Theory
of
doubly
resonant infrared-visible
sum-frequency
and difFerence-frequency
generation
from
adsorbed molecules
JungY.
HuangInstitute
of
Electro Op-tical Engineering, Chiao Tung University, Hsinchu, Taiwan, Republic ofChinaY.
R.
ShenDepartment
of
Physics, Universityof
California atBerkeley, Berkeley, California 94720 and Materials Sciences Division, Lawrence Berkeley Laboratory, Berkeley, California 94720(Received 20 December 1993)
We theoretically analyze doubly resonant infrared-visible sum-frequency (DR IVSFG)and diference-frequency generation (DR IVDFG) from a monolayer ofadsorbates at an interface. Our calculated re-sults with a model molecule indicate that the resonant amplitude ofnonlinear optical susceptibility of DR IVDFG and DRIVSFGprocesses conveys information about the electron-vibration coupling in ad-sorbates. Moreover, owing to a dephasing-rephasing procedure involved, DR IVDFG can also be developed into asensitive probe forinvestigating the coherent phase relaxation process in adsorbed mol-ecules without the complication ofinhomogeneous broadening.
PACSnumber(s): 42.50.Md, 68.35.Ja
I.
INTRODUCTIONMolecules at an interface often behave differently than they do in bulk. This may be ascribed to the fact that
molecules at surfaces are better oriented
[1]
and theirelectronic structures are modified by the interaction with substrate
[2].
Therefore probing the orientation andelec-tronic structures
of
molecules at an interface becomes an essential step for better understandingof
the surface dy-namics and chemistryof
adsorbates.Recently, second-order nonlinear optical effects have been demonstrated
to
be an effective and versatile probeof
the static [3,4] and dynamic [5,6] propertiesof
mole-cules and surfaces. During this investigation, various mechanismsof
resonance enhancement were used to im-prove the spectroscopic capabilityof
these techniques. In fact, fairly good agreement between the theoreticalcalcu-lation and experimental result
of
resonant second-harmonic generation (SHG) from molecular systems has been achieved[7].
Unfortunately, the line shapeof
reso-nant SHG is often too broad to be sensitive enough foridentifying molecular species. This, however, has been shown to be remedied by a newly developed technique called infrared-visible sum-frequency generation (IVSFG)
[3].
IVSFG
isalso a second-order nonlinear optical process in which two input laser beams, one at the infrared fre-quency ~& and the other at the visible frequency co2, in-teract and generate an output at the sum-frequency~,
=co,
+co2 in the visible spectrumof
light. TheIVSFG
signal from an interface can be resonantly enhancedif
co&approaches a surface vibrational resonance. The
enhanced
IVSFG
signai thus carries the vibrationalspec-troscopic information about the interface.
It
is notedthat the signal strength
of IVSFG
can be furtherin-creased by an electronic resonance when either the input visible frequency (co@)
or
output sum frequency isnear anelectronic transition. Because the electronic and vibra-tional transitions are excited simultaneously in this dou-bly resonant process,
IVSFG
near double resonance is ideal for investigating electron-vibration coupling inad-sorbates. Such acoupling is known
to
play an important role in the coherent evolutionof
a vibrational wave pack-etin anelectronic state.The ability toprobe the coherent evolution
of
asurfaceexcitation is crucial for understanding the dynamics and surface reaction
of
adsorbed molecules[8].
In the past,this was conducted by probing the phase relaxation pro-cesses through the measurement
of
the excitational linewidth[9].
Theoretical efFort to understand suchsur-face relaxation isthen focused on relaxation models that can produce the measured spectral line shape
[10].
How-ever, the widthof
aspectral line is often dominated by in-homogeneous broadening.To
find the homogeneous linewidth,i.
e, the inverseof
the dephasing time, it is necessary to resortto
spectroscopic techniques capableof
suppressing inhomogeneous broadening[11]
or
directly measure the phase relaxation rate in time domain with coherent optical processes such as photon echoes[12].
A one-to-one correspondence between each wave-mixing process capable
of
producing spectra with re-duced inhomogeneous broadening and photon-echopro-cesses was established
[13].
In a recent study, Shenfur-ther pointed out that doubly resonant difference-frequency generation
(DR
DFG)
in the time domain with properly time-ordered input pulses can yield an output in the formof
arephased echo[14].
