895
Journal of Biomolecular Structure & Dynamics, ISSN 0739-1102 Volume 28, Issue Number 6, (2011) ©Adenine Press (2011)
*Phone: +1-617-353-7123 E-mail: [email protected]; [email protected] (C.Y.-C. Chen)
Mao-Feng Sun
1,2Tung-Ti Chang
1,2Kai-Wei Chang
1Hung-Jin Huang
1Hsin-Yi Chen
3Fuu-Jen Tsai
3,4Jaung-Geng Lin
1Calvin Yu-Chian Chen
1,3,5,6*
1Laboratory of Computational and
Systems Biology, School of Chinese Medicine, China Medical University, Taichung, 40402, Taiwan
2Department of Acupuncture,
China Medical University Hospital, Taichung, Taiwan
3Department of Bioinformatics, Asia
University, Taichung, 41354, Taiwan
4Department of Medical Genetics,
China Medical University, Taichung, 40402, Taiwan
5Department of Systems Biology,
Harvard Medical School, Boston, MA 02115, USA
6Computational and Systems Biology,
Massachusetts Institute of Technology, Cambridge, MA 02139, USA
Blocking the DNA Repair System by Traditional
Chinese Medicine?
http://www.jbsdonline.com
Abstract
Non-homologous end joining (NHEJ) is a major DNA double strand breaks (DSBs) repair pathway that maintains genome integrity. However, this pathway may reduce radiotherapy efficacy by repairing DSBs on cancer cells. This research reported a computer-aided drug design (CADD) method to identify novel inhibitors from traditional Chinese medicine (TCM) that disrupt NHEJ. We aim to inhibit Ku86, the initiator of NHEJ. By integrating binding energy evaluation and molecular dynamics simulation methods, we reported glycyr-rhizic acid, macedonoside C, lithospermic acid, and salvianolic acid B as potential Ku86 inhibitors. All four TCM compounds show low binding energy and stable binding poses to Ku86. The carboxyl groups on a ligand are the major binding region by forming salt bridges at Ku86 binding sites. Additional features were defined by a carbonyl group or a dihydroxy-phenyl group that form additional hydrogen bond or pi-cation respectively with the ligand binding site on Ku86. These features strengthen the binding affinity between Ku86 and the potential TCM ligand. We reported all four TCM compounds are potential Ku86 inhibitors and may be used to enhance radiotherapy for cancer treatment.
Key words: Ku protein; Radiotherapy; Traditional Chinese Medicine; Docking; Molecular Dynamics.
Introduction
Radiotherapy is commonly used for the treatment of malignant cancer. The ionizing radiation kills targeted cells through inducing lesion in DNA, such as DNA double strand breaks (DSBs). Non-repaired DNA damages then can lead to cell death. Hence, radiation sensitivity of the target cancerous tumor cells plays an important role in efficacy of radiotherapy (1). Many researches suggest the disruption of non-homologous end joining (NHEJ) pathway, an important DNA repair mechanism, leads to increased radiation sensitivity (2-4). Therefore, target-specific inhibition of NHEJ pathway in cancer cells may lead to enhanced efficacy and specificity of radiotherapy.
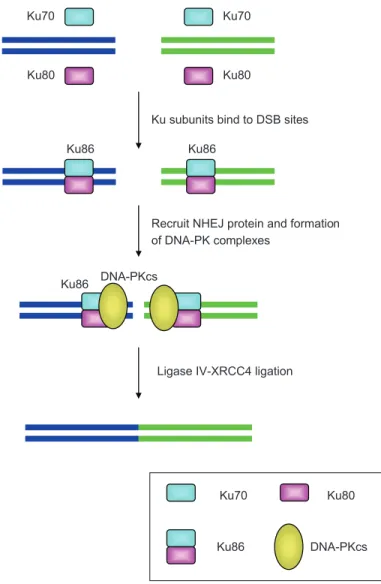
Ku protein (Ku86), is one of the key proteins involves in NHEJ pathway (5). Ku86 heterodimer recognizes a DSB site and binds to the broken DNA ends. As shown in Figure 1, the Ku86-DNA complex functions as a moecular scaffold that recruits DNA repair protein assembly, such as DNA-PKcs, XRCC4, and Ligase IV (6). NHEJ ends with the Ligase IV-XRCC4 ligation that rejoins the DSB ends. An active Ku protein contains two subunits, Ku79 and Ku80, which dimerize and form a ring-like structure that has high affinity to DNA broken ends (7, 8). This makes Ku86 not only involves in NHEJ, but also plays a role in telomere maintenance (9, 10). Ku86 deficiency has been linked to high radiosensitivity of cells. Mouse model suggests that deletion of Ku70 or Ku80 lead to hypersensitivity to ionization radiation under
896
Sun et al.
a p53 deficient condition (3). In addition, increased expression of Ku70 showed radioresistance in head and neck cancer cell lines (11). Hence, it is possible that inhibition of Ku protein function can abolish NHEJ and thus makes a cell more susceptible to ionizing radiation.
