國 立 交 通 大 學
生 物 科 技 學 院
生 化 工 程 研 究 所
碩 士 論 文
以血管內皮生長因子受器競爭物抑制血管新生與
B16/F10 腫瘤生長之活體研究
Inhibition of Angiogenesis and Tumor Growth of B16/F10 with A Novel
VEGF Receptor Antagonist in vivo
研 究 生:陳昱丞
指導教授:廖光文
以血管內皮生長因子受器競爭物抑制血管新生與
B16/F10 腫瘤生長之活體研究
Inhibition of Angiogenesis and Tumor Growth of B16/F10 with A Novel
VEGF Receptor Antagonist in vivo
研 究 生:陳昱丞
Student:Yu-Cheng Chen
指導教授:廖光文
Advisor: Kuang-Wen Liao
國 立 交 通 大 學
生 物 科 技 系 生 化 工 程 所
碩 士 論 文
A Thesis
Submitted to Institute of Biochemical Engineering
Department of Biological Science and Technology
National Chiao Tung University
For the Degree of Master
in
July 2008
以血管內皮生長因子受器競爭物抑制血管新生與 B16/F10 腫瘤生長之活體研究 學生:陳昱丞 指導教授: 廖光文 博士 國立交通大學生物科技系生化工程所 中文摘要 血管新生成是腫瘤發育時的一個重要階段,此外血管新生也參與腫瘤由良性 轉為惡性的過程。血管內皮細胞生長因子 (Vascular endothelial growth factor, VEGF)是促進內皮細胞生長與血管新生的必要因子,在先前的實驗中,本實驗
室建構了一嵌合蛋白 RBDV-Ig (Receptor binding domain of VEGF-Immuno-
glibin):是以人類血管內皮細胞生長因子(VEGF)中第 1 至 108 個胺基酸序列做為
指標區域 (targeting domain),其具有與受器,血管內皮細胞生長因子受器 (VEGF Receptor),結合的能力;加上人類免疫球蛋白 G1 (Immunoglobin G1) 的 Fc 片段
做為作用區域,以延長嵌合蛋白的半衰期。RBDV-Ig 已被證明可於細胞培養模式
中藉由專一性地與人類臍帶靜脈內皮細胞 (human umbilical vein endothelial cells, HUVECs) 表面的血管內皮細胞生長因子受器結合而阻斷血管內皮生長因子的 訊息傳導途徑,達到抑制人類臍帶靜脈內皮細胞增生與形成管狀結構。本研究欲
探討於小鼠模式中,嵌合蛋白 RBDV-Ig 是否可藉由抑制血管新生達到抑制腫瘤
發 展 的 目 的 。 人 類 血 管 內 皮 細 胞 生 長 因 子 之 受 器 結 合 區 域 (Human VEGF
receptor binding domain, hRBDV) 與小鼠血管內皮細胞生長因子之受器結合區域 (Mouse VEGF receptor binding domain, mRBDV) 有 92%相似度,顯示 RBDV-Ig
有可能可與小鼠血管內皮細胞生長因子結合,且細胞實驗證明 RBDV-Ig 可與已 證實有表現小鼠血管內皮細胞生長因子受器 2 (mVEGFR2) 的 MS1 細胞結合。 此外,RBDV-Ig 可抑制 SVEC4-10 內皮細胞的增生。在 C57/B6 小黑鼠活體實驗 方面,皮下注射 RBDV-Ig 與靜脈注射 RBDV-Ig 皆可顯著抑制 B16/F10 腫瘤生 長。以上結果顯示,RBDV-Ig 可能可藉由抑制血管新生而抑制腫瘤生長,是為一 種有潛力的癌症治療用藥。
Inhibition of Angiogenesis and Tumor Growth of B16/F10 with A Novel VEGF Receptor Antagonist in vivo
Student: Yu-Cheng Chen Advisor: Kuang-Wen Liao Institute of Biochemical Engineering of National Chiao Tung University
ABSTRACT
Angiogenesis is not just an important stage in tumor growth but also involved in transforming a tumor from a benignancy to a malignant stage. Vascular endothelial growth factor (VEGF) is an essential factor in promoting endothelial cell growth and angiogenesis. In our previous experiments, we created a novel fusion protein, RBDV-Ig, which has a targeting domain, containing the amino acid sequences of VEGF from 1 to 108 with the binding activity to the human VEGF receptor, and an effector domain with Fc region of a human IgG1, used to increase a half-life of the fusion protein. RBDV-Ig has proven to inhibit tube-formation and proliferation through blocking the VEGF signal pathway by targeting the VEGF receptor on human umbilical vein endothelial cells (HUVECs) in vitro. Whether the RBDV-Ig chimeric protein can suppress tumor progression in vivo by anti-angiogenesis effect in the mouse model was verified in this study. The human VEGF receptor binding domain, RBDV is 92.7% similar to the mouse VEGF binding domain suggesting that the RBDV-Ig may bind to mouse VEGF receptors and later it was exactly proven to bind mouse VEGF receptor 2 (VEGFR2). It was also shown to bind to surface of mouse cells such as MS1 which have been proven with the VEGFR2 expression. Moreover, RBDV-Ig inhibits the proliferation of mouse SVEC4-10 endothelial cells in a dose-dependent way, but not of B16/F10. The results revealed that the tumor growths of B16/F10 tumors in C57/B6 mice were dramatically suppressed by subcutaneously injected RBDV-Ig. Tumor therapy with i.v. injection of RBDV-Ig also showed a similar result. Based on the results above, RBDV-Ig was shown to have the potent ability to inhibit tumor progression through suppression of angiogenesis and can be as a potential therapeutic drug in cancer therapy.
Acknowledgements 寢室裡的老舊電風扇咿咿啊啊響著,讓這漫漫夜晚不那麼靜謐,還留有一口 生氣。回想過去多少個夜晚,在冷房裡煎熬只為那肉眼難見的蛋白質。又有多少 個夜晚,我坐在電腦前處理實驗數據、結果與這本論文,對於那些日子的努力與 今日的成果,我由衷地感動,更感謝許多幫忙我、指導我完成這篇論文研究的人。 感謝我的指導教授,廖光文博士,在科學研究上所給予的指導,他不知教導 我實驗技術上的技巧,更重要的是,老師所傳授的科學精神引領我進入科學研究 的世界。除了在科學上的知識傳授,我也從老師身上學會許多待人接物的道理, 感謝老師為科學與教育的奉獻,使我在學業、研究或是與人相處上都獲益良多。 而四年來的實驗室生涯裡,我也要感謝實驗室中同甘共苦的每一個夥伴。感 謝靜宜學姐在這四年所給予的指導與鼓勵,偶爾情緒低落時,學姐都會耐心傾聽 並給予中肯的建議;感謝于鈴學姐幫我進行一次又一次的動物實驗,不厭其煩地 陪我一起測量、給藥與解剖;感謝彥谷學長熱心地整理、管理實驗室,讓我做實 驗無後顧之憂;感謝上知學長、詩涵學姊與弘育學長在實驗室草創時,一起努力 建置;感謝懷堯學長犀利地而逗趣的話語,讓人不經意地大笑;感謝韻如學姊三 不五時的聊天、關心我們的實驗狀況;感謝侑松學長建立起嵌合蛋白的生產與分 析系統,讓我的研究能穩定且順利地進行。而我更不會忘記對實驗有拼命三郎精 神的其翰同學、對乙醇與肉有正向趨性的依穎同學,以及對生命抱持著未雨綢繆 的態度的源庭同學,因為你們,讓許多的歡笑與淚水長存於我回憶裡。感謝何姵 學妹在實驗上的支援,家弘學弟的搞笑演出,莉沂學妹的冷笑話、立筠學妹的中 肯評論、筑婷學妹的辛苦交接,更感謝學弟妹們幫忙打點口試時的茶點。 除此之外,我要感謝一群幫我分憂解勞的朋友們,感謝冠賢與華妏聽我碎碎 念,感謝室友振富的理性分析、宜君不定期的關心、庭瑋提供暑宿,因為你們的 關心讓我更能專注於自己的實驗。還有許多陪伴我的好朋友們,因為你們讓我總 能保持樂觀進取的態度,在研究的路上前進。 最後我要感謝我親愛的家人,感謝父母養育我、同理我、關心我,在我實驗 失敗時給予我溫馨的打氣,讓我得以完成研究及這本論文;感謝姐姐看重我、鼓 勵我,使我即使遇到實驗不順利,仍能保有前進的動力。更感謝鈺珊時時刻刻的 陪伴,分擔實驗失敗的悲傷、分享實驗成功的喜悅,提供我撰寫論文的工具書, 並幫忙指導、訂正論文裡的錯字、文法,使我的論文臻至完整。因為有你們適時 地關懷,讓我的研究與論文得以順利完成。 碩士生涯將告一段落,回顧那些辛苦的日子,換得今日小小的結果,讓人不 自覺地驚呼:「摁,我畢業了!」 昱丞 戊子 仲夏夜
CONTENTS
Abstract in Chinese i
Abstract ii
Acknowledgements iii
Contents iv
List of figures vii
Abbreviations viii
Chapter 1 Introduction
1.1 Tumor associated angiogenesis...1
1.2 Angiogenic factor...1
1.2.1 TGF-β...1
1.2.2 Interleukin-8...2
1.2.3 Basic Fibroblast Growth Factor...3
1.2.4 Platelet-Derived Endothelial Cell Growth Factor...4
1.2.5 Vascular Endothelial Growth Factor...4
1.2.5.1 Activities of VEGF...5
1.2.5.2 Tumor associated VEGF...5
1.2.5.3 VEGF gene and isoforms...6
1.2.5.4 Regulation of VEGF gene expression...8
1.3 VEGF Receptors...9
1.3.1 VEGFR-1...10
1.3.2 VEGFR-2...12
1.3.3 VEGFR-3 and Neuropilin...13
1.4 Negative regulators of angiogenesis and angiogenesis inhibitor...13
1.5 Anti-angiogenesis therapy through inhibiting VEGF signaling...15
1.5.1 Small molecules...15
1.5.2 Anti-VEGF antibody...16
1.5.3 Anti-VEGF receptors antibody...17
1.5.5 Peptides...18
1.5.6 Others...19
1.6 Research rationale and strategy...20
Chapter 2 Material and Methods
2.1 Material...222.1.1 Plasmid...22
2.1.2 Cell lines...22
2.1.3 Mice...22
2.2 Method...24
2.2.1 Amplification of pAAV-MCS/IgG1 Fc and pAAV-MCS /RBDV-IgG1 Fc.24 2.2.1.1 Transformation...24
2.2.1.2 Midi-preparation...24
2.2.1.3 Restriction enzyme digestion...26
2.2.2 Expression of chimeric proteins...26
2.2.2.1 Transfection of HEK-293T cell...26
2.2.2.2 Expression and purification of chimeric proteins...27
2.2.2.3 SDS-PAGE and Western blot...28
2.2.2.4 Stripping of Nitrocellulose membrane...29
2.2.3 Receptor binding assay in vitro...29
2.2.3.1 Biotin label of proteins...29
2.2.3.2 ELISA...29
2.2.4 Cell surface binding assay...30
2.2.5 SVEC 4-10 and B16/F10 cells proliferation assay...31
2.2.6 Tube formation assay...32
2.2.7 Cell migration assay...32
2.2.8 In situ tumor therapy...33
2.2.8.1 H&E staining...33
2.2.8.2 IHC staining...34
2.2.9 Tumor therapy with i.v. injection...35
Chapter 3 Results
3.1 Compare of receptor binding domain of VEGF between human and mouse...36
3.2 Expression and purification of chimeric proteins, IgG1 Fc and RBDV-IgG Fc....36
3.3 The activity of RBDV-IgG1 Fc binding to mouse VEGF receptor...38
3.4 The activity of RBDV-IgG1 Fc binding to cells surface...39
3.5 The suppressive potency and efficacy of RBDV-IgG1 Fc to SVEC4-10 cells proliferation in vitro...39
3.6 Effect of blockade of VEGF receptors on tube formation in vitro...41
3.7 RBDV-Ig Fc inhibited the endothelial cell migration in a Transwell system...42
3.8 In vivo suppression of tumor growth with in situ RBDV-Ig Fc treatment...42
