國立交通大學
國立交通大學
國立交通大學
國立交通大學
分子
分子
分子
分子醫學與
醫學與
醫學與生
醫學與
生
生
生物
物
物
物工程研究所
工程研究所
工程研究所
工程研究所
碩士論文
碩士論文
碩士論文
碩士論文
克雷白氏肺炎桿菌
克雷白氏肺炎桿菌
克雷白氏肺炎桿菌
克雷白氏肺炎桿菌 CG43 中尿嘧啶雙磷酸葡萄糖去氫
中尿嘧啶雙磷酸葡萄糖去氫
中尿嘧啶雙磷酸葡萄糖去氫
中尿嘧啶雙磷酸葡萄糖去氫
酶之
酶之
酶之
酶之酪胺酸
酪胺酸
酪胺酸
酪胺酸磷酸化的
磷酸化的
磷酸化的
磷酸化的角色
角色
角色
角色
Role of tyrosine phosphorylation on the UDP-glucose
dehydrogenase of Klebsiella pneumoniae CG43
研究生
研究生
研究生
研究生:
:
:葉家華
:
葉家華
葉家華
葉家華
(Jia-Hua Yeh)
學號
學號
學號
學號:
:
:
:9729503
指導教授
指導教授
指導教授
指導教授:
:
:彭慧玲
:
彭慧玲
彭慧玲
彭慧玲
博士
博士
博士
博士
(Hwei-Ling Peng)
中華民國九十
中華民國九十
中華民國九十
中華民國九十九
九
九
九年
年
年六月
年
六月
六月
六月
i 中文摘要 中文摘要中文摘要 中文摘要 克雷白氏肺炎桿菌為一伺機性感染的格蘭氏陰性菌,可藉其外部 包覆的厚重多醣體莢膜躲避細胞的吞噬作用及避免被血清因子所毒 殺。我們先前的研究證實克雷白氏肺炎桿菌蛋白質酪胺酸激酶(Wzc) 可藉由磷酸化尿嘧啶雙磷酸葡萄糖去氫酶(UDP-glucose
dehydrogenase, Ugd)來提升 Ugd 酵素活性,進而調控莢膜多醣體的生
成;而在 Ugd 上的 17 個酪胺酸殘基,已被選取作定點突變的 9 個酪 胺酸殘基的改變並未影響 Ugd 的磷酸化。為了確認 Ugd 上被磷酸化 的酪胺酸殘基,本研究將其他 8 個酪胺酸殘基,分別在 53、71、76、 85、203、252、302 和 380 的位置,經點突變技術換成苯丙胺酸。再 將這幾個定點突變後的蛋白質表現純化後,利用西方墨點法發現這些 突變株仍能在大腸桿菌體內被磷酸化,此結果顯示克雷白氏肺炎桿菌 不同於已報導的大腸桿菌與枯草桿菌,可能具有不只一個可受磷酸化 的酪胺酸殘基。同時,酵素活性與動力學特性的分析結果得知,Tyr71、
Tyr85、Tyr252 和 Tyr380 的突變明顯影響了 Ugd 酵素活性,而這些點
突變蛋白經由(circular dichroism, CD)圖譜分析,確認其二級結構組成 與野生型 Ugd 無顯著差異,暗示著這些酪胺酸殘基的改變可能因無 法被磷酸化而降低 Ugd 活性。
ii Abstract
Klebsiella pneumoniae, an opportunistic gram-negative bacterial
phathogen, is mostly encapsulated by a thick capsular polysaccharide (CPS) which acting to protect the bacterium from phagocytosis and prevent from damage by serum factors. Via an in vitro phosphorylation assay, we have previously demonstrated the protein-tyrosine kinase, Wzc, was capable of phosphorylating the enzyme UDP-glucose dehydrogenase (Ugd) to increase the enzymatic activity. The Ugd phosphorylation led to increase of the synthesis of CPS. Nine of the 17 tyrosine residues on
KpUgd have been substituted individually with phenylalanine by
site-directed mutagenesis. However, in vitro phosphorylation assay
revealed that none of the changes affected the Ugd phosphorylation. Here, we generate specific mutation on the rest of the tyrosine residues on Ugd. Interestingly, all of the mutant proteins, Y53F, Y71F, Y76F, Y85F, Y203F, Y252F, Y302F, and Y380F, isolated from E. coli appeared to be
phosphorylated. This suggested that KpUgd carried an additional tyrosine phosphorylation residue except the one reported for EcUgd-Y71 and
BsUgd-Y70. Nevertheless, the enzyme kinetics analysis revealed that
UgdY71F, UgdY85F, UgdY252F, and UgdY380F exhibit much lower activity than wild type Ugd. Circular dichroism analysis of the mutant Ugd indicated that the reduced activity was not due to structural alteration, implying the change of Y85, Y252, or Y380 impaired the subjection to phosphorylation leading to the decreased activity.
iii 致謝 致謝致謝 致謝 真的(* ̄▽ ̄)/‧★*"`'*-.,)我完成我的碩士論文哩!致謝對 我來說,是一篇論文完美的ending,同時也說明著一篇論文從開始到 結束所面臨的難關,以及對協助渡過難關的恩人們感謝。回顧這兩年, 歡樂與淚水還有在實驗室鬼吼鬼叫╰(‵。′*)╯失控狀態的交織下, 譜出了我永遠難忘的碩士生涯。 這篇論文得以順利完成,首先要感謝我的老闆-彭慧玲博士。老 師在我碩一11月漂泊無助時,將我拎回實驗室進行培養,加入彭家這 可愛的大家庭。很感謝老師總在我遇到困難時,帶領我一起"問問題 ",找出癥結與解決的辦法,提供我實質和精神上的幫助。古人云: 「一日為師,終身為母。」這句話用來形容我最不為過。老師~謝謝 您的照顧與關懷!請好好照顧您的身體,不要讓我們擔心哩! 再來,誠心感謝實驗室的成員。首先是平易近人的丸子太后:很 享受和你一起在早晨霸占實驗室的時光,把你餵飽飽看你開心我也跟 著快樂ㄟ( ̄▽ ̄ㄟ)。謝謝在實驗上你對我得教導,希望一同離開實 驗室的你可以找到幸福與喜歡的工作;謝謝建誠與小新學長對我實驗 上的幫忙與平日的照顧,並將實驗室運作得井然有序;謝謝美麗且腿 超級長的靜柔學姊,在我初進實驗室時,帶領我做實驗與熟悉環境, 沒有妳我沒辦法那麼快融入這家庭;謝謝雅雯跟顗峰像母雞帶小雞般
iv 的關心、照顧;謝謝寡言的其駿,大家齊聚你家烤肉的歡樂畫面如昨 日歷歷;謝謝可愛的品宣與酷酷的葳云,實驗室的活力就靠你們繼續 維持;謝謝老派的豪君,希望你能將我們磷酸化研究系列發揚光大; 超級特別感謝佩君與哲充,你們兩是我碩士生涯的一大部分,沒有你 們一定會黯淡無聊許多。我不會忘記哲充師傅使出渾身解數教我實驗 技巧,希望你能夠在心靈上不斷成熟,有朝一日變成能獨當一面的實 驗室一哥。謝謝我的好麻吉佩君((>(′︶ ‵*)<))),很榮幸能夠認 識那麼充滿歡笑氣質的你,一起實驗一起瘋狂、一起歡樂一起難過一 起畢業,希望哪天你能駕著黑頭車載我一起去吃貴婦下午茶。謝謝分 子調控實驗室歷來所有的成員,有你們才有我順遂的碩士生涯。 謝謝張晃猷老師與其實驗室成員在我研究上給予的建議與幫助。 謝謝楊裕雄老師能夠騰出時間擔任口試委員,指導論文的修正。 謝謝貼心的恒毅v( ̄︶ ̄)y陪在我身邊守護、鼓勵、照顧我! 最後,感謝我的家人給我的支持與關愛,我把碩士這份榮耀獻給 你們,我不會讓你們失望的。 當學生的日子終告一個段落,o(〒﹏〒)o能夠遇見大家真是太幸 運哩!希望哪天相聚時,大家依舊過的很幸福很快樂! 家華 謹致於 交通大學分子醫學與生物工程研究所 中華民國九十九年六月
v Contents Page Abstract in Chinese ………..………. i Abstract …………....……….... ii Acknowledgment ……… iii Contents ….……… v Table contents ………... vi
Figure contents ……….……….... vii
Abbreviation ………... viii
Introduction …..……… 1
Materials and methods ….………... 12
Results ……… 21 Discussion ………... 27 References ……….. 34 Table ………... 44 Figure ……….……. 49 Appendix ……… 67
vi
Table contents
Page Table I Bacterial strains used and constructed in this study ………...… 44 Table II Plasmids used and constructed in this study ………. 45 Table III Oligonucleotides used in this study ………. 47 Table IV Kinetic parameters of UgdWT and the derived mutants ….. 48
vii
Figure contents
Page Fig.1. Sequence comparison of Ugd from different bacteria ……... 49 Fig.2. Expression and purification of the recombinant Ugd proteins .... 51 Fig.3. In vitro phosphorylation of Ugd ………... 53 Fig.4. In vivo phosphorylation of Ugd and the derived mutants ... 54 Fig.5. Specific activity of the Ugd variants ... 55 Fig.6. Lineweaver–Burk plot of the wild type Ugd and the derived mutants at five different concentration of NAD+ ... 56 Fig.7. Lineweaver–Burk plot of the wild type Ugd and the derived mutants at five different concentration of UDP-glucose ... 59 Fig.8. Circular dichroism spectra of wild-type Ugd and mutant Ugd... 62 Fig.9. Three-dimensional structure of the Ugd protein ... 63 Fig.10. Construction and phenotype analysis of the specific ugd-deletion mutant ... 64 Fig.11. Expression of pET-30b-Ugd in E.coli and K. pneumoniae ….... 66 Appendix 1 Schematic diagram presentation of the Ugd catalytic reaction ... 67 Appendix 2 Activation of the UDP-glucose dehydrogenase activity of Ugd by phosphorylation …... 68 Appendix 3 Overview of the QuikChange site-directed mutagenesis method assay…………... 69 Appendix 4 In vitro phosphorylation ………….……... 70 Appendix 5 In vitro phosphorylation of His6-KpUgd and GST-KpUgd
viii
Abbreviation ATP adenosine triphosphate
BCIP 5- bromo-4-chloro-3-indolyl phosphate bp base pair
CFU colony forming unit(s)
CIAP calf intestine alkaline phosphatase CPS capsular polysaccharide
DNA deoxyribonucleic acid DTT dithiothreitol
EDTA ethylenediamine-tetraacetic acid EPS exopolysaccharide IPTG isopropyl-1-thio-β-D-galactopyranoside kb kilobase(s) kDa kiloDalton(s) LB Luria-Bertani µM micromolar mM millimolar
NAD+ nicotinamide adenine dinucleotide
NADH nicotinamide adenine dinucleotide (reduced form) NBT nitro blue tetrazolium chloride
ORF open reading frame
PAGE polyacrylamide gel electrophoresis PBS phosphate buffered saline
PCR polymerase chain reaction PTK protein-tyrosine kinase
ix
PTP protein-tyrosine phosphatase PVDF polyvinylidene difluoride rpm revolutions per minute SDS sodium dodecyl sulfate UDP uridine 5'-diphosphate UDP-Glc UDP-glucose
UDP-GlcUA UDP-glucuronic acid
1
Introduction
Klebsiella pneumoniae
Klebsiella pneumoniae, a member of the Enterobacteriaceae family,
is a non-motile, facultative anaerobic, and rod-shape bacterium. Their environmental habitats are surface water, sewage, soil and on plants (Brown and Seidler 1973; Seidler, Knittel et al., 1975). K. pneumoniae is responsible for a variety of diseases in humans and animals (Brisse et al., 2006). Most notoriously, it is a prominent nosocomial pathogen (causing community-acquired diseases including bacteremia, septicemia, urinary, respiratory and blood infections) and the agent of specific human