In the past, many potential drug-like compounds targeting proteins such as EGFR, PDE5, influenza hemagglutinin, HER2, HIV enzymes, and more were identified through computer-aid drug design (CADD) (12-27). This study was interested using CADD approaches for discovering novel drug leads that can disrupt NHEJ pathways and therefore enhance radiotherapy efficacy. The research focused on identifying potential Ku86 inhibitors from traditional Chinese medicine (TCM), from which many prospective anti-tumor com-pounds, such as gallic acid, curcumin, and quercetin, have been identified (28-30). Compounds recorded in TCM database (TCM Database@Taiwan, http://tcm.cmu.edu.tw) (31) were used for in silico screening. The selected TCM compounds were further investigated for their interaction with the Ku protein using molecular dynamics (MD) simulation. Both virtual screening and MD simulation were common CADD methods often used for evaluate protein-ligand interactions. Biomedical experiments will be applied to validate each identified Ku heterodimer inhibitors.
Ku70 Ku70
Ku80 Ku80
Ku86 Ku86
Ku subunits bind to DSB sites
Recruit NHEJ protein and formation of DNA-PK complexes
DNA-PKcs Ku86
Ligase IV-XRCC4 ligation
Ku70 Ku80
Ku86 DNA-PKcs
Figure 1: NHEJ pathway diagram. (A) Ku70 and Ku80 subunits form Ku86 protein after
dimeriza-tion. (B) Each Ku86 binds to broken DNA ends. (C) Ku86 acts as a scaffold for recruiting other NHEJ proteins and form DNA-PKcs. (D) Ligase IV-XRCC4 complx is recruited and ligate the broken DNA ends. DNA-repair proteins then diassociate.
897
Blocking the DNA Repair
System
Materials and Methods Protein Preparation
Ku heterodimer crystal structure was obtained from Protein Data Bank (www.pdb. org PDB ID: 1JEY) (7). Pure protein structural information was extracted from the original pdb file and the missing hydrogen atoms were added. The CHARMm force field was then applied to the Ku protein prior docking or simulation. The DNA binding interface was defined as that ligand docking site. Acetylation and mutagenesis studies on Lys282, Lys338, Lys539 and Lys542 at Ku70 have been demonstrated to suppress the DNA end-binding activity (32). In this study, Lys282 and Lys338 were the only two residues presents in the 1JEY crystal file, and hence were investigated further.
Docking
A total of 20,000 isolated products from TCM were obtained from the TCM Data-base@Taiwan (http://tcm.cmu.edu.tw/) (31), where each compound was ionized according to the physiological setting, using Discovery Studio 2.5. These com-pounds were also filtered using ADMET module in Discovery 2.5 to remove any potentially toxic compounds. LigandFit module in Discovery Studio 2,5 was applied to evaluate the binding energy between each TCM compound and DNA binding region of the Ku protein (33). Monte Carlo simulation was used to gen-erate ligand conformations using a shape-based matching method. The docking site was held rigid for trying out ligand binding poses. Ligand conformations were energetically minimized using Smart Minimizer. Ligands were ranked according to the Binding Energy, which represents the binding affinity. In addition, LigScore (LigScore1 and LigScore2) and piecewise linear potential (-PLP and -PLP2) were applied to evaluate binding affinities. Binding Energy evaluates the free energy in a protein-ligand conformation. LigScore computes softened van der Waal descrip-tor and polar surface area descripdescrip-tors (34). LigScore2 differs from LigScore by the introduction of desolvation penalty. The piecewise linear potential calculate hydrogen bond interactions between hydrogen bond donor, hydrogen bond accep-tor, and non-polar atom types (35). The PLP2 function further includes a scaling factor based on the angles between receptor atoms and ligand atoms. Top ranking TCM candidates were visually inspected to identify ligands that interact with key residues, Lys282 and Lys338 of Ku70.
