國
立
交
通
大
學
照明與能源光電研究所
碩
士
論
文
經由調整主動層厚度改善
固態有機白光電化學元件之效率
Improving Device Efficiencies of Solid-State White
Light-Emitting Electrochemical Cells by Adjusting the
Active-Layer Thickness
研 究 生:張元佩
指導教授:蘇海清 副教授
經由調整主動層厚度改善
固態有機白光電化學元件之效率
Improving Device Efficiencies of Solid-State White Light-Emitting
Electrochemical Cells by Adjusting the Active-Layer Thickness
研 究 生:張元佩 Student:Yuan-Pei Jhang 指導教授:蘇海清 Advisor:Hai-Ching Su 國 立 交 通 大 學 照 明 與 能 源 光 電 研 究 所 碩 士 論 文 A Thesis
Submitted to Institute of Lighting and Energy Photonics College of Photonics
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
In
Lighting and Energy Photonics June 2013
Tainan, Taiwan, Republic of China
經由調整主動層厚度改善固態有機白光電化學元件之效率
學生:張元佩 指導教授:蘇海清 國立交通大學照明與能源光電研究所碩士班 摘 要 固態有機白光電化學元件由於具有結構簡單、可溶液製程、可使用非 活性金屬電極與低功耗等優點,在白光照明的應用上極具潛力,因此為了 達到實際應用的需求必須更進一步提升固態有機白光電化學元件的效率, 而在改善固態有機白光電化學元件的效率方面,靠近電化學摻雜層的複合 區激子猝熄是一大障礙。本文使用了調整主動層厚度的方法來減少激子猝 熄,固態有機白光電化學元件在適當加厚主動層厚度的情形下,其複合區 可以更接近主動層的中央,因此緩解了激子猝熄而提昇元件效率。在優化 元件主動層厚度之後可得到大約 11 %的外部量子效率以及 20 流明/瓦的功 率效率,這是目前固態有機白光電化學元件所發表過文獻中效率最高的。 本文的結果證實了修改主動層厚度來避免激子猝熄藉此改善固態有機白光 電化學元件效率是一個可行的方法。Improving Device Efficiencies of Solid-State White Light-Emitting
Electrochemical Cells by Adjusting the Active-Layer Thickness
Student:Yuan-Pei Jhang Advisors:Dr. Hai-Ching Su
Institute of Lighting and Energy Photonics National Chiao Tung University
ABSTRACT
Exciton quenching in the recombination zone close to electrochemically doped regions would be one of the bottlenecks for improving device efficiencies of solid-state white light-emitting electrochemical cells (LECs). To further enhance device efficiencies of white LECs for practical applications, we adjust the active-layer thickness to reduce exciton quenching. In white LECs with properly thickened active-layer thickness, the recombination zone can be situated near the center of the active layer, rendering mitigated exciton quenching and thus enhanced device efficiencies. High external quantum efficiencies and power efficiencies of optimized devices reach ca. 11% and 20 lm/W, respectively, which are among the highest reported for white LECs. These results confirm that tailoring the thickness of the active layer to avoid exciton quenching would be a feasible approach to improve device efficiencies of white LECs.
致謝
研究所短暫的生活一轉眼就到尾聲了,在台南的這兩年我遇到了許多貴 人,第一個要感謝的就是我的指導老師蘇海清教授,老師嚴謹的教學態度 以及平易近人的處世作風都令我印象深刻;感謝實驗室的學長姐們,大學 長志騰、寶哥、阿凱、Panda、蘇盈,從我剛進實驗室完全不會做製程到學 會所有設備,都很有耐心的教導我且不嫌煩的讓我一直問問題,在日常生 活上,更是時常帶我們去吃好料、一起打遊戲、看電影;感謝實驗室同學 柏材、白姐,我們從同一所大學畢業來到這裡,就好像已經認識很久的好 朋友一樣一拍即合,並在這兩年裡互相打氣一起成長,成為好夥伴,這真 是得來不易的緣分;感謝實驗室學弟妹阿昇、瑞哥、櫻桃,為我碩二忙著 寫碩論的生活增添了許多有趣的事情,感謝 FDL 的大家,我愛 FDL! 再來我要感謝我身邊所有的朋友,感謝你們在我低潮的時候總是陪伴著 我以及鼓勵我,讓我有動力可以變得更好;最後,我要感謝我的家人,感 謝他們一直以來不辭辛勞的幫助我、鼓勵我,供給我所需要的一切,如果 沒有你們,就沒有今天的我,謝謝你們!目錄
中文摘要 ... i 英文摘要 ... ii 致謝 ... iii 目錄 ... iv 圖目錄 ... vi 表目錄 ... viii 第一章 緒論... 1 1.1 前言 ... 1 1.2 有機發光二極體起源 ... 1 1.3 有機發光電化學元件簡史 ... 4 1.4 有機白光電化學元件文獻回顧 ... 6 1.5 研究動機與目的 ... 15 第二章 理論基礎 ... 16 2.1 螢光與磷光發光原理 ... 16 2.2 主客體發光系統 ... 18 2.2.1 能量轉移(energy transfer)形式 ... 18 2.2.2 載子捕捉(carrier trapping)形式 ... 19 2.3 有機發光二極體發光機制 ... 19 2.4 有機發光電化學元件發光機制 ... 20 2.4.1 電化學架構(Electrochemical model) ... 20 2.4.2 電動力學架構(Electrodynamic model) ... 21 2.5 發光元件效率計算法 ... 23 2.6 白光元件的量化方法 ... 24 2.6.1 色彩座標(chromaticity coordinates) ... 242.6.2 相對色溫度(correlated color temperature, CCT) ... 25
2.6.3 演色性指數(color rendering index, CRI) ... 26
3.1 實驗設備簡介 ... 28 3.2 實驗製程步驟 ... 30 3.2.1ITO 玻璃基板製備與清洗步驟 ... 30 3.2.2 元件製程步驟... 31 3.3 實驗量測系統 ... 32 3.3.1 量測設備簡介... 32 3.3.2 量測步驟 ... 33 3.4 元件結構 ... 35 第四章 實驗結果分析 ... 38 4.1 材料簡介及光物理特性... 38 4.2 實驗參數及電性分析 ... 41 第五章 結論... 52 參考文獻 ... 53
圖目錄