This immediately sug-gests that doubly resonant infrared-visible difference-frequency generation(DR IVDFG)
be used to probe the coherent phase relaxationof
surface vibrations without resorting tohigher-order nonlinear optical processes[25].
In this paper, we consider this aspect
of
DR
IVDFG
andits use as the probe
of
the coupling between electronic state and vibrational modesof
adsorbed molecuies atsur-faces.
II.
THEORYA. Doubly resonant infrared-visible sum-frequency generation
The general theory
of
sum-frequency generation (SFG)in reQection from the surface
of
a nonlinear medium has been described in detail elsewhere[15].
As shown inFig.
1,we explore surfaceSFG
in refiection(E,
) from an in-terface between linear media 1 and 2 with a dielectricconstant
of
c.&and c2,respectively.We consider the case in which the visible frequency
(co@)in the
SFG
process is close to a vibronic transition from the ground electronic state manifold So[gb]
to the first excited stateSi
[evj and cubi in resonance with cob,[see
Fig.
2(a)]. By applying the diagrammatic techniqueof
Yeeand Gustafson[13,
16] toFig.
2(b}, the doubly res-onant nonlinear polarizability canbe expressed as(gb~rk ~ga )
(ga
~r, ~eu )(ever~ ~gb)
(roi rogbg
+iyb,
) (&0 rue„g+iy
g)Here
oi„,
=(E„Eg,
)—
If&isthe frequencyof
the transition ~ev)~~ga
).
A common Lorentzian linewidth is taken foreach Franck-Condon (FC}transition (i.e.,
y,
„g,
=y,
g). The above equation isconveniently rewritten by introducing the integral representationof
energy denominators: (a+ib)
'=(
i)
f
—
o"e"'+'
'dt for b)
0
[17].
After applying theclo-sure property
of
the vibrational manifolds,Eq.
(1}becomes(t)
(
.
)I
~ '~st Yegt+~~&~ Yba&
(( '~Hg q s ge( ) ~~He q s eg( }
—i&'Hg(q)ls
p(
}It'H (q)lfi)~d
$2 p
Here,
H
(q) andH,
(q) are the vibrational Hamiltoniansof
molecules in the electronic ground and excited states.erg'(q) d—enotes the ith component
of
the electronic transition moment from the ground to excited state, anderP(q)
de—notes the kth componentof
the electric di-pole momentof
the ground state. The double bracketin-dicates the thermal average
of
the operators involved.The 3N
—
6 normal modesof
a polyatomic moleculecan be separated into two groups: The first group is comprised
of
the totally symmetric normal modes thatare strongly Franck-Condon active in an optical
transi-tion; and the second is formed by the partially symmetric normal modes that are either
FC
inactive or weaklyac-tive in the optical transition.
For
atotally symmetric vi-bration, the excited-state potentials can be displaced in the equilibrium configuration relativeto
the ground-statesurface. The displaced excited-state surface exerts a net
force on the initially prepared wave packet, causing its mean position
to
move, hence the dynamic processof
the wave packet is an important factor in the resonantexcita-tion
of
a totally symmetric vibration.viS qo
(a)
lev~ Iev& Igb& ga& &gal Ks Igb& &gal to 2 Medium 1 ViP,
Interfacial Layer jE Medium 2 IR Iga&(b)
&galFICx. 1. geometry of sum-frequency
(I(,
) anddifference-frequency (Kd) generation from an interface in the reflected direction. The z direction is taken as zero at the interface be-tween the media and ispositive going into medium 2.
FIG. 2. (a) Schematic for doubly resonant infrared-visible sum-frequency generation process between the potential energy surfaces ofthe electronic ground state [So]and the first elec-tronic excited state
[S,
],
and (b) the corresponding double Feynman diagram.H,
(q)=Hs(q)+i
g
LI(a/
a/)+—
fico,f
(3)Here,
Lf
denotes the linear electron-phonon coupling coefficient, %co, is the electronic transition energy ap-propriate to the ground electronic state equilibrium configuration(q=O),
and a& andaI
are phononannihila-tion and creation operators. We also have Hs(q)
gf
Acof(a&taf+
g).Equation (3)can be conveniently converted to the fol-lowing form
[19]:
The random perturbation
of
the partially symmetric normal modes on the periodic motionof
an opticallyac-tive vibration is embedded in the radiative damping
con-stant
of
theFC
active vibration[18].
Similarly to thecase observed with resonance Raman scattering (RRS)
[17,19],
the electron-vibration coupling in each optically active mode will also contributeto
DR
IVSFG.