Molecular Dynamics Simulation
Molecular dynamics (MD) simulation was applied to the selected protein-ligand complexes using the simulation module in Discovery Studio 2.5. Each complex was solvated in a simulated water box with 0.9% NaCl concentration, which cor-responds to physiological solution. Periodic boundary condition was applied. The initial minimization process consisted 2000 steps of Steepest Descent and 2000 steps of Conjugate Gradient. The system was heated from 50 K to 310 K without constraint within 20 ps with a time step of 1fs. The equilibration step was run for 100 ps at 310 K. The production was run under constant temperature condition (NVT) for 10 ns at 310 K. SHAKE algorithm was then applied to constraint bind-ings to hydrogen atoms. Root mean square deviations (RMSDs) were calculated for both protein-ligand complexes and ligands.
Results
Identification of TCM Candidates from Screening TCM Database
Molecular docking was performed to screen for potential Ku protein inhibitors from the TCM database. Binding energy was employed to evaluate the protein-ligand
898
Sun et al.
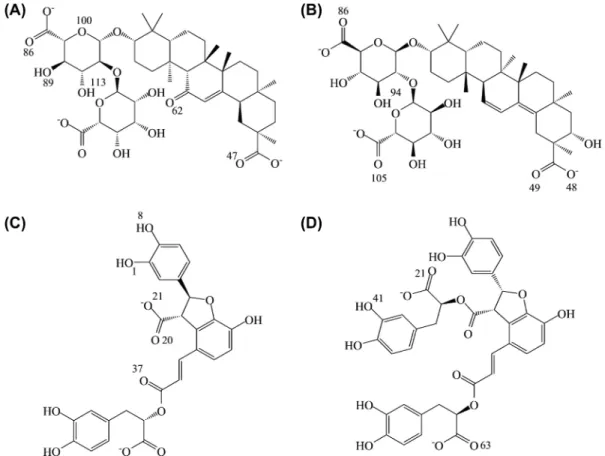
binding affinities. The screening results listed in Table 1 suggests that glycyrrhizic acid (top 1), macedonoside C (top 2), lithospermic acid (top 3), and salvianolic acid B (top 4) can form stable binding conformations at low energy stage, between -525.367 kcal/mol and -732.092 kcal/mol, with the DNA binding site on the Ku86 heterodimer. The ligands shared structural similarities between top1 and top2, and betweem top3 and top4 (Figure 2). For glycyrrhizic acid and macedonoside C, nega-tive charges of the carboxyl groups favored the bindings to the Ku86 DNA-binding domain, where most key residues contained positive charges (Figures 2 and 3). The binding conformations suggested that a hydrogen bond (H-bond) was the major intermolecular force in the protein-ligand binding (Figure 4). Glycyrrhizic acid and macedonoside C can be extracted from licorice (Glycyrrhiza glabra), which is a common TCM herb used in many formulations (36, 37). In the case of lithosper-mic acid, and salvianolic acid B, less carboxyl groups (Figure 2) implied fewer instances of H-bonds observed. Nevertheless, the negative electrostatic potential on the top 3 and the top 4 compounds still favored the alkaline residues in Ku86 ligand binding site (Figures 2 and 3). Intriguingly, the binding affinity between Ku86 and each of these ligands were strengthened by the pi-cation interactions (Figure 4). Lithospermic acid, and salvianolic acid B are extracts of Salvia miltiorrhiza, which is commonly used for circulation disorders in TCM.
Ligands Bind to Ku84 DNA Binding Site Through Formation of Salt Bridge
The protein-ligand binding conformation in Figure 4 and Table 2 suggested a binding hot spot on Ku86 heterodimer at Ku70: Arg403 and Ku70: Lys338. Fur-thermore, the binding conformation of glycyrrhizic acid, lithospermic acid, and salvianolic acid B on Ku86 shares similar binding position. The binding confor-mations revealed that the carboxyl groups on these ligands formed H-bond with residues Ku70: Lys338, Ku70: Glu335, and Ku80: Arg446. In addition, the H-bond and the electrostatic interaction between oxygen and nitrogen, which in combina-tion was defined as a salt bridge, made positive contribucombina-tion to the binding affinity at Ku70: Lys338, Ku70: Glu335 (Figure 4). However, macedonoside C bound to
Table I
TCM molecules docking result. The top 10 candi-dates are listed and calculated binding energy.
Name Binding energy (kcal/mol)
Glycyrrhizic acid -732.092 Macedonoside C -691.639 Lithospermic acid -527.754 Salvianolic acid B -525.367 Mumefural -514.916 Chicoric_acid -504.861 2-O-Feruloyl tartaric acid -492.562 Crocetin -449.829 Glutinic acid -424.322 Chebulinic acid -419.415
Figure 2: Chemical structures
of (A) Glycyrrhizic acid, (B) Macedonoside C, (C) Lithospermic acid, (D) Salvianolic acid B.