圖 1 典型多層有機發光二極體架構 ... 2 圖 2 [Ru(bpy)3] 2+ 的結構[9] ... 6 圖 3 BDOH-PF 的結構[51] ... 7 圖 4 EO-PF-DPO 結構[52] ... 8 圖 5 三原色共聚物化學結構(BGR 分別為藍綠紅)[53] ... 9 圖 6 MCP 的結構(m+n+o+p+q+r=1;R0,R2,R3,R4為增溶烷基,Ar1為芳基,R5與 R6為烷氧基)[54] ... 9 圖 7 MCP 鏈應用於有機發光二極體與有機發光電化學元件的示意圖[54] 10 圖 8 PF 合成不同離子及 2,1,3-Benzoselenadiazole 的化學結構[55] ... 10 圖 9 左: [Ir(dfppz)2(dtb-bpy)] + (PF6 ─ ),右: [Ir(ppz)2(biq)] + (PF6 ─ )[56] ... 11 圖 10 [Ir(dfppz)2(tp-pyim)]PF6 結構[57] ... 12 圖 11 雙重摻雜能階示意圖[58] ... 13 圖 12 藍:[Ir(dfppz)2(dtb-bpy)] + (PF6 ─ )、紅: [Ir(ppy)2(biq)] + (PF6 ─ )、橘: [Ir(ppy)2(dasb)] + (PF6 ─ )結構[58] ... 13 圖 13 DCJTB 結構[59] ... 14 圖 14 有機發光電化學元件結合色彩轉換層架構圖[59] ... 14 圖 15 SR101 結構[60] ... 15 圖 16 Jablonski 分子能階圖 ... 17 圖 17 電子電洞再結合所產生的激發態 ... 17 圖 18 Forster 與 Dexter 能量轉移示意圖 ... 18 圖 19 有機發光二極體多層膜能階及發光原理示意圖 ... 20 圖 20 (a)電化學架構示意圖(b)電動力學架構示意圖[61] ... 21 圖 21 兩種架構的電位變化示意圖(左:電化學架構,右:電動力學架構)[64] ... 22 圖 22 發光元件出光示意圖 ... 23 圖 23 外部量子效率推算示意圖 ... 23 圖 24 CIE 1931 色彩座標圖 ... 24圖 25 黑體輻射軌跡線 ... 25 圖 26 相對色溫表 ... 25 圖 27 計算 CRI 所需之八個測試光源頻譜 ... 26 圖 28 蒸鍍金屬電極所使用之遮罩... 32 圖 29 本實驗室所使用之量測平台示意圖 ... 34 圖 30 元件量測方式示意圖 ... 35 圖 31 本實驗之元件結構示意圖 ... 35 圖 32 PEDOT:PSS 之結構式 ... 37 圖 33 左:1 號錯合物、右:2 號錯合物之結構式 ... 39 圖 34 BMIM+ (PF6 ─ )之結構式 ... 39 圖 35 1 號錯合物及 2 號錯合物之能階排列示意圖 ... 40 圖 36 元件(a)I、(b)II 及(c)III 在驅動電壓 3.3 伏特隨時間變化的電致發光頻 譜圖 ... 43 圖 37 當元件 III 的複合區位置(以陰極為起始點開始測量)從 380 奈米移動 到 280 奈米的模擬電致發光頻譜圖 ... 45 圖 38 元件(a)I、(b)II 及(c)III 在 3.1 伏特、3.3 伏特及 3.5 伏特的穩定電致 發光頻譜 ... 47 圖 39 元件 II 在固定驅動電壓 3.1 伏特、3.3 伏特及 3.5 伏特下隨時間變化 的(a)亮度(實心符號)及電流密度(空心符號)和(b)外部量子效率(實心符 號)及功率效率(空心符號)圖 ... 48 圖 40 元件(a)I、(b)II 及(c)III 在驅動電壓 3.5 伏特之模擬(實心符號)及量測 (空心符號)的穩定電致發光頻譜。其模擬及量測電致發光頻譜的複合區 位置(Zi)如頻譜圖內的子圖所示。 ... 51
表目錄
表 1 有機發光二極體與有機高分子發光二極體的優缺點比較 ... 3 表 2 演色性指數與對應適用場所... 27 表 3 有機白光電化學元件電致發光特性總整理 ... 50
第一章 緒論
1.1 前言 無機半導體材料發光技術的研究已經持續了五十年以上,對於材料的改 進及製程技術皆趨於成熟,市面上商品化的產品比比皆是,幾乎已經取代 了前一代的冷陰極螢光燈管,成為新世代的光源技術,反觀有機半導體材 料發光技術從 1987 年鄧青雲博士等人發表的文獻受到矚目至今不過短短二 十幾年,以研究文獻及產業發展速度看來,有機材料發光技術是有過之而 無不及,重點就在於它能彌補無機半導體先天材料特性的不足,有機材料 分子結構具可塑性,可經由設計調變材料的熱穩定性、機械強度、發光特 性及導電特性,最大的優勢是可以在任何基板上成膜,甚至是做成可撓式 元件[4]。而其中又以白光有機發光元件的應用最為廣泛,同為自發光的無 機發光二極體屬於「點」光源,以照明應用來說,需要結構設計才能均勻 出光,而有機發光二極體本身就具有「面」光源特點,且容易製作大面積, 目前發光效率也慢慢接近市面上白光照明的水平,若未來能把成本再降低, 肯定能在照明市場佔有一席之地。除了照明上的應用,白光有機發光元件 在平面顯示器領域也有相當的競爭力,不管是當液晶顯示器的背光源或是 直接 加上 RGBW 彩 色 濾光 片 的全 彩 顯示 技 術製 作 出 有機 發 光二 極體 (organic light-emitting diode, OLED)顯示器,以上優點驅使全球學術界及業 界都開始默默在耕耘這項前途一片燦爛的明星產業。 1.2 有機發光二極體起源 有機材料電致發光現象的發現要追溯到 1963 年 Pope 等人所發表的文獻, 其實驗架構是利用昇華溶液的方式製作出 10 微米到 20 微米的單晶蔥 (anthracene)薄膜,並在薄膜兩端加上銀箔做為電極,實驗結果指出在施加 400 伏特的超高驅動電壓下,才觀察到微弱的藍光電致發光[1],礙於驅動 電壓太高以及發光效率不佳,在當時並未受到太多關注。直到 1987 年美國 柯 達 公 司 的 鄧 青 雲 博 士 等 人 將 有 機 螢 光 材 料 tris(8-hydroxyquinoline) aluminum(Alq3)與二胺(diamine)以熱蒸鍍的方式製作出雙層的有機發光二極體,元件架構為 Mg:Ag/Alq3/Diamine/ITO/Glass,其中銦錫氧化物(indium
tin oxide, ITO)做為陽極,鎂銀合金(Mg:Ag)做為陰極,實驗結果在小於 10 伏 特的驅動電壓下,可以得到外部量子效率 1 %(光子數/電子數)、功率效率 1.5 流明/瓦且亮度達 1000 燭光/平方公尺以上的綠光電致發光元件(強度最 大值在 550nm)[2],此文獻讓各國學者看見了有機發光元件的實用價值,並 且掀起了此領域的研究熱潮,然而 1989 年鄧青雲博士等人再提出第一篇由 加入客體摻雜物(dopant)的方式來調整光色的研究文獻,其內容是將發不同 光色的客體材料少量摻雜進入主發光體材料,再依摻雜的濃度來達到調整 元件光色及改善元件壽命等目的,同時少量摻雜也會提高外部量子效率, 因為它可將能量轉移給螢光效率較高的客體材料[3],此文獻提供了有別於 多層發光元件的製作方法,多摻雜發光元件亦是研究有機材料發光的重點 之一。 在鄧青雲博士發表了雙層有機發光二極體的文獻[2]後,多層有機發 光二極體的結構開始被廣泛的修正和優化,其目標在於要讓電子與電洞再 結合(electron-hole recombination)發光的區域可以遠離接觸電極,以避免激 子靠近電極的淬熄效應,而提高元件的發光效率,且在設計多層膜結構時, 也必須匹配各層間的能階差,才能使電子與電洞注入容易,降低元件驅動 電壓,基於以上條件,造就了現今多層膜有機發光二極體的基本結構,由 陰極電極、電子注入層(electron injection layer, EIL)、電子傳輸層(electron transport layer, ETL)、發光層(emissive layer, EML)、電洞傳輸層(hole transport layer, HTL)、電洞注入層(hole injection layer, HIL)及透明導電陽極所組成的 三明治結構,如圖 1 所示。
相較於美國柯達公司等人所使用的小分子有機材料以熱蒸鍍技術製作 的有機發光二極體,英國劍橋大學在 1990 年由 Burroughes 等人將共軛高分 子 有 機 材 料 poly(p-phenylene vinylene)(PPV) 以 溶 液 製 程 旋 轉 塗 佈 (spin coating)技術製作出第一 個有機高分子發光二極體(polymer light-emitting diode, PLED),其元件架構為 ITO/PPV/Al 的單層結構[5],因其製程簡單成 本低而受到學術以及產業界的矚目。有機發光二極體與有機高分子發光二 極體除了在發光原理上相似外,其優缺點可說是各有千秋,有機發光二極 體具有多層結構可得到良好的元件發光效率,但因為蒸鍍技術需要在真空 狀態進行才能保持膜層低雜質,因此有製程繁瑣、設備昂貴等問題;而有 機高分子發光二極體只要單層結構就能操作且可利用旋轉塗佈、噴墨或印 刷的技術製作,因此成本相對較低,且高分子材料熱穩定性高、可塑性良 好,適合做大尺寸以及可撓元件,但高分子材料的純化不易,容易發生不 良元件或元件壽命較短的問題。表 1 為有機發光二極體與有機高分子發光 二極體的比較整理。 有機發光二極體 有機高分子發光二極體 材料 分子量數百至數千 分子量達一萬以上 製程方法 真空熱蒸鍍 旋轉塗佈、噴墨、印刷 相對優點 效率高 材料純化容易 元件壽命較長 色彩控制容易且穩定 設備成本低 結構簡單 成膜性良好 熱穩定性與可塑性高 相對缺點 製程步驟繁瑣 設備昂貴 驅動電壓高 熱穩定性與機械強度低 易受水氧影響 材料合成與純化不易 彩色化不易 產業化步伐較慢 表 1 有機發光二極體與有機高分子發光二極體的優缺點比較