3.9 RBDV-Ig Fc-mediated in vivo therapy with i.v. injection of mice bearing B16/F10 tumors...43
Chapter 4 Discussion
...45References...80
List of figures
Figure 1. Role of the VEGF receptors...52
Figure 2. Sequence alignment of human and mouse receptor binding domain of VEGF...53
Figure 3. Gene construction of the chimeric proteins...54
Figure 4. Restriction enzyme digestion of the pAAV-MCS/IgG1 Fc and pAAV-MCS/RBDV-IgG1 Fc...55
Figure 5. The fluorescence expression in HEK-293 cell...56
Figure 6. Flow chart of procedure for the purification of chimeric proteins...57
Figure 7. SDS-PAGE analysis of recombinatant proteins...58
Figure 8. Characterization of purified chimeric proteins...59
Figure 9. Mouse VEGF Receptor 1 binding activities of purified RBDV IgG1 Fc....60
Figure 10. Mouse VEGF Receptor 2 binding activities of purified RBDV IgG1 Fc..61
Figure 11. Cell surface binding ability...62
Figure 12. RBDV-IgG1 Fc inhibits the VEGF-induced proliferation of SVEC4-10 cells in a dose-dependent manner...64
Figure 13. RBDV-IgG1 Fc inhibits the VEGF-induced proliferation of SVEC4-10 cells, but not of B16/F10 cells...65
Figure 14. Effect of RBDV-IgG1 Fc on in vitro tube formation, 100X magnification...66
Figure 15. Effect of RBDV-IgG1 Fc on in vitro cell migration, 100X magnification...67
Figure 16. In vivo suppression of tumor growth with in situ RBDV-Ig Fc treatment...68
Figure 17. Subcutaneous vascularizing of mouse dorsum...69
Figure 18. H&E staining of tumor sections, 200X magnification...70
Figure 19. In vivo tumor therapy with iv. injection of RBDV-Ig Fc...72
Figure 20.Survival rate of tumor therapy with iv. injection of RBDV-Ig Fc in vivo...73
Figure 21. H&E staining of kidney sections, 200X magnification...74
Figure 22. H&E staining of spleen sections, 400X magnification...76
List of abbreviations
ADCC Antibody dependent cell-mediated cytotoxicity
AMD Age-related macular degeneration
aFGF Acidic fibroblast growth factor
bFGF Basic fibroblast growth factor
CDC Cell dependent cytotoxicity
Cys Cysteine
ECM Extracellular matrix
EGF Endothelial cell growth factor
FGFR Fibroblast growth factor receptor
Flk Fetal liver kinase
Flt Fms-like tyrosine kinase
HIF-1 Hypoxia induicible factor-1
IFN-α Interferon alpha
Ig Immunoglobulin
IL-8 Interleukin 8
kDa Kilo dalton
KDR Kinase domain receptor
KO Knockout
NK Natural killer cell
NRP Neuropilin
NSCLC Non-small-cell lung cancer
PDGF Platelet-Derived Endothelial Cell Growth Factor
PDGFR-β Platelet-derived growth factor receptor-beta
PlGF Placenta growth factors
PI3 kinase Phosphotidylinositol kinase 3 kinase
RTK Receptor tyrosine kinase
RBDV Receptor binding domain of VEGF
RM Room temperature
sFlt-1 Soluble fms-like tyrosine kinase-1
TGF-β Transforming growth factor-beta
TNF-α Tumor necrosis factor-alpha
VEGF Vascular endothelial growth factor
Chapter 1 Introduction
1.1 Tumor associated angiogenesis
The history of tumor angiogenesis research can be traced back to over two centuries ago, pathologists in Germany observed that tumors are highly vascularized [1]. The development of tumor growth requires angiogenesis, the formation of new blood vessels [2]. In normal tissue, the blood vessels do not increase in size or number, because the endothelial cells that line theses narrow tubes do not divide [3]. In the tumor region, endothelial cells are abnormal and including multi-layer morphology, extension of bridging and splitting vessels, uncontrolled permeability, in addition, the endothelial cells undergo constant remodeling [4]. The angiogenic process is stimulated in part by the tumor cells and the complex microenvironment consisting of many different cell types [1, 5, 6].
1.2 Angiogenic factor
Some evidence shows that tumor cells are triggered to secrete several pro-angiogeneic molecules under a hypoxia environment, like TGF-β, VEGF, PDGF, bFGF, and IL-8 [1, 7, 8]. Moreover, these pro-angioneneic factors increase angiogenesis of endothelial cells has been associated with tumor growth in several human cancers [9-11].
1.2.1 TGF-β
Transforming growth factor (TGF)-β is a multifunctional protein that initiates its diverse cellular responses by binding to and activating its receptor, type I and type II serine/threonine kinase receptors. Although TGF-β is well known as an immunosuppressive growth factor [12], the importance of the signaling pathway in angiogenesis and vascular remodeling has been also highlighted during numerous recent studies [13].
In the studies of knockout (KO) mice and embryos, TGF- β and ΤβRII are critical for both formation of the primary vascular plexus (vasculogenesis) and the subsequent extension and remodeling into a complex network (angiogenesis) [14]. Furthermore, TGF- β was observed to be able to enhance the expression of VEGF, which could cause the excessive growth of endothelial cells. [15]
1.2.2 Interleukin-8
The chemokine interleukin-8 (IL-8), originally discovered as a chemotactic factor for leukocytes, has been shown to contribute to human cancer progression through its potential function as a mitogenic, angiogenic and motogenic factor [16]. IL-8 expression is regulated by the tumor microenvironment; tumor hypoxia and acid environment increase expression of IL-8. IL-8 may support tumor growth by direct or
indirect induction of angiogenesis. In a study of colon cancer, there was a strong correlation between constitutive expression of IL-8 and its receptors, CXCR1 and CXCR2, and increasing angiogenesis and metastasis [17]. In clinical studies, serum levels of IL-8 were significantly higher in human cancers, which was highly angiogenic and metastatic type [18, 19]. Thus, IL-8 may contribute to tumor progression and angiogenesis in several tumor types.
1.2.3 Basic Fibroblast Growth Factor
Basic fibroblast growth factor (bFGF) was a member of fibroblast growth factor and also named as FGF-2. FGFs are expressed in almost all tissues and play important roles in a variety of normal and pathological processes, including development, wound healing, and neoplastic transformation [20]. Moreover, bFGF was reported as a mitogen of endothelial cells, and promoter of tumor angiogenesis [21].
bFGF preferentially associates with the FGF recptor-3 (FGFR-3), which was also implicated in the developmentof a number of malignant tumors such as melanoma [22] orglioma [23]. FGFR-3 was expressed not only in endothelial cells but also tumor cells. bFGF and its receptor were reported to enhance the proliferation, migration, and angiogenesis of these cells [20]. Furthermore, antisense targeting of bFGF or its receptor in human melanoma inhibits tumor growth [22].
1.2.4 Platelet-Derived Endothelial Cell Growth Factor
Platelet-derived endothelial cell growth factor (PDGF), also known as thymidine phosphorylase, is another tumor angiogenic factor. In several cancer systems, PDGF has chemotactic activity for endothelial cells in vitro and angiogenic activity in vivo [24]. PDGF strongly induces neovascularization in the rat sponge model, and PDGF–transfected breast carcinoma cells exhibit accelerated growth in xenografts in mice [25]. In one study, Maeda and colleagues immunostained gastric cancer specimens for PDGF and microvessels and found a significantly higher microvessel density in tumors that expressed PDGF [26].