In the approximationof
the Born-Oppenheimer separationof
electronic and nuclear motions, a single-photon allowed vibronic transition is found
to
be described very well by a linearly coupled vibronic system. The linearly displaced vibrational Hamiltonianof
the excited state in this model has the form[19]
tion frequency. The electronic transition moment func-tion e—r's(q) for a totally symmetric normal mode can be replaced by
a
constant which isthe value evaluated atthe equilibrium configuration (q=O)
of
the groundelec-tronic state,
i.e.
,the Condon approximationer—
'
(q)= —
er's—
(q=0)
.
(sa)rss
=rss(0)+i
g
(a—
agq
f
f
This is a good approximation for strongly
FC
active modes that are knownto
be significantly affected by the dynamicsof
wave packets. Vibronic coupling between two excited electronic states can leadto
linear terms in the expansionof
er—
's(q) H.owever, this does not be-come a restriction in our molecular modelof
DR
IVSFG
because linear non-Condon terms can be included without much difficulty in calculating Eq. (2)[19].
Theground-state dipole moment
of
a molecule resonantlyex-cited by an infrared photon, however, is modulated by
the vibrations
of
the molecule: drssrs
(q)=
r(0}+
—
g
qIBgf
H,
(q)=e
H
(q)e+%cia,
(4)where
P
=i
gI
g/(a/+a~),
withg&=L//(hcoI).
Thequantity ttPs
=to,
—
g&gIto& is the zero-phonontransi-I
=r
(s0s)[l+D ].
Substituting Eqs. (4)and (5) into
Eq.
(2),we obtain(5b)
3
a,
'k( to„'F02,co—
i)=
r,"s(0)rjs'(0)rp(0)
f
e'
"
'f
(t, t')dt
dt',
0=((e
'"e
[1+D
(—
t')]&)
.
To
evaluatef
(t,t'),
the functionf
(A;t,
t')
~~a,
(—~')'.
'=((e
'"e
e s )) is defined and then reduced to aproduct
of
c
numbers through the useof
boson algebra identities[19,20].
If
we expand exp[ADs(—
t')
]to aterm linear in A, we can then setA=1
to recover the thermal averagef
(t,
t').
Thusa,
' k in finite temperatures can befound tohave the expression
E(N CO )f
ct(co)=i
f
(ala(t)&e'
"
""
"dt,
0(ala
(t) )
=exp
g
g
[(nI+1)(e
—
1)f
+nI(e
I
1)]—
(8) 3S/;
„=
(Ir, '(0)r'
(0)b,rP(0)
(7)««
l~(t)
&=exp[iH,
(q)t]la
)
describes the evolutionof
the vibrational wave packet in the excited electronic state, initially prepared by an optical transition from the ground state
lga).
The single-photon absorption spec-trumof
a molecule, o(co)/co, is equal to the imaginarypart
of 4(to)
by[17]
0'(Co) ~ I I i(t0 t0 )t—
r
—~t((t2ltt
(t) )e
's 'g dtX
[(nI+1}@(co,
—
co&)—
4(tt),
)],
=1m[A(a)
—
aP,)]
.
(9)where n&
=1/[exp(ficoI/kit
T)1],
andh—
rP(0}
—
=
[drp(q}/dq]
0 is the ground-state dipole moment
derivative. N(to) is the Fourier transform
of
the overlap functionof
the time-evolved wave packet[21]
Note that all the information about molecular electronic
structure has been absorbed into the time evolution behavior
of
the wave packet. This formalismof DR
IVSFG
allows us to skip the difficultyof
thesum-over-states approach and focus on key physical parameters. Equations (7)
—
(9) provide aconnection between the ab-sorption spectrum and theSFG
excitation profiles(Sf
vs co2)of
normal modesof
adsorbates.[(nf+1)4(co,
—
cof)—
4(co,
)],
which appears in the resonant amplitudeof
DR IVSFG,
is similar to that found in thetime-correlator formulation
of
resonance Raman scattering[18].
In Eq.(7),{gf,
cof,yf,
y,
,and co, I are used tode-scribe
DR IVSFG.
These parameters are also indispens-able for elucidating the dynamical propertiesof
mole-cules. We will show that these parameters can be deter-mined from the fitof
the experimentalDR
IVSFG
excita-tion profiles to
Eq.
(7)in the discussion section.B.