899
Blocking the DNA Repair
System
a distinct set of residues, which concentrated at Ku80 subunit, at Lys265, Arg271, and Lys332. Despite the distinct binding poses, the salt bridges formed between carboxyl group and the alkaline residues are the major interaction for the Ku-ligand binding affinity.
Stable RMSD Trajectories Suggest Stable Ku-ligand Bindings During MD Simulation
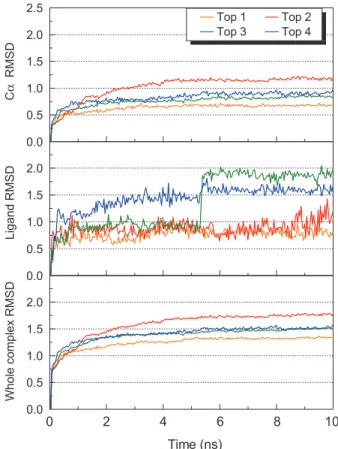
The molecular docking provided a static protein-ligand model for predicting a pre-ferred binding orientation. To further evaluate the binding poses in motion, we applied molecular dynamics (MD) simulation for 10ns on each of the selected Ku-ligand pair. The RMSD trajectories for Ku-ligand complex, C-alpha, and ligand of each binding pair were investigated and showed stable poses which the average atom movements were under 1Å range (Figure 5). This implied relatively stable bindings
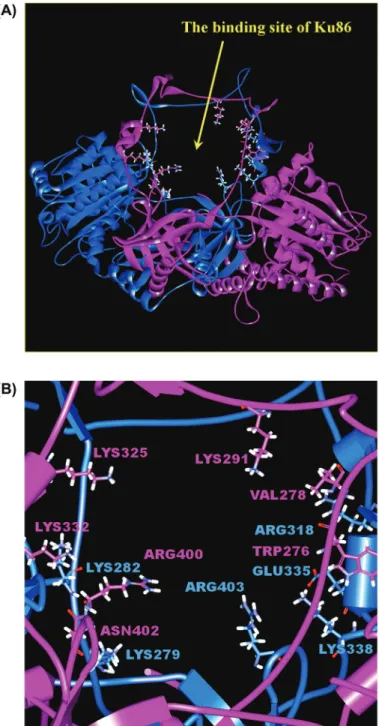
Figure 3: Ku heterodimer DNA-binding site, and presumed inhibitor binding
site. (A) The ring structure Ku heterodimer DNA-binding site. (B) Close look of binding site, where key binding site residues were shown. The Ku70 and the Ku80 subunits and residues are colored in blue and purple, respectively.
900
Sun et al.
between Ku86 and the selected ligands from molecular docking. Intriguingly, the ligand RMSD showed that lithospermic acid experienced a leap of change for over 1 Å at 5-6ns of the simulation period before stabilizing (Figure 5). However, the complex and protein RMSD displayed no significant change over the same period. This implied the change in the ligand pose of lithospermic acid had limited impact on protein-ligand interaction. By tracking the changes in total energy (Figure 6), glycyrrhizic acid and macedonoside C reached minimum energy state at around 5ns
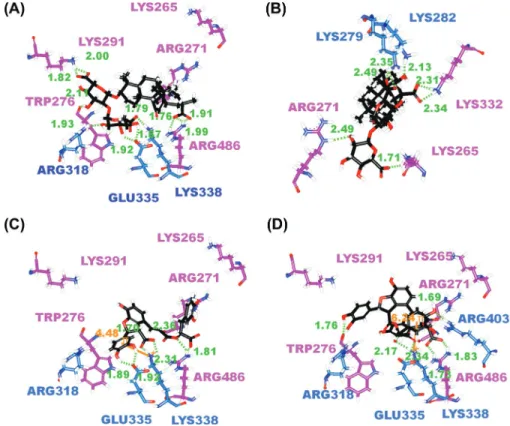
Figure 4: The docking poses of (A) Glycyrrhizic acid, (B) Macedonoside C, (C) Lithospermic acid, (D)
Salvianolic acid B in Ku 86 binding site. Residues on Ku70 and Ku80 are colored in blue and purple respectively. The pi-cation interaction is indicated in orange line.
Table II
Hydrogen bond frequency of top 4 TCM candidates during 10 ns simulation. Residues from Ku70 subunit and Ku80 subunits were included.