1.2.5 Vascular Endothelial Growth Factor
Vascular endothelial growth factor was first reported in 1989, and isolated a endothelial cell specific mitogen from medium condition by bovine pituitary follicular cells [27]. VEGF is potent, diffusible, and specific for vascular endothelial cells led to the hypothesis that this factor might play a role in the regulation of physiological and pathological growth of blood vessels. The role of VEGF in the regulation of angiogenesis has been investigating in two decades ago [21]. According to published literature, it has been indicates that new vessel growth are highly complex and
coordinated processes, requiring a serial of ligands binding to numerous receptors, which led to the proliferation, migration, and angiogenesis of endothelial cells [28].
1.2.5.1 Activities of VEGF
A well known in vitro activity of VEGF is a survival factor for endothelial cells derived from arteries, veins, and lymphatic. VEGF prevents endothelial apoptosis induced by serum starvation, which subsequently is mediated by the phosphatidy- linositol 3-kinase (PI3 kinase)/Akt pathway [29]. Moreover, VEGF could induce expression of the anti-apoptotic proteins Bcl-2 and A1 in endothelial cells [30].
VEGF induces a potent angiogenic response in a variety of in vivo models and angiogenesis in tridimensional in vitro models, inducing confluent microvascular endothelial cells to invade collagen gels and form capillary-like structures [31, 32]. Moreover, VEGF elicits a pronounced angiogenesis response in a variety of in vitro models including the matrigel plug in mice [33].
1.2.5.2 Tumor associated VEGF
In contrast to normal vessels, vessels in solid tumors are often abnormally enlarged, and blood flow in tumor vessels is often chaotic, slow and not efficient in meeting metabolic demands [34]. Furthermore, tumor cells usually represent the main source
of VEGF, though several studies have shown that tumor-associated stroma is also a site of VEGF production [35, 36]. Many tumor cell lines particularly secrete VEGF in
vitro, suggesting the possibility that this diffusible molecule may be a mediator of
tumor angiogenesis [37]. In situ hybridization studies have demonstrated that VEGF mRNA is expressed in many tumors, including lung, breast, gastrointestinal tract, renal, and ovarian carcinomas [32].
1.2.5.3 VEGF gene and isoforms
The VEGF-related gene family of angiogenic and lymphangiogenic growth factors comprises five secreted glycol- proteins referred to as VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placenta growth factors (PlGF) [38]. VEGF-A is the beststudied, has been most strongly associated with angiogenesis,and is the target of most current anti-VEGF treatments. VEGF-A signals through two receptor tyrosine kinases, VEGFR1and VEGFR2, and is the only member of the VEGF gene family foundto be induced by hypoxia [39]. VEGF-B selectively binds to VEGFR1and has a role in the regulation of extracellular matrix degradation,cell adhesion and migration [40]. Both VEGF-C and VEGF-D bind to VEGFR-2 and VEGFR-3 and regulate lymph- angiogenesis, and VEGF-C may alsobe involved in wound healing [41, 42]. PlGF selectively binds to VEGFR-1 and is the most abundantly expressed VEGF family
member in endothelial cells. PlGF may potentiate VEGF-A-induced endothelialcell proliferation, but on its own PlGF exerts only weak mitogenicity [39].
VEGF-A is a major angiogenic factor of the VEGF family, and is an important survival factor for endothelial cells, both in vitro and in vivo. VEGF-A was purified to be sequenced and cloned by Ferrara and collaborates. Alternative exon splicing of human VEGFs gene shows that it is comprised eight exons , denoted as: VEGFA121, VEGFA145, VEGFA165, VEGF165b, VEGFA189 and VEGFA206, gives rise to isoforms with different length of amino acids, and biological activities [43]. The properties of native VEGF closely correspond to those of VEGF165. VEGF121 is an acidic polypeptide that does not bind to heparin. VEGF189 and VEGF206 are highly basic and bind to heparin with high affinity. Whereas VEGF121 is a freely diffusible protein, VEGF189 and VEGF206 are almost completely sequestered in the extracellular matrix (ECM) [38].
VEGFA165 is a heparin-binding homodimeric glycoprotein of 46 kDa. Structurally, VEGFAs with intra-domain and inter-domain disulfide bonds between eight cysteine residues conserved positions. Anti-parrallel homodimer covalently linked by two disulfide bridges between Cys-51 and Cys-60 [44]. VEGFA165 is secreted, but significant fraction remains bound to the cell surface and extracellular matrix (ECM), by virtue of its heparin-binding properties. In the present studies,
VEGFA and its receptors is the best characterized signaling pathway in developmental angiogenesis. Furthermore, much research has also established the VEGFA in tumor angiogenesis, and VEGFA action constitutes a rate-limiting step in normal and pathological blood vessel growth [38].
1.2.5.4 Regulation of VEGF gene expression
VEGF gene expression is up-regulated by hypoxia. Hypoxia allows the stabilization of hypoxia-inducible factors 1 (HIF-1) that binds to specific promoter elements that are present in the promoter region of VEGFA. This region is a 28-base sequence in the 5’- promoter of human VEGF gene, which mediates hypoxia-induced transcription [45]. Importantly, another study has implicated the PI3 kinase/Akt pathway in the regulation of HIF-mediated responses in a hypoxia-independent manner. Mutations in Akt also results in increased activation of HIF-1 and increased VEGF transcription [46]. Specific transforming events also result in induction of VEGF gene expression. Oncogenic mutations or amplification of Ras leads to VEGF up-regulation, which indicates that mutant Ras-dependent VEGF expression is necessary for progressive tumor growth in vivo [47]. several major growth factors, including epidermal growth factor (EGF), TGF-α, TGF-β, FGF and PDGF, also up-regulate VEGF mRNA expression [48], suggesting that paracrine or autocrine release of such factors
cooperates with local hypoxia in regulating VEGF release in the microenvironment. Furthermore, inflammatory cytokines such as IL-8 also induce expression of VEGF in several cell types in agreement with the hypothesis that VEGF may be a mediator of angiogenesis in inflammatory disorders[49].
1.3 VEGF Receptors
VEGF binding sites were identified on the cell surface of vascular endothelial cells in
vitro and in vivo. Subsequently, it became apparent that receptors for VEGF also
occur on bone marrow-derived cells [50]. VEGF binds two highly related receptor tyrosine kinases (RTK), VEGFR-1 and VEGFR-2. Both VEGFR-1 and VEGFR-2 have seven immunoglobulin-like (Ig-like) domains in the extracellualar domain, a single transmembrane region, and a consensus tyrosine kinase sequence [51, 52]. VEGFR-3 (fms-like-tyrosine kinase (Flt)-4) is a member of the same family of RTKs, but is not a receptor for VEGF, binding instead to VEGF-C and VEGF-D [53]. In addition to these RTKs, VEGF interacts with a family of coreceptors, the neuropilins (NRP).
VEGFRs share similar regulatory mechanisms with well-characterized receptor tyrosine kinases, by which include receptor dimerization and activation of the tyrosine kinase. Moreover, VEGFRs perform cellular processes that are common to many
growth factor receptors such as cell survival and proliferation. The summary of VEGFRs shows in Figure 1, VEGF-A binds to VEGFR-1 and VEGFR-2, which are expressed in the cell surface of most blood ECs. In contrast, PLGF and VEGFB interact only with VEGFR-1. VEGFC and VEGFD bind VEGFR-2 and VEGFR-3, which is largely restricted to lymphatic EC. There is much evidence that VEGFR-2 is the major mediator of EC mitogenesis and survival, as well as angiogenesis and micro-vascular permeability. VEGFR-1 has an established signaling role in mediating monocyte chemotaxis. Also, in hematopoietic stem cells (HSC) or leukemic cells, both VEGFR-1 and VEGFR-2 may mediate a chemotactic and a survival signal [43].
1.3.1 VEGFR-1
VEGFR-1 or Flt-1 (fms-like-tyrosine kinase-1) was the first discovery of VEGF receptor [54]. The VEGFR-1 tyrosine kinase exhibits all the conserved motifs that are required for kinase activity. The crystal structure of part of the VEGFR-1 extracellular domain shows that the Ig domain-2 is the major ligand binding site on the receptor in physiology and pathology. VEGFR-1 expression is unregulated by hypoxia by a HIF-1 dependent mechanism [55].VEGFR-1 binds not only VEGF-A but also PLGF and VEGF-B [40, 56]. The crucial role of VEGFR-1, as mentioned above, was determined by Fong, 1995, which revealed that when disruption of VEGFR-1 gene in
mice resulted in embryonic lethality.
In addition to the full length of VEGFR-1, there is an alternatively spliced soluble form of VEGFR-1 (sFlt-1), which has been shown to be an inhibitor of VEGF activity [57]. Hence, not only the full length membrane bound form of VEGFR-1 but sFlt-1 as well could perform a decoy function, which sequesters VEGF and prevent its interaction with VEGFR-2 [58].
In some cases, VEGFR-1 is expressed by tumor cells and probably mediates a chemotatic signal, thus potentially extending the role of this receptor in cancer growth. For instance, Wu and collaborates indicated that VEGF-A autocrine growth activity is acquired by certain human breast tumor cell lines defined by expression of VEGFR-1 [59].
Anchoring of the extracellular domain of VEGFR-1 to the cell membrane is important, as 50% of the mice that lack both of the tyrosine kinase domains and the transmembrane domain died at embryonic stage, owing to vascular malformation [60]. This study indicates that endothelial cells develop but fails to organize in vascular channels. Excessive proliferation of angioblasts has been reported to be responsible for such disorganization and leathality, indicating that, at least during early development, VEGFR-1 is a negative regulator of VEGF action [61]. By contrarily, many evidence also indicated that VEGFR-1 is a positive regulator of monocyte and
macrophage migration [62, 63].