Doubly resonant infrared-visible difference-frequency generationIn addition to the
SFG
beam, a difference-frequency signal (Kd) also emerges from the interface, as shown inFig.
1.
The doubly resonant difference-frequency genera-tion isespecially interesting since it can suppress inhomo-geneous broadening, as pointed out by Shen[14].
To
fur-ther reveal its potential applications, we analyze the case in which the visible frequency in theDR
IVDFG
process is close to a transition from the electronic ground-state manifold So{ga]
tothe first excited stateS
1{ev] [seeFig. 3(a)]. The resonant nonlinear polarizability,ad,
'k,corre-sponding to
Fig.
3(b) can be written as(gbIr, ~ev
)
(evIrjIga )(gc2Irk gb )ad,
jk(
~'d ~2~1)
2gP
(~1 b 1Yb ) I.
(2
~1)
b+ir„]
(10)
Following the same procedure as detailed in Sec.
II
A,wecan readily obtain
(2) f,ij k
d,;jk(
—
d'»
i)=
X
(coi coj
1)'f
3
Df,
,
k=
gfrs'(0)r,
'
(0)b,rp(0)
assuming a single dominant inhomogeneous broadening channel. By substituting
Eq.
(8) into Eq.(11),
we can rewrite the nonlinear susceptibilityof DR IVDFG
asU
X
[4(cod+cof
)—
4(cod)](nf
+
I) .Note that at zero temperature the intensity profile
of
first-order Raman scattering from thefth
vibrational mode, If(co2), is proportional to I&(co2)—
4(co2 cof)I',which is essentially the same as the absolute square
of
Df
'jkof
Eq. (1 1). Thus the excitation profilesof DR
IVDFG
carry dynamical information about wave packetswhich is similar to what is widely recognized in reso-nance Raman scattering.
C. Inhomogeneous broadening
The resonant frequencies
of
a molecular system often depend on its local environment.To
account for this possible inhomogeneous broadening, the nonlinear sus-ceptibility should be convolved with a Gaussian distribu-tion funcdistribu-tionof
the resonant frequencies, givingviS qo lev& Igb& gQ& cgbl 1ev&
yd(2)(cod=co2
—
co,)=X
—
—
e ~ Io'[ad
(2)(cod,rj)]drI,
STY/0 (12) Vis 0 Iga& &gbl t0 2
where g represents a set
of
local parameters. The reso-nant frequenciesof
adsorbates in different localenviron-ments can be expressed as [22] Iga& &gal IR
=Q,
+b,
co,(g)
=0,
+greco,g,
cof cof
+
Ibcof(71 ) cof+
cjkcof(13) FIG.3. Schematic for (a)the energy level and (b)the
corre-sponding double Feynman diagram of doubly resonant
3
Xd,~'Jjk'( ~d ~1 ~2)' ' g22
Xkf
'j
1~
P
x
g
n,=O nN=0
+oo ], (q/go) dn
& 7T'gp (N) cof ')}Ecof ly
j
)N
[co2 o))
+
(cof+
rjkcof)(r—
jato,(t
+
Q,
g}—
g
n&( o)&+
rjht0& )+
iy,
(t]Q=1
N
[o)~
—
o),—
(rjtI),o),+Q,
e)
—
g
ng(oPg+rjb,tog)+iy,
]Q=1
(14)
Byusing the properties
of
the plasma dispersion function, Oudar and Shen [22]have shown that in adouble-resonancecase,
if
the imaginary partsof
the energy denominators in nonlinear optical susceptibility(y)
have opposite signs, theny
scanned over the resonance yields aspectral line with aLorentzian linewidth. Indeed, near the double resonance (i.e.
,co)
=o)f
ando)d=
—
o)f+Qs+
g&
n&oP& or o))=o)f
and o)d=Qe+
g&
n&co&) Eq. (14) exhibits a Lorentzian line shape with ahalf-widthof
yf
+y,
g. On the contrary, this isnot the case forDR
IVSFG
where the imaginary partsof
the denominators ing( 'have the same sign. Thus y( 'exhibits no singularity near the double resonance. Infact, at thedouble resonance, g( '(
—
o)„'o)),
o)z)exhibits awidth more than twice the widthof
inhomogeneous broadening.D. Transient effect
The reduced spectral linewidth via
DR IVDFG
can be better appreciated in the time domain. The coherence be-tween ~ev)
and (,gb~ [seeFig.
