Comparison of Genome-wide DNA Methylation in Urothelial Carcinomas of
Patients with and without Arsenic Exposure
Tse-Yen Yanga,b,c,d, Ling-I Hsub, Allen W. Chiue, Yeong-Shiau Puf, Sheng-Hsin Wangf,
Ya-Tang Liaob, Meei-Maan Wug, Yuan-Hung Wangh,i, Chin-Hao Changj, Te-Chang Leek,
Chien-Jen Chena,b,l*
Authors' Affiliations:
aGraduate Institute of Life Science, National Defense Medical Center, Taipei, Taiwan. bGenomics Research Center, Academia Sinica, Taipei, Taiwan.
cMolecular and Genomic Epidemiology Center, China Medical University Hospital,
Taichung, Taiwan.
dChina Medical University, Taichung, Taiwan
eCollege of Medicine, National Yang-Ming University, Taipei, Taiwan.
fDepartment of Urology, College of Medicine, National Taiwan University, Taipei, Taiwan. gGraduate Institute of Oncology, National Taiwan University, Taipei, Taiwan.
hDivision of General Surgery, Department of Urology, Shuang Ho Hospital, Taipei Medical
University, New Taipei City, Taiwan.
iGraduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University,
Taipei, Taiwan.
jDepartment of Medical Research, National Taiwan University Hospital, Taipei, Taiwan. kInstitute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan.
Taipei, Taiwan
Abbreviations: UC, urothelial carcinoma; AsUC, arsenic related urothelial carcinoma; non-AsUC, non-arsenic related urothelial carcinoma; CAE, cumulative arsenic exposure; SAM,
S-adenosyl methionine; , mean β value; , difference in mean β value; rS, Spearman Rank
correlation coefficients; DAVID, Database for Annotation, Visualization and Integrated
Discovery; KEGG, Kyoto Encyclopedia of Genes and Genomes; GOTerm_BP, Gene
Ontogeny Term Biological Processes
*Corresponding Author:
Professor Chien-Jen Chen, Genomics Research Center, Academia Sinica, No. 128, Academia
Road Section 2, Nankang, Taipei 11529, Taiwan
Tel.: +886 2 2789-9402; fax: +886 2 2785-3208
E-mail address: chencj@gate.sinica.edu.tw
Potential conflicts of interest: No
The sources of grant support:
Grants from the National Science Council (NSC-100-2314-B-001-004-MY3) and Academia
Sinica, Taipei, Taiwan
Word counts: Abstract 294; Text: 3600 Total number of Tables/Figures: 3/2
Abstract
Background: Arsenic is a well-documented carcinogen of human urothelial carcinoma (UC)
with incompletely understood mechanisms.
Objectives: This study aimed to compare the genome-wide DNA methylation profiles of
arsenic-induced UC (AsUC) and non-arsenic-induced UC (Non-AsUC), and to assess
associations between site-specific methylation levels and cumulative arsenic exposure.
Methods: Genome-wide DNA methylation profiles in 14 AsUC and 14 non-AsUC were
analyzed by Illumina Infinium methylation27 BeadChip and validated by bisulfite
pyrosequencing. Mean methylation levels ( ) in AsUC and non-AsUC were compared by
their ratio ( ratio) and difference ( ). Associations between site-specific methylation
levels in UC and cumulative arsenic exposure were examined.
Results: Among 27,578 methylation sites analyzed, 231 sites had ratio >2 or <0.5 and 45
sites had >0.2 or <-0.2. There were 13 sites showing statistically significant (q<0.05)
differences in between AsUC and non-AsUC including 12 hypermethylation sites in
AsUC and only one hypermethylation site in non-AsUC. Significant associations between
cumulative arsenic exposure and DNA methylation levels of 28 patients were observed in
nine CpG sites of nine gens including PDGFD (Spearman rank correlation, 0.54), CTNNA2
(0.48), KCNK17 (0.52), PCDHB2 (0.57), ZNF132 (0.48), DCDC2 (0.48), KLK7 (0.48),
FBXO39 (0.49), and NPY2R (0.45). These associations remained statistically significant for
CpG sites in CTNNA2, KLK7, NPY2R, ZNF132 and KCNK17 in 20 non-smoking women after
adjustment for tumor stage and age.
Conclusions: Significant associations between cumulative arsenic exposure and methylation
urothelial carcinoma. Arsenic exposure may cause urothelial carcinomas through the
hypermethylation of genes involved in cell adhesion, proteolysis, transcriptional regulation,
neuronal pathway, and ion transport. The findings of this study, which is limited by its small
sample size and moderate dose-response relation, remain to be validated by further studies
with large sample sizes.
Keywords:
1. Introduction
Arsenic has been well documented as a Group 1 human carcinogen by the International
Agency for Research on Cancer (IARC, 2004, 2012). A significant dose-response relation
exists between arsenic in drinking water and risk of non-melanoma skin cancers and internal
cancers, including urothelial carcinoma (Tseng, 1977; Chen et al., 1985, 1988, 1990, 1992).
A relation between arsenic and DNA methylation has been suggested based on the
observation that arsenic biotransformation and DNA methylation share the same methyl
group donor, S-adenosyl methionine (SAM) (Zhao et al., 1997). The DNA methyltransferase
(DNMT) transfers the methyl group from SAM to cytosine (Jair et al., 2006). DNA
methylation hotspots in mammals are located in the CpG islands of genes, especially in the
promoter regions, which affect genomic stability and regulation of gene expression (Yoder et
al., 1997; Ting et al., 2006). Competition between arsenic biotransformation and DNA
methylation for available methyl groups can lead to differential DNA methylation distribution
in arsenic-induced diseases including urothelial carcinoma (Wilhelm-Benartzi et al., 2010).