有機發光二極體還有一些要克服的問題,像是有機材料的電子電洞遷移 率都比無機材料相對低非常多,導致導電性不佳,而影響到元件發光效率, 若能由元件結構來改善載子注入及載子平衡方能解決此問題,目前有機發 光元件的研究面向相當廣泛,諸如材料合成、改善元件壽命、改善製程、 提高效率等,應用方面不管是固態照明、全彩顯示器、太陽能電池、生醫 等,都可以看見有機材料極大的潛力,並且朝向商品化的方向發展,相信 再過不久,有機材料發光技術就能趕上無機半導體的腳步,為新世代的光 源應用推向另一座高峰。 1.3 有機發光電化學元件簡史 有機發光二極體和有機高分子發光二極體的多層結構需要各層的能階 差相互匹配,才能減少載子注入能障,達到降低驅動電壓的效果,且陰極 必需使用低功函數材料,但低功函數材料有高度化學活性的問題,導致容 易氧化造成元件損壞,因此在1995年Pei等人提出了一種新樣式的有機發光 元件想要嘗試改善上述的問題,命名為有機發光電化學元件(light-emitting electrochemical cells, LEC),其元件架構是使用旋轉塗佈的方法製作出簡單
的 單 層 元 件 ITO/MEH-PPV+PEO(LiCF3SO3)/Al, 材 料 是 使 用 共 軛 高 分 子
poly[5-(2´-ethylhexyloxy)-2-methoxy-1,4-phenylene vinylene)(MEH-PPV)摻入
鹽 類 LiCF3SO3與 固 態 電 解 質 poly(ethylene oxide)(PEO) , 並 使 用 乙 腈
(acetonitrile)做為溶劑,再加入少量的水增加MEH-PPV與PEO的溶解性,在 功用上MEH-PPV為主要發光材料,LiCF3SO3為提供離子,而PEO的摻雜是 為了改善MEH-PPV與LiCF3SO3因極性差異的相分離造成元件損壞,此文獻 提出的電化學發光機制是當施加驅動電壓時,電解質(PEO+LiCF3SO3)解離 出正負離子往兩邊的電極移動做補償使得有機層材料產生氧化還原反應, 形成p-i-n的摻雜結構,使載子從陽極與陰極分別注入p型與n型摻雜層並在i 型區結合放光,而一旦正負離子形成完整的p-n接面,載子注入位障就會大 幅下降,所以較沒有電極與有機層能階匹配的問題,因此可使用空氣中穩 定的金屬當做電極,最小驅動電壓也只需要發光材料的最低未填滿分子軌 域(lowest unoccupied molecular orbital, LUMO)至最高填滿分子軌域(highest
occupied molecular orbital, HOMO)的化學電位差(eVon=Eg),且因離子性材料 具有可逆氧化還原反應,其驅動元件的方式可以是順向偏壓、逆向偏壓甚 至是交流電[6][7]。有機發光電化學元件的發光機制到現今為止尚未定論, 目前最具有公信力的理論除了上述Pei團隊所提出的電化學模型,還有 Slinker團隊所提出的電動力學模型,其詳細的理論探討將在第二章再繼續。 早期具有電化學特性的材料所製造出來的發光元件在效率與壽命方面 比較少有顯著改善,直到1996年Lee等人提出第一篇用離子性過渡金屬錯合 物(ionic transition metal complexes, iTMC)做成的單層發光元件,以過渡金屬
釕 元 素 (Ruthenium) 的 錯 合 物 Ru(phen´)3 2+做 為 發 光 材 料 , 其 中 phen´為 1,10-phenanthroline,製程技術為旋轉塗佈(spin-coating)技術,這些離子性過 渡金屬錯合物材料本身可以提供載子注入、載子傳輸及載子複合發光等三 種過程,如此一來這些元件內部過程都由單一材料來主導,就可以避免高 分子有機發光電化學元件因材料極性不同造成的相分離現象,其初步研究 成果指出此材料的確有提昇元件穩定性的功用[8],1999年Handy等人發表的 文 獻 中 再 將 釕 錯 合 物 進 行 改 良 成 [Ru(bpy)3] 2+ (PF6 ─ )2, 其 中 bpy 是 2,2´-bipyridine[9],而[Ru(bpy)3] 2+的結構如圖2,在施加電壓的情況下,錯合 物的陰離子(PF6 ─ )2會被解離出來,並且受到內部電場作用而往陽極移動,而 剩下的正離子[Ru(bpy)3] 2+由於空間結構太大導致較不易被內部電場影響而 產生移動,在陰離子一直漂移到陽極堆疊的情況下,正離子也就像是被推 往陰極進行堆疊,接下來就跟上述有機發光電化學元件的發光機制是一樣 的,因此本材料保有有機發光電化學元件能改善載子注入的優點。此外, 離子性過渡金屬錯合物材料與一般螢光元件相比,可以有效利用到三重激 發態能階所釋放出來的磷光,使得元件效率較高,若其材料的光致發光量 子產率(photoluminescence quantum yield, PLQY)可以達到100 %,則內部量 子效率就有機會接近100 %[10],文獻指出此元件在很低的驅動電壓2.5 伏 特下可發出橘紅光,其外部量子效率提升至1 %以及1.4 流明/瓦的功率效率, 且在驅動電壓5 伏特時,可得到1000 燭光/平方公尺的亮度,如此的高效率、 高亮度及高穩定性,吸引了許多團隊開始進行對於離子性過渡金屬錯合物 材料的相關研究[11-49]。
有機發光電化學元件雖然具有比傳統有機發光二極體與有機高分子發 光二極體有更佔優勢的優點,諸如低功耗、低製程成本、可使用低活性金 屬電極以及高效率等;但也有需要改進的缺點,諸如反應速度慢、材料相 分離導致元件壽命減短等,因此若要應用於固態照明及平面顯示器,上述 的缺點是未來主要的研究指標。 1.4 有機白光電化學元件文獻回顧 固態有機白光元件具有自發光、低功耗、廣視角、可製作大型面板及可 撓曲等特性,在固態照明及顯示器應用極具潛力。最早期的白光有機發光 二極體的文獻是由日本山形大學 Kido 教授等人在 1994 年所發表,元件結
構 為 Glass/ITO/doped poly(N-vinyl carbazole)(PVK)/1,2,4-triazole
derivative(TAZ)/Alq3/Mg:Ag , 其 中 doped PVK 層 摻 雜 三 種 螢 光 染 料
1,1,4,4-tetraphenyl-1,3-butadiene(TPB)、發綠光的香豆素(Coumarin6)、發橘 光的 DCM1 以溶液塗佈製程做為發光層,而 TAZ、Alq 屬於小分子材料可 與陰極 Mg:Ag 一樣使用熱蒸鍍製程製作成膜,其發光原理是利用高分子材 料 PVK 為主體材料以能量轉移(energy transfer)的方式讓三種染料發光而得 到白光[50],在此之後白光有機發光二極體的研究便開始蓬勃發展,至今已 廣泛應用於許多科技產品當中,但白光有機發光二極體有先天缺陷,諸如 製程複雜導致成本高、電極材料不穩定等,因此研究學者開啟了另一條有 機白光電化學元件的研究之路,欲將固態有機白光元件變得更有價值。 圖 2 [Ru(bpy)3] 2+的結構[9]
有機白光電化學元件以材料差別大致上可區分為兩種系統,第一種是利 用 修 改 高 分 子 材 料 的 側 基 (side group) 或 接 上 發 不 同 光 色 的 共 聚 單 體 (comonomer)使材料具有促進離子傳導特性及發出白光[51-55],通常會再加 入鹽類(如 LiCF3SO3)來改善元件的點亮時間,第一篇文獻是在 1997 年 Yang 等 人 所 發 表 , 他 們 使 用 高 分 子 材 料 聚 芴 (polyfluorene) 的 衍 生 物 poly[9,9-bis(3,6-dioxaheptyl)-fluorene-2,7-diyl](BDOH-PF) 摻 入 鹽 類
LiCF3SO3 做 成 元 件 結 構 為 ITO/BDOH-PF+LiCF3SO3/Al 的 單 層 結 構 ,
BDOH-PF 的結構如圖 3 所示,實驗結果發現在低驅動電壓(3~4 伏特)的情 況下,可得到外部量子效率 4 %、功率效率 12 流明/瓦及亮度超過 1000 燭 光/平方公尺的高效率藍綠光元件,並且再多加 poly(ethylene oxide)(PEO)到 發光層後,PEO 與 BDOH-PF 產生相分離現象,光色卻因此位移成頻寬較 大的白光,在驅動電壓 4 伏特下得到 2.4 %的白光外部量子效率,且 PEO 的加入也使光色更加穩定[51]。 在第一篇基於高分子材料的有機白光電化學元件發表後,隔了許久才在 2010 年由 Sun 等人再度發表文獻,其內容是利用 fluorene-oxadiazole 的共聚 物(copolymer)做為發光層,此材料稱為 EO-PF-DPO,它是第一篇單一種高 分子材料製作成有機白光電化學元件的文獻,由分子堆疊而形成活化雙體 (excimer)來形成多波段的 EL 頻譜[52]。圖 4 為 EO-PF-DPO 結構。 圖 3 BDOH-PF 的結構[51]
2011 年,Tang 等人提出利用三原色(trichromatic)的共聚物(copolymer)
混合離子傳輸材料 trimethylolpropane ethoxylate(TMPE)與鹽類 LiCF3SO3,
以溶液製程方式製作出單層白光元件,實驗結果指出此元件的電流效率達 3.1 燭光/安培及功率效率 1.6 流明/瓦,且點亮時間非常短(約 5 秒亮度可 達 100 燭光/平方公尺), 還具有良好的元件壽命(亮度大於 100 燭光/平方 公尺可達 17 小時)及穩定性[53],這是未來要應用在產品上最不可或缺的因 素,圖 5 為三原色共聚物化學結構。 而 同 樣 的 Tang 團 隊 在 2013 年 又 發 表 了 使 用 多 螢 光 共 軛 共 聚 物 (multifluorophoric conjugated copolymer,MCP)為發光層材料,混合離子傳輸