3(b)],which is set up byE(co,
) and E(co&), can directly emit the output radiation atcod=(vz
—
o),if
transition between ~ev}
and (gb~ is single-photon allowed. The polarization set up by pulsed excitationshas difFerent phases fordifferent molecules in difFerent local environments. The resulting dephased polarization can be expressed as
&d"(tod=to2
to„t)—
=N
f
n(rj)Tr[
«pd"—
(t, rj)]&rj
=(
e)Nf
n(g)
—
g g
&gb[r[ev)pd,
'„(,
(t,
7(I)drj+c.c.
b u
(15)
where n
(rj}
is the distribution functionof
inhomogeneous broadening and Nthe surface density. Byfollowing the di-agrammatic rulesof
the time-dependent density matrix, we canwrite(2) e (()g2
—
)g() r(g)—t'[n,g+(tt—1)tdf}(r,
gg+r,g(2—rf((2 &,)—
pd,gtt(tb ttrj—
2ex
zptm(evlrlga)(g
lrlg &af ' tte' '"
'+
"'
(t )'dr"tttft'
et'
'"d(ltt
tdt, e a(16)
assuming that each level in the vibrational manifolds
of
the excited and ground electronic states isequally affected by the inhomogeneous broadening Here.
Hd(t,rj)=
Ij(gto,g(rj)t
—
[j(go),g(rj)t
&+bee&,(rj}(t
2t,
)]] denote—s the phase differenceof
the induced nonlinear polarization at the local environment specified by g;andA;(t;)
is the envelopeof
the exciting pulsed field which has a carrier frequency at o);and peaks at t; As noted.
fromEq.
(16),all the moleculesat the surface will generate the maximum coherent output whenever the phase difference vanishes
[13,
14],i.
e., Od(t,g}=0,
(17) This indicates that with properly time-ordered input pulses (t2&
t)
},the transientDR
IVDFG
will give riseto a
pho-ton echo
of
frequency o)d att
=t,
which is later than both t2 andt,
[14]
[seeFig.
4(a)].
The amplitudeof
theDR
inho-mogeneous broadening. Thus in frequency domain
DR
IVDFG
exhibits a reduced linewidth which contains theintrin-sicdynamical information about molecules.
Byapplying similar analysis to
DR IVSFG,
we finde ~(kz+k() r(.&) (—(Q, +U~f)t r—
, (+r,
i,
r—f(t,
t'(—) iI[0
—co +(v—1)co jIt&&
gp',
,
(ever)gb)(gb~r~ga)I
e"
'
'
'A2(t)
)dt'sJ
e ''gi(ti)dtie
a(18}
where
8,
(t,
rt)=
Ibrp,g(rt)t [pro—,
g(rt)tz ~rambo(rt)( zt,
)]
j—.
The time at which 6),=0
is always less than t2 [see Fig. 4(b}]. ThereforeDR
IVSFG
does not involve adephasing-rephasing process
of
photon echoes and its spectral line will be affected by inhomogeneous broaden-ing.III.
NUMERICAL RESUI.TSAND DISCUSSIONThe chemisorption
of
molecules onasubstrate not onlycan perturb the electronic states
of
the substrate but also change the potential energy surfaces and vibrational structuresof
adsorbates[2].
These changes, once they have been measured, could yield valuable information about the static and dynamical behaviorsof
adsorbates. In this section, we will show that such measurements canbe fulfilled effectively by the use
of DR IVSFG
andDR
IVDF G
processes.It
isnoted that Eqs.(7) and (9)allow the connection be-tween the absorption spectrum andDR
IVSFG
ampli-tudesof
normal modesof
adsorbates. This relation is similar to the transform theoryof
resonance Raman scattering[17],
which seeks to determine the Raman exci-tation profile (REP)from the absorption cross section. In an indirect approachof RRS
model potentials are used tocalculate the time cross correlation function
((b~a(t))
)which can then be transformed to the frequency domain.
The parameters
of
the potential energy surfaces can be adjusted in order to get a good fitof
the experimental profile. In this respect, adirect inversionof
resonantRa-Vis rephase
DF dephase = = =, Echo
I
man excitation profiles to yield time domain information is more attractive. Recently, a direct inversion scheme has been proven tobe feasible
[23].
In the case
of
an interfacial system,DR
IVSFG
andDR IVDFG
can be used to determine the parametersof
the vibrational structures and potential energy surfacesof
adsorbed molecules, which are indicated by[gf
cof yf,
y,
, happ, and rlpkcpj.