1.3.2 VEGFR-2
At the same time VEGFR-1 was discovered, Shalaby et al. found VEGFR-2, another receptor for VEGF, and proposed that VEGFR-2 had similar characteristics and continuously shows today. Biochemical analyses showed that the second and third Ig-like domains in VEGFR-2, also known as kinase domain region (KDR), is important for the determination of ligand binding specificity for VEGF [64]. VEGFR-2 undergoes RTK dimerization and strong ligand-dependent tyrosine phosphorylation in intact cells.
There are many studies indicating that VEGFR-2 is the major mediator of the mitogenesis, survival, and permeability enhancing effects of VEGF-A in endothelial cells [32]. VEGF binds to VEGFR-2 and stimulates activation of Ras in HUVECs. Besides, Ras activation has been coupled to an angiogenic phenotype of endothelial cells [65]. The early finding that the binding of VEGF to VEGFR-2 is enhanced by heparin has been confirmed by recent studies, which shows that heparin amplifies signalling by VEGF. Byzova et al. have reported that VEGFR-2 activation by VEGF results in PI3 kinase/Akt-dependent activation of several integrins, which indicates that VEGF enhanced cell adhesion, migration, soluble ligand binding [66]. Moreover,
VEGFR-2 activation has been shown to be required for the antiapoptotic effects of VEGF for human umbilical vein endothelial cells (HUVECs) [29].
1.3.3 VEGFR-3 and Neuropilin
VEGFR-1 and VEGFR-2 are expressed on the surface of blood endothelial cells. By contrast, VEGFR-3 (Flt-4) is largely restricted to lymphatic endothelial cells. In recent studies, VEGFR-3 is important for lymphatic endothelial cell development and function [53].
Neuropilin-1 (NRP1), as its name suggests, is a molecular that is implicated in neuronal guidelince and had been previously shown to bind the collapsin/semaphorin family. NRP1 is a cell surface glycoprotein that lacks intrinsic catalytic activity, a receptor for the heparin-binding isoforms of VEGF, and seems to present VEGF165 to VEGFR-2 in a manner that potentiates VEGFR-2 signaling [67]. This result shows that neuropilin stabilizes the VEGF/VEGFR-2 signaling complex when expressed on adjacent cells.
1.4 Negative regulators of angiogenesis and angiogenesis inhibitor
In a normal physiological condition, angiogenesis is a regulated process and seems to be under the control of both positive and negative regulatory factors. Several
fragments of large proteins have been discovered as endogenous inhibitor of angiogenesis; i.e, thrombospondin, endostatin, tumstatin and vasostatin. [68-71], but relatively little is known about their role in the physiological regulation of angiogenesis. The precise mechanism of action of these proteins remains to be more clearly defined, although several hypotheses have been proposed [72].
The similar concept was also used in cancer therapy. In 1971, Folkman proposed that inhibition of angiogenesis was a strategy to treat cancer, and ever since then it initiates the inhibitors that associated with angiogenesis. In 1989, interferon alpha (IFN-α), the first clinical trial of an antiangiogenic agent which decreases production of the angiogenic protein FGF made by tumors, was began for the treatment of life-theatening hemangioma. Furthermore, The United States Food and Drug Administration (FDA) approved TNP-470, a low molecular weight agent which selectively inhibits proliferation and migration of endothelial cells, to be used in clinical trials for a wide variety of cancers in clinics. From then on, there are many strategies toward this field, for example, inhibition of the influx of calcium into cells to suppress proliferation of endothelial cells [73], or inducement of inflammation in tumors, which destroying growing capillaries [74].
Antiangiogenesis therapy does not directly target tumors. Instead, it interferes with the expanding network of blood vessels and attacks blood supply in tumors to
suppress proliferation, and angiogenesis. So far, many angiogenesis inhibitors are proved successfully in vivo and in clinical trials, inhibitors based on anti-VEGF were major.
1.5 Anti-angiogenesis therapy through inhibiting VEGF signaling
VEGF is now general considered a central in the process of angiogenesis. Apart from the molecular interactions that are discussed above, several different strategies have been designed to target VEGF/VEGFR signal transduction, including small molecules inhibiting VEGFR signal transduction, humanized anti-VEGF monoclonal antibody, anti-VEGFR-1 antibody, anti-VEGFR-2 antibody, and a VEGFR chimeric protein [75].
1.5.1 Small molecules
Lots of small-molecule receptor tyrosine kinase inhibitors targeting the VEGF receptors have been developed. Such as SU5416 [76], ZD4190 [77], and PTK787/ZK2284 [78] are all small molecules as ATP mimics that target VEGFR-2 tyrosine kinase and have been shown to inhibit angiogenesis and tumorigenesis in animal models. The most advanced are BAY43-9006 and SU11248 [79]. BAY43-9006, also named Sorafenib, is a multi-targeted kinase inhibitor, including
VEGFR-2, PDGFR-β, Flt-3 and c-kit, which have been tested in large phase III clinical trials for treatment of metastatic kidney cancer. The FDA-approved SU11248, which named as Sunitinib, also targets multiple kinases, which inhibits VEGFRs, PDGFR, c-kit and Flt-3, for treatment of gastro-intestinal stoma tumor.
1.5.2 Anti-VEGF antibody
Tumor growth inhibition has been demonstrated by numerous laboratories using many anti-VEGF approaches. One of these is inhibiting angiogenesis by treatment with antibodies against VEGF. This reported anti-VEGF monoclonal antibodies exert a potent inhibitory effect on the growth of several tumor cells lines in nude mice, whereas the antibody had no effect on the tumor cells in vitro [80]. Subsequently, many other tumor cells lines were found to be inhibited in vivo by anti-VEGF monoclonal antibodies [81, 82] .
One of the most well-known monoclonal antibody to VEGF is bevacizumab (Avastin) which was a humanized antibody and approved for the treatment of metastatic colorectal cancer by US Food and Drug Administration (FDA) [83]. Furthermore, bevacizumab has applicability to other tumor types, such as non-small-cell lung cancer (NSCLC), prostate cancer, renal cell cancer [84]. Willett and his colleagues had shown that VEGF blockade with bevacizumab decreases
tumor perfusion, vascular volume, microvascular density, interstitial fluid pressure and the number of viable circulating endothelial and progenitor cells in colorectal cancer patients [85]. The strategy of anti-VEGF acts by blocking tumor associated angiogenesis, which appears to be widely required by many different types of tumors. Therefore, this strategy may be usefully against different types of cancer.
1.5.3 Anti-VEGF receptors antibody
As anti-VEGF antibody, anti-VEGF receptors antibody was produced to inhibit angiogenesis through VEGF signaling transduction. Several anti-VEGFR-2 inhibitors are being clinically pursued. In 1999, Prewett and colleagues reported that anti-Flk (fetal liver kinase 1, VEGFR-2 in mouse) monoclonal antibody, and results showed that blockade of the Flk-1 receptor by systemic administration of the monoclonal antibody inhibits angiogenesis in animal models and the growth of several mouse and human tumors [86]. Skobe et al. have shown previously in a malignant keratinocyte invasion model that anti-Flk-1 monoclonal antibody treatment inhibits endothelial cell proliferation and induces endothelial cell apoptosis that leads to vessel regression. Furthermore, the use of anti-VEGFR-2 therapy in combination with conventional chemotherapy, radiotherapy or immunotherapy may improve the efficacy of anti-VEGFR-2 antibody [87].
1.5.4 Soluble VEGF receptors
Initial attempts to block VEGF by using a bevacizumab are beginning to show promise in human cancer patients, underscoring the importance of targeting VEGF pathway. Therefore, this clinical promise of humanized monoclonal antibody highlights the need to optimize blockade of this pathway. In 2002, Holash and collaborates engineered a soluble VEGF receptor, VEGF-Trap. The parental VEGF-Trap was created by fusing the first three Ig domains of VEGFR-1 to the Fc region of human IgG. Also, their variant, a chimeric soluble receptor consisting of domain 2 fused with domain 3 of VEGFR-2, was also created to determine the requirements to maintain high affinity while extending in vivo half-life [88]. Furthermore, it has been proposed that this fusion between the Fc region of human IgG1 and the Ig domain of VEGFRs represent an advantage over antibodies because they can result in higher binding affinity [89]. Luckily, VEGF-trap is also undergoing clinical development as an anti-cancer agent and is in Phase II/III trials for ovarian cancer.
1.5.5 Peptides
antagonist of VEGF to its receptor [90-92]. Besides, a cyclic peptide corresponding to amino acids 79-93 of the VEGF sequence was reported to inhibit angiogenesis [93]. Moreover, the chemotherapeutic drugs combined with some small peptides were used currently cancer therapy. Peptide-mediated delivery of the drugs selectively to tumor and tumor associated neo-vessels [94].
1.5.6 Others
Another strategy to antagonist VEGF signaling transduction is aptamers. Aptamers were nucleic acids that can be highly potent antagonists of enzyme catalysis or of specific protein-protein interactions. In 1998, Ruckman proposed that an aptamer that interacts with the heparin-binding domain of VEGFA165. They have already isolated and modified 2’F-pyrimidine RNA nucleotide ligands by using the SELEX (systematic evolution of ligands by exponential enrichment) process. The results show that the binding ability of these aptamers to human VEGFA165 as well as murine VEGF164. Also, these aptamers potently inhibit the binding of VEGF to the human VEGF receptors, VEGFR-1 and VEGFR-2, expressed by transfected porcine aortic endothelial cells. Successfully, this VEGF-neutralizing aptamer, pegaptanib or Mucugen, became the anti-VEGF inhibitor to be approved by the FDA for the treatment of age-related macular degeneration (AMD), which is an effective at
slowing vision loss.