Compound Ligand Atom Subunit Amino Acid H-Bond Occupancy
Glycyrrhizic acid O100 Ku70 ARG318 : HE 75.60%
H113 Ku70 GLU335 : OE2 52.00%
O47 Ku70 LYS338 : HZ3 43.20%
O62 Ku70 ARG403 : HH12 86.80%
O89 Ku80 VAL278 : HN 77.60%
O86 Ku80 LYS291 : HZ1 65.60%
Macedonoside C O105 Ku70 LYS279 : HZ1 67.20%
O48 Ku80 LYS325 : HZ3 63.20%
O49 Ku80 LYS325 : HZ3 61.60%
O86 Ku80 LYS332 : HZ1 77.20%
H94 Ku80 ARG400 : O 100.00%
Lithospermic acid H8 Ku70 GLU335:OE2 98.00%
O1 Ku70 ARG403:HH22 71.20%
O21 Ku70 LYS338:HZ1 42.40%
O37 Ku80 ARG271:HH11 88.00%
O20 Ku80 ARG486:HH22 88.80%
Salvianolic acid B O63 Ku70 ARG403:HH21 77.20%
O21 Ku70 LYS338:HZ1 62.40%
O21 Ku70 LYS338:HZ3 86.00%
901
Blocking the DNA Repair
System
and 6ns simulation time respectively. All four selected TCM compounds reached minimum energy after 8ns, and hence implied stable Ku-ligand complexes.
MD Simulation show Stabilization of Ku-ligand Interaction Through Persistent Hydrogen Bonds
To further investigate the binding poses during MD simulation, we compared the structure snapshots at a set of selected time points. For glycyrrhizic acid, the
0.0 0 2 4 6 8 10 0.5 1.0 1.5 2.0 0.0 0.5 1.0 1.5 2.0 0.0 0.5 1.0 1.5 2.0 2.5 W ho le c om pl ex R M SD Time (ns) Li ga nd R M SD C α R M SD Top 1 Top 2 Top 3 Top 4
Figure 5: The RMSD value (Å) of Ku-ligand complexes for top 4
TCM candidates. Top 1 to Top 4 compounds represent glycyrrhizic acid, macedonoside C, lithospermic acid, and salvianolic acid B respectively.
–53250 0 2 4 6 8 10 –53000 –52750 –52500 –52250 –52000 –51750 –51500 –51250 –51000 –50750 To ta l e ne rg y (k ca l/m ol ) Time (ns) Top 1 Top 2 Top 3 Top 4
Figure 6: Total energy of each Ku-ligand complex for selected TCM candidates. Top 1 to Top 4
compounds represent glycyrrhizic acid, macedonoside C, lithospermic acid, and salvianolic acid B respectively.
902
Sun et al.
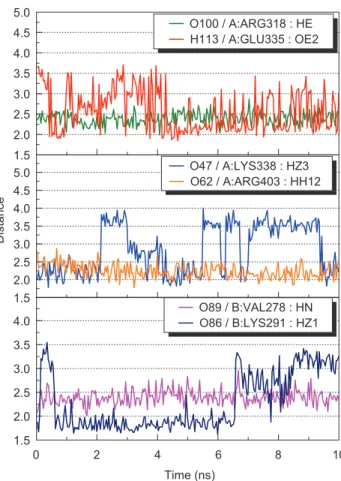
carbonyl group O62 moved close to Ku70: Arg403 at range of 2.0-2.5 Å during MD simulation (Figures 4A, and 7). Furthermore, the calculated H-bond occu-pancy was 86.80% (Table II). These observations implied formation of a new H-bond that was not observed from molecular docking. Glycyrrhizic acid also periodically formed H-bonds with Glu335 and Lys338 at Ku70 subunit (Figure 7, supplementary video 1) during the 10 ns MD simulation, suggesting possible presence of polar interactions that maintain the binding conformation.
Macedonoside C has a distinct binding conformation to Ku86, and the ligand favored interaction with Ku80 subunit. During MD simulation, all three carboxyl groups on macedonoside C formed salt bridges with the lysine residues, Ku70: LYS279, Ku80: LYS325, and Ku80: LYS332 at Ku86 DNA-binding site (Figure 4, Table II). It is possible that the missing carbonyl O62 on macedonoside C decreased the ligand binding affinity to Ku70 subunit, and resulted in a distinct binding con-formation compare to Ku-glycyrrhizic acid complex. Nevertheless, most H-bonds in Ku-macedonoside C remained stable with occupancies maintained at range 61.60%-77.20% (Table II) A notable stable H-bond was observed at Ku80: Arg400, which maintained a 100% H-bond occupancy to the ligand, in spite of conformation change on the same residue (supplementary video 2). The hydrogen bonding inter-action on each alkaline residue stabilized near the end of MD simulation (Figure 8). These analysis presented stable molecular interactions between ligand and the key residues.