Gene promoter hypermethylation has been observed in urothelial carcinomas.
Specifically, arsenic-induced urothelial carcinomas (AsUC) were found to be associated with
the hypermethylation of the gene promoter of protein kinases (Marsit et al., 2006a, 2006b;
Chen et al., 2007). Previous in vitro and in vivo studies have shown that arsenic may induce
differential DNA methylation in transcription factors, cell cycle mediators, tumor suppressor
genes, and oncogenes (Chai et al., 2007; Jensen et al., 2009). Moreover, arsenic may induce
alteration in DNA methylation at target sites, such as RAS association domain family 1A
(RASSF1A), trypsin family of serine proteases 3 (PRSS3), death-associated protein kinase
(DAPK), cyclin-dependent kinase inhibitor 2A (CDKN2A/p16), tumor protein p53 (TP53),
and tumor suppressor genes (Marsit et al., 2006a, 2006b; Chen et al., 2007; Chai et al., 2007;
expression and transcriptional regulation through gene-specific DNA methylation, it is
hypothesized that a differential DNA methylation pattern exists between AsUC and
non-arsenic-induced urothelial carcinomas (non-AsUC).
The specific aims of this study on the genome-wide DNA methylation in urothelial
carcinoma were to (1) compare the DNA methylation patterns between AsUC and non-AsUC,
(2) examine the association between cumulative arsenic exposure and site-specific
methylation level, and (3) identify a possible biological pathway for AsUC.
2. Materials and methods
2.1. Enrollment of Patients affected with AsUC and non-AsUC
In total, 28 urothelial carcinomas were obtained from 14 matched pairs of patients with
and without exposure to arsenic through drinking artesian well water. They were enrolled
from two medical centers, Chi-Mei Hospital and National Taiwan University Hospital. The 14
patients affected with AsUC had been living in arseniasis-endemic areas of southwestern
Taiwan for more than 10 years, another 14 patients affected with non-AsUC had never lived
in arseniasis-endemic areas. Their urothelial carcinomas were confirmed by pathological
examinations (Hsu et al., 2008). These 14 pairs of AsUC and non-AsUC patients were
matched by age, gender, cigarette smoking and tumor stage.
The cumulative arsenic exposure (CAE, in ppm-years) was defined as the sum of
products, derived by multiplying the arsenic concentration in well water (in ppm) by the
duration of water consumption (in years) during consecutive periods of living in different
villages of southwestern arseniasis-endemic areas (Hsu et al., 2008). Written informed
consent was obtained from all patients after a complete description of the study. Sample
collection and laboratory examinations were approved by the institutional review board of
2.2. Tumor collection and DNA extraction
Urothelial carcinoma tissues were frozen in liquid nitrogen immediately after their
surgical removal, and then stored in a freezer at -80 °C. Tumor tissues were examined by
pathologists at Chi-Mei Hospital or National Taiwan University Hospital. The DNA from
each urothelial carcinoma was extracted using the TALENT genomic DNA Extraction kit
(TALENT) or the Quick-gDNA™ MiniPrep kit (Zymo Research, Irvine, CA, USA), and then
stored in a freezer at -80 °C.
2.3. DNA Methylation analysis
The commercialized method, Illumina Infinium Methylation27 BeadChip (Illumina Inc.,
San Diego, CA, USA) containing 27,578 methylation sites, was used for the analysis of
genome-wide DNA methylation. Bisulfite conversion of DNA specimens was performed
using the EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA) in accordance with the
manufacturer’s recommended protocol. DNA methylation levels were assessed using the
Infinium Methylation27 BeadChip following the standard protocol of the manufacturer.
Bisulfite-converted DNA was used for whole genome amplification, enzymatic digestion was
performed to obtain fragmented DNA, and followed by a DNA clean-up process and
application to hybridization of Infinium Human Methylation27 BeadChip. The hybridization
steps were based on a single-base extension, using the DNA as a template to incorporate
fluorescently labeled nucleotides of Cy3 and Cy5 dyes, each pairing with the cytosine
(methylated) or uracil (unmethylated) identity of the bisulfite-converted DNA at a specific
site. The Illumina GenomeStudio program with a methylation module was used to analyze
Infinium Human Methylation27 BeadChip data to derive DNA methylation β-values for each
compared to negative controls from both the methylated and unmethylated signals. The ratio
of the methylated signal to the sum of both methylated and unmethylated signals was
calculated and defined as the β-value. The β-value was a continuous variable between 0 and 1
(Bibikova et al., 2009).
The detection p-values reflecting the strength of DNA hybridization over the
background were calculated by comparing the CpG-intensity with the intensities of negative
control probes. Non-significant detection p-values indicated bad probe design, bad
hybridization or possible chromosome abnormalities (like mutations and insertion-deletions)
at the probe matching locations (Du et al., 2008). The detection p-value reported by
GenomeStudio denoted the probability that the signal from a given probe was greater than the
average signal from negative controls, which targeted bisulfite-converted sequences that did
not contain CpG dinucleotide. Assay probes were randomly permutated and should not
hybridize to the DNA template. The mean signal of these probes defined the system
background. For Illumina Infinium Methylation27 BeadChip, the intensities from both
channels/beads for each CpG site were added. The detection p-value for CpG locus j was
given by pj = 1-Φ[(Ij-μneg)/σneg], where Ij was the sum of intensities from Cy3 and Cy5 (or bead
A and bead B for Infinium), whereas μneg and σneg were the mean and standard deviation of
signals of internal negative controls and Φ[ ] was the normal cumulative probability
distribution function (Kuan et al., 2010).