1.6 Research rationale and strategy
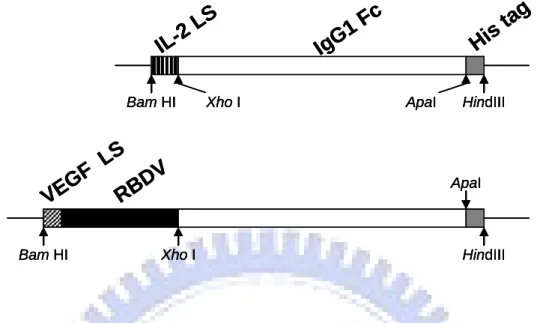
Here we report a novel antibody-like molecule, RBDV-IgG1 Fc chimeric proteins, constructed by fusing the receptor binding domain of VEGF (RBDV) with the constant region of human IgG1.
VEGF is an important growth factor of promoting specific mitogen and formation of blood vessels in pathological angiogenesis of tumor growth. The biological properties make VEGF as an important therapeutic target, and it has been shown that anti-VEGF signal pathway can inhibit tumor growth in vivo. At molecular level, VEGF activity is mediated by its interaction with two distinct receptors, VEGFR-1 and VEGFR-2. Charge reversal and alanine scanning mutagenesis have allowed identification of the receptor-binding domain of VEGF for VEGFR-2, which amino acids lined between 8 and 109 of full VEGFA165 [44]. Most importantly, the loss of the heparin-binding domain, the amino acids 111-165, results in a reduction in the mitogenic activity of VEGF [95].
The IgG1 subclass was selected because it is arguably the most active in the immune systems, being able to engage receptors on cytotoxic effective cells, called FcγR, and to activate complement and destroy target cells, effectively [96]. The Fc
component contains the hinge region, CH2 domain, and CH3 domain, but not the CH1 domain of IgG. The hinge and Fc regions form a conventional scaffold and flexibility for presenting functional RBDV domain. Furthermore, Fc provides a ready means for detection of the expression of chimeric receptors.
We expected RBDV as an antagonist to suppress VEGF signaling, and addition of constant region of human IgG1 could promote structure and stability of the fusion protein [97]. In previous data, RBDV-IgG1 Fc can bind to human VEGF receptors, VEGFR-1 and VEGFR-2, and be as an antagonist that inhibits the physiological interactions between VEGF to its receptors in vitro. Moreover, we determined that RBDV-IgG1 Fc could also be effective at inhibiting tumorigenesis and angiogenesis of B16/F10 melanoma cells inoculated into immunocompetent mice in vivo.
Chapter 2 Material and Methods
2.1 Material
2.1.1 Plasmid
PAAV-MCS/IgG1 Fc and pAAV-MCS /RBDV-IgG1 Fc were constructed by Yo-shong Chung and the map and full sequence of two plasmids are in the Appendix1.
2.1.2 Cell lines
Human epithelial kidney (HEK) 293T cells, mouse melanoma cells (B16/F10), and mouse vascular epithelium cells (SVEC4-10) were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Gaithersburg, MD) containing 10% FBS (Fetal bovine serum qualified; Invitrogen, Gaithersburg, MD) and 1% penicillin- streptomycin amphotericin B (PSA; Biological industries, USA). Mouse endothelial cells (MS1) were cultured in DMEM supplemented with 5% FBS and 1% PSA. All the cells were incubated in tissue culture incubator with 5% CO2 at 37℃. Except for SVEC4-10 was obtained from Dr. Tsai Nu-man, other cells were obtained from BCRC (Food Industry and Development Institute, Taiwan).
2.1.3 Mice
Taiwan University Laboratory Animal Center (Taipei, Taiwan, R.O.C.) and housed in the Animal Maintenance Facility of National Chiao Tung University (Hsin-chu, Taiwan, R.O.C.).
2.2 Method
2.2.1 Amplification of pAAV-MCS/IgG1 Fc and pAAV-MCS /RBDV-IgG1 Fc
2.2.1.1 Transformation
The competent cell, E. coli Top 10 strain (Invitrogen, USA), was used for transformation. Each plasmid was diluted with sterile water to 1µg/ml. One vial of stored competent cells (100 µl) was thawed and kept on ice. One µl of the diluted plasmid was first mixed into each vial containing the competent cells by stirring and then incubated for 30 minutes on ice. Then, the vials were heat shocked for exactly 90 seconds in a 42°C water-bath and placed on ice for additional 2 min. 250 µl of LB broth medium (1% tryptone, 0.5% yeast extract, 1% NaCl) were added to each tube and the vials were incubated at 37°C for 1 hour at 230 rpm shaking. The vials with the transformed cells were placed on ice after shaking. 100 µl of each transformation were spread out evenly on the agar plates with ampicillin (50µg/ml). Plates were then inverted and incubated at 37°C for 16 hours.
2.2.1.2 Midi-preparation
For further midi-preparation experiments, single colony was picked from each transformation, placed into 100 ml of LB broth medium containing ampicillin (50µg/ml) and incubated at 37°C for 20 hours at 230 rpm shaking incubator. Then, LB
broth medium with bacteria were centrifuged at 8,000 rpm for 15 min, the supernatant was discarded, and the bacterial pellet was resuspended in 4 ml of Buffer S1 (50 mM Tris-HCl, 10mM EDTA, 100μg/ml RNase A, pH8.0). Four ml of Buffer S2 lysis buffer (200 mM NaOH, 1% SDS) was added, the sample was gently mixed by inverting 6-8 times, and the bacteria were allowed to lyse for 3 min at room temperature. Four ml of ice-cold Buffer S3 neutralization solution (2.8 M KAc, pH 5.1) was added. The sample was gently mixed by inverting 6-8 times, before being incubated on ice for 5 min. Then, the mixture was centrifuged at 10,000 rpm for 30 min at 4°C. The clear solution was applied onto NucleoBond ion-exchange resin, which was pre-equilibrated with 5 ml Buffer N2 (100 mM Tris base, 15% ethanol, 900 mM KCl, 0.15% Triton X100, adjusted to pH 6.3 with H3PO4). The column was washed 2 times with 10 ml Buffer N3 (100 mM Tris, 15% ethanol, 1M KCl, adjusted to pH6.3 with H3PO4). Plasmid DNA was eluted from the column with 5 ml high-salt Buffer N5 (100 mM Tris base, 15% ethanol, 1M KCl, adjusted to pH 8.5 with H3PO4). The plasmid DNA was precipitated with 0.7 volumes isopropanol, kept on ice for 10 min and centrifuged at 13,000 rpm for 30 min at 4°C in 1.5 ml tube. The DNA pellets were washed with ice-cold 70% ethanol, air-dried and dissolved in 20 µL sterile water. The concentration of the DNA was determined by measuring the absorbance at 260/280 nm.
2.2.1.3 Restriction enzyme digestion
Restriction enzyme digestion was used for checking plasmids. Restriction enzyme digestion of IgG1 Fc and RBDV-IgG1 Fc fragment were carried out in 20 µl volumes using the appropriate restriction enzyme buffer, as recommended by the manufacturer (Promega, WI, USA), supplied with the enzyme and incubated at 37°C. Digestion of plasmid DNA was fractionated by electrophoresis through a 1.5% agarose gel and was analyzed with UV photography system (EZlab, USA) after ethidium bromide (EtBr) staining.
2.2.2 Expression of chimeric proteins
2.2.2.1 Transfection of HEK-293T cell
HEK 293T cells were plated and distributed evenly at 4×106 cells per 100 mm tissue culture plate in 10 ml of DMEM growth medium. After 12 hours of incubation, 70-80% confluent of the plates. Pipette 10 μl of desired plasmid solution (1μg/ml) into a 10 ml glass tube containing 1 ml of 0.25M CaCl2 solution and mix gently. At the same time, pipette 1ml of 2×HBS buffer (280 mM NaCl, 1.5 mM Na2HPO4 and 50 mM HEPES, pH7.1) into a second 10 ml glass tube. Then, the 1.01 ml plasmids/CaCl2 mixture was pipetted into the second tube containing 2×HBS solution.
All the reagents were mixed by repeated pipetting, immediately adding the mixture of plasmids/CaCl2/HBS to the cell on the plate, and swirling gently to distribute the plasmid suspension evenly in the growth medium. After 6 hours of incubation, the medium was removed from the plate and replaced with 25 ml of fresh DMEM growth medium.
2.2.2.2 Expression and purification of chimeric proteins
The generated vectors were then transfected into HEK-293 cells using the calcium-phosphate method as described in the instruction manual of pAAV helper-free system (Stratagene, La Jolla, CA, USA). Supernatants from plasmid transfected HEK-293 cells were harvested (0.45 filtered) after a 48-hour incubation upon adding the fresh DMEM growth medium added. All supernatants were passed over a 2 ml protein G agarose affinity chromatography column (Upstate, Lake Placid, NY, USA). The column was next washed with TBS buffer (50 mM Tris-HCl, and 150 mM NaCl, pH 7.4). Bound proteins were eluted with TBS buffer, pH 2.7, and neutralized immediately with the neutralization buffer, pH 8.0 (1 M Tris-HCl, 1.5 M NaCl, and 1 mM EDTA). Following this neutralization, the eluted fractions from protein G column were loaded onto 1 ml nickel-charged HisTrap HP affinity column (Amersham Biosciences, Piscataway, NJ, USA), washed with binding buffer (20 mM
NaH2PO4, 0.5 M NaCl, 30 mM imidazole, pH7.4), and eluted with elution buffer (20 mM NaH2PO4, 0.5 M NaCl, 500 mM imidazole, pH7.4). The buffer was desalted by using sephadex G-25 (Amersham Biosciences). The recombinant proteins in PBS were concentrated by the Centricon centrifugal filter unit (Millipore, MA, USA). Protein concentration was determined by comparing it to a BSA standard curve.