From the experimental pointof
view, this approach can be done by first scanning the in-frared beam (c0)) through each vibrational mode with afixed visible frequency. Equation (7}isthen used to fitthe observed resonant nonlinear optical susceptibility. The
fit determines cof,
yf,
andSf.
In the second step, we let cu&=cof and measureSf
as a functionof
cu2. Thismea-surement yields a set
of
excitation profilesof
DR
IVSFG.
In an indirect approach, we can fit these
DR IVSFG
profiles to Eq. (7) with the helpof
model potential sur-faces to determine the rest parameters(gf
y g 7/pktp g,and to, ). We will elucidate this procedure in detail by using a model molecule with linearly displaced harmonic potential surfaces along three
FC
active normal modecoordinates. The vibrational frequencies and disp1ace-ment parameters
of
these vibrational modes aredescribed in TableI.
Figure 5shows the calculated absorption spectra
of
the model molecule, for which the short-dashed, solid, and dotted curves are the calculated results obtained with the displacement parameters[gf
j taken from sets 1,2, and 3,respectively. In the calculations, we model the homo-geneous broadening
of
an electronic transition by adamping constant,
y,
,and the inhomogeneous broaden-ing by the parameterof
rtb,to, [see Eq.(13)].
In Fig. 5, these two parameters are assumed to be (a)y,g=300
cm',
lb,r,
~=00 cm',
(b)y,g=600
cm',
tlhcp,g=0
cmi;and{c) y,g=300cm
i,n~~,g=300cm-i.
Asex-pected, the higher vibronic features in the absorptionspectra become more distinctive when the linear electron-phonon coupling is increased
($3=0.
7:
short-Vis TABLE
I.
The frequencies,cd, and displacement parame-ters, gf, of three Franck-Condon active vibrational normal modes ofamodel molecule.
I
t'
2
FIG.
4. Pulse sequences for (a) transient infrared-visible difference-frequency generation, and (b) sum-frequency genera-tion processes. Mode no. cd (cm')
900 1200 1500 Set 1 0.3 0.6 0.7f
Set 2 0.3 0.6 1.0 Set 3 0.3 0.6 1.5~rt 0 0 t gf 4 0 20 . 10-0 -4000 20-10. 0 -4000 2000 2000 (ug-(ueg (Cm 1) 8000 20 10-0 8000-4000
(c)
2000 Mg—Greg (Cm-1 ) 8000FIG.
5. Calculated absorption spectra for linearly displaced harmonic potential energy surfaces along Franck-Condon active normal mode coordinates. The vibrational frequencies and displacement parameters of this model molecule are described in TableI.
The calcu-lated spectrum with the displacement parame-ters, {g~),taken from set 1 in TableI
is indi-cated by the short-dashed curve; solid curve is from set 2, and dotted curve for set 3. In addi-tion, the electronic damping constanty,
g and inhomogeneous broadening parameter gpss,co,gare assumed to be (a)
y,
g=300
cmgphco,g
=0
cm';
(b)y,
g=600
cm 'gpkco g 0 cm ', and (c) Z~=
300 cm gphco,g=
300cm (b)mode 2 4 ~IS/ C4 0 -4000 2000 (a)mode 1 8000FIG.
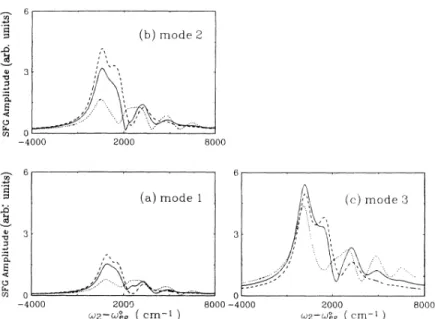
6. The resonant amplitude,Sf(
—
co&—
cof,co&, cof ),of DR IVSFG for each vibrational normal mode is plotted as a func-tion ofthe visible frequency (co&—
co~). Theabsorption spectra ofthe molecule are shown in Fig.5(a). 4-0 -4000 2000 GfS
-
(deg (cm-
1) 0 8000-4000 2000 1dg 1Ieg (Cm 1) ~ ~ 8000 (b)mode 2 0 -4000 2000 (a)mode 1 8000FIG.