材料 TMPE 與鹽類 LiCF3SO3製作成單層且單一材料發白光元件,其發白光 的原理是利用離子傳輸材料 TMPE 將 MCP 鏈相互隔開,抑制住激子(exciton) 在分子間(或分子內)所進行的擴散及 Forster 能量轉移,使 MCP 鏈有因分離 (isolated)得到頻寬較大的藍光和綠光以及因聚集(aggregated)得到的紅光,因 此產生一個寬頻的白光,單一發光(single-emitter)的有機白光電化學元件也 解決了因為相分離或多發光(milti-emitter)有機白光電化學元件所導致的色 彩偏移,提昇了穩定性,其實驗結果指出此白光的演色性(color rendering index, CRI)達 82 以及電流轉換效率 3.8 燭光/安培[54]。圖 6 為 MCP 的結構, 圖 7 為 MCP 鏈應用於有機發光二極體與有機發光電化學元件的示意圖。 圖 4 EO-PF-DPO 結構[52]
圖 5 三原色共聚物化學結構(BGR 分別為藍綠紅)[53]
圖 6 MCP 的結構(m+n+o+p+q+r=1;R0,R2,R3,R4為增溶烷基,Ar1為芳基,R5
同樣在 2013 年,Tsai 等人也提出了單一成分(single-component)發白光 的有機高分子電化學元件,其利用聚芴(polyfluorene, PF)電解質在離子側鏈 C-9 號位置接上 Br─,BF4─或 PF6─陰離子,再合成 2,1,3-Benzoselenadiazole 到聚合物主鏈形成發白光的元件,在高分子材料接上陰離子的方法可提供 可移動離子,因此不需再額外摻雜鹽類,且接上不同陰離子也會讓材料的 熱穩定性與光學特性跟著改變,實驗結果得到在驅動電壓 3 伏特時功率效 率達 1.56 流明/瓦,其所發白光的 CIE 座標接近(0.33,0.33)以及 CRI 高達 85 以上[55]。圖 8 為 PF 合成不同陰離子及 2,1,3-Benzoselenadiazole 的化學結 構。 圖 7MCP 鏈應用於有機發光二極體與有機發光電化學元件的示意圖[54] 圖 8PF 合成不同離子及 2,1,3-Benzoselenadiazole 的化學結構[55]
另一種有機白光電化學元件的系統是使用離子性過渡金屬錯合物材料, 此類材料的優點在於發光機制是磷光系統,材料發光效率比螢光高分子材 料還要高,因此製作出來的白光效率也比高分子材料來得好,且由於摻雜 的主客體材料本身就具有離子傳導特性,因此不需再額外摻入離子傳輸材 料 PEO , 其 第 一 篇 文 獻 是 2008 年 由 Su 等 人 所 發 表 , 以 藍 綠 光 材 料 [Ir(dfppz)2(dtb-bpy)] + (PF6 ─ ) ( 其 中 dfppz 為 1-(2,4-difluorophenyl)pyrazole , dtb-bpy 為 [4,4´-di(tert-butyl)-2,2´-bipyridine]) 做 為 主 體 加 入 少 量 紅 光 材 料 [Ir(ppz)2(biq)] + (PF6 ─ ) (其中ppz為1-phenylpyrazole,biq為2,2´-biquinoline)做為 客 體 的 主 客 體 摻 雜 型 白 光 元 件 , 並 且 再 加 入 適 量 鹽 類 1-Butyl-3-methylimidazolium hexafluorophosphate(縮寫為BMIM+(PF6 ─ ))做為 額外添加離子使p-n 接面建立更快速,讓反應時間減短,製程方法是以旋轉 塗佈主動層的方法製作成結構為ITO/主動層(100 奈米)/銀(150 奈米)的單 層元件,此元件在驅動電壓2.9~3.3 伏特時得到CIE座標從(0.45,0.40)到 (0.35,0.39)的白光及CRI達80以上,且在驅動電壓2.9 伏特時可達到外部量子 效率4 %、電流效率7.2 燭光/安培及功率效率7.8 流明/瓦的高效率白光[56]。 圖9為[Ir(dfppz)2(dtb-bpy)] + (PF6 ─ ) 及[Ir(ppz)2(biq)] + (PF6 ─ ) 的結構。
在 2010 年 He 等 人 所 發 表 的 文 獻 是 利 用 修 改 藍 綠 光 材 料 的 環 金 屬 基 (cyclometalated ligand)及輔助配位基(ancillary ligand)製作成大側基團的材料
並命名為[Ir(dfppz)2(tp-pyim)]PF6(其中dfppz為1-(2,4-difluorophenyl)-1H-pyr-
azole,tp-pyim為2-(1-(4-tritylphenyl)-1H-imidazol-2-yl)pyridine),文獻指出具 有大側基團的主體藍綠光材料有更好的立體阻礙,藉此可降低分子間的自 我猝熄(self-quenching)而達到提升發光效率的效果,再摻入客體紅光材料
[Ir(ppy)2(qlbi)]PF6(全名為bis(2-phenylpyridinato-N,C
2´ )iridium(III)[3-(1-phen- yl-1H-benzo[d]imidazol-2-yl)-isoquinoline] hexafluorophosphate)及BMIM+PF6 做成白光,其實驗結果得到CIE座標(0.37,0.41)及CRI達80以上的白光,並且 外 部 量 子 效 率 提 升 至 5.6 % 以 及 功 率 效 率 11.2 流 明 / 瓦 [57] 。 圖 10 為 [Ir(dfppz)2(tp-pyim)]PF6 的結構。 主客體摻雜型元件的材料能階差會引起載子捕捉(carrier trapping),且 LUMO與HOMO的能階差不一致更是使載子遷移率(carrier mobility)變差, 導致元件效率下降,為了改善此問題,2011年Su與Shen等人發表的文獻提 出了雙重摻雜(double-doped)的固態有機白光電化學元件,其利用多摻入一 能 階 介 於 藍 綠 光 材 料 [Ir(dfppz)2(dtb-bpy)] + (PF6 ─ ) 與 紅 光 材 料 [Ir(ppy)2(biq)] + (PF6 ─ ) 的 橘 光 材 料 [Ir(ppy)2(das1b)] + (PF6 ─ )( 其 中 ppy 為 2-phenylpyridine,dasb為4,5-diaza-9,9´-spirobifluorene)來減少載子捕捉現象, 圖 10[Ir(dfppz)2(tp-pyim)]PF6 結構[57]
使載子遷移率變好而讓元件效率更提昇,實驗結果在驅動電壓3.1 伏特時得 到外部量子效率7.4 %及功率效率15 流明/瓦[58]。圖11為雙重摻雜能階示意 圖,圖12為此文獻中三種發光材料的化學結構。
圖 11 雙重摻雜能階示意圖[58]
圖12 藍:[Ir(dfppz)2(dtb-bpy)]+(PF6─)、紅: [Ir(ppy)2(biq)]+(PF6─)、
然而,主客體摻雜型白光元件在低驅動電壓時,激子容易受到載子捕捉 現象的影響,停留在能階較低的客體材料上的比例增加,先前的文獻裡也 指出經常當成客體摻雜的紅光離子性過渡金屬錯合物,它的光致發光量子 產率低於0.2(激發出的光子數/施加的光子數)[56-58],因此主客體摻雜型元 件有著對不同電壓下發出的光色位移以及客體材料導致元件發光效率下降 等缺點,對於上述缺點,在2012年Su與Wu等人提出將紅光染料DCJTB(全名 為4-(Dicyanomethylene)-2-tert-butyl-6-(1,1,7,7-tetramethyljulolidin-4-yl-vi- nyl)-4H-pyran) 旋 轉 塗 佈 在 載 玻 片 上 做 為 色 彩 轉 換 層 (color conversion
layers,CCL),讓有機主動層只有主體藍綠光材料[Ir(dfppz)2(dedaf)] + (PF6 ─ )及 鹽類BMIM+ (PF6 ─ ),實驗結果得到最高外部量子效率達5.93 %及功率效率 15.34 流明/瓦,且證明了利用色彩轉換層製作白光的方法,可消除載子捕 捉現象使不同電壓下都有穩定的白光頻譜,在高效率與穩定白光光色上都 是極具潛力的固態有機白光電化學元件[59]。圖13為DCJTB結構,圖14為有 機發光電化學元件結合色彩轉換層架構圖。 圖 13DCJTB 結構[59] 圖 14 有機發光電化學元件結合色彩轉換層架構圖[59]
最近的一篇使用離子性過渡金屬錯合物的有機白光電化學元件的文獻 是2012年由Su與Chen等人所發表的利用磷光敏化系統所得到的高效率固態 有 機 白 光 電 化 學 元 件 , 內 容 是 用 磷 光 主 體 藍 綠 光 材 料 [Ir(dfppz)2(dedaf)] + (PF6 ─ )摻雜螢光客體紅光染料sulforhodamine 101(SR101), 原理是基於磷光材料具有單重激發態(singlet state)與三重激發態(triplet state) 都可有效釋放出光子的特性,使磷光主體材料在Forster能量轉移到螢光客體 材料時,螢光客體材料可獲得主體單重激發態與三重激發態的激發光子能 量,且激發態壽命較短的螢光客體材料可能減少了磷光主客體摻雜系統的 三重態-三重態消滅(triplet-triplet annihilation)的現象,因此可得到與磷光主 客體摻雜系統的元件相近的元件效率,實驗結果得到高達7.9 %的外部量子 效率及15.6 流明/瓦的功率效率,是目前固態有機白光電化學元件文獻的最 高效率記錄,且文獻中發現了元件結構具有微共振腔(microcavity)效應,剛 好在主體材料偏綠光的部分形成破壞性干涉,造成波峰藍移,再摻入紅光 後,可得到CIE座標接近(0.33,0.33)的標準白光[60]。圖15為SR101。 1.5 研究動機與目的 有機發光電化學元件的發光原理在於當元件處在一固定驅動電壓下發 光層會因正負離子往兩旁電極移動而形成p型摻雜層/本質層(複合區)/n型摻 雜層(p-i-n)結構使激子可以從摻雜層注入本質層並在此發光[6][7],且本質 層厚度會由於摻雜層的延伸而減少,此時若本質發光層太靠近電極或摻雜 層將會導致激子猝熄而使元件效率變差,因此本文提出一調整主動層厚度 的方法使複合區可以遠離摻雜層與電極,藉此來緩解激子猝熄並改善元件 效率。 圖 15 SR101 結構[60]