Control panel in the GenomeStudio program showed good intensity for staining
(above 10,000), clear clustering for the hybridization probes, and satisfactory target removal
intensity (<1000) and bisulfite conversion (Shen et al., 2012).
The bisulfite-converted DNA used for pyrosequencing was prepared using EpiTect
Bisulfite kits. The primers for PCR amplification and pyrosequencing were designed using
the PyroMark Assay Design v2.0 software (Qiagen, Hilden, Germany).
The primer sequences of 13 sites are shown in Supplementary Table S1.
Bisulfite-converted DNA (1 μL) was amplified using Hot-Start Taq-polymerase. Amplicons were
analyzed on the PyroMark Q24 pyrosequencer as specified by the manufacturer, and the
percentage of methylation was quantified as a ratio of C (methylated C) to C+T (methylated C
+ unmethylated C) using PyroMark Q24 software. PCR amplification of target sequences
were included with these significant CpG sites from Illumina Infinium Methylation 27
BeadChip (Supplementary Table S1).
2.5. Statistical analysis
The scatter plot was first generated using Illumina GenomeStudioTM software to illustrate
the mean methylation levels at all sites in AsUC and non-AsUC. Based on the literature
review of epigenetic studies using Illumina Infinium Methylation 27 BeadChip, two criteria
were used to identify the methylation sites with differential DNA methylation patterns
between AsUC and non-AsUC. First criterion was the ratio of mean β-values between AsUC
and non-AsUC indicated as ratio (Fackler et al., 2011). Second criterion was the
difference in mean β-values between the AsUC and non-AsUC indicated as . The
methylated sites with a ratio >2 or <0.5 and a >0.2 or <-0.2 were considered the
differential methylation sites between AsUC and non-AsUC.
The statistical significance of the difference in at each site between AsUC and
non-ASUC was further assessed by the Wilcoxon signed-rank test using SAS/JMP genomics 5
R software (R Development CT, Vienna, Austria) in order to obtain an estimated false
discovery rate (Wei et al., 2009). A q-value <0.05 indicated the probability of false discovery
of methylated sites was less than 5%. The methylation sites showing a q < 0.05, a ratio >2
or <0.5, and a >0.2 or <-0.2 were considered as arsenic-associated methylation sites.
The consistency of methylation levels detected by both BeadChips and pyrosequencing
methods at these arsenic-associated sites were assessed by pairwise correlation coefficients.
The methylation levels at sites with low pairwise correlation coefficients were considered
invalid.
The correlations between methylation level (β) at arsenic-associated sites and cumulative
arsenic exposure (CAE) in 28 patients were further examined by Spearman’s rank correlation
coefficient (rS) using SAS/JMP genomics 5. The log-transformed linear regression analyses
using SAS/JMP genomics 5 were carried out to examine associations between
log-transformed methylation levels and CAE after adjustment for other methylation-related
factors including age, gender, cigarette smoking and tumor stage.
2.6. Gene functional classification
The official gene symbols of the arsenic-associated methylation sites were put into the
Database for Annotation, Visualization and Integrated Discovery (DAVID)
(http://david.abcc.ncifcrf.gov) for the classification of gene functions. The DAVID consists of
an integrated biological knowledgebase and analytical tools aimed at systematically extracting
biological meaning from a large list of genes (Leshchenko et al., 2010). DAVID requires
uploading a gene list, containing any number of common gene identifiers, followed by
analysis using one or more text and pathway mining tools, such as Gene Functional
Genomes (KEGG) pathway or Gene Ontogeny Term Biological Processes (GOTerm_BP) by
the web-based tool from DAVID, version 6.7 (Huang et al., 2009a, 2009b).
3. Results
3.1. Characteristics of AsUC and non-AsUC patients
Patients affected with AsUC and non-AsUC were matched on gender, age, smoking
status and tumor stage. Among 14 patient pairs, 4 male patient pairs were all cigarette
smokers and 10 female patient pairs were all non-smokers. Their age at enrollment ranged
55-76 years with mean age ± standard derivation of 67.3±7.05 and 68.1±5.77 years, respectively,
for AsUC and non-AsUC patients. There were 5 patient pairs with tumor stage Ta, 8 patient
pairs with T1, and 1 patient pair with T4. In other words, most patients were affected with
noninvasive urothelial carcinoma (92.9%). There was a significant difference in cumulative
arsenic exposure (CAE) (p < 0.001) between AsUC and no-AsUC patients. The range of CAE
was 0.25–20.08 ppm-years in AsUC patients.
3.2. Differential DNA methylation patterns between AsUC and non-AsUC
All 27,578 methylation sites examined by the Illumina Infinium Methylation 27 BeadChip
met the quality control criteria in all samples. Among them, 24,694 methylation sites in
13,599 genes had a detection p-value <0.05 in all samples as shown in Figure 1. The scatter
plot in Figure 2A shows the average methylation level in non-AsUC patients by the average
methylation level in AsUC patients for these 24,694 sites. Striking differences indicated by
ratio >2 or <0.5 were observed at 231 methylation sites in 213 genes. There were 208 sites
with higher mean methylation levels ( ratio >2) in AsUC patients, and 23 sites with higher
difference in average methylation level ( ) by the average methylation level of AsUC at
24,694 methylation sites. There were 45 sites in 42 genes had >0.2 or <-0.2, including 44
sites with higher mean methylation levels in AsUC ( >0.2) and only one site with a higher
mean methylation level in non-AsUC ( <-0.2).