2.2.2.3 SDS-PAGE and Western blot
The high-purity proteins, IgG1 Fc and RBDV-IgG1 Fc, were separated with 10% SDS-polyacrylamide gel electrophoresis, before being transferred to Nitrocellulose membrane (PALL, FL, USA). Membranes were blocked with 5% of skim milk in phosphate buffered saline (PBS, 137 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4, pH7.4). Protein bands were all recognized by using horseradish peroxidase (HRP)-conjugated antibodies, which are anti-human IgG antibody (AbD Serotec, Raleigh, NC, USA) and anti-his tagged antibody (Novus Biologicals, Littleton, CO, USA). The membrane was developed with enhanced chemiluminescence (ECL) system (Pierce Biotechnology, Inc., Rockford, IL, USA) and exposed to X ray film (Midsci, St. Louis, USA). PBST (0.05% Tween-20 in PBS) were used for all washing steps.
2.2.2.4 Stripping of Nitrocellulose membrane
The membrane was stripped with stripping buffer (20mM glycine, 3.5mM SDS, 1% Tween20 in PBS, pH2.2) twice, and washed with PBST twice. Finally, the membrane was washed with PBS, and ready to be blocked with skim milk.
2.2.3 Receptor binding assay in vitro
2.2.3.1 Biotin label of proteins
Five hundred μl of proteins were reacted with 2 μl of No-WeightTM Sulfo-SBED (Sulfosuccinimidyl [2-6-(biotinamido)-2-(p-azidobenzamido)-hexanoamido]-ethyl- 1,3´-dithiopropionate; PIERCE, USA) in room temperature for 30mins. Nonreacted Sulfo-SBED was removed by using sephadex G-25 column, and the proteins were monitored in the fraction by mixing 10 μl of each fraction with 200 μl of BCATM Protein Assay Reagent (PIERCE, USA) in 96-well microtiter plate (Nunc, Denmark).
2.2.3.2 ELISA
Enzyme link immuno-sorbent assays (ELISA) were carried out to examine the binding activities of the RBDV-IgG1 Fc. 0.5 μg/well of the extracellular domain of mouse VEGFR-1 (R&D systems, MN, USA) or the extracellular domain of mouse
VEGFR-2 (R&D systems, MN, USA) in PBS were first coated onto the 96-well microtiter plate (Nunc, Denmark) at room temperature overnight. Wells were blocked by incubating with blocking buffer (2% skim milk, and 0.05% Tween-20 in PBS) for 1 hour. The plates were washed with PBST three times, and then the biotin-labeled proteins were added into the coated plates. After washing the plates three times with PBST, the plates were then incubated with HRP-conjugate streptavidin (R&D systems, MN, USA) at room temperature for an additional 20 minutes. The reactions were developed by the addition of TMB substrate (KPL, Gaithersburg, MD, USA), and the colorimetric reactions were stopped with 1N HCl after 10 min. The absorbance was measured by ELISA reader (TECAN, Austria) at 450nm.
2.2.4 Cell surface binding assay
Flow cytometry was used to analyze the cell surface binding ability. MS1, B16/F10 and SVEC4-10 cells were seeded at a density of 5×106 in a T75 flask. Viable cell counts were determined by trypan blue dye exclusion. MS1, B16/F10 and SVEC4-10 cells were washed with PBS, detached by Versine (0.2% EDTA in PBS, pH=7.4), washed, and resuspended in flow cytometry buffer (1% bovine serum albumin in PBS, pH 7.4). Aliquots of 100 μl containing 5×105 cells were distributed in 15 ml centrifuge tube (Corning Inc., Corning, NY, USA). Cells were pre-incubated with 10
μg/ml of purified recombinant proteins for 1 hour at 4°C, followed by 1 hour of incubation with goat anti-human IgG fluorescein isothiocyanate (FITC) conjugate (Acris Antibodies GmbH, Hiddenhausen, Germany). The cells were washed three times with ice-cold flow cytometer buffer after incubation. Cell pellets were suspended in 1 ml flow cytomer buffer and analyzed with a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). Negative controls were the cells incubated with anti-goat FITC, with no prior exposure to recombinant proteins.
2.2.5 SVEC 4-10 and B16/F10 cells proliferation assay
Five thousand SVEC 4-10 and B16/F10 cells were seeded onto the flat-bottomed well of a 96-well plate (Corning Inc., Corning, NY, USA) in DMEM growth medium, allowed to settled for 16 hours, incubated with recombinant proteins for 2 hours, and then challenged for 70 hours with human VEGF165 (Upstate Inc., Lake Placid, NY, USA) at a final concentration of 10 ng/ml. The proliferative response was measured by adding 1.9 mg/ml of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carbo-xymetho xyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (Promega, WI, USA) to each well followed by a further 2 hour incubation and spectrophotometric analysis at 492nm. The % of survival was calculated as the ratio of average OD values in wells containing recombinant proteins-treated cells to average OD of wells containing only
cells with MTS containing medium.
2.2.6 Tube formation assay
Fifty microliter of growth factor reduced matrigel (BD Biosciences, San Jose, CA, USA) was added to each well of a 96-well plate (Corning Inc., Corning, NY, USA), and allowed to be polymerized for 2 hours at 37℃. A suspension of 3×104 SVEC 4-10 cells were incubated with recombinant proteins for 1 hour and passed into each well containing 16 ng/ml of VEGF. As the positive control, SVEC 4-10 cells were incubated with 16 ng/ml of VEGF (Upstate Inc., Lake Placid, NY, USA) alone. Cells were incubated for a further 16 hours at 37°C and photographed using a Olympus microscope (40× magnification). The total number of network formations was counted.
2.2.7 Cell migration assay.
8-µm pore Transwell (Costar, Corning, NY) was used. SVEC4-10 cells were pre-cultured with 10 µg/mlprotein for 1 h (37 °C, 5% CO2), then washed with sterile PBS. These cells were next resuspended in DMEM growth medium, and plated (30,000 cells/well) in the upper well. The SVEC4-10 cells wereallowed to migrate toward DMEM growth medium with 10ng/ml VEGF for 6 hrs (37°C, 5% CO2), cells
on the top of the filter were removedwith a cotton swab, and migrated cells on the underside werefixed with methanol, stained with Propidium Iodide (PI, 50μg/ml) and counted with fluorescence microscope.
2.2.8 In situ tumor therapy.
Female C57/BL6 (6-8 week age) was inoculated with 1×106 B16/F10 cells subcutaneously in 200μl PBS. When the tumor average volume was up to 50 mm3, mice were injected with 150μg protein in situ. Tumor volume was measured every 2 days after injecting with proteins. Mice were sacrificed on the final day of the experiment, and tumors were removed, fixed by paraformaldehyde (4% paraformaldehyde in PBS, pH=7.4), and examined by microscopic H&E staining and IHC (immune- histochemistry) staining.
2.2.8.1 HE staining
For the histological HE staining of the B16/F10 melanoma tumor tissues (s.c. melanoma tumors with or without RBDV treatments), the tumors were harvested and fixed with 10% neutral formalin. The tissues were dehydrated using graded alcohols, cleared with xylene, and infiltrated with paraffin wax. The tissue was subsequently embedded with paraffin wax in molds, which facilitate tissue sectioning. Collect
sectioned tissues (4 μm/ section) from paraffin-embedded blocks on clean glass slides and dehydrated in an oven for 30 minutes at 60oC. Prior to staining, tissue slides were deparaffinized and rehydrated, then stained with Mayer’s hemotoxylin and Eosin Y solution for 3 minutes, respectively. After air-dry, tissue slides were mounted with mounting media and visualized under a Nikon light microscope camera system.
2.2.8.2 Immunohistochemical staining (IHC staining)
The Paraffin-embedded sections (7 μm/ section) were obtained from the tumors and were processed for immune-histochemical staining. A rat anti-mouse CD31 monoclonal antibody (1/150 dilution; Novus Biologicals, Littleton, CO, USA) were used in immune-histochemical studies for 4oC overnight incubation. Briefly, the slides were treated with 3% hydrogen peroxide in 1×PBS for 10 min to block endogenous peroxidase activity after dewaxed and rehydrated processes. Next, the sections were washed three times with PBS-T (1×PBS containing 0.05% tween-20) for 5 min each time and non-specific reactions were blocked by 10% FBS in PBS for 10 min at room temperature. The sections were incubated with the anti-CD31 first antibody for 4oC overnight, biotin–conjugated anti-rat IgG secondary antibodies (1/1000 dilution) for 1 hour at RT, and the immune complexes were visualized using the horseradish peroxidase–conjugated straptavidin. LSAB2 system (DAKO, Carpinteria, CA) was
used to visualize the immune complexes, and incubated with 0.5mg/ml diaminobenzidine and 0.03% (v/v) H2O2 in PBS for 10 min. Finally, sections were counterstained with hematoxylin, mounted and observed under a light microscope at magnifications of × 400 and photographed.
2.2.9 Tumor therapy with i.v. injection.
The female C57/BL6 (6-8 week age) was inoculated with 1×106 B16/F10 cells subcutaneously in 200μl PBS. When the tumor average volume was up to 30 mm3, the mice were intravenously (i.v.) injected with 150μg/kg proteins. The tumor volume was measured every 2 days after injecting with proteins. Mice were sacrificed when tumor grew up to 2,500mm3. Tumors and organs were obtained, fixed by paraformaldehyde, and examined by microscopic H&E staining and IHC staining.
2.2.10 Statistic analysis
Statistical analyses were done using SPSS statistics software (SPSS Inc., Chicago, IL, USA). The t-test was used when comparing two independent samples and ANOVA test was used when comparing multiple samples. A value of p < 0.05 was considered significant.