infections including Friedla¨nder’s pneumonia, rhinoscleroma and the emerging disease pyogenic liver abscess (PLA), occurring particularly in immunocompromised patients (Podschun and Ullmann, 1998; Brisse et
al., 2009). Other K. pneumoniae infections that are severe but more rarely
reported include meningitis, necrotizing fasciitis and prostatic abscess (Lu et al., 2002; Kohler et al., 2007). Finally, granuloma inguinale (donovanosis) (Richens, 1985) is caused by uncultivated bacteria, which may belong to K. pneumoniae (Grimont, 2005).
Factors that are implicated in the virulence of K. pneumoniae strains include the capsular serotype, lipopolysaccharide, iron-scavenging
systems, and fimbrial and non-fimbrial adhesins (Regue et al., 2001; Chou et al., 2004; Ma et al., 2005). Most Klebsiella strains are encapsulated by a polysaccharidic capsule of considerable thickness responsible for the glistening and mucoid colonies on agar plates. The abundant polysaccharidic capsule protects against the bactericidal action
2
of serum and impairs phagocytosis, and may be regarded as the most important virulence determinant of K. pneumoniae.
K. pneumoniae could be classified into 77 serological K antigen
types according to the diverse structure of the capsular polysaccharides (Ørskov and Ørskov, 1984). The K1/K2 strains were found to be
especially pathogenic in causing PLA, a common intra-abdominal
infection with 10-30% high mortality rate worldwide (Fung et al., 2002). Other serotypes showed little or no effectin PLA pathogenesis (Mizuta et
al., 1983). The highly virulent clinical isolates are often carry large
capsules as an important virulence factor to protect the bacteria from opsonophagocytosis and prevent from complement-mediated killing (Simoons-Smit et al., 1986).
Capsular polysaccharides and cps genes regulation
K. pneumoniae CG43, showing a strong virulence to Balb/c mice
with 50% lethal dose of 10 CFU, is a highly encapsulated clinical isolate of K2 serotype (Chang et al., 1996). The structure of the K2 capsular polysaccharides (CPS) has been determined as
[→)4-Glc-(1→3)-α-Glc-(1→4)-β-Man-(3←1)- α-GlcA)-(1→]n
(Wacharotayankun et al., 1992), which is made from a similar
biosynthetic pathway to that of Escherichia coli group 1 CPS (Whitfield and Roberts, 1999). In E. coli, four groups of CPS biosynthesis pathways could be identified. Group 1 and 4 CPS synthesis requires a Wzx/Wzy dependent pathway: the repeat unit oligosaccharide is transferred to a lipid carrier, undecaprenyl phosphate (und-P), forming und-PP-linked
3
repeat units, which flips across the inner membrane in a process requiring Wzx.The polymer is formed in Wzy-dependent polymerization, as the growing chain is transferred to und-PP-linked unit. Then, polymer is translocated by Wza, the outer membrane protein. The biosynthesis of group 2 and 3 CPS is ATP-binding-cassette (ABC) transporter-dependent, of which the diacylglycerophosphate-linked repeat unit is produced by glycosyltransferase followed by the export of polymer across inner membrane via ABC transporter (Whitfield, 2006).
The K2 cps (capsular polysaccharide synthesis) gene cluster of K.
pneumoniae Chedid has been determined, which contains 19 open
reading frames (ORFs) organized into 3 transcriptional units (Arakawa et
al., 1995). Among these genes, orf3 to orf6, a highly conserved
gene-block, are counterparts of E. coli wzi-wza-wzb-wzc (Rahn et al., 1999). Wzi is an outer membrane protein and wzi mutant showed a significant reduction in cell-bound CPS polymer with a corresponding increase in cell-free material. This proposed that Wzi plays in the process that links high-molecular-weight capsule to the cell surface (Alvarez et
al., 2000; Rahn et al., 2003). Wza is a periplasmic protein involved in
forming a multimeric putative translocation channel. The Orf5, KpWzb, is a low molecular weight protein tyrosine phosphatase (PTP) and Orf6,
KpWzc, a protein tyrosine kinase (PTK) that participated in high-level
polymerization of capsular polysaccharide. Enzymatic activities of the two proteins, KpWzb and KpWzc, have already been demonstrated (Preneta et al., 2002).
4
Protein phosphorylation on tyrosine in bacteria
Protein phosphorylation is one of the most important
post-translational covalent modifications and has gained recognition as a key device in the pleiotropic regulation of multiple cellular functions in eukaryotic organisms (Hunter, 2000; Pawson and Scott, 2005). It is best known that protein phosphorylation is a reversible enzyme-catalyzed process which controlled by various kinases and phosphatases. The first two protein-phosphorylating systems, ‘‘two-component systems (2CS)’’ and ‘‘phosphotransferase system (PTS)’’ have been recognized as the hallmark of bacterial signaling (Deutscher et al. 2006; Klumpp and Krieglstein, 2002). The third phosphorylating system in bacteria closely resembles the ‘‘classical’’ ATP/GTP-dependent system in eukaryotes (Cozzone, 1998; Shi et al., 1998). In this system, proteins are
phosphorylated on serine and/or threonine or tyrosine. Recently,
accumulating evidences suggest that Ser/Thr/Tyr phosphorylations also contribute to the regulation of a diverse range of cellular responses and physiological processes in prokaryotes (Cozzone, 2005). Modification at serine/threonine is usually more frequent than modification at tyrosine (3-15%), but both types of modification appear to coexist in nearly all bacterial species (Sun et al., 2010).
Tyrosine phosphorylation is a key device in numerous cellular functions in eukaryotes, but in bacteria this protein modification was largely ignored until mid-1990s (Grangeasse et al., 2007). Recent data have shown that a variety of cellular processes essential for bacterial survival and virulence are regulated by the phosphorylation of certain
5
endogenous proteins (Cozzone, 2009). This reaction is catalyzed by autophosphorylating ATP-dependent protein-tyrosine kinases which use homologues of Walker motifs of nucleotide-binding proteins for their catalytic mechanism (Grangeasse et al., 2007). These kinases exhibit similar but not identical structural and functional features with their eukaryotic counterparts (Olivares-Illana et al., 2008; Lee et al., 2008). For antibacterial therapy, this is important because it opens the way to molecular characterization of specific ligands that would selectively block the enzymatic activity of bacterial kinases (Cozzone, 2009). Based on these features, most of them have been recently unified in a new enzyme family called Bacterial tyrosine kinase (BY-kinase), a typical BY-kinase containing a catalytic domain, Walker A and B motifs, and C-terminal tyrosine cluster. (Grangeasse et al., 2007).