7. The resonant amplitudeSf(
—
co&—
&of,'co&,~f)
of DR IVSFG for each vibrational normal mode is plotted as a func-tion ofthe visible frequency (co&—
co,g). The absorption spectra ofthe molecule are shown inFig.5(b). Q '0 74 4 CO 2-0 -4000 2000 G)P-fdeg (Cm 1) 0 8000-4000 2000 (dP-(de (crn- ) 8000(b)mode 2 Q '0 A. E U 0 -4000 6 3 A. E U 0 -4000 I 2000 (a)mode 1 2000 (cm-i ) 8000 0 8000-4000 I 2000 cu2-~,g (Cm-I ) 8000
FIG.
8. The resonant amplitudeSf
ofDRIVSFG for each vibrational normal mode is plotted as a function ofthe visible frequency (co&
—
co,gj. The absorption spectra ofthemol-ecule are shown in Fig. 5(c).
dashed line;
(3=
1.
0:
solid line; and(3=1.
5:
dotted). Bycomparing
Fig.
5(b} with 5(c),we note that the separationof
the line broadening into the homogeneous and inho-mogeneous parts leads to different absorption strength.But the difference in the absorption line shape between two cases cannot be detected clearly.
The displaced difference operation on
4
gives rise tothe sensitive dependence
of
Sf
ongf.
This can be clearly seen inFig.
6, where the resonant amplitudeSf
'jk( cop cof cog cof)of
DR
IVSFG
foreachvibration-al normal mode is plotted as a function
of
the visible fre-quency (co~—
co,).
The corresponding molecular absorp-tion spectra are depicted inFig.
5(a).It
is interesting tonote that the position
of
the second peak (—
1825 cm')
in
Fig.
6 coincides with thatof
the first vibronic peakof
the absorption spectra [seeFig. 5(a}j.
Furthermore, the frequency differenceof
the first and second spectral peaks in Fig. 6is identical to the corresponding normal mode frequency. In addition, g3 not only changes the line shape(S3) of
mode 3 but also affects the profilesof
theother two normal modes. Particularly noticeable, the second peak height
of
Sf
is found to decrease as g3in-creases. At sufftciently large g3, this peak disappears completely. The molecule with larger broadening exhib-its a similar trend, which is depicted in Figs. 7 and
8.
From the comparison between Figs. 7 and 8,we can find that
Sf
can resolve the effectof
the homogeneous and in-homogeneous broadening with higher accuracy than thatwith absorption spectrum. Moreover, with the immunity
of
DR
IVDFG to
inhomogeneous broadening, we can in principle measure the homogeneous broadening directly from the spectral profilesof
DR
IVDFG.
Even by usingDR IVSFG
only, agood fitof
the excitation profiles pro-vides a very strict test for the parameters used. Thesimulations show that
Sf
asa functionof
co2indeedaccu-rately reflects the electron-vibration coupling in an
adsor-bate.
Recently, the application
of
the resonant third-harmonic generation (THG) technique to investigate thevibronic structures
of
all-trans P-carotene in solution has been reported by van Beck, Kajzar, and Albrecht [24]. Their results indicate that a suitable fitof
the nonlinear third-harmonic susceptibility dispersion could beaccom-plished using fewer
FC
active normal modes, but allFC
active normal modes in an electronic transition are re-quired to correctly fit the observed resonance Ramanex-citation profiles. The higher sensitivity
of
REP
to the vi-brational structuresof
molecules can be attributed tothe selective excitationof
molecular vibrations, which ap-pears inRRS
but not inTHG.
The resonant vibrational transitions induced by the infrared photons inDR
IVSFG
andDR IVDFG
processes warranty the sensitivi-ty to the vibronic structuresof
molecules. In addition, bothDR IVSFG
andDR IVDFG
belong to alower-order wave-mixing process with double-resonance enhancement, therefore they are capable
of
generating stronger signal than that viaRRS
and other higher-order wave-mixing processes [25,26j.
Furthermore,DR
IVDFG
andDR
IVSFG
are surface specific, thus are ideally suited for surface studies.In summary, an analytic expression
of
doubly resonant infrared-visible difference-frequency and sum-frequency susceptibilities in termsof
the overlap functionof
the wave packet in an excited electronic state has been de-rived. Our results show that these second-order non-linear optical effects can be developed into an effective probe forthe electron-vibration coupling in molecules ad-sorbed at surfaces. In time domain,DR IVDFG
isfoundto go through a dephasing-rephasing process
of
photonechoes, therefore it can become asensitive technique for
the investigation
of
coherent dynamical processes appear-ing at surfaces.ACKNOWLEDGMENT
The author
(J.