Using the Wilcoxon signed-rank test to examine the statistical significance of the
differences in mean methylation levels between AsUC and non-AsUC (multiple comparison q
value <0.05), we found AsUC and non-AsUC had significantly different mean methylation
levels at 34 methylation sites in 33 genes with >0.2 or <-0.2 and at 75 methylation sites
in 70 genes with ratio >2 or <0.5. In combination of both criteria of ratio and ,
there were 13 methylation sites in 13 genes showing significantly different between AsUC
and non-AsUC (q value <0.05). Among these 13 sites, 12 sites had higher mean methylation
levels in AsUC than non-AsUC. They were located in CYP1B1, KCNK17, PDGFD, NPY2R,
CTNNA2, DCDC2, KLK7, HSPA2, SIPA1, ZNF132, HSPA2, and FBXO39. The only one
methylation site with a higher in non-AsUC than AsUC was in ATP5G2.
3.3. Bisulfite pyrosequencing for validation of methylation levels detected by Illumina Infinium Methylation27
The DNA methylation levels of 13 sites with significant differences between AsUC and
non-AsUC were further validated using bisulfite pyrosequencing. The methylation levels of
specific sites detected by the Illumina Infinium Methylation27 and bisulfite pyrosequencing
were compared in 28 DNA samples. Bisulfite pyrosequencing data were very consistent with
above 0.85 at 11 sites (PCDHB2, CTNNA2, KCNK17, ZNF132, PDGFD, NPY2R, KLK7,
HSPA2, FBXO39, DCDC2, and CYP1B1) as shown in Table 1. There were only two sites
with lower correlation coefficients (<0.7) in SIPA1 and ATP5G2.
3.4. Correlation between CAE and gene-specific DNA methylation level in AsUC
The DNA methylation level was significantly correlated with CAE in 9 of 11 sites with
consistent methylation levels detected by the Illumina Infinium Methylation27 and bisulfite
pyrosequencing. The nine methylation sites significantly associated with CAE were in
PDGFD (Spearman rank correlation, 0.54), CTNNA2 (0.48), KCNK17 (0.52), PCDHB2
(0.57), ZNF132 (0.48), DCDC2 (0.48), KLK7 (0.48), FBXO39 (0.49), and NPY2R (0.45).
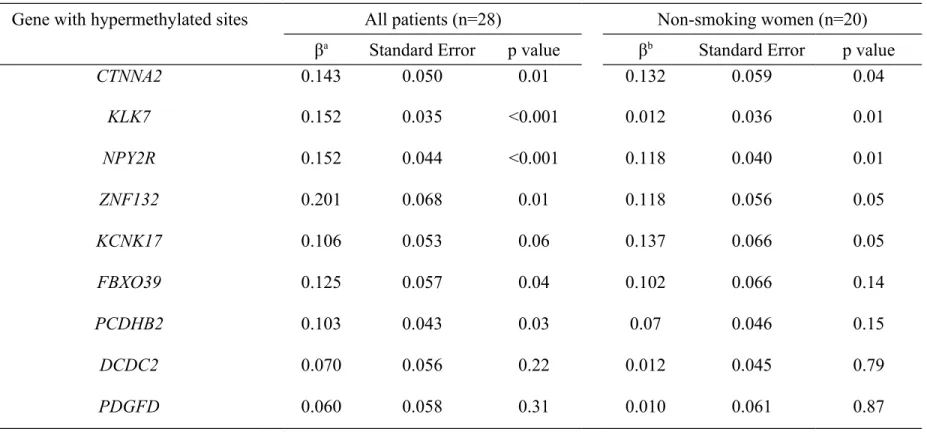
3.5. Log-transformed linear regression analysis of associations between gene-specific DNA methylation and CAE in AsUC
The DNA methylation levels of the nine methylation sites were log-transformed to further
assess for their associations with CAE after adjustment for age, cigarette smoking habit and
tumor stage in linear regression analyses as shown in Table 3. The DNA methylation levels of
seven methylation sites in CTNNA2, FBXO39, KLK7, NPY2R, PCDHB, ZNF132 and
KCNK17 were significantly associated with CAE after adjustment for age, cigarette smoking
habit and tumor stage. These seven associations were further examined in non-smoking
women after adjustment for age and tumor stage. In non-smoking women, five methylation
sites in CTNNA2, KLK7, NPY2R, ZNF132 and KCNK17 remained significantly associated
with CAE.
3.6. Associated pathways of genes with differential methylation levels in AsUC and non-AsUC
The gene functional classifications of the 5 methylation sites in 5 genes were queried
using KEGG pathway and Gene Ontology database. There were 5 methylation sites located
within CpG islands of promoter regions with distances from the transcriptional start site
(TSS) less than 500 bp. Using the DAVID web-based tool, we classified these 5 genes
according to their gene functions. These genes were involved in cell adhesion (CTNNA2),
proteolysis (KLK7), transcriptional regulation (ZNF132), neuronal pathways (NPY2R), and
ion transport (KCNK17).
4. Discussion
Epigenetic changes in individuals with arseniasis have been reported in several recent
studies (Chanda et al., 2006; Smeester et al., 2011). However, the DNA methylation patterns
in AsUC and non-AsUC has never been compared using genome-wide screening previously.
In this exploratory study on a small number of patients affected with AsUC and non-AsUC,
matching method (Heller et al., 2009) was used to select 14 patient pairs in order to control
potential confounding effect of age, gender, cigarette smoking and tumor stage. We compared
the genome-wide DNA methylation patterns in AsUC and non-AsUC, and identified 13 sites
with differential methylation levels. Most (12/13) of them were hypermethylated in AsUC in
comparison to non-AsUC. Unmatched analysis was used to examine the associations between
methylation levels and CAE after adjustment of potential confounding factors. Significant
associations between methylation level and CAE were observed at 5 hypermethylation sites in
CTNNA2, KLK7, NPY2R, ZNF132 and KCNK17, respectively, in 20 non-smoking female
patients.