Chapter 3 Results
3.1 Compare of receptor binding domain of VEGF between human and mouse.
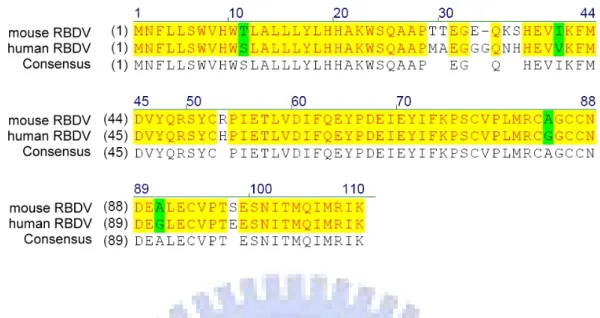
The mature VEGF165 of human has an 88.5% similarity to the VEGF164 of murine. Besides, many studies have already shown that the signal transduction of VEGF and its receptors can cross over between human and murine [98, 99]. It could not be confirmed whether the same part of VEGF had the same function between human and murine. Because of this doubt, the amino sequence of the receptor binding domain of VEGF (RBDV) between human and mouse was necessary to be determined.
The amino acid sequence of mouse VEGF was obtained from the NCBI protein database (NCBI No: NP_001020421). Two sequences was compared to find out the mouse receptor binding domain of VEGF (mRBDV), and the highest similarity was obtained when RBDV was compared with 179-288 amino acids of mouse VEGF. There was a 92.7% similarity between RBDV and mRBDV (Figure 2). According to the result in silico, it was thought that RBDV-Ig Fc might bind to mouse VEGF receptors in vitro.
3.2 Expression and purification of chimeric proteins, IgG1 Fc and RBDV-IgG Fc.
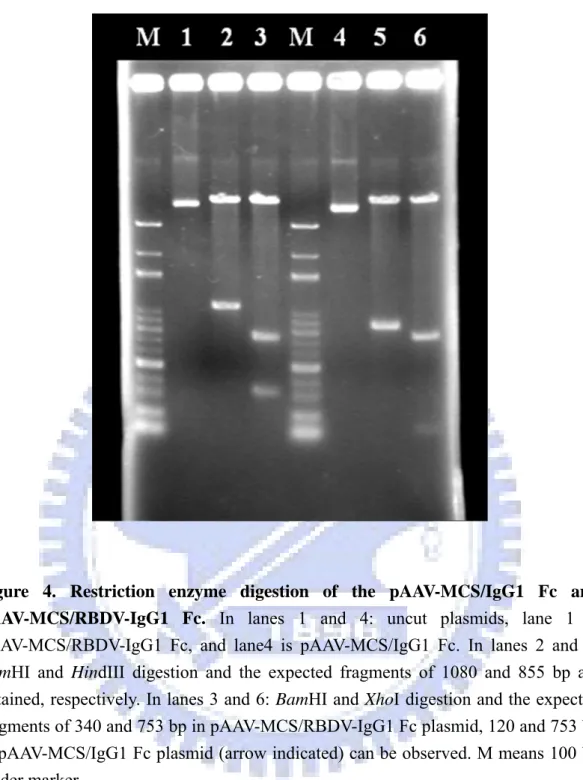
The IgG1 Fc and RBDV IgG1 plasmids were amplified by transforming into the E.
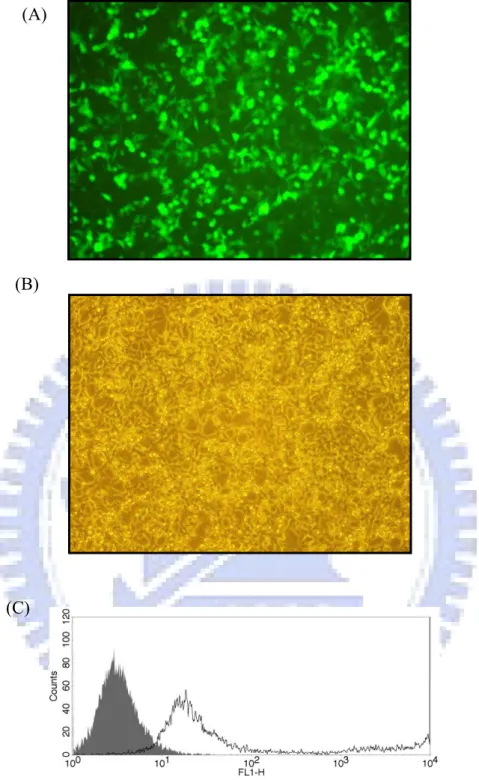
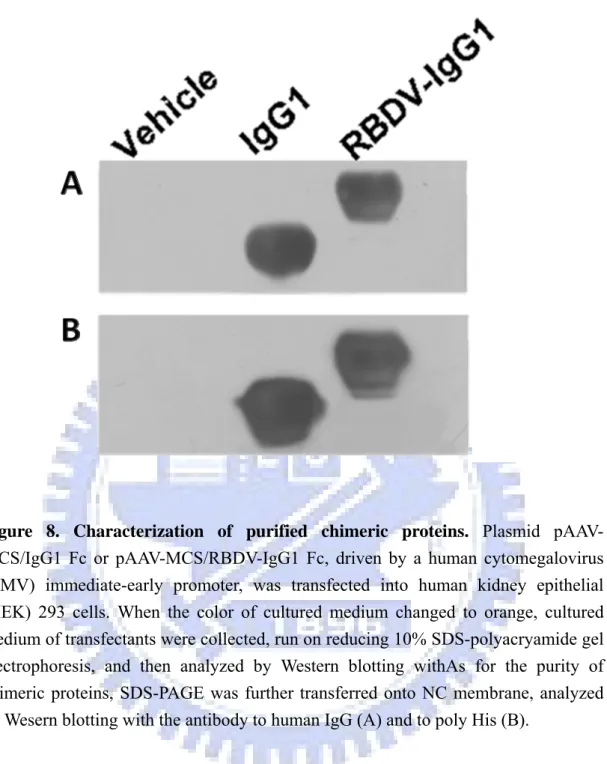
recombinant plasmid was confirmed by restriction enzyme digestion (Figure 4). The resulting constructs, pAAV-MCS/IgG1 Fc and pAAV-MCS/RBDV-IgG1 Fc, were transiently transfected into HEK-293 cells using calcium phosphate based methods as described in section 2.2.2.1. The efficiency of transfection of this method had been confirmed by pAAV-hrGFP vector (Strategene, USA; Figure 5). Expression of the secreted IgG1 Fc and RBDV-IgG1 Fc fusion proteins were confirmed by directly harvesting the culture supernatant followed by SDS-PAGE under reducing conditions, and transferred and blotted as described in 2.2.2.3. IgG1 Fc and RBDV-IgG1 Fc had immunoreactivity against both anti-his tag antibody; or an anti-human IgG antibody was detected (data none shown), where no signal could be detected in the vehicle group.
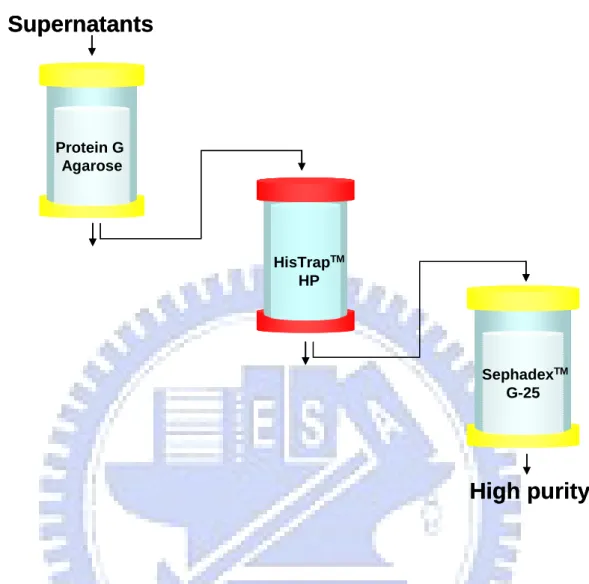
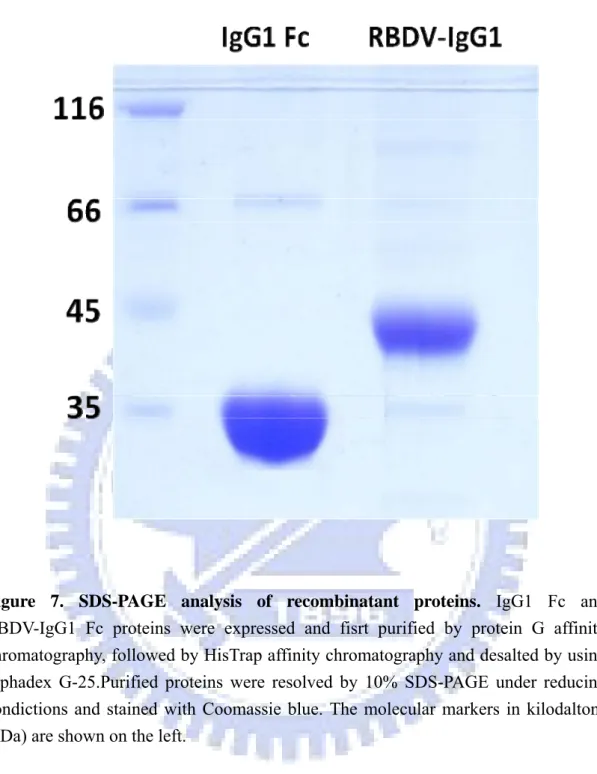
To purify the IgG1 Fc and RBDV-IgG1 Fc fusion proteins, the cultured supernatants were harvested after the initial transfection. The resulting supernatants were first loaded into a protein G affinity column. Elutes of the protein G affinity chromatography were subsequently applied onto a nickel-charged HisTrap affinity chromatography and were desalted by using sephadex G-25. Figure 6 is the flow chart of the purification. To determine the purity, coomassie-stained SDS-PAGE was performed (Figure 7). The protein was a single band at ~38 kDa for IgG1 Fc and at ~48 kDa for RBDV-IgG1 Fc with >95% of purity. The purified protein was also
confirmed by using the western blot with anti- human IgG antibody (Figure 8A) or an anti- his tag antibody (Figure 8B), the stripping buffer was used between recognition of antibodied.