Although BY-kinases from Gram-negative and Gram-positive bacteria exhibit significant sequence similarity, they possess different domain organizations. BY-kinases of Gram-negative bacteria are usually large proteins (~80 kDa) composed of an N-terminal transmembrane domain and a C-terminal cytosolic PTK domain containing the active site Walker motifs A and B (Doublet et al., 1999, 2002). When expressed separately, the soluble C-terminal domain of E. coli Wzc still exhibits autophosphorylation activity (Grangeasse et al., 2002). By contrast, PTKs of Gram-positive bacteria are naturally separated into two distinct
proteins, i.e. a transmembrane protein with limited similarity to the
N-terminal domain and a soluble protein with significant similarity to the C-terminal domain of PTKs from Gram-negative bacteria. The soluble
6
protein containing the Walker motifs A and B also autophosphorylates at a tyrosine cluster located in its C terminus (Macek et al., 2003). The different domain organization of PTKs between Gram-negative and Gram-positive bacteria has suggested a different regulation of these enzymes.
In terms of genomic organization, the genes encoding a
protein-tyrosine kinase and a protein-tyrosine phosphatase in bacteria are most often located next to each other on the chromosome. In addition, these genes are generally part of large operons that direct the coordinate synthesis of proteins involved in the production or regulation of
exopolysaccharides and capsular polysaccharides. Recent data provide evidence that there exists a direct relationship between the reversible phosphorylation of proteins on tyrosine and the production of these polysaccharidic polymers, which are involved in the early steps of the infection process and are considered potent virulence factors (Cozzone et
al., 2004).
In the past, functional roles of the critical components involved in protein phosphorylation used to be defined by biochemical and genetic approaches (Cozzone, 2005). A salient gap exists between the growing number of identified protein-tyrosine kinases/phosphatases and the relative paucity of the protein substrates characterized to date (Lin et al., 2009). Nevertheless, in the past few years, high-performance techniques (based on gel-free peptide analysis and mass spectrometry) have been developed in association with systematic determination of genomic sequences for a fast identification of hundreds of phosphoproteins and
7
corresponding phosphorylation sites. They have been applied so far to the phosphoproteomes (i.e. the entire complement of phosphorylated proteins expressed by a cell) of B. subtilis (Macek et al., 2007), E. coli (Macek et
al., 2008), Lactococcus lactis (Soufi et al., 2008), Pseudomonas sp.
(Ravichandran et al., 2009) and Klebsiella pneumoniae (Lin et al., 2009). These endogenous protein substrates are present in a wide array of metabolic and regulatory processes from genetic competence and gene transcription to morphogenesis and stress. They also participate in cell division and differentiation, motility, biofilm formation, sporulation, central metabolism, protein biosynthesis and antibiotic resistance
(Cozzone, 2009). This discovery contributes to the emerging picture that bacterial tyrosine phosphorylation, in addition to the classical
serine/threonine kinases, and the 2CS and PTS, is an important regulatory arsenal of bacterial physiology beyond its sole implication in
pathogenesis.
The first known kinase substrates are the kinases themselves because they are autophosphorylating enzymes. The later identified protein is UDP-glucose dehydrogenases (Ugd) (Grangeasse et al.,2003; Mijakovic
et al., 2003). Phosphorylation of the enzyme significantly increases the
dehydrogenase activity thereby stimulates formation of the precursors for polysaccharide production, lipopolysaccharide modification for the
resistance to cationic peptides or polymyxin-type antibiotics or phosphate metabolism in E.coli and B. subtilis (Breazeale et al., 2002; Soldo et al., 1999).
8 UDP-glucose dehydrogenase (Ugd)
UDP-glucose dehydrogenase (Ugd) catalyzes a two-step
NAD+-dependent oxidation of UDP-glucose to produce UDP-glucuronic acid (UDP-GlcUA). The mechanism of Ugd proceeds through an initial oxidation of UDP-glucose with transfer of the C6” pro-R hydride (HR in
Appendix 1) to the si face (B face) of NAD+ to form NADH and the aldehyde intermediate (Feingold and Franzen, 1981). Covalent catalysis proceeds with nucleophilic attack of Cys 260 on the aldehyde to give a thiohemiacetal that is oxidized to a thioester intermediate by transfer of the remaining hydride (HS in Appendix 1) to a second molecule of NAD
+
. In the final step of the normal enzymatic reaction, the thioester
intermediate is irreversibly hydrolyzed to give UDP-GlcUA. The importance of UDP-GlcUA is apparent considering the downstream polymers that utilize this compound or its derivates, such as UDP-xylose, UDP-arabinose, and UDP-galacturonic acid in a variety of organisms ranging from mammals to bacteria (Seifert, 2004).
In mammals, UDP-GlcUA is the substrate for UDP-glucuronosyl transferases in the liver that catalyze the formation of glucuronide conjugates with various substances such as bilirubin and thereby aid in their excretion (Dutton, 1980). UDP-GlcUA is also essential for the biosynthesis of hyaluronan and various glycosaminoglycans such as chondroitin sulfate and heparan sulfate (Roden, 1980). Mutation of the Ugd gene of Drosophilia melanogaster (designated sugarless) disrupts biosynthesis of the heparan sulfate side chains on proteoglycan core proteins and is identical in phenotype to the classical wingless mutation
9
(Binari et al., 1997). In plants, Ugd may be an important regulatory enzyme in the carbon flux toward cell wall and glycoprotein biosynthesis due to feedback inhibition from UDP-xylose (Dalessandro and Northcote, 1977).
In many pathogenic bacteria, UDP-GlcUA is a precursor and an essential component for the biosynthesis of exo- polysaccharides (EPS) and lipopolysaccharides (LPS). EPS or LPS is critical virulence factor which enables the bacteria to evade attacks by host immune system, such as phagocytosis (Cross, 1990; Moxon and Kroll, 1990; Watson and
Musher, 1990; Wessels et al., 1994). UDP-GlcUA also participates in the production of UDP-4-amino-4-deoxy-Larabinose (L-Ara4N) which is a crucial element in bacterial resistance to antibiotics such as polymyxin and cationic peptides of the innate immune system (Breazeale et al., 2003).
Recent studies demonstrate that transcription of the Salmonella ugd is controlled by three regulatory systems that respond to different signals (Mouslim et al., 2003). Ugd mutation in the pathogenic fungus
Cryptococcus neoformans alters the cell integrity and nucleotide sugar
pool. The cells also become temperature-sensitive and fail to grow in an animal model (Griffith et al., 2004). Pseudomonas aeruginosa PAO1 encodes two Ugd, PA2022 and PA3559. The PA2022 mutant and
PA2022-PA3559 double mutant, but not the PA3559 mutant, are more
susceptible to chloramphenicol, cefotaxime, and ampicillin. The PA3559 mutant, however, shows a reduced resistance to polymyxin B compared with wild type PAO1(Hung et al., 2007). In E.coli, phosphorylation of
10
Ugd by Wzc plays a role in the regulation of the amount of the EPS colanic acid, whereas Etk-mediated Ugd phosphorylation participates in the resistance to the polymyxin (Lacour et al., 2008).
Activity of other dehydrogenases, including Bacillus subtilis Ugd (Mijakovic et al., 2004) and the UDP acetyl-mannosamine
dehydrogenase CapO of Staphylococcus aureus (Soulat et al., 2007), have recently been shown to be regulated by tyrosine-phosphorylation. It is hence speculated that tyrosine phosphorylation of this class of enzymes is a common regulatory mechanism found in bacteria.
Identification of the phosphotyrosine residues
Although a lot of protein kinases and phophatases have been predicted and identified in a variety of bacterial species, classical
biochemical approaches have so far revealed only a few substrate proteins and even fewer phosphorylation sites. In previous work, we have
provided in vitro evidence that the Wzc of K. pneumoniae CG43 (KpWzc) was able to phophorylate Ugd the phosphorylation appeared to enhance the Ugd activity (Appendix 2, Zhi-Kai Li, 2006). To advance our
knowledge of the underlying mechanism in capsule formation for the development of new therapeutic strategies, it is crucial to identify the phosphorylation site on Ugd. The previously employed mass
spectrometry analysis (MALDI-TOF) failed to identify the Ugd phosphorylated tyrosine probably because of the low occupancy of
bacterial phosphorylation sites (Macek et al., 2006). Nevertheless, a most recently study of the Bacillus subtilis phosphoproteome indicated that
11
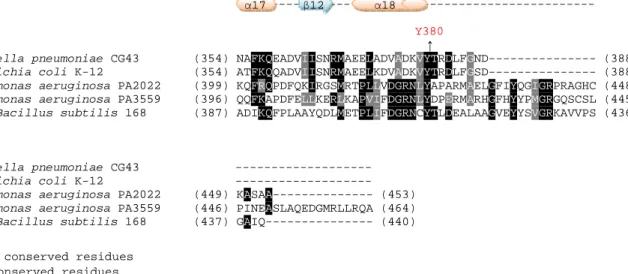
Ugdwas phosphorylated in vivo on tyrosine 70 (Macek et al., 2007). Tyrosine 71, the counterpart residue of EcUgd was later demonstrated as the specific phosphorylation residue (Lacour et al., 2008). As shown in Fig. 1, multiple sequence alignment suggested the Tyrosine 71 (Tyr71) of
KpUgd sequence is the phosphorylation site.