In this study, we used a conservative method (two criteria of ratio and with a q
value <0.05) to detect possible methylation sites associated with arsenic exposure. It is
words, there may be type II error when we tried to narrow down the false positives in order
to detect the genuine differences in site-specific methylation levels between AsUC and
non-AsUC groups.
Other toxicants such as cigarette smoke might affect the site-specific DNA methylation.
Potential confounding effect of cigarette smoking was controlled through matching method,
but we could not rule out effects of many other toxicants in the environment. The lack of
information on exposures to other environmental factors which may have effects on
site-specific DNA methylation is another limitation of this study.
We hypothesized that arsenic-related DNA methylation patterns may exist in AsUC after
earlier long-term exposures to arsenic. A previous study showed that exposure to famine early
in life may cause persistent changes in the DNA methylation levels of several genes with
diverse biological functions, and that the association between early-life environmental
exposure and health outcomes later in life can be mediated by epigenetic changes (Tobi et al.,
2009). Another study had documented that DNA methylation may persist in target organs and
tissues after exposure to the external environment, and this methylation may even be
maintained throughout life (Heijmans et al., 2008). AsUC patients in this study had been
exposed to high levels of arsenic in drinking water after birth. We identified five novel
arsenic-associated hypermethylated sites in five genes which are involved in cell adhesion
(CTNNA2), proteolysis (KLK7), transcription regulation (ZNF132), neuronal pathways
(NPY2R), and ion transport (KCNK17).
A previous study showed that promoter hypermethylation of PRSS3 and RASSF1A was
significantly associated with invasive tumor stage and high toenail arsenic level in tumor
tissue (Marsit et al., 2006a, 2006b). The finding suggests that DNA methylation of these two
genes may be involved at late stage of carcinogenesis of the bladder, and arsenic exposure
study, most patients were affected with tumors at a non-invasive stage, which might explain
why the nine genes we identified are inconsistent with those identified in the previous study
(Marsit et al., 2006a, 2006b).
CTNNA2 (alpha N-catenin), a protein of the vinculin family, is a membrane-cytoskeletal
protein in focal adhesion plaques that is involved in the linkage of integrin adhesion
molecules to the actin cytoskeleton (Geiger et al., 1979). Its sequence is 20-30% similar to
α-catenin, which serves a similar function (Burridge et al., 1980). A lack of vinculin may
decrease cell adhesion by inhibiting focal adhesion assembly and preventing actin
polymerization, while overexpression of vinculin may restore adhesion and spreading by
promoting the recruitment of cytoskeletal proteins to the focal adhesion complex at the site
of integrin binding (Ezzell et al., 1997).
KLK7 belongs to the kallikrein subfamily of serine proteases, which are involved in a
variety of enzymatic processes (Gan et al., 2000). Dysregulation of KLK7 has been linked to
several skin disorders, including atopic dermatitis, psoriasis, and Netherton syndrome. These
diseases are characterized by excessively dry, scaly, and inflamed skin, due to a disruption of
skin homeostasis and correct barrier function (Descargues et al., 2005). A recent study
showed that non-synonymous single nucleotide polymorphisms (ns-SNPs) of KLK7 might
be associated with arsenic-induced carcinogenesis (Isokpehi et al., 2010).
NPY2R (neuropeptide Y receptor Y2) plays an important role in the neuromodulation of
ureteral motility and erectile function (Prieto et al., 1997, 2004; Rose et al., 1995). DNA
methylation of NPY2R in urine sediment was shown to be significantly associated with
bladder tumors (Chung et al., 2011). ZNF132 is a member of the zinc finger protein family,
which is essential in transition metal ion binding and might be involved in transcriptional
regulation (Klug et al., 1987). The mechanism for the association of ZNF132 expression and
KCNK17 (potassium channel subfamily K member 17) is a member of the
alkaline-activated subfamily of tandem pore potassium channels, which are open at all membrane
potentials and contribute to cellular resting membrane potential (Suzuki et al., 2004).
KCNK17 is documented to exhibit arsenic-related gene expression patterns in lymphocytes
(Andrew et al., 2008). The genetic and epigenetic changes of KCNK17 might be associated
with the development of AsUC.
The matched-pair exploratory study would improve the balance of covariates for
regression analyses, especially in the subgroup of non-smoking female patients. The
matching-based analysis does not assume linearity and is robust to outliers with no danger of
extrapolation (Heller et al., 2009). However this exploratory study with a small sample size
and moderate dose-response relation could not exclude the possibilities that false positive
may have still existed and important differences may be missed using the conservative
method.
The findings of this study need to be validated by a study with a large sample size and
detail information on environmental toxicants other than arsenic and cigarette smoke.
Moreover we also need to assess if a synergy exists between the effects of arsenic and other
environmental factors.
5. Conclusions
In this study, the DNA methylation levels were found to be significantly different at 13
sites between AsUC and non-AsUC. Nine of them showed significant associations between
site-specific methylation level and CAE in 28 patients. Methylation levels at five sites in
CTNNA2, KLK7, NPY2R, ZNF132 and KCNK17 remained significantly associated with CAE
sample size and detail information on environmental toxicants other than arsenic and cigarette
smoke.
Acknowledgments
This work was supported by grants from the National Science Council
References
Andrew, A., et al., 2008. Drinking-water arsenic exposure modulates gene expression in
human lymphocytes from a U.S. population. Environ. Health Perspect. 116, 524-531.
Bibikova, M., et al., 2009. Genome-wide DNA methylation profiling using Infinium®
assay. Epigenomics 1, 177-200.
Burridge, K., et al., 1980. Microinjection and localization of a 130K protein in living
fibroblasts: a relationship to actin and fibronectin. Cell 19, 587-595.