第二章 理論基礎
2.1 螢光與磷光發光原理
有機材料具有吸收和放光特性,以量子力學的觀點,這些特性是由分子 軌域所決定,根據包利不相容理論(Pauli exclusion principle),每個分子軌域 最多只能填滿兩個電子,且同一層能階的兩個電子自旋方向必須相反,當
分 子 的 電 子 處 在 基 態 S0 時 , 稱 為 最 高 填 滿 分 子 軌 域 (highest occupied
molecular orbital, HOMO),而激發能量的振動頻率與分子某個能階差一致時 會產生共振,此時激發能量被分子吸收,使電子跳到較高能階處於激發態, 處於激發態的電子會很快以內轉換或振動鬆弛回到最低能量的軌域,稱為 最低未填滿分子軌域(lowest unoccupied molecular orbital, LUMO),在激發態 電子回到基態的過程,依電子自旋對稱方式不同會產生兩種情況:一種是 激發態電子與基態電子自旋方向相反,此激發態稱為單重激發態(singlet
excited state, Sn),這時能量若以輻射形式釋放稱為螢光(fluorescence),螢光
待在單重激發態時間較短,大約 10-5 ~10-8 秒,所以螢光是較好觀察到的; 另一種是激發電子經由系統間跨越到能階較低的三重激發態(triplet excited state, Tn),此時激發電子與基態電子自旋方向相同,這時能量若以輻射型式 釋放稱為磷光(phosphorescence),使電子留在三重激發態的時間較長,大約 10-4秒以上,因此能量容易以非輻射形式來釋放,所以磷光在一般情況下較 難觀察。 激發態電子除了由螢光及磷光的輻射形式釋放能量外,其餘都是由非輻 射形式所釋放,其中包括內轉換(internal conversion)、振動鬆弛(vibration relaxation)、系統間跨越(intersystem crossing),如圖 16 所示之 Jablonski 分 子能階圖。有機發光元件是利用電子與電洞再結合而發光,而電子與電洞 再結合而引起的單重激發態與雙重激發態的比例為 1:3,意謂著單重激發 態所發出螢光的比例只佔全部的 25 %,三重激發態則佔了 75 %,但卻受限 於選擇定律,使得三重激發態的電子回到基態是不被允許的,導致在三重 激發態的分子有足夠時間以非輻射方式釋放能量,因此對於講求發光效率 的發光元件而言,設法讓有機材料的三重激發態能以磷光形式放光是重要
的課題,而近年來發現使用重原子的錯合物,分子的自旋軌域偶合作用 (spin-orbital coupling)會變大,使得單重激發態分子經系統間跨越到三重激 發態變的更快速,所以分子停留在三重激發態的時間大幅縮短,若停留時 間短於非輻射形式釋放能量所需時間,將可得到能量以輻射方式釋放的磷 光[4]。圖 17 為電子電洞再結合所產生的激發態。 圖 16 Jablonski 分子能階圖 圖 17 電子電洞再結合所產生的激發態
2.2 主客體發光系統 主客體發光系統是目前有機發光元件最常用來製作白光元件的架構,其 原理是在能階較大的主體發光材料少量摻雜能階較小的客體發光材料,藉 以達到主體材料傳遞能量給客體材料發光的目的,所以可經由摻雜不同客 體材料或不同濃度來調控光色,且少量客體材料的摻雜能減少客體材料的 濃度猝熄(concentration quenching)而提升元件發光效率及元件壽命。而在磷 光元件的主客體系統中,有下列兩種發光形式: 2.2.1 能量轉移(energy transfer)形式 能量轉移形式可再細分為兩種方式,分別為 Forster 能量轉移及 Dexter 能量轉移,如圖 18 所示。Forster 能量轉移是由距離較長(約 10 奈米)的偶 極-偶極間產生共振耦合來傳遞能量,若主體材料的放光頻譜與客體材料的 吸收頻譜相互重疊,且雙方的躍遷是允許的,則此主客體系統將產生快速 且非輻射的能量轉移現象。Dexter 能量轉移則是由距離較短(約 1~1.5 奈米) 的電子交換方式來傳遞能量,而在電子交換時須遵守 Wigner-Witmer 選擇定 則,即兩者的自旋參數在交換過程前後是保持固定的 [4]。 圖 18Forster 與 Dexter 能量轉移示意圖
2.2.2 載子捕捉(carrier trapping)形式 這是另一種讓客體材料發光的機制,簡單來說就是電子與電洞直接在客 體材料的同分子內形成激子而使客體材料發光,此機制發生在客體材料的 HOMO 與 LUMO 能階都被包含在主體材料內,當主客體材料間能隙過大時 ,電子與電洞不容易注入到主體材料而直接注入客體材料進行再結合發光, 若客體材料的 HOMO 與 LUMO 能階只有其中一個包含在主體材料當中, 則須觀察客體材料激子是否處在較低能態,如果是則形成客體材料激子而 發光,如果不是則形成主客體間的電子電洞對,因而不利發光[4]。 通常能量轉移與載子捕捉形式在主客體系統中是同時存在的,只是依照 不同情況,其中一項形式會成為主要的發光機制,一般來說,在低摻雜濃 度或電壓夠大(高電流密度)的情況,能量轉移是主要的發光機制;反之,則 是載子捕捉為主要的發光機制[4]。 2.3 有機發光二極體發光機制 有機發光二極體的發光機制是經由外加電壓驅動時電洞從陽極端注入 有機層的 HOMO 能階而電子則從陰極端注入有機層的 LUMO 能階,當電 子與電洞進入到發光層再結合形成激子(exciton),此激發態分子回到基態 (ground state)時能量將以光的形式釋放,因此設計多層結構必須考量各層間 匹配使激子得以在有效發光層內發光。 陰極電極為了降低驅動電壓,必須 減少電子注入有機層的能障,因此選用功函數(work function)較低的金屬, 如鈣(Ca)、鉀(K)、鋰(Li)等,然而低功函數的金屬在大氣中穩定性差,容易 氧化及剝離,因此可以利用低功函數金屬與抗腐蝕金屬的合金來提高其穩 定性,電子注入層(EIL)是幫助電子注入電子傳輸層,因此要選用 LUMO 能 階可與陰極功函數緩衝匹配的材料;電子傳輸層(ETL)是幫助電子傳輸至發 光層並阻擋電洞穿過電子傳輸層,使電子與電洞能在發光層(EML)內再結合 發光,因此要選用電子遷移率高且 HOMO 能階低於發光層(EML)的材料, 而電洞傳輸層(HTL)與電子傳輸層的功能類似,是幫助電洞傳輸至發光層並 阻擋電子穿過電洞阻擋層,因此選用電洞遷移率高且 LUMO 能階高於發光 層的材料;電洞注入層(HIL)與電子注入層的功能類似,是幫助電洞注入電
洞傳輸層,因此選用 HOMO 能階可與陽極功函數緩衝匹配的材料,而陽極 位於玻璃基板上方,為了使光從陰極反射再於陽極端透射出來,因此一般 使用透明導電的銦錫氧化物做為陽極材料[4],圖 19 為有機發光二極體多層 膜的能階及發光示意圖。 2.4 有機發光電化學元件發光機制 至今為止,有機發光電化學元件發光機制在學術界尚有兩大學說:電化 學架構(Electrochemical model)和電動力學架構(Electrodynamic model),此兩 種架構各自有其證據佐證,因此實際原理尚未定論。 2.4.1 電化學架構(Electrochemical model) 根據 Pei 團隊提出,在外加驅動電壓下,有機材料與鹽類會解離出正負 離子分別往陰極和陽極移動而形成氧化或還原的分子,此時為了補償氧化 還原反應,使得在陽極端可接受電洞,陰極端則接受電子,並且由有機層 與電極所交會之處漸漸往有機層中心形成如 p 型摻雜與 n 型摻雜的區域, 此摻雜層與電極形成類似歐姆接觸(ohmic contact)的低阻抗界面,因此可促 進載子注入,且不易受電極材料的功函數所影響,當兩摻雜層相當靠近時, 會形成一絕緣摻雜交界,稱為複合區,意即電子與電洞複合發光的區域, 而複合區位置會由於有機材料的載子注入以及載子遷移率不平均而偏向某 一邊電極[61-64]。 圖 19 有機發光二極體多層膜能階及發光原理示意圖
2.4.2 電動力學架構(Electrodynamic model)
根據 Slinker 團隊提出,在外加驅動電壓下,有機層內的離子會漂移且 做些微的重新分布,陽極端吸引陰離子(anion)進行聚集,陰極端則吸引陽離 子(cation)進行聚集,這些離子的游離會直到整個有機層內的區塊達到穩定 態而形成一個自由場區,此時有足夠多的離子靠近兩邊電極而形成電雙層 (electric double layer, EDL),而電雙層將造成有機層與電極間的電位差,促 使載子注入變得容易,其注入的電子與電洞則漂移至內部自由場區進行再 複合發光[61-64]。圖 20 為電化學與電動力學架構示意圖。