The sequence alignments of Ugd from K. pneumoniae, E. coli K-12,
S. pyogenes, B. subtilis, E. amylovora, and S. pneumoniae revealed nine
conserved tyrosine residues Tyr10, Tyr91,Tyr150, Tyr210, Tyr217,
Tyr242, Tyr249, Tyr265 and Tyr335. We have previously tried to generate site-directed mutation, tyrosine changed to phenylalanine, on Ugd
targeting to the 9 residues (Mei-Ju Li, 2008). However, the in vitro phosphorylation assays indicated that all the mutated proteins were able to be phosphorylated by the catalytic domain of Wzc, Wzccyto, although
some of the mutations had slightly changed the Ugd activity,
Here, the rest of the eight tyrosine residues on KpUgd (Tyr53, Tyr71, Tyr76, Tyr85, Tyr203, Tyr252, Tyr302, and Tyr380) were subjected to site-directed mutagenesis. Phosphorylation assay was then employed to analyze the mutant proteins carrying with each of the alterations. In the mean time, the enzymatic activity determined if Tyr71 is the only phosphorylation site and if any of the tyrosine residues plays a role in influencing the Ugd activity.
12
Materials and methods Bacterial Strains, Plasmids, and Growth Conditions
Genomic DNA prepared from K. pneumoniae CG43 was used as template for PCR amplification of the ugd gene. Bacterial strains and plasmid used in this study are listed in Table 1. K. pneumoniae CG43 is a clinical isolates recovered from Chang Gung Memorial Hospital, Linkou. All strains were routinely cultured at 37°C in Luria-Bertani (LB; 10 g/l tryptone, 5 g/l yeast extract, 10 g/l sodium chloride) broth or on LB agar. The following antibiotic concentrations were used: Kanamycin 25 µg/ml, Ampicillin 100 µg/ml, Tetracycline 20 µg/ml and Streptomycin 50 µg/ml. The plasmid generated and primers used are listed in Table 2 and Table 3, respectively.
Recombinant DNA manipulation
All recombinant DNA experiments were carried out by standard procedures as described (Sambrook et al., 2001). Restriction
endonucleases and DNA modifying enzymes were purchased from MBI Fermentas (Hanover, MD) or New England Biolab (Beverly, MA, USA), and were used according to the recommendation by the suppliers.
Plasmids were purified by using the High-Speed Plasmid Mini kit (Geneaid, Taiwan). PCR amplifications were performed with Taq DNA polymerase (MDBio, Inc, Taiwan). PCR products and DNA fragments were purified using the Gel/PCR DNA Fragments Extraction kit (Geneaid, Taiwan). The primers used in this study were synthesized by MDBio, Inc, Taiwan.
13
Construction for the overproduction of His6-tagged Ugd and the
derived mutants
The Ugd mutants were generated using QuikChange site-directed mutagenesis method (Stratagene). The procedure utilized the yT&A vector with an insert of ugd and two synthetic oligonucleotide primers containing the desired mutation. The oligonucleotide primers, each complementary to opposite strands of the vector, were extended during temperature cycling by Finnzymes’ Phusion™ High-Fidelity DNA polymerase. Incorporation of the oligonucleotide primers generated a mutated plasmid containing staggered nicks. Following temperature cycling, the product is treated with Dpn I. The Dpn I endonuclease (target sequence: 5´-Gm6ATC-3´) is specific for methylated and hemimethylated DNA and is used to digest the parental DNA template and to select for mutation-containing synthesized DNA. The nicked vector DNA
containing the desired mutations is then transformed into E.coli JM109 (Appendix 3).
The plasmid was then subcloned into the pET-30b expression vector at EcoRⅠ/SalⅠrestriction sites to be in-frame with the His6 tag at the N
terminus of the protein. The constructs were then introduced into E. coli NovaBlue (DE3) by the heat shock method, and the transformants were selected on LB agar containing 25 µg/ml Kanamycin. The DNA was sequenced to verify the correctness of the cloned gene and the reading frame fusion with the His6 tag. The resulting plasmids were named
14
pET30UgdY203F, pET30UgdY252F, pET30UgdY302F,
pET30UgdY380F and the mutants Ugd proteins were expressed in E.coli NovaBlue (DE3).
Overproduction and purification of the His6-Ugd and the derived
mutants
The bacterial cells were incubated in 100 ml of LB medium
supplemented with Kanamycin at 37°Cwith shaking until OD600 reached
0.5~0.6. Isopropyl-1-thio-β-D-galactopyranoside (IPTG) was then added to a final concentration of 1 mM and the growth was continued for 4 h at 37°C. Subsequently, the cells were harvested by centrifugation at 5000 rpm for 10 min, resuspended in binding buffer (20 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole, pH 7.9), and the cell suspension disrupted by sonication and then the cell debris removed by centrifugation at 13000 rpm for 20 min. Finally, the His6-tagged proteins were purified from the
supernatant via affinity chromatography using His-Bind resin (Novagen), and the elution was carried out with elute buffer (20 mM Tris-HCl, 500 mM NaCl, 1 M imidazole, pH 7.9). Aliquots of the collected fractions were analyzed by SDS-PAGE and the fractions containing most of the purified His6-tagged Ugd were dialyzed against the buffer containing 50
mM Tris-HCl (pH7.5), 100 mM NaCl, 1 mM EDTA, and 10% (v/v) glycerol.
15
Overproduction and purification of His6 tag fusion KpWzc
cytoplasmic domain
E.coli BL21-RIL cells were transformed with pET30-KpWzcE23
(Table 2), which expressing a mutated KpWzc cytoplasmic domain,
His6-KpWzc (Arg451-Lys722). Overproduction for His6-KpWzc was carried
out with similar conditions for Ugd except IPTG was added to a final concentration of 0.5mM. Subsequently, the cells were harvested by centrifugation at 5000 rpm for 10 min, resuspended in binding buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, 10% (v/v) glycerol, pH 8.0), and the cells disrupted by sonication and then the cell debris removed by centrifugation at 13000 rpm for 20 min. Finally, the His6-KpWzc were purified from the supernatant via affinity
chromatography using His-Bind resin (Novagen), and the elution was carried out with elute buffer (50 mM sodium phosphate, 300 mM NaCl, 100 mM imidazole, 10% glycerol, pH 8.0). Aliquots of the collected fractions were analyzed by SDS-PAGE and the fractions containing most of the purified His6-KpWzc were dialyzed against the buffer containing
50 mM sodium phosphate (pH 8.0), 150 mM NaCl, 5 mM MgCl2, 5 mM
dithiothreitol and 10% (v/v) glycerol.
SDS-polyacrylamide gel electrophoresis
Protein preparation were treated for 10 min at 95°C in loading buffer (0.0626 M Tris-HCl buffer pH 6.8, 2% (v/v) SDS, 10% (v/v) glycerol, 0.01% (v/v) bromophenol blue,and 100 mM dithiothreitol). Twenty microliters of sample was applied to a 12.5% (v/v) SDS polyacrylamide
16
slab gel. Electrophoresis was carried out at room temperatureuntil the tracking dye ran off the bottom of the slab gel. The gel was stained for 5 min using solution containing 2.5% (v/v) Coomassie Blue R250, 45% (v/v) methanol, and 10% (v/v) aceticacid, and destained in destain buffer (40% (v/v) methanol and 10% (v/v) acetic acid) for 30 mins.
In vitro phosphorylation assay
The phosphorylation assay was carried out essentially as described (Appendix 4, Grangeasse et al., 2003). Briefly, the 20 µl reaction mixtures contains about 2 µg of the purified wild-type Ugd or mutant Ugd, 2 µg kinase and 10 µM ATP in 25 mM Tris-HCl (pH 7.0), 1 mM DTT, 5 mM MgCl2, 1 mM EDTA was incubated at 37 °C for 1h. The
reaction was stopped by addition of the sample buffer and heated at 95°C for 5 min. After electrophoresis (12.5% SDS-PAGE), the gel was
analyzed by western blotting or stained with Pro-Q○R
Diamond Phosphoprotein fluorescent dye (Invitrogen, catalog # P33300) for detection of the phosphorylated proteins and the result visualized using Amersham Typhoon™ 9200 Imager.
Western blot analysis of the phosphotyrosine proteins
The purified proteins were analyzed by SDS-PAGE and the resolved proteins were transferred to a polyvinylidene difluoride (PVDF)
membrane electrophoretically in the transfer buffer containing 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4and 20% methanol.
17
antibody (Upstate, catalog # 05-321) and the secondary antibody, an anti-mouse IgG alkaline phosphatase conjugated antibody (Sigma), was then applied and the bound complex was detected by using nitro blue tetrazolium chloride (NBT)/5-bromo-4-chloro-3-indolyl phosphate (BCIP) as the substrates.