3.3 The activity of RBDV-IgG1 Fc binding to mouse VEGF receptor
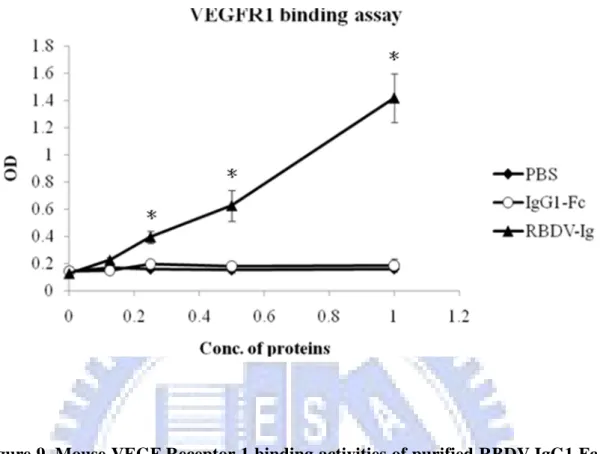
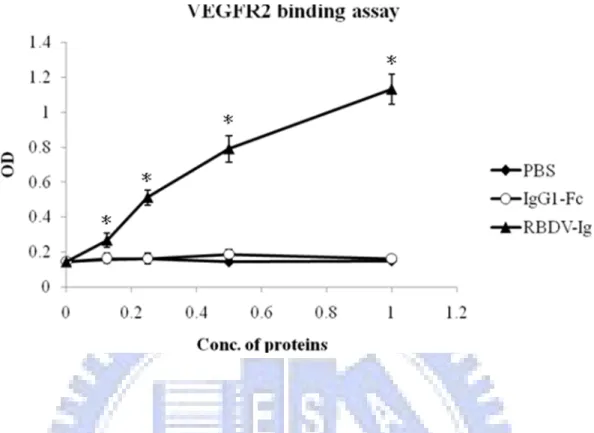
To directly confirm that RBDV-IgG1 Fc can also bind to mouse VEGF receptors as well as to human VEGF receptors, the sandwich enzyme-linked immune sorbent assay (ELISA) was used here. Because the recombinant extracellular domain of mouse VEGFR-1 and VEGFR-2 were fused with human Ig Fc domain and poly His, it was difficult to assay the binding activity with the ELISA. The sandwich ELISA was modified by coupling the biotin to the chimeric proteins, and detecting the binding activity of RBDV-IgG1 Fc with HRP-conjugate streptavidin. The immobilized mouse VEGFR-1 and VEGFR-2 extra- cellular domains were incubated with the RBDV-IgG1 Fc biotin-labeled protein, respectively. The results showed that the binding activity of the RBDV-IgG1 Fc to the immobilized VEGFR-1 (Figure 9) and VEGFR-2 (Figure 10) were strong and in a dose dependent manner. In contrast, the human IgG1 Fc could not bind to the mouse VEGF receptors, either in the VEGFR-1 or VEGFR-2.
3.4 The activity of RBDV-IgG1 Fc binding to cells surface
RBDV-IgG1 Fc was proven and confirmed to have a binding activity to the mouse VEGF receptors with ELISA. To further characterize the in vitro properties of the chimeric proteins, its ability to target the VEGF receptors expressed on the cell surface of MS1, B16/F10 and SVEC4-10 cells was tested for. MS1 cells were strongly probed with RBDV-Ig Fc (Figure 11A), because of the high expression of VEGFR-2 [100]. The B16/F10 and SVEC4-10 cells were less bound with RBDV-Ig Fc compared with MS1 cells (Figure 11B, C). The B16/F10 and SVEC cells had also been demonstrated to express VEGFR-1 and VEGFR-2. [100, 101]. According the results, we demonstrated that RBDV-Ig Fc can bind to the mouse VEGFR-1 and VEGFR-2 expressed on mouse cells.
3.5 The suppressive potency and efficacy of RBDV-IgG1 Fc to SVEC4-10 cells
proliferation in vitro.
The VEGF signal transduction through the VEGFR-2 does indeed stimulate the proliferation of endothelial cells (Ferrara, 2003). After confirming the binding ability of RBDV-Ig Fc to mouse VEGFRs, in vitro proliferation assay was used as described in section 2.2.5 to determine the bio-function of RBDV-Ig Fc. The effect of RBDV-IgG1 Fc on SVEC4-10 cells was examined fist to confirm whether the specific
binding activity could suppress post-receptor signal through the VEGF, which enhances proliferation of endothelial cells. The SVEC4-10 cells (5×103 cells/well in a 96-well plate) were incubated in the presence of IgG1 Fc, RBDV-IgG1 Fc or PBS, as a negative control, for 72 hours and the proliferation assay was determined by MTS. Pervious data showed that the presence of RBDV-IgG1 Fc (2.5 μg/ml), HUVECs did not show any significance in proliferation, as well as IgG1 Fc (2.5 μg/ml) (Appendix
2, Figure 1A), but it was different on mouse endothelial cells. The results show that
RBDV-Ig Fc could inhibit proliferation of SVEC4-10 cells with dose-dependence (Figure 12A), but there was no significant variation in efficiency by treating it with IgG1 Fc (Figure 12B). According to the previous results, we observed that RBDV-Ig Fc could inhibit the proliferation of HUVECs with VEGF stimulating in vitro (Appendix 2, Figure 1B). The SVEC4-10 cells were also treated with RBDV-Ig Fc in the condition of adding of the VEGF (10 ng/ml) to determine the proliferation of SVEC4-10 cells. As expected, the proliferative response of endothelial cells was significantly enhanced by the addition of 10 ng/ml of VEGF (Figure 12A). Besides, when the VEGF was co-incubated with different concentrations of RBDV-IgG1 Fc, cellular proliferation was reduced dose-dependently, whereas an equivalent concentration of the control, IgG1 Fc, had no significant effect on the cell growth, indicating this response was specific to the VEGF and an antagonistic action of
RBDV-IgG1 Fc.
Because of the binding ability of RBDV-Ig Fc to B16/F10 (Figure 11B), we also wondered whether RBDB-Ig Fc had the ability to inhibit proliferation of B6/F10. We treated B16/F10 cells with 2.5μg/ml RBDV-Ig Fc. No matter whether presenting VEGF stimulation or not, the proliferation of B16/F10 showed no significant difference from the negative group (Figure 13B). At the same time, the SVEC4-10 cells were also treated in the same condition (Figure 13A).
3.6 Effect of blockade of VEGF receptors on tube formation in vitro
To investigate the angiogenesis mechanism in vitro, by which VEGFR-1 promotes capillary morphogenesis, the effect of RBDV-IgG1 Fc on capillary-like structures in SVEC4-10 cells was examined. SVEC4-10 cells were cultured with the presence of IgG1 Fc (10 μg/ml), RBDV-IgG1 Fc (10 μg/ml) or PBS for 1 hour before being given stimulation with VEGF (10 ng/ml), and allowed to incubate at 37℃ for 6 hours. The RBDV-IgG1 Fc strongly inhibited the VEGF-induced tube formation and capillary connections compared with VEGF alone or IgG1 Fc within VEGF stimulation. (Figure14 A, B, and C). In the presence of RBDV-IgG1 Fc, SVEC4-10 cells could not extend morphology to form a tube-like structure, and accumulated in aggregates. In contrast, IgG1 Fc alone did not cause a significant decrease in the VEGF-induced
tube and network formation (Figure 14B). These data showed that capillary connections were apparently blocked by the RBDV-IgG1 Fc. The number of capillary network connections in each group was also calculated (Figure 14D).
3.7 RBDV-Ig Fc inhibited the endothelial cell migration in a Transwell system
RBDV-Ig Fc (10 μg/ml) induced a significant inhibition of VEGF-stimulated endothelial cell migration from the upper chamber to the lower one through the membrane (Figure 15C) whereas IgG1 Fc had no any suppressive effect on migration ability of SVEC4-10 cells (Figure 15B). This result indicates that RBDV-Ig Fc could suppress the angiogenic factors-stimulated cell locomotion in responding to chemo-attractive surrounding.
3.8 In vivo suppression of tumor growth with in situ RBDV-Ig Fc treatment.
After confirming the function of RBDV-Ig Fc to inhibit proliferation, tube formation, and migration of endothelial cells, the efficiency of RBDV-Ig Fc by in situ injection was first determined. Female C57/BL6 (6-8 week age) was inoculated with 1×106 B16/F10 cells to right dorsum in 200μl PBS subcutaneously. When the tumor average volume was up to 50 mm3, mice were injected with 150 μg proteins in situ. Tumor volume was measured every 2 days after being injected with proteins. After being
treated with 150 μg of RBDV Ig-Fc, the tumor growth was significantly inhibited in 2 days (Figure 16). Moreover, RBDV-Ig Fc could suppress the B16/F10 tumor growth under 500 mm3 while tumor growth of other groups was up to 2500mm3.
After scarifying mice, their dorsal skin was regularly cut from them. The vessels were obvious in the negative (Figure 17A) and the Ig-G1 Fc (Figure 17B) groups, but not in the RBDV-Ig group (Figure 17C), regardless of whether the fountainhead of new vessels formed were from axilla. The results showed that RBDV-Ig Fc could be an antagonist of VEGF and inhibit B16/F10 tumor growth through antiangiogenesis.
Based on tumor sections of the H&E staining, the tumor associated blood vessels and around tumor were disrupted (Figure 18C) with the RBDV-Ig Fc treatment. Moreover, the tumor cells were also damaged randomly with the IgG1 Fc group (Figure 18B), which did not show any destruct effects in the negative group (Figure
18A).
3.9 RBDV-Ig Fc-mediated in vivo therapy with i.v. injection of mice bearing
B16/F10 tumors.
After confirming the suppressive ability of RBDV-Ig Fc to tumor growth with in situ treatment, the mice bearing B16/F10 were treated with an i.v. injection of RBDV-Ig