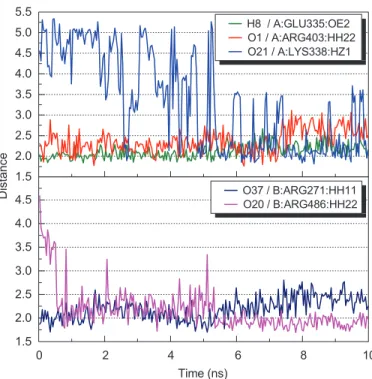
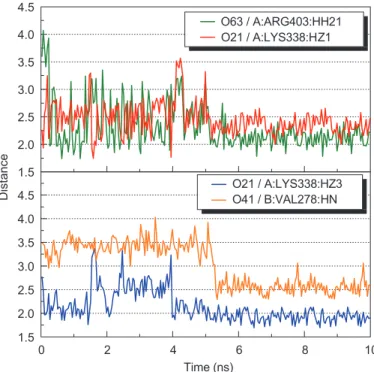
The MD simulation on lithospermic acid and salvianolic acid binding conforma-tions revealed that Ku70: Lys338 and Ku70: Arg403 were potential binding resi-dues on the Ku86 DNA-binding site. Both alkaline resiresi-dues were found to form salt bridges with the carboxyl groups on each ligand. The high H-bond occupan-cies observed (Table II) and the decreasing hydrogen bonding distances over time
1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 1.5 0 2 4 6 8 10 2.0 2.5 3.0 3.5 4.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 O100 / A:ARG318 : HE H113 / A:GLU335 : OE2 Distance Time (ns) O89 / B:VAL278 : HN O86 / B:LYS291 : HZ1 O47 / A:LYS338 : HZ3 O62 / A:ARG403 : HH12
903
Blocking the DNA Repair
System
during MD simulations (Figures 9 and 10) implied stable molecular interactions on these potential residues. We further investigated the RMSD leaps observed around 5-6ns with regard to the lithospermic acid MD simulation (Figure 5). The confor-mation analysis (Figure 11, supplementary video 3) revealed that the Ku80: Arg486
1.5 2.0 2.5 3.0 3.5 4.0 1.5 2.0 2.5 3.0 3.5 4.0 1.5 0 2 4 6 8 10 2.0 2.5 3.0 3.5 O105 / A:LYS279 : HZ1 Distance O48 / B:LYS325 : HZ3 O86 / B:LYS332 : HZ1 Time (ns) O86 / B:LYS332 : HZ1 H94 / B:ARG400 : O
Figure 8: Hydrogen bond distances of Macedonoside C during 10 ns simulation.
1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 1.5 2.0 2.5 3.0 3.5 4.0 4.5 D is ta nc e H8 / A:GLU335:OE2 O1 / A:ARG403:HH22 O21 / A:LYS338:HZ1 Time (ns) 0 2 4 6 8 10 O37 / B:ARG271:HH11 O20 / B:ARG486:HH22
904
Sun et al.
1.5 2.0 2.5 3.0 3.5 4.0 4.5 1.5 0 2 4 6 8 10 2.0 2.5 3.0 3.5 4.0 4.5 Distance O63 / A:ARG403:HH21 O21 / A:LYS338:HZ1 Time (ns) O21 / A:LYS338:HZ3 O41 / B:VAL278:HNFigure 10: Hydrogen bond distances of Salvianolic acid B during 10 ns simulation.
Figure 11: Snapshots of lithospermic acid in Ku binding site at (A)
5.31 ns and (B) 5.39 ns. Residues of Ku70 and Ku80 are colored in blue and purple, respectively.
pose was altered and pulled the ligand carboxyl group closer and formed more H-bonds that resulted conformation change on lithospermic acid within the binding site. In addition, the conformation change moved the ligand dihydroxyphenyl group closer to the Ku70: Lys338, from 5.32 Å to 4.83 Å, and result high pi-cation binding affinity. Hence, the observed RMSD leap may imply a conformation change that further stabilize the protein-ligand binding.