Enzyme activity measurement and the kinetics characterization UDP-glc dehydrogenase activity was determined by monitoring the change in absorbance at 340 nm that accompanies the reduction of NAD+ to NADH using a spectrophotometric assay as described (Pagin et al., 1999). The enzyme assay was performed at room temperature in 100 mM Tris-HCl (pH 9.0), 100 mM NaCl, 2 mM DTT, 2 mM NAD+ and 5 mM UDP-glc. The Km and Vmax for UDP-glc and NAD+ were determined
independently using standard assay conditions. Kinetic study for UDP-glc as the substrate was performed with a fixed concentration of NAD+ and the concentration of UDP-glc varied in the range from 0.01 to 5 mM. Similarly, NAD+ kinetic measurement was made by holding UDP-glc concentration and varying NAD+ from 0.005 to 2 mM. Km and Vmax were
calculated by fitting the data to the equation (V = Vmax [S]h/ ([S]h + Kmh)),
where h is the Hill coefficient, and assuming a single binding site each for substrate and cofactor.
Circular dichroism spectrum analysis
The interaction of a chiral molecule with polarized light is very
18
small molecule and macromolecular structures (Fasman, 1996).
Essentially, one type of measurements commonly made to determine the effects of polarized light on asymmetric molecules is circular dichroism (Hammes, 2005), which is defined as the difference in absorption of left-hand and right-hand circularly polarized light with optically active compounds.
Protein secondary structure can be determined by CD spectroscopy in the ‘far-UV’ spectral region (190–250 nm). At these wavelengths, the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment (Kelly and Price, 1997). As determined, far-UV-CD of random coil is positive at 212 nm (n → π*) and negative at 195 nm (π → π*). Far-UV-CD of β-sheet is negative at 218 nm (π → π*) and positive at 196 nm (n → π*). For α-helix, the exciton coupling of the
π → π* transitions leads to positive (π → π*) perpendicular at 192 nm,
negative (π → π*) parallel at 208 nm, and negative at 222 nm is red shifted (n → π*), respectively (Manavalan et al., 1987; Kelly and Price, 1997). The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analysing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type.
Secondary structures of wild-type and mutant Ugd were assessed by CD spectroscopy on an Aviv 62A DS CD spectrophotometer with a 1-mm path length cell, 0.5 nm wavelength step, and an averaging time of 3 × 10-1 s. The measurements were performed on 2.5 µM of protein in 10 mM Tris-HCl (pH 7.4). The CD spectra signals were collected from 195 nm to
19
260 nm at 25 °C and averaged over three scans (Coligan, 2003). The results were expressed as molar ellipticity, [θ] (degree cm2 dmol-1) which was determined as [θ] = (θ× 1000)/(cl), where c is the protein
concentration in mmole/ml, l is the light path length in millimeters, and θ is the measured ellipticity in degrees at wavelength λ.
Software
Homology sequences were found from Protein database at NCBI (http://www.ncbi.nlm.nih.gov/) and alignment were performed with ClustalW2 program (Thompson et al., 1994) in EMBL-EBI
(http://www.ebi.ac.uk/). The multiple alignments resulting from ClustalW analysis was used as input for BOXSHADE program
(http://www.ch.embnet.org/software/ BOX form.html) to indicate residue similarity. The SWISS-MODEL (http://swissmodel.expasy.org/)
comparative protein modeling server (Guex and Peitsch, 1997) was employed to generate a 3D model of the KpUgd protein based on the structural alignment of its sequence with the highest scoring template structure, with Pymol (http://www.pymol.org/) as a molecular viewer.
Construction of the specific ugd-deletion mutant
K. pneumoniae CG43 mutants disrupted specifically at ugd genes
were constructed by the allelic exchange strategy. The primer sets used for PCR amplification of the DNA fragments flanking sequence are ugd001 and ugdM04 (Table 3). The generated DNA fragments were
20
University of New Hampshire), and the resulting plasmids, pHY034, were then mobilized to K. pneumoniae CG43-S3 through conjugation from E. coli S17-1 λ pir.The transconjugants were selected with
Ampicillin/Kanamycin on minimal medium (M9 minimal salts, Sigma). Some of the Ampicillin/Kanamycin resistant transconjugants was picked and then spread onto a LB plate supplemented with Streptomycin. When the occurrence of a double cross-over, the streptomycin-resistant and Ampicillin/Kanamycin-sensitive colonies were isolated, and the deletion of ugd was verified by PCR.
Construction of the Ugd and the UgdY71F-complemented strains To obtain the complement of ugd, full length ugd including their promoter region was amplified from K. pneumoniae CG43-S3 with the primer pairs ugdNTUpo3 and ugdR (Table 3) and the DNA fragment was ligated into yT&A vector to generate yT&A-pugd. The yT&A-pugdY71F was then constructed using QuikChange site-directed mutagenesis
method (Stratagene). DNA fragments containing full length ugd with their promoter region were excised from yT&A-pugd and
yT&A-pugdY71F, respectively, with HindIII and XbaI.The DNA fragments were ligated respectively, into a HindIII/XbaI -digested plasmid pRK415, a broad host range plasmid (Keen et al., 1998), to generate the ugd complementation plasmids, pRK415-pugd and pRK415-pugdY71F.
21 Results
K. pneumoniae Ugd (KpUgd)
As shown in Fig. 1, alignment of the Ugd sequence of K. pneumoniae CG43 (Dr. S.-F Tsai, unpublished results), E. coli K-12 (NCBI accession No. NP_416532), P. aeruginosa PAO1 PA2022 (NCBI accession No. NP_250712) and PA3559 (NCBI accession No. NP_252249), and B.
subtilis 168 (NCBI accession No. CAB15640) revealed that KpUgd and EcUgd, sharing 82% identity and 93% positives, are most closely related.
The alignment also shows that, KpUgd contains a NAD+
dinucleotide-binding domain, GXGXXG “fingerprint” of the Rossmann fold (Rossmann, 1981), at the N-terminal region and a nucleotide
sugar-binding domain at the C-terminal part. In addition to the conserved signature of nucleotide sugar dehydrogenase, KpUgd also contains
flanking sequence GGXCXXXD known to participate in catalysis. Finally, like the other Ugd, KpUgd contains an Arg at the position corresponding to Arg244 on SpUgd, which is one of the determinants of the substrate specificity of nucleotide sugar dehydrogenase (Campbell et al., 2000).
Selection for the phosphotyrosine residues on KpUgd
Recently, a B. subtilis phosphoproteome study reported that BsUgd was phosphorylated in vivo on a specific tyrosine residue Tyr70 (Macek
et al., 2007). The counterpart residue Tyr 71of EcUgd was subsequently
demonstrated to be the phosphorylation site by both tyrosine kinase
EcWzc and EcEtk. The phosphorylation of EcUgd-Tyr71 appeared to be
22
complementation analysis (Lacour et al., 2008). Therefore, we assumed that the residue Tyr71 of KpUgd is also subject to phosphorylation.
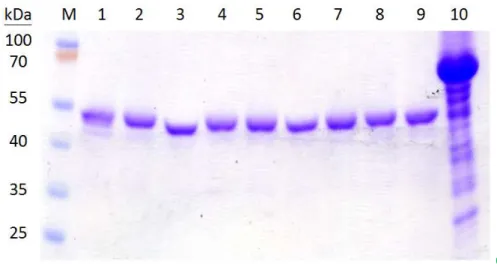
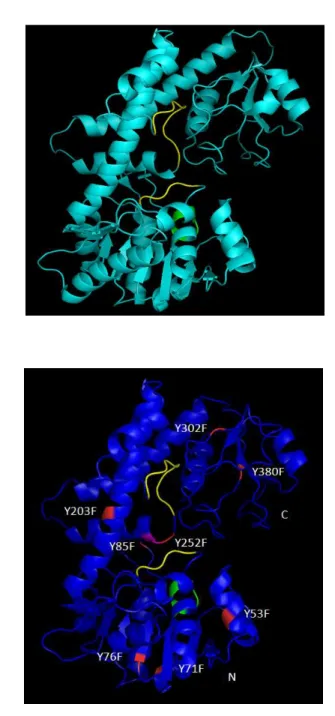
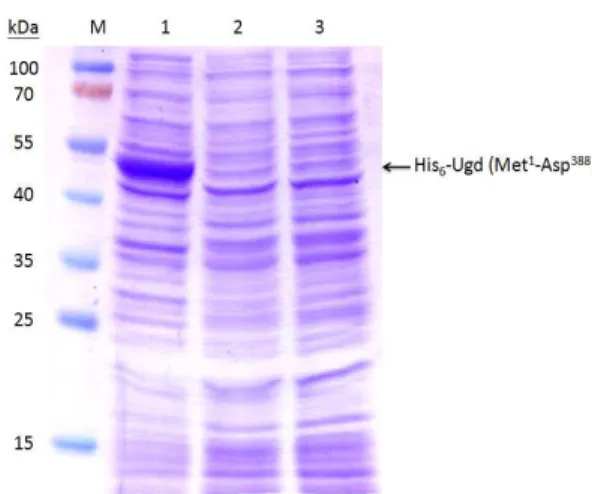
Construction, expression and purification of the KpUgd mutants In addition to Tyr71, 7 tyrosine residues of KpUgd (Y53, Y76, Y85, Y203, Y252, Y302, and Y380), which were not studied previously were selected for phenylalanine substitution (Fig. 1). After the site-specific substitution was confirmed by nucleotide sequencing, the mutant Ugd were cloned in pET-30b and expressed in E. coli NovaBlue (DE3), and the proteins purified through Ni+2-NTA-agarose matrix. As shown in Fig. 2A, all the recombinant proteins could be overexpressed in E. coli
NovaBlue (DE3). Most of the recombinant proteins appeared to be soluble and could be obtained in the supernatant fraction (Fig. 2B). In general, about 3.6 mg of Ugd and the derived mutant proteins of high purity (> 95%) could be obtained from a 100 ml culture (Fig. 2C). Interestingly, the IPTG-induced total protein lysates of pET-UgdY71F was less soluble compared to the others. Thus, about 500 ml cultured cells is needed to produce 2 mg of UgdY71F.
