek, Israel
Japan Glycine Amresco, Solon, Ohio HindIII Fermentas, Burlington, Canada Hydroboric acid (H3PO4) Riedel-de Haën, Seelze, Germany Hydrochloric acid (HCl) Scharlau, Barcelona, Spain Isopropanol C-Echo, Taiwan, ROC Kanamycin MDBio, Taiwan, ROC LipofectamineTM 2000 Invitrogen, Leek, The Netherlands
Luria Bertani (LB) agar
Amresco, Solon, Ohio
Luria Bertani (LB) broth
Scharlau, Barcelona, Spain
Methanol C-Echo, Taiwan, ROC MluI Fermentas, Burlington, Canada Ninhydrin Sigma, St. Louis, MO
Penicillin-streptomycin amphotericin B (PSA)
Biological
Industries, Kibbutz Beit Haemek, Israel
Pfu polymerase MDBio, Taiwan, ROC Polyethyleneimine Aldrich, St. Louis, MO
Potassium acetate (KOAc)
Showa, Tokyo, Japan
Potassium chloride (KCl)
Showa, Tokyo, Japan
Propidium iodide (PI)
Sigma, St. Louis, MO
Sephacryl S-200
GE Healthcare, Chalfont St., UK
Sodium azide (NaN3)
Showa, Tokyo, Japan
Sodium dihydrogenphosphate dihydrate (NaH2PO4.
2H2O) Showa, Tokyo, Japan Sodium chloride Amresco, Solon, Ohio
Sodium hydrogen carbonate (NaHCO3)
MP Biomedicals, Irvine, CA
Sodium hydroxide (NaOH)
Showa, Tokyo, Japan T4 kinase buffer Fermentas, Burlington, Canada T4 ligase (2U) Epicentre, Madison, WI T4 ligase (10U) Epicentre, Madison, WI
WI
T4 polynucleotide kinase NEB, Hitchin, UK
Taq polymerase
BioKit, Taiwan, ROC
Taq DNA polymerase XL
Protech, Taiwan, ROC Tris base MDBio, Taiwan, ROC Tris-HCl MP Biomedicals, Irvine, CA
Trypan blue stain
Gibco, Grand Island, NY
Trypsin
Gibco, Grand Island, NY
Tryptone CONDA, Spain
Tween 20
MP Biomedicals, Irvine, CA
XhoI (restriction enzyme)
Fermentas,
Yeast extract
Conda, Madrid, Spain
2.1.5 Antibodies
Table 5: Antibodies used in this study
Antibody Description Company
Anti-HA-fluorescein, high affinity (3F10)
Recognizing the HA peptide sequence [YPYDVPDYA] Roche, Basel, Switzerland Polyclonal rabbit anti-mouse IgG/HRP Secondary antibody recognizing the mouse IgG
DakoCytomatio n, Glostrop, Denmark
2.1.6 Kits
Table 6: Kits used in this study
Kit Company Used in
Geneaid gel/PCR DNA fragments extraction kit
Geneaid, Taiwan, ROC
DNA extraction, clean-up
Duran, Germany SuperSignal West Pico
Chemiluminescent Substrate Pierce, Rockford, IL The substrate of HRP in dot blot
2.1.7 Buffers
Table 7: Buffers used in this study
Buffer Description Used in
1X PBS 137 mM NaCl, 10 mM Na2HPO4, 2.7
mM KCl, 1.8 mM KH2PO4, pH7.4
Cell culture
5% Blocking buffer
5%(w/v) non-fat powdered milk in 1X PBS buffer
Dot blot
50X TAE buffer 48.4 g Tris base, 0.5 M EDTA (pH8.0) 20 ml, 11.42 ml acetic acid. dd H2O was
added to 200 ml.