Conclusion
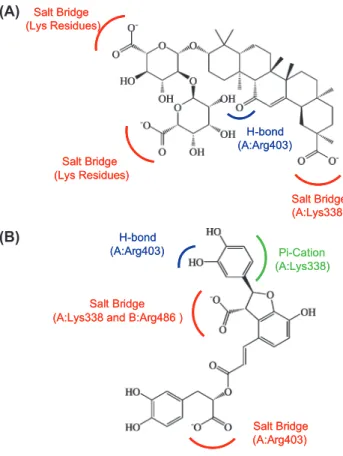
The alkaline residues on Ku86 DNA binding site plays important role in ligand binding. In particular, the Lys338 and Arg403 on Ku70 subunit were the binding hot spots for interacting with glycyrrhizic acid, lith-ospermic acid, and salvianolic acid B. On the other hand, macedonoside C lacked a carbonyl group, which was required to form H-bond with Ku70: Arg403, and results stronger interaction with Ku80 subunit of the Ku heterodimer. Nevertheless, the three carboxyl groups on macedono-side C form stable H-bonds with the alkaline residues on Ku80 subunit, which results stable binding conformation. Based on the CADD design, an efficient Ku86 inhibitor is expected to form strong binding affinity with both Lys338 and Arg403 on Ku70 subunit. Figure 12 demonstrated that the carboxyl groups played a major role in protein-ligand interac-tion. Moreover, the carbonyl group (Figure 12A) and the dihydroxy-phenyl group (Figure 12B) that formed additional H-bond and pi-cation interaction respectively were the key features for stable Ku86-ligand binding. The selected TCM compounds, glycyrrhizic acid, macedonoside C, lithospermic acid, and salvianolic acid B, demonstrated low binding energy and formation of stable Ku86-ligand complexes. We conclude that all four compounds are potential enhancers for radiotherapy.
Supplementary Material
Supplementary videos of the MD simulations are displayed at the website of the article at jbsdonline.com.
905
Blocking the DNA Repair
System
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC 99-2221-E-039-013-), China Medical University (CMU98-TCM, CMU99-S-02) and Asia University (CMU98-ASIA-09). This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH100-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH100-TD-C-111-005). We are grateful to the National Center of High-performance Computing for computer time and facilities.
References
J. Thoms and R. G. Bristow.
1. Semin Radiat Oncol 20, 217-222 (2010).
Z. E. Karanjawala, N. Adachi, R. A. Irvine, E. K. Oh, D. Shibata, K. Schwarz, C. L. Hsieh, 2.
and M. R. Lieber. DNA Repair (Amst) 1, 1017-1026 (2002).
D. S. Lim, H. Vogel, D. M. Willerford, A. T. Sands, K. A. Platt, and P. Hasty.
3. Mol Cell Biol
20, 3772-3780 (2000).
T. Leong, M. Chao, S. Bassal, and M. McKay.
4. Br J Cancer 88, 1251-1255 (2003).
M. H. Yun and K. Hiom.
5. Nature 459, 460-463 (2009).
B. L. Mahaney, K. Meek, and S. P. Lees-Miller.
6. Biochem J 417, 639-650 (2009).
J. R. Walker, R. A. Corpina, and J. Goldberg.
7. Nature 412, 607-614 (2001).
A. Rivera-Calzada, L. Spagnolo, L. H. Pearl, and O. Llorca.
8. EMBO Rep 8, 56-62 (2007).
H. L. Hsu, D. Gilley, E. H. Blackburn, and D. J. Chen.
9. Proc Natl Acad Sci USA 96,
12454-12458 (1999).
G. B. Celli, E. L. Denchi, and T. de Lange.
10. Nat Cell Biol 8, 885-U162 (2006).
H. W. Chang, S. Y. Kim, S. L. Yi, S. H. Son, Y. Song do, S. Y. Moon, J. H. Kim, E. K. Choi, 11.
S. D. Ahn, S. S. Shin, K. K. Lee, and S. W. Lee. Oral Oncol 42, 979-986 (2006). A. M. Andrianov.
12. J Biomol Struct Dyn 26, 445-454 (2009). A. M. Andrianov and I. V. Anishchenko.
13. J Biomol Struct Dyn 27, 179-193 (2009).
M. T. Cambria, D. Di Marino, M. Falconi, S. Garavaglia, and A. Cambria.
14. J Biomol Struct Dyn 27, 501-509 (2010). (A) H-bond (A:Arg403) Salt Bridge (Lys Residues) Salt Bridge (Lys Residues) Salt Bridge (A:Lys338) H-bond (A:Arg403) Salt Bridge (Lys Residues) Salt Bridge (Lys Residues) Salt Bridge (A:Lys338) (B) Salt Bridge (A:Arg403) Salt Bridge
(A:Lys338 and B:Arg486 ) H-bond (A:Arg403) Pi-Cation (A:Lys338) Salt Bridge (A:Arg403) Salt Bridge
(A:Lys338 and B:Arg486 ) H-bond
(A:Arg403) Pi-Cation
(A:Lys338)
Figure 12: Key features for Ku86 drug design. The common features of (A) top1 and top2
com-pounds; and (B) top 3 and top 4 compounds were included. Salt bridges (red), H-bonds (blue), and Pi-Cation (green) interactions were demonstrated.