Chai, C., et al., 2007. Arsenic salts induced autophagic cell death and hypermethylation of
DAPK promoter in SV-40 immortalized human uroepithelial cells. Toxicol. Letters 173,
48-56.
Chanda, S., et al., 2006. DNA hypermethylation of promoter of gene p53 and p16 in
arsenic-exposed people with and without malignancy. Toxicol. Sci. 89, 431-437.
Chen, W.T., et al., 2007. Urothelial carcinomas arising in arsenic-contaminated areas are
associated with hypermethylation of the gene promoter of the death-associated protein
kinase. Histopathology 51, 785-792.
Chen, C.J., et al., 1985. Malignant neoplasms among residents of a Blackfoot
disease-endemic area in Taiwan: high-arsenic artesian well water and cancers. Cancer Res. 45,
5895-5899.
Chen, C.J., et al., 1988. Arsenic and cancers. Lancet 1, 414-415.
Chen, C.J., et al., 1990. Ecological correlation between arsenic level in well water and
age-adjusted mortality from malignant neoplasms. Cancer Res. 50, 5470-5474.
Chen, C.J., et al., 1992. Cancer potential in liver, lung, bladder and kidney due to ingested
inorganic arsenic in drinking water. Brit J Cancer 66, 888-892.
Chung, W., et al., 2011. Detection of bladder cancer using novel DNA methylation
Descargues, P., et al., 2005. Spink5-deficient mice mimic Netherton syndrome through
degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet 37, 56–65.
Du, P., et al., 2008. lumi: a Bioconductor package for processing Illumina microarray.
Bioinformatics 24, 1547-1548.
Ezzell, R., et al., 1997. Vinclin promotes cell spreading by mechanically coupling integrins
to the cytoskeleton. Experimental Cell Research 231, 14-26.
Fackler, M., et al., 2011. Genome-wide methylation analysis identifies genes specific to
breast cancer hormone receptor status and risk of recurrence. Cancer research 71,
6195-6207.
Gan, L., et al., 2000. Sequencing and expression analysis of the serine protease gene cluster
located in chromosome 19q13 region. Gene 257, 119-130.
Geiger, B.,1979. 130K Protein from Chicken Gizzard - Its Localization at the Termini of
Microfilament Bundles in Cultured Chicken-Cells. Cell 18, 193-205.
Heijmans, B., et al., 2008. Persistent epigenetic differences associated with prenatal
exposure to famine in humans. Proc. Natl. Acad. Sci. U.S.A 105, 17046-17049.
Heller, R., et al., 2009. Matching method for observational microarray studies.
Bioinformatics 25, 904-909.
Hossain, M., et al., 2012. Environmental arsenic exposure and DNA methylation of the
tumor suppressor gene p16 and the DNA repair gene MLH1: effect of arsenic
metabolism and genotype. Metallomics 4, 1167-1175.
Hsu, L.I., et al., 2008. Comparative genomic hybridization study of arsenic-exposed and
non-arsenic-exposed urinary transitional cell carcinoma. Toxicol. Appl. Pharmacol. 227,
229-238.
Huang, D., et al., 2009a. Systematic and integrative analysis of large gene lists using
Huang, D., et al., 2009b. Bioinformatics enrichment tools: paths toward the comprehensive
functional analysis of large gene lists. Nucleic acids research 37, 1-13.
International Agency for Research on Cancer, 2004. IARC Monographs on the Evaluation
of Carcinogenic Risks to Humans. Some Drinking-water Disinfectants and
Contaminants, including Arsenic. Lyon: IARC, 1-512.
International Agency for Research on Cancer, 2012. IARC Monographs on the Evaluation
of Carcinogenic Risks to Humans. A Review of Human Carcinogens: Arsenic, Metals,
Fibres, and Dusts. Lyon: IARC, 1-469.
Isokpehi, R.D., et al., 2010. Candidate single nucleotide polymorphism markers for arsenic
responsiveness of protein targets. Bioinform. Biol. Insights 11, 99-111.
Jair, K.W., et al., 2006. De novo CpG island methylation in human cancer cells. Cancer
Res. 66, 682-692.
Jensen, T.J., et al., 2009. Epigenetic mediated transcriptional activation of WNT5A
participates in arsenical-associated malignant transformation. Toxicol. Applied
Pharmacol. 235, 39-46.
Klug, A., et al., 1987. Zinc fingers: a novel protein motif for nucleic acid recognition.
Trends in Biochemical Sciences 12, 464-469.
Kuan, P.F., et al., 2010. A statistical framework for Illumina DNA methylation arrays.
Bioinformatics 26, 2849-2855.
Leshchenko, V.V., et al., 2010. Genomewide DNA methylation analysis reveals novel
targets for drug development in mantle cell lymphoma. Blood 116, 1025-1034.
Marsit, C.J., et al., 2006a. Carcinogen exposure and gene promoter hypermethylation in
bladder cancer. Carcinogenesis 27, 112-116.
Marsit, C.J., et al., 2006b. Carcinogen exposure and epigenetic silencing in bladder cancer.
Prieto, D., et al., 1997. Distribution and functional effects of neuropeptide Y on equine
ureteral smooth muscle and resistance arteries. Regul. Pept. 69, 155-165.
Prieto, D., et al., 2004. Heterogeneity of the neuropeptide Y (NPY) contractile and relaxing
receptors in horse penile small arteries. Br. J. Pharmacol. 143, 976-986.
Rose, P.M., et al., 1995. Cloning and functional expression of a cDNA encoding a human
type 2 neuropeptide Y receptor. J. Biol. Chem. 270, 22661-22664.
Shen, J., et al., 2012. Genome-wide DNA methylation profiles in hepatocellular carcinoma.