Gel
electrophores is
Buffer S1 50 mM Tris-HCl, 10mM EDTA, 100μg/ml RNase A, pH8.0
Midi
preparation Buffer S2 200 mM NaOH, 1% SDS Midi
Buffer S3 2.8 KAc, pH 5.1 Midi
preparation Buffer N2 100 mM Tris,15% ethanol, 900 mM KCl,
0.15% Triton X100, adjusted to pH 6.3 with H3PO4
Midi
preparation
Buffer N3 100 mM Tris, 15% ethanol, 1M KCl, adjusted to pH6.3 with H3PO4
Midi
preparation Buffer N5 100 mM Tris, 15% ethanol, 1M KCl,
adjusted to pH 8.5 with H3PO4
Midi
preparation EDTA-trypsin 2.5 g trypsin, 0.1 M EDTA (pH8.0) in 1L
1X PBS, pH7.4, 0.2 μm filtered
Cell culture
PBST 0.05% Tween 20 in 1X PBS Dot blot Solution I 50mM Tris-HCl, 10mM EDTA , 10mg/ml
RNase A, pH=8.0
Mini
preparation Solution II 0.2M NaOH, 1%(w/v)SDS Mini
preparation Solution III 2.8M potassium acetate, pH=5.1 Mini
preparation Staining buffer 1% BSA , 0.05% NaN3 in 1X PBS
Versene 0.2g EDTA in 1L 1X PBS Cell culture
2.1.8 Media
Table 8: Media used in this study
Media Description Used in
DMEM growth medium
10% FBS, 1% PSA in Dulbecco’s Modified Eagle’s Medium
Cell culture
LB (Luria-Bertani) broth
1% tryptone, 0.5% yeast extract, 1% NaCl Bacteria culture LB (Luria-Bertani)/Ampi cillin agar
1% tryptone, 0.5% yeast extract, 1% NaCl, 1.5% agar, 50μg/ml ampicillin
Bacteria culture
LB
(Luria-Bertani)/Ampi cillin broth
1% tryptone, 0.5% yeast extract, 1% NaCl, 50μg/ml ampicillin Bacteria culture LB (Luria-Bertani)/Kana mycin agar
1% tryptone, 0.5% yeast extract, 1% NaCl, 1.5% agar, 30μg/ml kanamycin
Bacteria culture
(Luria-Bertani)/Kana mycin broth
NaCl, 30μg/ml kanamycin culture
Opti-MEM I Medium without serum Cell culture
SOB broth
2% tryptone, 0.5% yeast extract, 0.05% NaCl, %0.0186 KCl, 10mM MgCl2
Bacteria culture
2.1.9 Equipment
Table 9: Equipment used in this study
Equipment Company
–20°C low temperature refrigerator
Frigidaire, Pittsburgh, PA
–80°C low temperature refrigerator
Nuaire, Caerphilly UK 4°C refrigerator MINI KINGCON, Taiwan, ROC Biophotometer DPU-414 Eppendorf, Hamburg, Germany Centrifuge 5415D Eppendorf, Hamburg, Germany
Centrifuge 5804 R
Eppendorf, Hamburg, Germany
DNA electrophoresis unit Gel Mate 2000 Toyobo, Japan
Dot-blot machine Bio-East, Taiwan, ROC Econo column 737-0722 Bio-Rad, Taiwan, ROC
Flow cytometer, FACScan
Becton Dickinson, Moutain View, CA
Heating plate Firstek, Taiwan, ROC
Inverted research microscope, IX71
Olympus, Tokyo, Japan
Biological safety cabinet, Forma Class II, A2 Thermo, USA
Lead blocker Okamoto, Fukuyama, Japan Microscope, CX31 Olympus, Tokyo, Japan
Orbital Shaking incubator OS1500R TKS
Thermal cycler
Eppendorf, Hamburg, Germany
Uni-photo gel image system EZ lab, Taiwan, ROC Water bath Firstek, Taiwan, ROC
2.2 Methods
2.2.1 Construction of transcription factor-based synthetic promoter
(TSP)
The pD5-hrGFP was obtained by replacing the CMV promoter of pAAV-MCS-hrGFP with TSP. Briefly, the vector pAAV-MCS-hrGFP was double digested by MluI and ClaI (Fermentas, Burlington, Canada) to eliminate the CMV promoter. TSP was obtained by direct ligation of insert1 and insert2. The insert1 and insert2 were obtained by primer annealing. The primers 5' TSP1 (5’-CGC GTG GGA CTT TCC GCT GGG GAC TTT CCG CTG GGG ACT TTC CGC TGT GAC GTC AGA GAG-3’) and 3’ TSP2 (5’-TCA GCT CTC TGA CGT CAC AGC GGA AAG TCC CCA GCG GAA AGT CCC CAG CGG AAA GTC CCA-3’) were heated to 95°C for 5 minutes and then cooled down to room temperature. The insert2 was obtained by annealing of 5’ TSP2 (5’- CTG ACG
GTA CGT GAT-3’) and 3’ TSP1 (5’- CGA TCA CGT ACA CAC ACG TAC ACA CAC GTA GCT CTC TGA CGT CAG CTC TCT GAC G -3’) as described above. The primers contained three copies of the binding sites of NF-kB (underlined), CREB (bold), and HIF-1 (dotted). Each binding site was separated by at least a 4-nucleotides spacer according to the commercial design (Stratagene, Cedar Creek, TX). The 5’ end of insert1 was designed as a MluI protruding end and the 3’ end of insert2 was designed as a ClaI protruding end. The inserts were phosphorylated by T4 polynucleotide kinase (NEB, Hitchin, UK) according to the manufacturer’s protocol. The vector and inserts (insert1 and insert2) were then ligated with a molar ratio 1:5:5 or 1:10:10 at 16°C for 16 hours. After 16 hours incubation, the ligation products were transformed into DH5α competent cells by heat shock method. The colonies were picked and checked by restriction enzyme digestion. The correct clone pD5-hrGFP was then obtained and sequenced.