In vivo phosphorylation of the mutants with a substitution at a
tyrosine residue
To avoid the use of radioactive isotope for the detection of the phosphorylation reaction, we used anti-phosphotyrosine monoclonal antibody 4G10 (Fig. 3A) or Pro-Q○R
Diamond Phosphoprotein
23
phosphorylate Ugd (Grangeasse et al., 2003). We have also shown that
EcWzc could phosphorylate KpUgd (Appendix 4), and the recombinant
Ugd purified from E. coli has been phosphorylated (Zhi-Kai Li, 2006). As shown in Fig. 3A or 3B, both KpWzc and KpUgd exhibited
phosphorylation signals indicating phophatase has to be applied to remove the endogenous phosphorylation by EcWzc prior to the phosphorylation reaction.
In case that the phosphatase application may interfere the following phosphorylation, an in vivo phosphorylation without adding KpWzc was carried out to assess the tyrosine residue mutation effect on the KpUgd phosphorylation. As shown in Fig. 4, either the UgdWT or the derived site-specific mutants exhibited a phosphosignal of 46 kDa indicating none of the tyrosine mutation impaired the phosphorylation of KpUgd by
EcWzc.
Determination of kinetic parameters of the UgdWT and the derived mutants
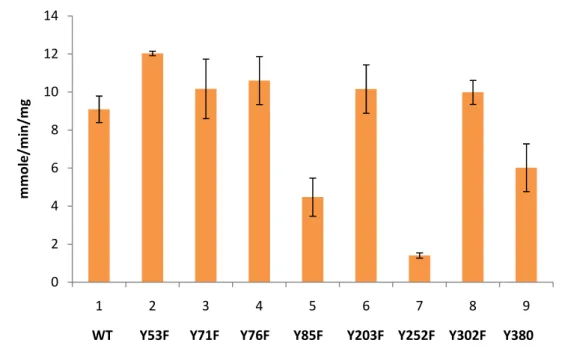
The initial velocity of the reaction by measuring the NADH
absorbance at 340 nm was determined. As shown in Fig. 5, most of the mutant proteins (UgdY53F, UgdY71F, UgdY76F, UgdY203F, and
UgdY302F) except Y85F, Y252F and Y380F mutants, when subjected to an enzyme specific activity measurement, performed just like the wild type. The specific activity of UgdY85F decreased to 0.49-fold,
UgdY380F to 0.66-fold and UgdY252F to 0.15-fold of that of the UgdWT (Fig. 5).
24
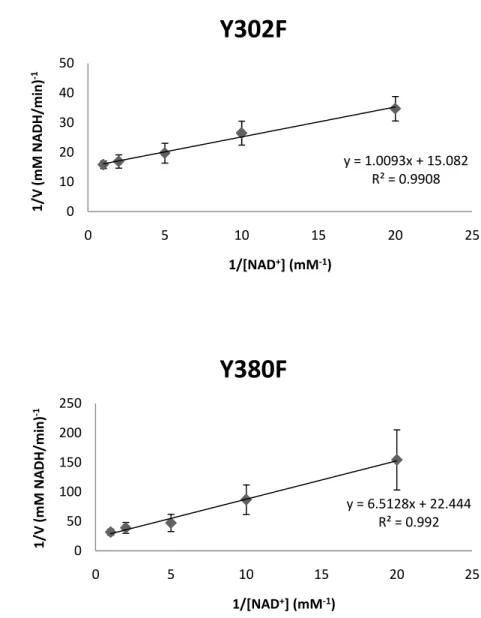
Typical Lineweaver-Burk plots were obtained when 1/[v] was plotted against 1/[S]. Kinetic parameters, Km and Vmax, were estimated by linear
regression from Lineweaver-Burk plots. The Vmax values were converted
to kcat by assuming that the molar mass was 49416.38 g·mol -1
. The kinetic constants of His6-tagged wild-type and mutant forms of Ugd for
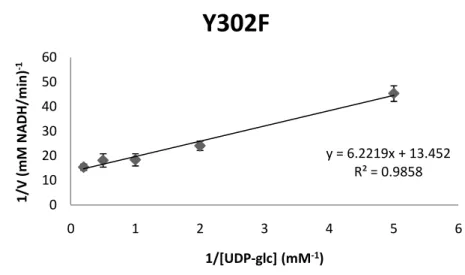
NAD+ were calculated by fixing the concentration of UDP-glc with various concentrations of NAD+ (Fig. 6). On the other hand, varying the concentration of UDP-glc with a constant concentration of NAD+ was also used to measure the activity (Fig. 7).
As summarized in Table 4, the Ugd mutants (Y71F, Y252F and Y380F) could not efficiently utilize UDP-Glc and NAD+ as demonstrated by the Km and kcat values. Interestingly, the specific activity obtained for
UgdY71F was similar to that of wild-type. Moreover, the Y71F mutation had moderate effects on the kinetics of catalysis (15.4- and 10.3-fold above Km values using UDP-glc and NAD+ as substrates). The Y252F
Ugd mutant exhibited undetectable Km values to UDP-glc and NAD +
, while Y380F showed undetectable Km values to UDP-glc but 3.5-fold
increase of Km values to NAD +
. The kcat value of UgdY85F, Y252F
orY380F for UDP-glc was much lower than that of the wild type Ugd. For NAD+, the kcat value of UgdY85F was slightly decreased (0.69-fold
of wild type) while undetectable in Y262F.
Secondary structure analyses of Ugd and the derived mutants according to the circular dichroism spectrum.
25
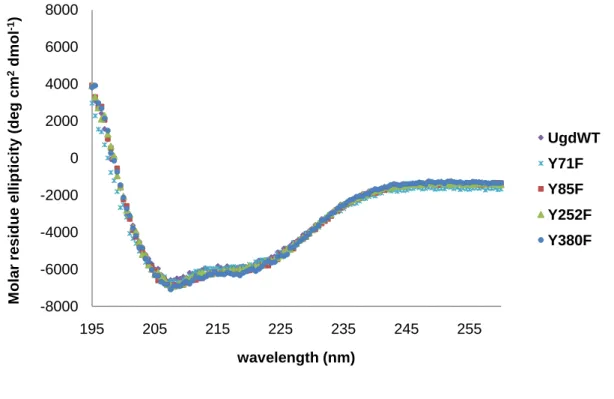
CD can be a good technique to compare between native and modified forms (Tafreshi et al., 2007; Hadizadeh et al., 2007). The CD spectra were therefore used as a measure of the relative quantities of changes made in the derived mutants by site-directed mutagenesis.
As shown in Fig. 8, the far UV CD spectra of Y71F, Y85F, Y252F, Y380F and wild-type Ugd in Tris-HCl (pH 7.4) appeared to be identical and all showing a ‘w’-shaped spectra with minimum point at 208 nm and 222 nm indicated the presence of high α-helix. This revealed that there is no major alteration of the secondary structures of the mutant proteins.
Structure modeling of KpUgd
The Streptococcus pyogenes Ugd (SpUgd) molecular structure has been solved and reported previously (Campbell et al., 2000). On the basis of homology (54.2% identity), comparative structural modeling was used to predict three-dimensional structure for KpUgd using the structure of
SpUgd (PDB ID: 1DLIA) (Fig. 9A) as a template. By superimposing the
context of the Ugd crystal structure of S. pyogenes, Cys253 in KpUgd was found at the position equivalent to catalytic nucleophile Cys260 in
SpUgd (Fig. 9B).
Characterization of KpUgd deletion mutant
The ugd gene-specific deletion strain was constructed using the allelic exchange strategy to determine the functional roles of Ugd in K.
pneumoniae physiology. As shown in Fig. 10A, the specific deletion was
26
CG43 a change of morphotype from mucoid, fatty and shiny appearance to small and dull colonies (Fig. 10B). As assessed by the sedimentation assay shown in Fig. 10C, the ugd mutant appeared to be more readily precipitated via centrifugation in comparing with the wild type CG43-S3 suggesting the deletion of ugd reduced the synthesis of CPS.
However, introducing the complementation plasmids pRK415-Ugd or pET30b-Ugd (Fig. 11) into the ugd deletion mutant failed to restore the phenotype. The possibility that an impaired Ugd expression in the ugd mutant could be verified using pQE30 expression vector in the future.