Hepatology 55, 1799-1808.
Smeester, L., et al., 2011. Epigenetic changes in individuals with arsenicosis. Chem. Res.
Toxicol. 24, 165-167.
Suzuki, Y., et al., 2004. Sequence Comparison of Human and Mouse Genes Reveals a
Homologous Block Structure in the Promoter Regions. Genome Res. 14, 1711–1718.
Ting, A.H., et al., 2006. Differential requirement for DNA methyltransferase 1 in
maintaining human cancer cell gene promoter hypermethylation. Cancer Res. 66,
729-735.
Tobi, E.W., et al., 2009. DNA methylation differences after exposure to prenatal famine are
common and timing- and sex-specific. Human Molecular Genetics 18, 4046-4053.
Tseng, W.P., 1977. Effects and dose--response relationships of skin cancer and Blackfoot
disease with arsenic. Environ. Health Perspect. 19, 109-119.
Wei, Z., et al., 2009. Multiple testing in genome-wide association studies via hidden
Markov models. Bioinformatics 25, 2802-2808.
Wilhelm-Benartzi, C.S., et al., 2010. DNA methylation profiles delineate etiologic
heterogeneity and clinically important subgroups of bladder cancer. Carcinogenesis 31,
Yoder, J.A., et al., 1997. DNA (cytosine-5)-methyltransferases in mouse cells and tissues:
studies with a mechanism-based probe. J. Mol. Biol. 270, 385-395.
Zhao, C.Q., et al., 1997. Association of arsenic-induced malignant transformation with
DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U.S.A. 94,
Figure legends
Figure 1. Workflow for the identification of significant differences in site-specific methylation levels between arsenic-induced urothelial carcinomas (AsUC) and
non-arsenic-induced UC (non-AsUC) and the differential sites which methylation levels were significantly
associated with cumulative arsenic exposure (CAE). The default control probe in Illumina
Infinium Methylation27 BeadChip was used for quality control. In comparison with the mean
signal of the negative control which target bisulfite-converted sequences that did not contain
CpG dinucleotides (i.e., the system background), those specific sites without a significant
difference (detection p-value > 0.05) were filtered out. The mean β value ( ) is the average
methylation level of AsUC or non-AsUC; mean β difference ( ) is the difference in
between AsUC and non-AsUC; and the mean β ratio ( ratio) is the ratio in between
AsUC and non-AsUC.
Figure 2. Comparison of genomic DNA methylation profiles in arsenic-induced urothelial carcinomas (AsUC) and non-arsenic-induced UC (non-AsUC). In Panel A, red line indicates
the differential methylation levels equal to two-fold. Blue dots indicate methylated sites at
which the mean β ratio ( ratio) between AsUC and non-AsUC was >2 (208 sites) or <0.5
(23 sites). In Panel B, red line indicates the differences in methylation levels equal to 0.2.
Blue dots indicate methylated sites at which the difference in mean β value ( ) between
Figure 1.
27,578 methylation sites per sample in Illumina Infinium Methylation 27 BeadChip (All 28 samples had passed the quality control criteria)
24,694 methylation sites (in 13,599 genes) showing detection p value less than 0.05 in all samples
45 methylation sites (in 42 genes) with mean β difference ( ) >0.2 or <-0.2 (Figure 2B)
34 methylation sites (in 33 genes) with significant difference in β value (Wilcoxon Signed Rank test with a multiple comparison q<0.05)
13 methylation sites (in 13 genes) with a significant differential methylation level (Table 1)
231 methylation sites (in 213 genes) with mean β ratio >2 or <0.5 (Figure 2A)
75 methylation sites (in 70 genes) with significant difference in β value (Wilcoxon Signed Rank test with a multiple comparison q<0.05)
11 methylation sites were examined for their Spearman Rank correlation (rS) with cumulative arsenic exposure (CAE) in 28 UC samples (Table 2)
9 methylation sites were examined for their associations with CAE after adjustment for other covariates using log transformed linear regression method (Table 3)