Y.
H.
)acknowledges the financial supportfrom the National Science Council
of
ROC under Grant[1)
K.
Bhattacharyya, A. Castro,E.
V.Sitzmann, andK. B.
Eisenthal,J.
Chem. Phys. 89, 3376 (1988).[2)W. Brenig, S.Kuchenhoif, and H. Kasai, Appl. Phys. A 51, 115 (1990).
[3]See, for example, Y.
R.
Shen, Nature (London) 337, 519 (1989),and references therein.[4]T.
F.
Heinz, C.K.
Chen, D.Ricard, and Y.R.Shen, Phys. Rev.Lett. 48,478(1982).[5]P.Saeta,
J.
-K.Wang, Y.Siegal, N. Bloembergen, andE.
Mazur, Phys. Rev.Lett. 67,1023(1991);X.
D.Ziao,X.
D.Zhu, W.Daum, and Y.
R.
Shen, ibid. 66,2352(1991).[6]A. L.Harris, L.Rothberg, L.H. Dubois, N.
J.
Levinos, andL.
Dahr, Phys. Rev. Lett. 64,2086(1990);P. Guyot-Sionnest, ibid. 66,1489(1991);67,2323(1991).[7]S.H. Lin,
R.
G. Alden, A. A. Villaeys, and V. Pfumio, Phys. Rev.A48, 3137 (1993).[8]G. A. Somorjai, Chemistry in Two Dimensions: Surfaces (Cornell University Press, Ithaca, 1981),p. 360;
J.
W. Gad-zuk, Appl. Phys. A51,108(1990).[9]
B.
N.J.
Persson and M.Persson, Solid State Commun. 36, 175(1980);Surf. Sci. 97, 609 (1980).[10]
J.
W.Gadzuk and A.C.Luntz, Surf.Sci.144, 429(1984).[11]W. Demtroder, Laser Spectroscopy (Springer-Verlag, Ber-lin, 1981).
[12] N. A. Kurnit,
I.
D. Abella, and S.R.
Hartmann, Phys. Rev.Lett. 13,567(1964).[13]
P.
X.
YeandY. R.
Shen, Phys. Rev. A 25,2083(1982). [14]Y.
R.
Shen, Phys. Rev. A 45,446(1992).[15]
P.
Guyot-Sionnest, W.Chen, and Y.R.
Shen, Phys. Rev.B33,8254(1986);P.Guyot-Sionnest and Y.
R.
Shen, ibid. 35,4420(1987);38,7985(1989),and references therein.[16]
T. K.
Yee andT.
K.
Gustafson, Phys. Rev. A 18, 1597 (1978).[17]
J.
B.
Page and D.L.
Tonks,J.
Chem. Phys. 75, 5694 (1981).[18]
R.
M.Shelby, C.B.
Harris, and P. A.Cornelius,J.
Chem. Phys. 70,34(1979);B.
S.Neporent and V.S.Yarunin, Zh. Eksp. Teor. Fiz.99,447 (1991) [Sov.Phys. JETP 72, 249(1991)].
[19]C.
K.
Chan,J.
Chem. Phys. 81,1614 (1984).[20]W. H. Louisell, Quantum Statistical Properties
of
radiotion (Wiley, New York, 1973).
[21] Z.Deng and S.Mukamel,
J.
Chem. Phys. 85,1738(1986). [22]J.
-L.Oudar andY.
R.
Shen, Phys. Rev. A 22,1141 (1980). [23]F.
Remacle andR. D.
Levine,J.
Chem. Phys. 99,4908(1993).
[24]
J.
B.
van Beck,F.
Kajzar, and A. C.Albrecht,J.
Chem. Phys. 95, 6400 (1991).[25]
X.
D.Zhu and Y.R.
Shen, Appl. Phys. B50,535(1990);P.Guyot-Sionnest, Phys. Rev.Lett. 67,2323(1991).
![FIG. 2. (a) Schematic for doubly resonant infrared-visible sum-frequency generation process between the potential energy surfaces of the electronic ground state [So] and the first elec-tronic excited state [S, ], and (b) the corresponding double Feynman d](https://thumb-ap.123doks.com/thumbv2/9libinfo/7956521.157990/2.918.531.747.495.1023/schematic-infrared-frequency-generation-potential-electronic-corresponding-feynman.webp)