906
Sun et al.
C. Y. Chen.
15. J Biomol Struct Dyn 27, 627-640 (2010).
C. Y. Chen, Y. H. Chang, D. T. Bau, H. J. Huang, F. J. Tsai, C. H. Tsai, and C. Y. C. Chen. 16.
J Biomol Struct Dyn 27, 171-178 (2009).
E. F. F. da Cunha, E. F. Barbosa, A. A. Oliveira, and T. C. Ramalho.
17. J Biomol Struct Dyn
27, 619-625 (2010).
L. I. D. Hage-Melim, C. H. T. D. da Silva, E. P. Semighini, C. A. Taft, and S. V. Sampaio. 18.
J Biomol Struct Dyn 27, 27-35 (2009).
H. J. Huang, K. J. Lee, H. W. Yu, C. Y. Chen, C. H. Hsu, H. Y. Chen, F. J. Tsai, and C. Y. 19.
C. Chen. J Biomol Struct Dyn 28, 23-37 (2010).
H. J. Huang, K. J. Lee, H. W. Yu, H. Y. Chen, F. J. Tsai, and C. Y. Chen.
20. J Biomol Struct
Dyn 28, 187-200 (2010).
A. K. Kahlon, S. Roy, and A. Sharma.
21. J Biomol Struct Dyn 28, 201-210 (2010).
C. Koshy, M. Parthiban, and R. Sowdhamini.
22. J Biomol Struct Dyn 28, 71-83 (2010).
S. Mohan, J. J. P. Perry, N. Poulose, B. G. Nair, and G. Anilkumar.
23. J Biomol Struct Dyn 26,
455-464 (2009). J. Sille and M. Remko.
24. J Biomol Struct Dyn 26, 431-444 (2009). Y. Tao, Z. H. Rao, and S. Q. Liu.
25. J Biomol Struct Dyn 28, 143-158 (2010).
L. H. Zhong and J. M. Xie.
26. J Biomol Struct Dyn 26, 525-533 (2009).
T. T. Chang, H. J. Huang, K. J. Lee, H. W. Yu, H. Y. Chen, F. J. Tsai, M. F. Sun, and 27.
C. Y. Chen. J Biomol Struct Dyn 28, 309-321 (2010).
C. C. Su, J. S. Yang, C. C. Lu, J. H. Chiang, C. L. Wu, J. J. Lin, K. C. Lai, T. C. Hsia, 28.
H. F. Lu, M. J. Fan, and J. G. Chung. Phytother Res 24, 189-192 (2010).
C. Lo, T. Y. Lai, J. H. Yang, J. S. Yang, Y. S. Ma, S. W. Weng, Y. Y. Chen, J. G. Lin, and 29.
J. G. Chung. Int J Oncol 37, 377-385 (2010).
S. H. Wu, L. W. Hang, J. S. Yang, H. Y. Chen, H. Y. Lin, J. H. Chiang, C. C. Lu, J. L. Yang, 30.
T. Y. Lai, Y. C. Ko, and J. G. Chung. Anticancer Res 30, 2125-2133 (2010). C. Y. C. Chen.
31. PLoS ONE 6, e15939 (2011).
C. S. Chen, Y. C. Wang, H. C. Yang, P. H. Huang, S. K. Kulp, C. C. Yang, Y. S. Lu, 32.
S. Matsuyama, C. Y. Chen, and C. S. Chen. Cancer Res 67, 5318-5327 (2007). C. M. Venkatachalam, X. Jiang, T. Oldfield, and M. Waldman.
33. J Mol Graph Model 21,
289-307 (2003).
A. Krammer, P. D. Kirchhoff, X. Jiang, C. M. Venkatachalam, and M. Waldman.
34. J Mol
Graph Model 23, 395-407 (2005).
D. K. Gehlhaar, G. M. Verkhivker, P. A. Rejto, C. J. Sherman, D. B. Fogel, L. J. Fogel, and 35.
S. T. Freer. Chem Biol 2, 317-324 (1995).
B. Ploeger, T. Mensinga, A. Sips, W. Seinen, J. Meulenbelt, and J. DeJongh.
36. Drug Metab
Rev 33, 125-147 (2001).
H. Hayashi, E. Miwa, and K. Inoue.
37. Biol Pharm Bull 28, 161-164 (2005).
Date Received: November 24, 2010