此兩種結構若由電位來分析比較是有一些相同與不同之處,如圖 21 所 示,其中的黑線部分為電位隨位置不同所產生的變化,而兩者相同之處在 於靠近電極的區域由於正負離子的累積而造成電雙層以促進載子注入;而 兩者不同之處在於有機層中間的電位差,電化學架構指出不只在電雙層會 有電位差,在有機層中間也會有一高電位區,而電動力學則指出只有電雙 層部分屬於高電位區[61-64]。 圖 21 兩種架構的電位變化示意圖(左:電化學架構,右:電動力學架構)[64]
2.5 發光元件效率計算法
有機電致發光元件的發光效率一般稱為元件的外部量子效率(external quantum efficiency, EQE),依照各種物理機制可將其分成幾個部分來討論, 其算式為: ηeqe = α‧γ‧ηex‧φpl α:光透過有機層及玻璃基板等層到達量測儀器的比例係數,其等式為 1/(2n2),而上述的各層經由計算後的總折射率 n 約為 1.6,因此該係數之值 大約為 20 %,如圖 22 為發光元件出光示意圖。 γ:載子注入至主動層內的再複合機率,若元件內的電子與電洞注入達到平 衡狀態,其再複合機率可視為 100 %。 ηex:激子發光效率,因前文所提及的電子激發態屬於單重激發態或三重激 發態而有所區分,若是純螢光元件屬於單重激發態,激子發光機率佔 25 %, 故 ηex為 25 %;而磷光元件則屬於三重激發態,激子發光機率佔 75 %,再 加上單重激發態的 25 %,因此 ηex為 100 %。 φpl:螢光或磷光元件的光致發光量子產率,與材料內部能量傳輸路徑有關。 根據上述四個部分,可推算出螢光元件的外部量子效率理論極限值為 5%,而磷光元件則是 20 %。圖 23 為外部量子效率推算示意圖。 圖 22 發光元件出光示意圖
2.6 白光元件的量化方法
2.6.1 色彩座標(chromaticity coordinates)
為了使人類所感知的顏色可以被清楚的表述,在 1931 年國際照明委員 會(Commission Internationale d´Eclairage, CIE)提出一個使用數學方式來定 義色彩空間的方法,並制訂色彩座標系統的國際標準和技術交流平台。由 於人類眼睛的視錐細胞對於短、中、長波長的光有感測的效果,所以基本 上只要三個參數就能描述出顏色,因此色彩座標系統可分別以(x,y,z)標示, 如圖 24,圖上僅有 x 和 y 座標,由恆等式 x+y+z=1 可導出 z。在色度座標 圖中馬蹄型範圍內為可見光譜的所有顏色,馬蹄型的邊緣是飽和的單色波 長[4],中央部分則統稱為白光,理論上座標在(0.33,0.33)的點稱為最白光, 但是在白光的實際應用上,各種類型的白光皆有其用途及愛用者。 圖 24CIE 1931 色彩座標圖
2.6.2 相對色溫度(correlated color temperature, CCT) 根據 Planck 的黑體輻射理論,將一具有完全吸收和放射能力的標準黑 體加熱,溫度逐漸升高時光色亦隨之改變,在 CIE 色彩座標中間由右上衍 生至左下的曲線即為黑體輻射軌跡線,如圖 25,在該線上的所有色座標即 為絕對色溫。黑體加熱到出現與光源相同或接近光色時的溫度,定義為該 光源的相對色溫(簡稱色溫),以絕對溫度 K 為單位。相對色溫亦是用來描述 光色相對於白的程度,因此相對色溫低時,光色越偏紅,稱為暖白光;而 相對色溫高時,光色越偏藍,稱之為冷白光,圖 26 為相對色溫表。 圖 25 黑體輻射軌跡線 圖 26 相對色溫表
2.6.3 演色性指數(color rendering index, CRI) 光源對物體的顯色能力稱為演色性,意即物體在參考光源下的感受與在 標準光源(白熾燈或晝光)下的感受的真實度比較。由於在色彩座標上同樣光 色的光可以是由不同的光譜所混合而成,故寬廣光譜所混合的光有機會和 僅由兩單色光譜的混合光光色是相同的,但在物體上呈現的物體真實色彩 卻是有差異的,因此定義此為演色性指數來衡量光源的顯色能力,主要是 用來衡量白光的照明品質,該指數的數值評定法是分別以參考光源及待測 光源照在 DIN6169 所規定的八個色樣,如圖 27 所示,並逐一做比較再以下 列式子量化其差異性: 當平均演色性指數 Ra 越高代表差異性越低,即代表待測光源之演色性 越佳,而平均演色性指數最高為 100,即可以讓物體所呈現的各種顏色如同 參照光源所照射的顏色,太陽光為非常寬廣的白光光譜,因此演色性指數 為 100,而螢光燈為 60-85,螢光粉白光 LED 為 60-90,雙色白光 LED 為 10-60。一般數值在 85 以上即可以當作白光光源使用,而不同數值的光源也 有適合使用的場所及用途,如表 2 所示。 圖 27 計算 CRI 所需之八個測試光源頻譜
指數(Ra) 等級 CRI 評價 一般應用 90~100 1A 優良 需要色彩精確比對與檢核之場所 80~89 1B 優良 需要色彩正確判斷及討好表觀之場所 60~79 2 普通 需要中等演色性之場所 40~59 3 普通 演色性的要求較低,唯色差不可過大之場所 20~39 4 較差 演色性不重要,明顯色差亦可接受之場所 表 2 演色性指數與對應適用場所
第三章 實驗內容與製程步驟
3.1 實驗設備簡介 (1)鑽石刀玻璃切割平台 具有鑽石切割刀與滑輪軌道的切割平台,其刀片的升降高度可以細微調 整至 1 毫米。使用方法是將 ITO 玻璃正面朝上放在平台並對準平台上的量 尺,接著調整刀片至可以在 ITO 玻璃上割出一條痕跡的高度,再輕微施予 外力即可折斷,比起筆型手動鑽石切割刀,玻璃切割平台較能有效避免玻 璃大小不一或切割面不平整的狀況,進而提昇 ITO 玻璃的使用效率。 (2)超音波洗淨機 在清洗槽內加入純水(RO 水)並且藉由洗淨機產生的 43 千赫茲的超音 波傳導至清洗槽內,使得在液體中發生因為壓力差而產生微小氣泡的現象, 稱為空穴效應(cavitation),利用此現象所產生的小氣泡與待洗物表面接觸後, 在小氣泡消滅時一併帶走待洗物上面的汙垢,主要是用來清洗人類肉眼看 不到的一些微小粒子。 (3)紫外光臭氧清洗機 紫外光臭氧清洗技術是一種乾式清洗技術,不需加入任何溶劑,其原理 是利用高能量的紫外光照射待洗物,使上面的有機汙染分子(如光阻、油脂、 清潔劑殘留物等)變成激發狀態分子,同時氧分子經紫外光照射也分解成氧 原子,若氧原子與另一個氧分子結合則形成臭氧(O3),而剩餘的氧原子則與 有機汙染分子反應結合,形成可揮發的分子而離開待洗物表面,因而達到 清洗的效果,一般應用於 ITO 玻璃表面的清洗,同時也能達到增加 ITO 功 函數使得電洞注入變好。 (4)旋轉塗佈機 應用於一般濕式成膜或脫水,其中間軸心部分會有一真空吸盤吸住其上 方的試片,再利用旋轉所產生的離心力讓滴在試片表面的溶液瞬間成一均 勻薄膜,其薄膜的均勻度易受機器旋轉轉速、溶液的濃度、溶液的黏著度、 溶液的表面張力或試片大小等而影響。 (5)加熱攪拌平台 此平台可提供攝氏兩百度以上的高溫,應用範圍可以是加熱清洗用溶劑或是加熱使旋轉塗佈後元件上的溶劑揮發而達到快速固化的效果,同時該 台機器具有加入磁石旋轉攪拌的功能,能夠幫助一些在正常情況下難溶於 溶劑的有機材料有更好的溶解度。 (6)微量電子磅秤 本實驗室溶液製程所使用的溶液重量很少,因此為了確保有機材料濃度 的精準性,需使用秤量重量尺度極小的微量電子磅秤,該機器最精準可量 到小數點後第一位,而小數點後第二位為估計值。 (7)真空蒸鍍機 由真空腔體(chamber)、真空系統、一組隨時監控腔體真空程度的儀表以 及控制蒸鍍速率、厚度等參數的控制器所組成,其真空系統包括各級真空 幫浦、真空閥門及壓力計。蒸鍍步驟首先將蒸鍍源(金屬或固態有機材料) 置於耐高溫且導電的鎢舟上並固定於真空腔體內的導電支座上並蓋上腔體, 接著打開機械幫浦將腔體抽至中度真空(約 5x10-2 托爾),再開啟擴散幫浦 使腔體壓力下降到高度真空(約 1x10-5 托爾),而擴散幫浦的原理是利用高 分子量高速運動的蒸氣分子,經由碰撞把動能轉移給待抽氣體,賦予待抽 氣體單一方向的動量,並將之排往較高壓力之區域再經由機械幫浦帶出, 此時可在擴散幫浦加入液態氮使其因熱而產生的蒸氣分子被凝結,再由擴 散幫浦抽出而達到更低的壓力條件(約 6x10-6 托爾),此時腔體內的狀態已 達到雜質極低的蒸鍍條件。達到蒸鍍條件後,通以直流電壓將置於鎢舟上 的蒸鍍源加熱至昇華狀態,使其蒸鍍源氣體附著於基板表面上而形成一薄 膜,而一般蒸鍍源為製作陰極的金屬或者小分子的有機材料。 (8)超低水氧手套箱 由於本實驗室的所製作之有機發光電化學元件並未經過封裝製程,而有 機材料又容易與大氣中的水氧起作用,而造成元件壽命減短或衰壞,且陰 極金屬材料若為低功函數材料則在大氣中也易氧化而導致絕緣,使元件的 電性變差,因此在元件製作完成後,要以最短時間放入手套箱,減少元件 因水氧而變質的機會,而本實驗室的整套量測系統也都置於低水氧手套箱 當中,因此也可確保在元件量測結束之前,不會有元件衰壞的因素存在。