7 methylation sites significantly associated with CAE in linear regression analysis (Table 3)
5 methylation sites significantly associated with CAE in non-smoking women
2 methylation sites non-significantly associated with CAE in non-smoking women were excluded
2 methylation sites non-significantly associated with CAE in multiple linear regression analysis were excluded 2 methylation sites with non-significant Spearman Rank correlation coefficient (rS) were excluded
2 methylation sites were with low correlation (r <0.85) between Illumina Infinium Methylation 27 BeadChip and pyrosequencing were excluded
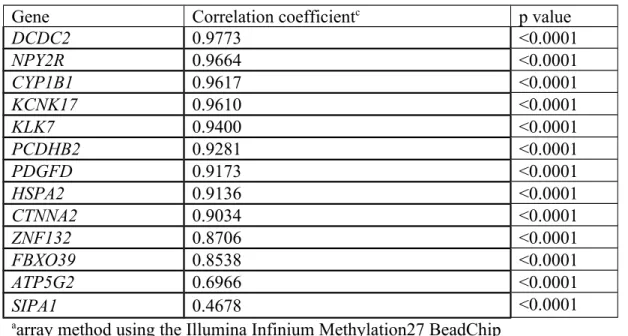
Table 1. CpG site-specific correlations of DNA methylation levels detected by arraya and pyrosequencingb methods in 28 tumor tissues
Gene Correlation coefficientc p value
DCDC2 0.9773 <0.0001 NPY2R 0.9664 <0.0001 CYP1B1 0.9617 <0.0001 KCNK17 0.9610 <0.0001 KLK7 0.9400 <0.0001 PCDHB2 0.9281 <0.0001 PDGFD 0.9173 <0.0001 HSPA2 0.9136 <0.0001 CTNNA2 0.9034 <0.0001 ZNF132 0.8706 <0.0001 FBXO39 0.8538 <0.0001 ATP5G2 0.6966 <0.0001 SIPA1 0.4678 <0.0001
aarray method using the Illumina Infinium Methylation27 BeadChip bbisulfite pyrosequencing
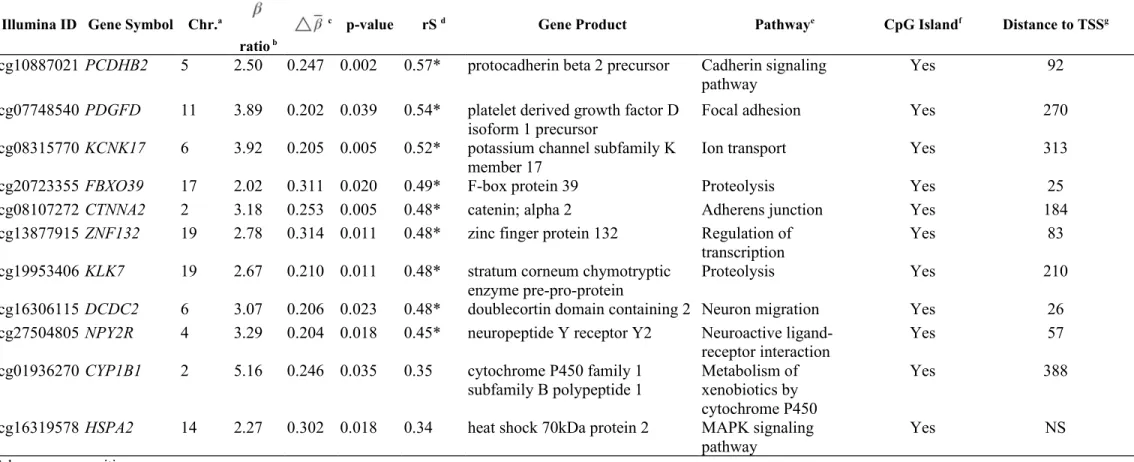
Table 2. Eleven genes with 11 methylation sites showing significant differences in mean methylation levels between induced and non- arsenic-induced urothelial carcinoma ( q value <0.05) with a mean β difference ( ) >0.2 and a mean β ratio >2
Illumina ID Gene Symbol Chr.a
ratio b
c p-value rS d Gene Product Pathwaye CpG Islandf Distance to TSSg
cg10887021 PCDHB2 5 2.50 0.247 0.002 0.57* protocadherin beta 2 precursor Cadherin signaling
pathway
Yes 92
cg07748540 PDGFD 11 3.89 0.202 0.039 0.54* platelet derived growth factor D
isoform 1 precursor
Focal adhesion Yes 270
cg08315770 KCNK17 6 3.92 0.205 0.005 0.52* potassium channel subfamily K
member 17
Ion transport Yes 313
cg20723355 FBXO39 17 2.02 0.311 0.020 0.49* F-box protein 39 Proteolysis Yes 25
cg08107272 CTNNA2 2 3.18 0.253 0.005 0.48* catenin; alpha 2 Adherens junction Yes 184
cg13877915 ZNF132 19 2.78 0.314 0.011 0.48* zinc finger protein 132 Regulation of
transcription
Yes 83
cg19953406 KLK7 19 2.67 0.210 0.011 0.48* stratum corneum chymotryptic
enzyme pre-pro-protein
Proteolysis Yes 210
cg16306115 DCDC2 6 3.07 0.206 0.023 0.48* doublecortin domain containing 2 Neuron migration Yes 26
cg27504805 NPY2R 4 3.29 0.204 0.018 0.45* neuropeptide Y receptor Y2 Neuroactive
ligand-receptor interaction
Yes 57
cg01936270 CYP1B1 2 5.16 0.246 0.035 0.35 cytochrome P450 family 1
subfamily B polypeptide 1
Metabolism of xenobiotics by cytochrome P450
Yes 388
cg16319578 HSPA2 14 2.27 0.302 0.018 0.34 heat shock 70kDa protein 2 MAPK signaling
pathway
Yes NS
achromosome position.
bthe ratio of mean methylation levels between arsenic-induced and non-arsenic-induced urothelial carcinomas. cthe difference in mean methylation levels between arsenic-induced and non-arsenic-induced urothelial carcinomas.
fsites located within a CpG island are shown as “Yes”.
gtranscription start site is abbreviated as “TSS”, and the distance indicates the number of nucleotides between the start codon and the specific CpG site. NS is “Not Shown” in gene information of Illumina BeadChip.
Table 3. Multivariate regression analysis of association between log-transformed DNA methylation levels in urothelial carcinomas and cumulative arsenic exposure
Gene with hypermethylated sites All patients (n=28) Non-smoking women (n=20)
βa Standard Error p value βb Standard Error p value
CTNNA2 0.143 0.050 0.01 0.132 0.059 0.04 KLK7 0.152 0.035 <0.001 0.012 0.036 0.01 NPY2R 0.152 0.044 <0.001 0.118 0.040 0.01 ZNF132 0.201 0.068 0.01 0.118 0.056 0.05 KCNK17 0.106 0.053 0.06 0.137 0.066 0.05 FBXO39 0.125 0.057 0.04 0.102 0.066 0.14 PCDHB2 0.103 0.043 0.03 0.07 0.046 0.15 DCDC2 0.070 0.056 0.22 0.012 0.045 0.79 PDGFD 0.060 0.058 0.31 0.010 0.061 0.87
aadjusted for age, cigarette smoking habits and tumor stage badjusted for age and tumor stage