2.2.1.1 Restriction enzyme digestion
The restriction enzyme digestion of DNA was performed following the manufacturer’s protocol (Fermentas, Burlington, Canada). Generally, 1 μg DNA was digested with 5 unit of restriction enzyme in a 10 μl volume reaction at 37°C overnight.
2.2.1.2 DNA extraction
After digestion by restriction enzyme, the DNA was cleaned up by Geneaid gel/PCR DNA fragments extraction kit (Geneaid, Taiwan, ROC) following the manufacturer’s protocol.
Briefly, the digestion product was spun at 13,000 rpm for 30 seconds in the spin column. The filtrate in the collection tube was discarded. 700 μl Washing buffer (Geneaid, Taiwan, ROC) was added and the solution was spun at 13,000 rpm for 1 minute. This step was repeated twice. The filtrate was discarded by centrifugation at 13,000 rpm for 3 minutes to remove residual trace of ethanol. The column was additionally incubated at 65°C for 5 minutes to evaporate ethanol. The DNA was eluted by 30 μl ddH2O in a new tube and stored at -20°C.
2.2.1.3 Ligation
The ligation reaction was performed following the manufacturer’s protocol (Epicentre, Madison, WI). Briefly, 500 μg vector was used in a 10 μl volume reaction with 1mM ATP. The molar ratio of the vector and the inserts (insert1 and insert2) was 1:5:5 or 1:10:10. The mixture was then incubated at 16°C for 16 hours.
2.2.2 Transformation of E. coli
Single colony of E. coli was inoculated in 3 ml of LB broth and grew for 12 hours at 37°C with agitation until the OD600 was between 0.35~0.45 (about 12
hours). 1 ml of the overnight culture was transferred into 100 ml LB broth and was then incubated at 37 °C with agitation until the OD600 was between 0.35~0.45. The
cells were havested by centrifugation at 4100 rpm for 10 minutes and then re-suspended in 30 ml ice-cold 0.1M CaCl2. The cells were pelleted by
centrifugation at 4100 rpm for 10 minutes. The pellet was re-suspended in 2 ml 0.1M CaCl2 containing 10% glycerol. The cells were dispensed at 100 μl per tube
and stored at -80°C.
2.2.2.2 Transformation of competent cell by heat shock method
Stored competent cells were thawed on ice. 1 ng DNA was mixed with 100 μl competent cells and was then stored on ice for 30 minutes. The mixture was incubated in a preheated 42°C heating block for 90 seconds and quickly placed on ice for 2 minutes. Then 250 μl of LB broth was added to the cells. The culture was incubated at 37°C with shaking for 50 minutes. 100 μl of the culture was plated on the LB agar plate with 50μg/ml ampicillin or 30μg/ml kanamycin. The plate was incubated at 37°C for 16 hours later.