3.2 實驗製程步驟 3.2.1 ITO 玻璃基板製備與清洗步驟 1. 將大片 ITO 玻璃正面朝上放置於切割平台上,並對準量尺切割成長寬皆 為 1.9 公分的元件,此長寬比是為了符合蒸鍍時使用的遮罩(mask)而定, 切完之後需確認元件切面平整及 ITO 面有無刮痕。 2. 準備好五個乾淨的震洗壺,依序裝入介面活性劑、去離子水、丙酮兩壺 及甲醇,並確保溶液量足夠讓元件在震洗期間能夠完全浸入裡面。 3. 清洗步驟第一步是將清洗用的鑷子以丙酮先洗淨,接著夾起元件,利用 沾有介面活性劑的棉花棒清潔元件的雙面及切面,再使用去離子水沖洗 掉元件上的污垢和玻璃碎屑,最後用氮氣槍斜吹元件除去殘留的水分, 此步驟要注意盡量不要在元件上留下水痕,以免造成旋轉塗佈時的膜會 不均勻。重複以上步驟直到肉眼看不見汙垢及水痕即可放入步驟 2 所準 備的介面活性劑,並置於超音波洗淨機震洗 10 分鐘。 4. 步驟 3 震洗完後,將元件取出並放入去離子水再震洗 10 分鐘,此時將 一壺丙酮及甲醇放置於加熱平台上加熱至攝氏 150 度等待使用,去離 子水震洗完後,再將元件取出並放入未加熱丙酮並同樣震洗 10 分鐘。 5. 步驟 4 的震洗完成後,將元件取出並且放入已經沸騰的丙酮當中並煮 15 分鐘以上,此步驟可利用丙酮蒸發所產生的氣泡將元件上肉眼看不 到的微小粒子帶走。 6. 步驟 5 煮完丙酮後,再將元件取出放入另一壺沸騰的甲醇裡煮 30 分鐘, 此步驟為玻璃清潔的最後一步,使用甲醇是為了沸騰甲醇可以快速蒸發 的特性,讓待會取出元件時,可以減少水痕留在元件上的機會。 7. 使用無塵紙沾乙醇以擦拭的方式清潔紫外光臭氧清洗機載台及燈管,並 且用氮氣槍將乙醇吹至揮發。 8. 將元件緩慢由沸騰甲醇裡取出,緩慢的動作可以使元件上的污垢及水痕 隨著甲醇慢慢蒸發,若元件完整取出後還有殘留水分,可以使用氮氣槍 稍微吹乾即可,之後再將元件以正面朝上的方式放置於紫外光臭氧清洗 機的載台上並蓋上遮罩,照射 UV 光 30 分鐘。
3.2.2 元件製程步驟 1. 在元件照射 UV 光的同時,開始調配 poly(3,4-ethylenedioxythiophene) poly(styrenesulfonate) (PEDOT:PSS)溶液準備進行旋轉塗佈,首先準備一 個乾淨的不透光樣品瓶,並將調配好的 PEDOT:PSS 溶液以 0.2 微米濾 徑的尼龍(Nylon)過濾頭過濾到乾淨不透光樣品瓶中,此步驟是為了減少 PEDOT:PSS 溶液在放置一段時間後會呈現的結塊現象以及濾掉雜質。 2. 設定好旋轉塗佈機的轉速及時間,接著從紫外光臭氧清洗機取出元件並 且 正 面 朝 上 快 速 放 置 於 塗 佈 機 的 真 空 吸 附 平 台 上 , 再 用 滴 管 吸 取 PEDOT:PSS 溶液,其量只需均勻滴滿元件即可,最後要注意沒有氣泡 在元件上就可以開始進行旋轉塗佈。 3. 塗佈完成後,將元件從真空吸附平台上取下並快速放入乾淨的培養皿當 中,以減少微小粒子又附著於元件表面,再將培養皿放置於加熱平台上 加熱至攝氏 150 度烤 30 分鐘以烤乾元件上多餘的水分。 4. 烤完 30 分鐘後,可將培養皿放置室溫中退溫約 15 分鐘,此時可以使 用微量電子磅秤調配塗佈有機層薄膜所需之溶液,並且一樣要經過過濾 頭過濾雜質,才能避免微小粒子影響薄膜的均勻性。 5. 設定好有機層所需的轉速及時間,與步驟 2 一樣將元件快速放置於真空 吸附平台上,但此時要注意,在要將滴管內的有機溶液滴到元件的步驟, 必須在滴完之後馬上按下旋轉塗佈的開關,以減少有機溶劑侵蝕上一層 薄膜的時間,而此有機薄膜相對的也會比較均勻。 6. 塗佈完有機層後,用一沾有丙酮的棉花棒將元件的切面以及表面的一角 擦拭,使之做為 ITO 面的陽極端接觸點,接著將元件放回培養皿,並快 速的載入超低水氧手套箱中的加熱平台以低溫約攝氏 60 度烤 6 小時將 有機層的溶劑烤乾即可。 7. 烤完六小時後,開始進行蒸鍍電極的步驟,將元件及放在手套箱內的金 屬錠及鎢舟一併取出,根據蒸鍍機的步驟進行蒸鍍,並將元件正面朝下 放置於如圖 28 的金屬遮罩(mask)中,當腔體抽氣抽至高度真空狀態時, 以適當穩定的輸出電流操控鍍率來鍍上平坦均勻的電極薄膜,最後便完 成了一片具有數十顆金屬陰極的完整的有機發光元件。
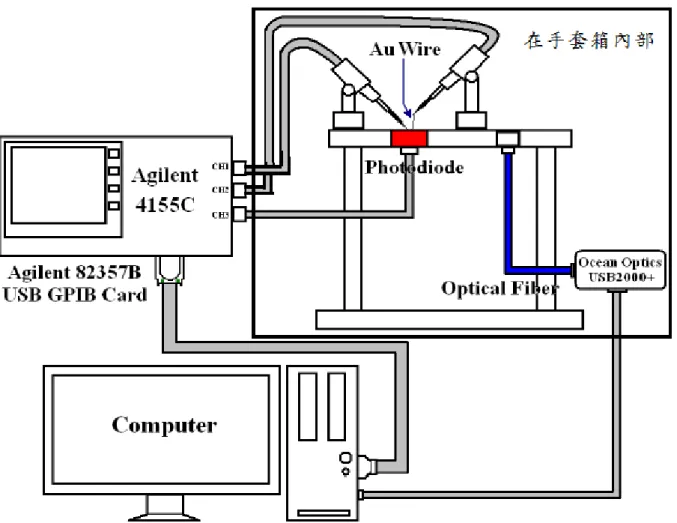
3.3 實驗量測系統 3.3.1 量測設備簡介 1. 半導體參數分析儀(Aglilent 4155C) 為一台桌上型數據分析儀,可達約 10-14 安培及 10-7 伏特的量測解析度, 可透過特定的 USB 傳輸裝置連接一般電腦使用圖形介面軟體 Desktop EasyEXPERT 來操作儀器以及做數據分析。
2. 微型光纖光譜儀(Ocean Optics USB2000+ )
是由一個 2MHz 模數(A/D)轉換器、可程式編輯模塊、2048 像素(pixel) 的 CCD 陣列探測器以及 USB2.0 接頭所組成,可提供高達 0.35 奈米的辨別 率(FWHM),當與電腦連接後,搭配軟體 OOIBase32 將可以以每毫秒擷取 並儲存一幅完整的光譜圖至電腦儲存設備。 3. 光檢測器(photodiode) 是一種可將光的訊號轉為電訊號的半導體元件,能夠測量接收到光子的 能量再傳至數據分析儀做分析,如此可用來計算元件的發光強度、發光效 率、壽命及點亮時間等。 3. 橢圓偏光儀(Alpha-SE) 是一種利用光譜反射的快速薄膜厚度量測儀器,屬於非破壞性的量測方 式,主要可以量測薄膜的膜厚及折射率(n)、消光係數(k)等,本實驗室所使 用之橢偏儀(Alpha-SE)提供的波長在380~900 奈米,並有65/70/75/90 度的 量 測 角 度 可 供 調 整 , 單 點 量 測 最 快 約 3 秒 即 可 取 得 數 據 , 搭 配 軟 體 CompleteEASE使用,軟體內建多種數學函數來描述吸收膜層的特性:如 Lorentz、Gaussian、Harmonic等。 圖 28 蒸鍍金屬電極所使用之遮罩
3.3.2 量測步驟 1. 將製作完成的元件放到低水氧手套箱中的量測平台上,並且要注意 ITO 面要朝上,本實驗室所使用之量測平台如圖 29 所示。在量測平台上有 兩個小孔,分別為測量元件的電性及發光頻譜的特製裝置,倘若要量測 電性,需將元件對準下方為光檢測器的接收孔;而量測發光頻譜的話, 則需將元件對準下方為光纖光譜儀的接收孔。 2. 在量測平台上,有兩個由半導體參數分析儀的頻道 1(CH1)與頻道 2(CH2) 連接電線到手套箱中的磁性基座,分別做為陽極與陰極,陽極部分使用 金屬探針接觸元件被丙酮擦拭過所露出的 ITO 面,陰極部分由於需避免 金屬探針刺穿金屬薄膜,因此使用較柔軟的純金導線(99.95)以銲接方式 銲在探針上,如此一來便能以金線輕輕接觸金屬薄膜達到量測的目的, 圖 30 為元件量測方法示意圖,而頻道三(CH3)則連接到量測電性使用的 光檢測器。 3. 將元件固定且確認陽極與陰極都有接觸到元件後,即可關掉手套箱的照 明光源,並且蓋上符合量測平台的特製金屬箱以隔離外在光源干擾光檢 測器或光譜儀的收光。
4. 接著就可以使用電腦內 Desktop EasyEXPERT 軟體透過 USB 介面的 GPIB 卡來遙控 Agilent 4155C,並設定施加的電壓或訊號值,其元件的 電流、光電流及效率比值可從此軟體的圖形介面上顯示並手動存檔,若 是量測發光光譜,則需另搭配 OOIBase32 軟體設定時間及光子數積分 參數以擷取發光光譜的圖片。 5. 透過步驟 4 可以獲得相同電壓下光電流與電流隨時間變化的電性圖以 及隨時間變化的發光頻譜,即可搭配數據分析軟體 Matlab 及 Origin 計 算出元件的發光亮度、電流密度、功率效率以及外部量子效率等數值, 此時需注意的是發光效率的計算與光強度有關係,而不同波長的光其光 強度也不同,因此若元件的發光頻譜會隨時間產生位移,則需要將不同 波長的發光頻譜與其相對時間點之電性圖做效率計算,再利用拼接的方 式計算出元件的完整發光效率,而元件發光頻譜位移的情況一般發生在 有機材料老化或元件複合區移動而產生的微共振腔效應。
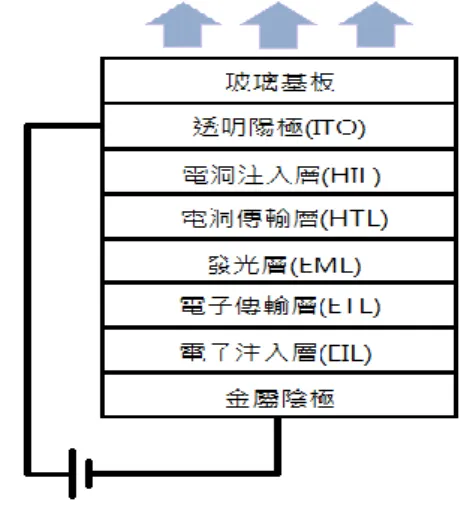
3.4 元件結構 本文所製作的有機發光電化學元件皆為單一主動層之結構,其結構為: ITO/PEDOT:PSS(約 40 奈米)/主動層(厚度為本實驗討論主旨)/銀電極(100 奈米),其元件結構示意圖如圖 31 所示,ITO 為元件的陽極端,PEDOT:PSS 為電洞傳輸層以及使 ITO 表面更加平坦化的作用,而銀電極則做為陰極端 鍍在主動層與 PEDOT:PSS 之間,形成單層三明治結構元件,且可利用銀薄 膜的反射特性反射主動層所發出的光至朝下,故本實驗室之發光元件屬於 下發光元件。本文所主要調變的實驗參數為主動層的厚度,藉由改變主動 層厚度來優化其元件效率以及探討其元件物理。 圖 30 元件量測方式示意圖 圖 31 本實驗之元件結構示意圖
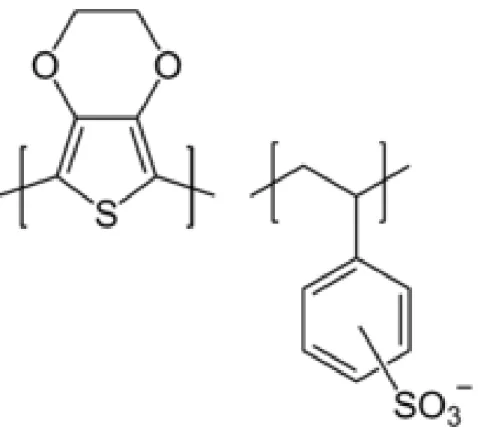