27 Discussion
KpUgd has more than one tyrosine-phosporylation residue.
Capsular polysaccharide biosynthesis is controlled by
phosphorylation at two levels: the assembly and export of the CPS (Wzc phosphorylation) and the synthesis of the CPS repeat unit (Ugd
phosphorylation). We have previously established that Wzc-mediated phosphorylation of Ugd of K. pneumoniae CG43 influences the
production of UDP sugar, the precursor for the bacterial CPS production. We have also shown that the phosphorylation of Ugd resulted in a
significant increase of its dehydrogenase activity and the dephosphorylation by CIAP reduced its enzyme activity.
In order to identify the specific phosphotyrosine residue, nine of the 17 tyrosine residues have been chosen for site-directed mutagenesis study previously (Mei-Ju Li, 2008). However, all the Ugd site-directed mutants as well as the wild-type protein were phosphorylated by KpWzc,
indicating the specific phosphoylation tyrosine residue on KpUgd has not yet identified. Here, an in vivo system to explore the
tyrosine-phosphorylated residue of Ugd by EcWzc was used to avoid the interference of the CIAP treatment. Again, we found that all the mutants could be phosphorylated suggesting KpUgd has more than one tyrosine phosporylation site. The result is in agreement with the previous study showing that Ugd has multiple tyrosine phosphorylation sites detected by isotope autoradiography (Appendix 5).
The subject comes to identification of the second or third tyrosine residue. As shown in Appendix 5, the fragmentsGST-KpUgd2 (His68 to
28
Ala167) and GST-KpUgd3 (Glu168 to Gly300) showed intensive signals. Since Tyr71 is contained in GST-KpUgd2 (His68 to Ala167), we reason that the additional phosphotyrosine residue could be in GST-KpUgd3. Lacour and his co-workers have excluded the tyrosines Tyr10, Tyr150, Tyr249, Tyr335 and Tyr380 of EcUgd as phosphorylation site (Lacour et
al., 2008). Thus, the Tyr252 of KpUgd of which the Phe replacement has
caused changes of the kinetic properties was selected as the second phosphorylated site. The Ugd mutant with double site mutation (Tyr71 and Tyr252) will be generated to validate the hypothesis.
The Tyr residues participate in the KpUgd activity.
As reported by Mei-Ju Li (Mei-Ju Li, 2008), the activities of UgdY91F and UgdY210F were higher than that of the wild-type Ugd while UgdY10F, UgdY242F and UgdY249F exhibited a lower activity than that of the wild-type Ugd. In the study, three mutants with tyrosine to phenylalanine substitutions at positions 85, 252, and 380 had much lower activity than wild type. In addition, the kcat/Km value of UgdY71F,
UgdY252F, and UgdY380F appeared much smaller than that of wild type. The single substitutions do not affect protein folding and have equivalent conformations as demonstrated via circular dichroism spectrum analysis. Tyr71 has been demonstrated as the phosphorylation site for EcUgd and Tyr70 for BsUgd. In analogy, we expected the purified UgdY71F would lose the phosphorlation signal and hence the enzyme activity. However, the tyrosine replacement only reduced slightly the kcat/Km value.
29
molecule to a hydroxyl group of tyrosine residue that can turn a
hydrophobic portion of a protein into a polar and extremely hydrophilic portion of molecule. In this way it introduces a conformational change via interaction with other hydrophobic and hydrophilic residues in the protein. Consequently, the entrance of catalytic pocket may be slightly moved to let the substrates contact the catalytic pocket more easily. The lower affinity to the substrates detected for UgdY71F may be due to the change of the phosphorylation state. This further supports the results of Appendix 2 that the phosphorylation of the Tyr71 of KpUgd plays a regulatory role for the enzymatic activity, which is different from the essential role of tyrosine phosphorylation on EcUgd.
The UDP-glc binding pocket can be divided into two regions: the UMP binding pocket composed solely of the residues from the C-terminal domain, and the glucose 1-phosphate binding pocket consisting primarily the residues from the N-terminal domain. The UMP binding pocket was lined with a stretch of coil (Tyr 242~Gly 250) that makes three main chain hydrogen bonds, two side chain hydrogen bonds, and a π-edge stacking interaction of Tyr242 with the UMP moiety. The glucose
1-phosphate binding pocket was found at the dimer interface, limited to a small region (Phe 142-Glu 145) between α7 and α8 of the N-terminal domain that forms three main hydrogen bonds to the glucose 1-phosphate moiety (Campbell et al., 2000). Nevertheless, Fig. 1 also revealed that central α-helix (α10) serving as the core of the dimer interface could be important. There are a total of 24 hydrogen bonds to stabilize the dimer interface, though none of the amino acids involved are strictly conserved.
30
The aromatic residues including Phe199, Tyr203, Tyr210, Tyr217, and Tyr265 were found to be located within the dimer interface.
On the basis of the structure modeling of KpUgd, Tyr85 is located beside the dimer interface and also close to the catalytic pocket.
According to the enzymatic analysis, Y85F had effect on the specific activity, but not the kinetic properties. This suggested that Y85F may destroy the hydrogen bonds that stabilize the dimer interface without influencing the affinity between Ugd and substrates. The possibility could be verified using gel filtration chromatography to study whether a size difference is present between the preparation of Ugd and UgdY85F. Tyr252 is situated beside the catalytic site, Cys253, and very close to the interface of NAD+ and UDP-glc which bound to the structurally pivotal active site of the enzyme. The activity abolished by substitution of
Tyr252 to Phe may be attributed to the deletion of the phenolic hydroxyl group leading to impair the proton conductance pathway which in turn prevent from the oxidation of UDP-glc. Since Tyr380 was found to be strictly conserved among Ugds, if the alteration affects Ugd activity via influencing the binding to substrate or cofactor is unknown.
Overall, whether the altered enzymatic or kinetic effect is due to missing of the tyrosine phosphorylation or the change from tyrosine to phenylalanine per se remains to be investigated.
Bacterial tyrosine phosphorylation: novel targets for antibacterial therapy
31
mechanism for biosynthesis of the CPS, a vital determinant for the initial stages of infection. Ugd has been considered as a crucial drug target for its critical role in EPS synthesis (Campbell et al., 2000). The kinetics study revealed some of the tyrosine residues are important for Ugd activity. Moreover, the presence of multiple tyrosine phosphorylation sites on KpUgd makes Ugd as a drug target more attractive. Further investigation on how Ugd affects drug susceptibility in bacteria may reveal new implications for future drug development.
Conclusion
To validate if KpUgd has more than one tyrosine-phosporylation residue, double-site mutations (Tyr71 and Tyr85, Tyr252, or Tyr380) or triple mutation will be generated. Phosphorylation assay and enzymatic measurement will then be performed to confirm the phosphorylation role of the tyrosine residues.
32 Reference
1. Alvarez, D., Merino, S., Tomas, J.M., Benedi, V.J., and Alberti, S. (2000) Capsular polysaccharide is a major complement resistance factor in lipopolysaccharide O side chain-deficient Klebsiella
pneumoniae clinical isolates. Infect Immun 68: 953-955.
2. Arakawa, Y., Wacharotayankun, R., Nagatsuka, T., Ito, H., Kato, N., and Ohta, M. (1995) Genomic organization of the Klebsiella
pneumoniae cps region responsible for serotype K2 capsular
polysaccharide synthesis in the virulent strain Chedid. J Bacteriol 177: 1788-1796.
3. Bai, Ping-Hui (2004) Capsular polysaccharide production and protein-tyrosine phosphorylation in Klebsiella pneumoniae CG43. Master thesis, Department of Biological Science and Technology, National Chiao Tung University.
4. Binari RC, Staveley BE, Johnson WA, Godavarti R, Sasisekharan R, Manoukian AS. (1997) Genetic evidence that heparin-like
glycosaminoglycans are involved in wingless signaling. Development 124(13):2623-32.
5. Breazeale SD, Ribeiro AA, Raetz CR. (2002) Oxidative decarboxylation of UDP-glucuronic acid in extracts of
polymyxin-resistant Escherichia coli. Origin of lipid a species modified with 4-amino-4-deoxy-L-arabinose. J Biol Chem 277(4):2886-96.
6. Breazeale SD, Ribeiro AA, Raetz CR (2003) Origin of lipid A species modified with 4-amino-4-Deoxy-L-Arabinose in polymyxin resistant
33
mutants of Escherichia coli:An aminotransferase (ArnB) that generates UDP-4-amino-4-deoxy-Larabinose. J Biol Chem 278: 24731–24739.
7. Brisse S, Grimont F, Grimont PAD (2006) The genus Klebsiella. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, eds. The Prokaryotes.A Handbook on the Biology of Bacteria. 3rd edition ed. New York: Springer.
8. Brisse S, Fevre C, Passet V, Issenhuth-Jeanjean S, Tournebize R, Diancourt L, Grimont P. (2009) Virulent clones of Klebsiella
pneumoniae: identification and evolutionary scenario based on
genomic and phenotypic characterization. PLoS One 4(3):e4982. 9. Brown, C. and R. J. Seidler (1973). "Potential pathogens in the
environment: Klebsiella pneumoniae, a taxonomic and ecological enigma." Appl Microbiol 25(6): 900-4.