2.2.3.1 Minipreparation method
A single colony of E. coli was inoculated in 3 ml of LB broth (with antibiotics) and allowed to grow overnight at 37°C with agitation. 1 ml culture was recovered by centrifugation at 13,000 rpm for 1 minute and then re-suspended in 200μl ice-cold Solution I buffer in a new tube. 250 μl Solution II buffer was added and mixed gently. After 3 minutes, 250 μl Solution III buffer was added to the mixture and mixed gently until a homogeneous suspension containing an off-white flocculate was formed. The mixture was incubated on ice for 5 minutes and then spun at 13,000 rpm for 5 minutes at 4°C. The supernatant was transferred to a fresh tube. Equal volume of phenol: chloroform (700 μl) was added. The organic and aqueous phases were mixed by vortex and then the emulsion was centrifuged at 13,000 rpm for 3 minutes at 4°C. The aqueous upper layer was transferred to a fresh tube. Nucleic acids from the supernatant were precipitate by adding 0.7 volumes of isopropanol at room temperature. The solution was mixed completely and incubated for 2 minutes at room temperature. The precipitated DNA was collected by centrifugation at 13,000 rpm for 20 minutes at 4°C. The supernatant was removed by gentle aspiration. 1 ml of 70% ethanol was added to the pellet and the DNA was recovered by centrifugation at 13,000rpm for 5 minutes at 4°C. The supernatant was removed by gentle aspiration and the tube was incubated opened at room temperature to evaporate ethanol (7 minutes). The DNA was dissolved in 50
μl ddH2O and vortexed gently for few seconds. The products were stored at -20°C.
2.2.3.2 Midipreparation method
The midipreparation was performed by NucleoBond PC 100 kit (Macherey-Nagel, Duran, Germany) following the manufacturer’s protocol. Briefly, a single colony of E. coli was inoculated in 100 ml of LB broth (with antibiotics) and grew overnight at 37°C with agitation. The cells were recovered by centrifugation at 8,000 rpm for 15 minutes at 4°C. The pellet was collected, and 4 ml buffer S1 (Macherey-Nagel, Duran, Germany) was added to dispense the pellet. Then 4 ml buffer S2 (Macherey-Nagel, Duran, Germany) was added to the suspension. The lysate was mixed gently and incubated at room temperature for 3 minutes (no more than 5 minutes). The pre-cooled 4 ml buffer S3 (Macherey-Nagel, Duran, Germany) was then added to the solution and mixed gently until a homogeneous suspension containing an off-white flocculate was formed. The mixture was incubated on ice for 5 minutes and then spun at 13,000 rpm for 25 minutes at 4°C. The supernatant was loaded onto the NucleoBond AX 100 Midi column which was equilibrated with 2.5 ml buffer N2 (Macherey-Nagel, Duran, Germany). The flow-through was emptied by gravity flow and discarded. 10 ml buffer N3 (Macherey-Nagel, Duran, Germany) was added to wash the column twice. The DNA was eluted by 5 ml buffer N5 (Macherey-Nagel, Duran, Germany).
Then 3.5 ml isopropanol was added to precipitate the DNA. The mixture was incubated on ice for 10 minutes and recovered by centrifugation at 13,000 rpm for 30 minutes at 4°C. 6 ml 70% ethanol was added to the pellet and the solution was spun at 13,000 rpm for 5 minutes. Finally, the pellet was dissolved in appropriate amount of ddH2O and stored at -20°C.
2.2.4 Cell culture
All cells were cultured following the ATCC’s instructions. Generally, cells were cultured in DMEM (Sigma, St. Louis, MO) supplemented with 10% FBS (Biological Industries, Kibbutz Beit Haemek, Israel) and 1% PSA (Biological Industries, Kibbutz Beit Haemek, Israel). Cells were incubated in tissue culture incubator with 5% CO2 at 37°C. All cells were subcultured to ensure that the
confluency was no more than 80%.
2.2.4.1 Procedures of subculture
All cells were passaged following the ATCC’s instructions. Generally, the culture medium was removed and discarded. The cell layer was briefly rinsed with 1X PBS to remove all traces of serum that contains trypsin inhibitor. 3 ml Trypsin-EDTA solution was added to the flask for about 5 minutes until the cell layer was dispersed. The cells were centrifuged at 1,200rpm for 5 minutes at 4°C
and were re-suspended by 2 ml growth medium. Appropriate aliquots of the cell suspension were added to a new culture vessel. Cells were incubated in tissue culture incubator with 5% CO2 at 37°C.