1. 銀薄膜陰極(Ag cathode) 一般有機元件需要使用低功函數金屬材料(如鈣、鎂等)當陰電極才能幫 助電子注入主動層,但低功函數材料常因為高活性而容易水氧,造成元 件易損壞,若要避免此問題則需使用合金的方式製作電極,因此提高了 製程的複雜度,而本實驗所使用的有機發光電化學元件之載子注入好壞 並不是取決於電極的功函數,因此本實驗中使用低活性的銀(Ag,功函 數 4.52-4.74 電子伏特)做為金屬陰極材料,便能一次解決易水氧及製程 難度的問題。 2. 銦錫氧化物陽極(ITO anode) 本實驗室所使用之 ITO 玻璃為市面上的商用材料,是將銦錫氧化物附著 於玻璃上做為有機發光元件主要基板的技術,一般當作有機元件的陽極 使用,其功函數與有機材料的 HOMO 能階匹配,本實驗的主要要求在 於導電度、化學穩定度都要夠高,且需要配合發光元件出光方向,因此 透明度也是重要的考量。 3. PEDOT:PSS 層 全名為 poly(3,4-ethylenedioxylthiophene):poly(styrenesulfonate),為導 電高分子聚合物的水溶液,由 PEDOT 和 PSS 兩種聚合物以特定比例混 合而成,其化學結構如圖 32,此材料的能階剛好能當作幫助電洞由陽 極 ITO 注入主動層的媒介,在有機發光二極體的多層結構裡是屬於電洞 注入層,而在有機電化學發光元件中電極功函數與載子注入的影響不大, 因此在本實驗中使用 PEDOT:PSS 主要因素是要讓 ITO 表面更平整並減 少元件短路的機率。 4. 主動層 本文的主動層是採用多摻雜發光層,以主客體摻雜的方式製作出白光有 機電化學發光元件,材料是用曾在文獻發表過的兩種離子性過渡金屬錯 合物材料做為主客體材料[24][56]再混入離子性鹽類 BMIM+(PF6 ─ ) (見 Page.11) 水 溶 液 的 發 光 層 , 為 了 方 便 起 見 以 下 將 主 體 材 料 [Ir(dfppz)2(dtb-bpy)] + (PF6 ─ ) (見 Page.12)表示為 1 號錯合物,而客體材料 [Ir(ppy)2(biq)] + (PF6 ─ ) (見 Page.12)表示為 2 號錯合物。
第四章 實驗結果分析
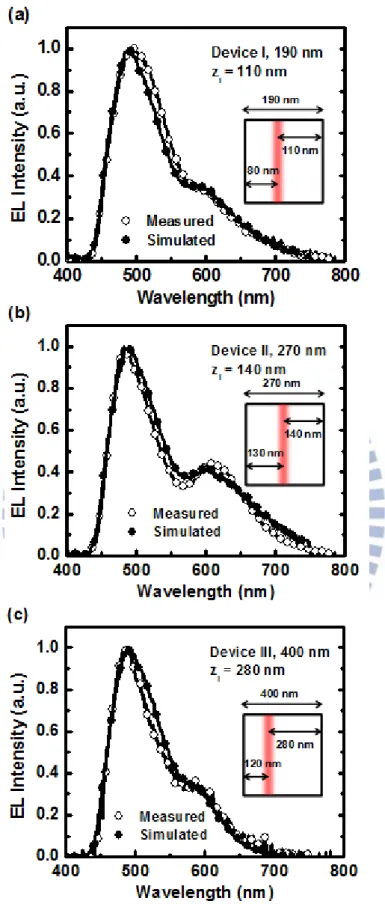
4.1 材料簡介及光物理特性 以下為本文之主動層所使用的材料介紹: A. 1 號錯合物[Ir(dfppz)2(dtb-bpy)] + (PF6 ─ ):為發藍綠光含銥(Ir)的離子性過 渡金屬錯合物,其中的 dfppz 為 1-(2,4-difluorophenyl)pyrazole,dtb-bpy 為[4,4-di(tert-butyl)-2,2´-bipyridine],此材料在 2005 年 Tamayo 等人的文 獻中所發表[24],主要做為主體發光材料,如圖 33 左所示。 B. 2 號錯合物[Ir(ppy)2(biq)] + (PF6 ─ ):為發紅光含銥(Ir)的離子性過渡金屬錯 合物,其中的 ppy 為 2-phenylpyridine,biq 為 2,2´-biquinoline,此材料 在 2008 年 Su 等人的文獻中所發表[56],主要做為客體發光材料,如圖 33 右所示。 C. 離子性鹽類 BMIM+(PF6 ─ ):為一種黏稠狀、透明且不溶於水的離子性鹽 類液體,可用來提供有機發光電化學元件額外的離子以減少離子建立起 p-n 接面的時間,因此少量摻雜具有使元件點亮時間變短以及提昇元件 發光效率的作用。其中 BMIM 為 1-butyl-3-methylimidazolium,PF6 為 hexafluorophosphate。如圖 34 所示。 由於 1 號錯合物的聯吡啶配位基(bipyridine ligand)有兩個巨大的立體 tert-butyl 基團[24],使分子間距拉大而減少分子間作用力,藉此減少了材料 在薄膜態的分子自我猝熄現象,使相較稀溶液的光致發光量子產率(1.00), 在薄膜態仍保持相當高的光致發光量子產率(0.75)[58],在已發表的文獻中 發現 1 號錯合物有很突出的載子平衡和外部量子效率大於 14 %的高效率 [59],因此非常適合做為有機白光電化學元件的主體材料;而 2 號錯合物不 僅在薄膜態及溶劑中皆可發出相當飽和的紅色光致發光頻譜,但由於其薄 膜態的光致發光量子產率不高(0.09)[58],所以適合做為少量摻雜在有機白 光電化學元件的客體材料,經過循環伏安法(cyclic voltammetry)所得知的主 客體材料能階排列圖如圖 35 所示[58]。另外加入離子性鹽類 BMIM+(PF6 ─ ) 除了可以提供額外離子降低 p-n 接面建立時間使元件點亮時間變短之外, 在過去文獻中發現加入 BMIM+(PF6 ─ )會抑制分子間的交互作用使得元件的 光致發光頻譜些微藍位移同時提高少許的光致發光量子產率[26]。N
N
Ir
N
PF
6-F
F
F
F
+
N
N
N
N
N
Ir
N
N
PF
6-+
圖 33 左:1 號錯合物、右:2 號錯合物之結構式 圖 34BMIM+(PF6─)之結構式2
-5.66 eV
-3.43 eV
1
-6.05 eV
-2.97 eV
圖 35 1 號錯合物及 2 號錯合物之能階排列示意圖4.2 實驗參數及電性分析 本實驗所使用的主動層皆是經由上述三種材料以 1 號錯合物(79.85 wt.%):2 號錯合物(0.15 wt.%):離子性鹽類 BMIM+(PF6 ─ )(20 wt.%)的重量百 分比例再加入乙腈(acetonitrile)溶劑所調配而成,其內容是製作三個不同主 動層厚度的元件分別稱為 I、II 和 III,其中改變的製程參數為材料在溶液中 的濃度分別為 120、150、150 毫克/毫升與旋轉塗佈(spin-coating)之轉速分 別為 3000、3000、1500 每分鐘轉速,藉此得到相同材料比例而不同主動層 厚度的發光元件分別為 190 奈米、270 奈米及 400 奈米,此厚度是由橢圓 偏光儀量測並計算得知。 在施予驅動電壓 3.3 伏特時元件 I、II 和 III 隨時間變化的電致發光頻譜 如圖 36(a)、(b)及(c)所示,其中頻譜已對藍光波段做歸一化以強調紅光波段 隨時間變化的趨勢,在圖 36(a)的元件 I 指出紅光會隨著時間明顯的下降, 這主要是因為主客體材料的能階差異所造成的載子捕捉現象。在基於主客 體摻雜的有機白光電化學元件方面,其貢獻客體發光的形式有兩種:一種 是在主體材料形成激子再經由主客體能量轉移形式將能量轉移到客體材料 發光;另一種是直接在客體材料形成激子的載子捕捉現象,通常會發生在 客體材料的 HOMO 及 LOMO 能階都包含在主體材料內的情況。而在有機 發光電化學元件剛開始施加電壓的前期,其元件內的 p-n 接面尚未建立完 全,導致載子要注入主體材料仍然存在高能障,因此客體發光的形式會傾 向於載子捕捉現象,即直接注入在較小能階的客體材料形成激子,所以在 圖 36(a)可以看到紅光在操作前期 50 分鐘內紅光是較為突出的,而在摻雜 層建立完全後,載子注入主體材料的能障就能顯著減少而使激子在主體材 料形成,而隨後客體發光的形式將由載子捕捉現象變成由主客體能量轉移 為主,因此藍光隨著時間相對增加,最後隨著摻雜層建立完全其電致發光 頻譜也會接近穩定(時間大於 175 分鐘)。而圖 36(b)的元件 II 在同樣驅動電 壓 3.3 伏特的電致發光頻譜也顯示出類似上述隨時間變化的趨勢,但元件 II 的主動層厚度比起元件 I 的厚,因此在施加相同電壓下,元件 II 的電場 相對減少,導致激子更容易在客體材料上形成,使其電致發光頻譜穩定時 紅光相對較高,而光色未穩定的時間下紅光相對變化即元件差異性所致。
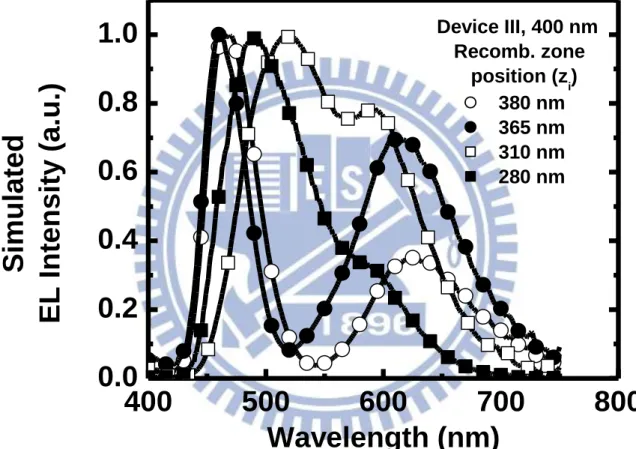
在主動層厚度最厚(400 奈米)的元件 III 方面,其電致發光頻譜隨時間演 變的趨勢與元件 I 和元件 II 有很明顯的差異,在操作時間小於 200 分鐘時, 由主體材料所發出的藍光最大值藍位移且半高寬(full width at half maximum, FWHM)變窄;而操作時間在 400 分鐘時,其頻譜的藍光產生紅位移且紅光 產生藍位移,因此頻譜的光色接近綠光區域且半高寬變寬;最後在操作時 間 700 分鐘時得到一個白光電致發光頻譜,但紅光部分相對於元件 I 和元 件 II 都還要低,如圖 36(c)所示。這並不符合上述主動層越厚則穩定頻譜下 相對紅光較高的理論,所以元件 III 隨時間演變發光頻譜的異常現象不應該 歸因於載子捕捉現象,且在頻譜演變過程中不只有藍色與紅色發光,其半 高寬也有明顯的變化,這些現象是元件具有微共振腔效應的典型特徵[65], 通常在厚度較厚的元件才會發現[60][66]。
400 500 600 700 800 0.0 0.5 1.0 1.5 2.0 2.5 (a) Device I, 190 nm 25 min 50 min 100 min 175 min EL Int ensit y (a.u .) Wavelength (nm) 400 500 600 700 800 0.0 0.5 1.0 1.5 (b) Device II, 270 nm 25 min 50 min 150 min 200 min EL Int ensit y (a.u .) Wavelength (nm) 400 500 600 700 800 0.0 0.2 0.4 0.6 0.8 1.0 (c) Device III, 400 nm 100 min 200 min 400 min 700 min EL Int ensit y (a.u .) Wavelength (nm) 圖 36 元件(a)I、(b)II 及(c)III 在驅動電壓 3.3 伏特隨時間變化的電致發光頻 譜圖

![圖 5 三原色共聚物化學結構(BGR 分別為藍綠紅)[53]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8639763.193103/19.892.162.778.128.1031/圖5三原色共聚物化學結構BGR分別為藍綠紅53.webp)