10.Campbell RE, Mosimann SC, van De Rijn I, Tanner ME, Strynadka NC. (2000)The first structure of UDP-glucose dehydrogenase reveals the catalytic residues necessary for the two-fold oxidation.
Biochemistry 13;39(23):7012-23.
11.Chang, H.Y., Lee, J.H., Deng, W.L., Fu, T.F., and Peng, H.L. (1996) Virulence and outer membrane properties of a galU mutant of
Klebsiella pneumoniae CG43. Microb Pathog 20: 255-261.
12.Chien, HS (2008)ManC, Gnd and KpUgd are phosphorylated by tyrosine kinase, KpWzc of Klebsiella pneumoniae-Kinetic analysis of ManC and Gnd, identification of phosphotyrosine residues of KpUgd and search for other phosphorylation targe.Master thesis, Institute of
34
Molecular Medicine, National Tsing-Hua University.
13.Chou HC, Lee CZ, Ma LC, Fang CT, Chang SC, et al. (2004) Isolation of a chromosomal region of Klebsiella pneumoniae
associated with allantoin metabolism and liver infection. Infect Immun 72: 3783–3792.
14.Coligan, J.E. (2003) Current Protocols in Protein Science, John Wiley & Sons Inc., Brooklyn, NY, Unit 7.6.
15.Cozzone AJ (1998) Post-translational modification of proteins by reversible phosphorylation in prokaryotes. Biochimie 80:43–48 16.Cozzone, A. J., Grangeasse C, Doublet P, Duclos B. (2004) Protein
phosphorylation on tyrosine in bacteria. Arch Microbiol 181(3): 171–81
17.Cozzone, A. J. (2005) Role of protein phosphorylation on
serine/threonine and tyrosine in the virulence of bacterial pathogens. J
Mol Microbiol Biotechnol 9, 198-213.
18.Cozzone, A. J. (2009) Bacterial tyrosine kinases: novel targets for antibacterial therapy? Trends Microbiol 17(12):536-43
19.Cross, A. S. (1990) Curr. Top. Microbiol. Immunol. 150, 87–95 20.Dalessandro G, Northcote DH. (1977) Changes in enzymic activities
of nucleoside diphosphate sugar interconversions during
differentiation of cambium to xylem in sycamore and poplar. Biochem
J 162(2):267-79.
21.Deutscher J, Francke C, Postma PW (2006) How phosphotransferase system-related protein phosphorylation regulates carbohydrate
35
22.Doublet, P., Vincent, C., Grangeasse, C., Cozzone, A. J., and Duclos, B. (1999) On the binding of ATP to the autophosphorylating protein, Ptk, of the bacterium Acinetobacter johnsonii.FEBS Lett 445, 137–143 23.Doublet, P., Grangeasse, C., Obadia, B., Vaganay, E., and Cozzone, A.
J. (2002) Structural organization of the protein-tyrosine autokinase Wzc within Escherichia coli cells. J. Biol. Chem 277, 37339–37348 24.Dutton GJ (1980) Acceptor substrates of UDP-glucuronosyltransferase
and their assay, in Glucuronidation of Drugs and Other Compounds (Dutton GJ ed) pp 69–78, CRC Press, Boca Raton.
25.Fasman G.D. (1996) Circular Dichroism and the Conformational Analysis of Biomolecules. New York, USA: Plenum Publishing Corp. 26.Feingold, D. S. and Franzen, J. S. (1981). Pyridine nucleotide-linked
four- electron transfer dehydrogenase. Trends Biochem. Sci. 6, 103-105.
27.Fung, C. P., Chang, F. Y., Lee, S. C., Hu, B. S., Kuo, B. I., Liu, C. Y., Ho, M., and Siu, L. K. (2002) A global emerging disease of Klebsiella
pneumoniae liver abscess: is serotype K1 an important factor for
complicated endophthalmitis? Gut 50, 420-424.
28.Grangeasse, C., Doublet, P., and Cozzone, A. J. (2002) Tyrosine phosphorylation of protein kinase Wzc from Escherichia coli K12 occurs through a two-step process. J. Biol. Chem 277, 7127–7135 29.Grangeasse C, Obadia B, Mijakovic I, Deutscher J, Cozzone AJ, et al.
(2003) Autophosphorylation of the Escherichia coli protein kinase Wzc regulates tyrosine phosphorylation of Ugd, a UDP-glucose dehydrogenase. J Biol Chem 278: 39323–39329.
36
30.Grangeasse C, Cozzone AJ, Deutscher J, Mijakovie I. (2007) Tyrosine phosphorylation: an emerging regulatory device of bacterial
physiology. Trends Biochem Sci. 32(2):86-94
31.Griffith, C. L., Klutts, J. S., Zhang, L., Levery, S. B., and Doering, T. L. (2004) J. Biol. Chem 279, 51669–51676
32.Grimont PAD, Grimont F (2005) Genus Klebsiella. In: Brenner DJ, Krieg NR, Staley JT, eds. Bergey’s manual of Systematic
Bacteriology Volume 2: The Proteobacteria, Part B: The
Gammaproteobacteria. New York: Springer-Verlag. pp 685–693. 33.Guex,N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-
PdbViewer: an environment for comparative protein modeling.
Electrophoresis, 18, 2714-2723.
34.Hadizadeh Shirazy N., Ranjbar B., Hosseinkhani S., Khalifeh K., Riahi Madvar A., Naderi-Manesh H. (2007) Critical role of Glu175 on stability and folding of bacterial luciferase: stoppedflow fluorescence study. J Biochem Mol Biol 40:453–458.
35.Hammes G.G. (2005) Spectroscopy for the Biological Sciences. New York, USA: John Wiley & Sons, Inc.
36.Hung RJ, Chien HS, Lin RZ, Lin CT, Vatsyayan J, Peng HL, Chang HY. Comparative analysis of two UDP-glucose dehydrogenases in
Pseudomonas aeruginosa PAO1. J Biol Chem 282(24):17738-48.
37.Hunter T (2000) Signaling—2000 and beyond. Cell 100:113–127 38.Kelly S.M., Price N.C. (1997) The application of circular dichroism to
studies of protein folding and unfolding. Biochim Biophys Acta 1338:161–185.
37
39.Keen NT, Tamaki S, Kobayashi D, Trollinger D. (1998) Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria.
Gene 70(1):191-7
40.Kh. Tafreshi N., Hosseinkhani S., Sadeghizadeh M., Sadeghi M., Ranjbar B., Naderi-Manesh H. (2007) The influence of insertion of a critical residue (Arg356) in structure and bioluminescence spectra of firefly luciferase. J Biol Chem 282:8641– 8647.
41.Klein G, Dartigalongue C, Raina S (2003) Phosphorylation-mediated regulation of heat shock response in Escherichia coli. Mol Microbiol 48: 269–285.
42.Klumpp S, Krieglstein J (2002) Phosphorylation and
dephosphorylation of histidine residues in proteins. Eur J Biochem 269:1067–1071
43.Kohler JE, Hutchens MP, Sadow PM, Modi BP, Tavakkolizadeh A, et al. (2007) Klebsiella pneumoniae necrotizing fasciitis and septic
arthritis: an appearance in the Western hemisphere. Surg Infect (Larchmt) 8: 227–232.
44.Lacour S, Bechet E, Cozzone AJ, Mijakovic I, Grangeasse C. (2008) Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS One 3(8):e3053.
45.Lee DC, Zheng J, She YM, Jia Z (2008) Structure of Escherichia coli tyrosine kinase Etk reveals a novel activation mechanism. EMBO J. 46.Li, Zhi-Kai (2006) Functional analysis of the core elements, Wza,
38
biosynthesis in Klebsiella pneumoniae CG43. Master thesis,
Department of Biological Science and Technology, National Chiao Tung University.
47.Li, Mei-Ju (2008) Identification of the phosphotyrosine residues in UDP-glucose dehydrogenase of Klebsiella pneumoniae CG43. Master thesis, Department of Bioengineering, National Chiao Tung
University.
48.Lin MH, Hsu TL, Lin SY, Pan YJ, Jan JT, Wang JT, Khoo KH, Wu SH. (2009) Phosphoproteomics of Klebsiella pneumoniae NTUH-K2044 reveals a tight link between tyrosine phosphorylation and virulence.
Mol Cell Proteomics 8: 2613-2623
49.Lu CH, Chang WN, Chang HW (2002) Klebsiella meningitis in adults: clinical features, prognostic factors and therapeutic outcomes. J Clin
Neuro sci 9: 533–538.
50.Ma LC, Fang CT, Lee CZ, Shun CT, Wang JT (2005) Genomic heterogeneity in Klebsiella pneumoniae strains is associated with primary pyogenic liver abscess and metastatic infection. J Infect Dis 192: 117–128.
51.Macek B, Mijakovic I, Olsen J, Gnad F, Kumar C, et al. (2006)
Deciphering the prokaryotic Ser/Thre/Tyr phosphoproteome: the case of Bacillus subtilis. Seattle, WA, USA.
52.Macek B, Mijakovic I, Olsen JV, Gnad F, Kumar C, et al. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium
Bacillus subtilis. Mol Cell Proteomics 4: 697–707.