2.3 Transcription factors and TSP activity assay
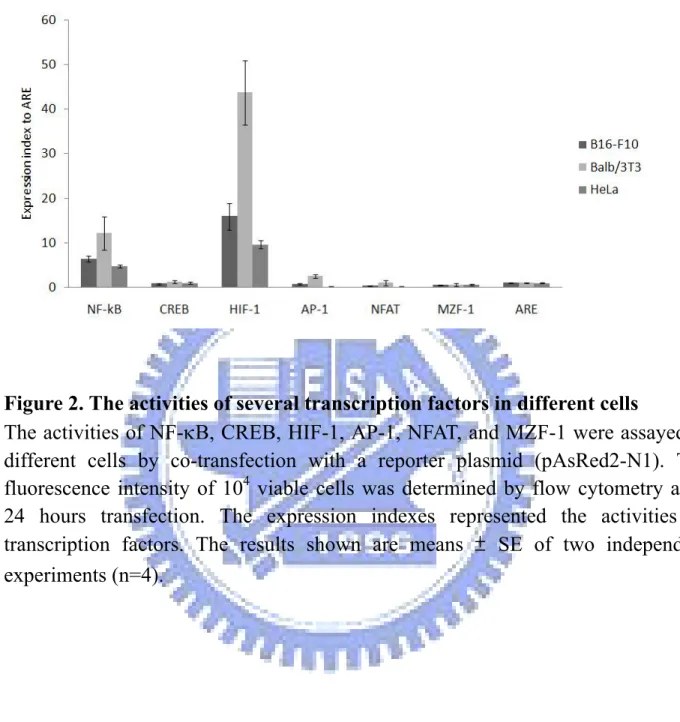
The transcription factors assay were performed to measure the activities of several transcription factors and TSP in different cells. Briefly, the assay plasmids (pAP-1-hrGFP, pCRII-hrGFP, pCRE-hrGFP, pD5-hrGFP, pNF-κB-hrGFP, pNFAT-hrGFP , MZF-1-hrGFP) and a control plasmid (pARE-hrGFP) were co-transfected with a reporter plasmid (pAsRed2-N1) into B16-F10, Balb/3T3, and HeLa cells. The assay plasmids pAP-1-hrGFP, pCRII-hrGFP, pCRE-hrGFP, pNF-κB-hrGFP, pNFAT-hrGFP, and MZF-1-hrGFP each contained 7, 7, 4, 5, 4, and 3 copies of the responding binding site respectively. ARE was a binding site of a prokaryotic transcription factor ampR and it was used as a negative control group. In addition, the reporter plasmid (pAsRed2-N1) with a transgene encoding the red fluorescent protein driven by CMV promoter was used to normalize the transfectant efficiency between each sample. 24 hours after transfection, the gene expressions were measured by FACScan flow cytometry (Becton Dickinson, Moutain View, CA).
2.3.1 Transfection of mammalian cells
2.3.1.1 Seeding cells
The culture medium was removed and discarded. The cell layer was briefly rinsed with 1X PBS and then 3 ml Trypsin-EDTA solution was added to the flask for about 5 minutes until the cell layer was dispersed. The pellet was recovered by centrifugation at 1,200rpm for 5 minutes at 4°C. The supernatant was discarded. Cells were re-suspended by 2 ml growth medium. Certain amount of cells was stained by trypan blue and calculated by a bright-line chamber (Marienfeld, Germany). Appropriate cells were plated in 6-well or 24-well plate and incubated in tissue culture incubator with 5% CO2 at 37°C.
2.3.1.2 Polyethyleneimine transfection
105 cells were plated in 24-well plate to be approximately 50% confluent at the time of transfection. Cells were transfected with different plasmid DNA by polyethyleneimine (PEI). Briefly, 1 μg plasmid DNA and 6 μl of 5μM PEI (Aldrich, St. Louis, MO) were each diluted into 50 μl of 150mM NaCl and vortexed. The PEI solution was added into DNA solution after 5 minutes (Notice: not the reverse order), and thenvortexed. After 20 minutes, the cells were rinsed and supplemented with 200 μl Opti-MEM I Medium (Gibco, Grand Island, NY). The PEI-DNA
mixture was gently added to each well. After 18 hours incubation, 700 μl fresh growth medium were added into each well. After 24~48 hours, the gene expressions were measured by FACScan flow cytometry (Becton Dickinson, Moutain View, CA).
2.3.1.3 LipofectamineTM 2000 transfection
2×105 cells were plated in 24-well plate to be approximately 80% confluent at the time of transfection. Cells were transfected with different plasmid DNA by LipofectamineTM 2000 (Invitrogen, Leek, The Netherlands). The transfection procedure was performed according to the manufacturer’s protocol. Briefly, 3 μg DNA was diluted in 250 μl Opti-MEM I Medium (Gibco, Grand Island, NY) and mixed gently. 10 μl LipofectamineTM 2000 was gently mixed with 250 μl Opti-MEM I medium and incubated for 5 minutes at room temperature. The diluted DNA was combined with the diluted LipofectamineTM 2000 for 20 minutes at room temperature. The medium in the cells were discarded and cells were gently washed with Opti-MEM I medium twice. The DNA- LipofectamineTM 2000 mixture was added into each well gently. 500 μl Opti-MEM I medium was added into each well gently and the cells were incubated at 37°C in a CO2 incubator for 12 hours. 2 ml
transfection. After 24 hours, the gene expressions were measured by FACScan flow cytometry (Becton Dickinson, Moutain View, CA).
2.3.2 Measurement of reporter gene expression by flow cytometry
After 24 hours transfection, cells were harvested to measure the gene expression. Briefly, the medium was discarded and each well was rinsed with 1 ml PBS. 1ml versene or trypsin was then added, and the cells were incubated at 37°C for 5 minutes. 1 ml growth medium was added into each well and the cells were recovered by centrifugation at 1,500rpm for 5 minutes at 4°C. The supernatant was discarded and the pellet was re-suspended by 1ml staining buffer in FACS tube. The reporter gene expression was measured by FACScan flow cytometry (Becton Dickinson, Moutain View, CA). Fluorescence intensities were analyzed with CELLQUEST software (Becton Dickinson).
2.4 RGD-4C-HA binding assay
The culture medium was removed and discarded. The cell layer was briefly rinsed with 1X PBS to remove all traces of serum that contains trypsin inhibitor. 5 ml versene was added to the flask (Notice: the use of trypsin might digest the ligands on cell membrane). After centrifugation to remove versene and the
measurement for the cell number, 2×105 cells were suspended in staining buffer (1% BSA, 0.05% NaN3 in 1X PBS) containing different concentrations (2μM,
200nM or 20nM) of RGD-4C-HA peptide (CDCRGDCFCGGGYPYDVPDYAGGGDDDEC which was purchased from
MDBio, Taiwan, ROC) at 4°C for 1 hour. After 1 hour incubation, cells were washed, suspended in staining buffer with anti-HA-FITC and incubated at 4°C for 1 hour. After 1 hour incubation, cells were washed and the surface immunofluorescence was measured by FACScan flow cytometer (Becton Dickinson, Moutain View, CA). Fluorescence intensities were analyzed with CELLQUEST software (Becton Dickinson, Moutain View, CA).
2.5 The absorption of RGD-4C-HA to PEI assay
2.5.1 Separating PEI-peptide complex with un-absorbed PEI and RGD-4C-HA by gel filtration column S-200
The PEI and RGD-4C-HA were incubated at room temperature for 30 minutes at a molar ratio 1:1. The PEI-peptide complex was applied into a gel filtration column which was packed with Sephacryl S-200 (GE Healthcare, Chalfont St., UK) following the manufacturer’s protocol to separate the un-absorbed products. Briefly, the S-200 gel was first equilibrated to room temperature, and gently shaken to make slurry. The homogeneous suspension was poured into an empty glass column.
The column was packed following two steps:
STEP 1: The column was packed at 0.5 ml/min for 2 hours. STEP 2: Increased the flow rate to 0.9 ml/min for 1 hour.
The elution products were collected 0.5 ml per fraction by fraction collector.
2.5.2 Ninhydrin test
The elution products were put onto a thin layer chromatography (TLC) plate. The 15% ninhydrin solution (solved in methanol) was then put onto the TLC plate. After 10 minutes incubation, the TLC plate was pictured.
2.5.3 Dot-blotting
The elution products were applied onto the nitrocellulose (NC) paper (Pall, USA) which was rinsed with 1X PBS buffer on a dot-blot machine (Bio-East, Taiwan). Samples were gently for 10 minutes. The NC paper was blocked by 5% of skin milk at 4°C overnight or 37°C for 3 hours with shaking. The paper was then washed with 1X PBS containing 0.05% Tween 20 three times at room temperature for 5 minutes. The anti-HA antibody (Roche, Basel, Switzerland) were diluted 200X in 5% blocking buffer (5% skin milk in 1X PBS buffer) and applied onto the NC paper gently at room temperature for 1 hour with shaking. The mixture was then discarded. The NC paper was washed with 0.05% PBST three times at room