行政院國家科學委員會專題研究計畫 成果報告
生物系統內分子交互作用及生化路徑之大規模分析--(子計
畫三)建立大型分子交互作用資料庫系統與資料探索(3/3)
研究成果報告(完整版)
計 畫 類 別 : 整合型 計 畫 編 號 : NSC 99-2627-B-009-003- 執 行 期 間 : 99 年 08 月 01 日至 100 年 07 月 31 日 執 行 單 位 : 國立交通大學生物科技學系(所) 計 畫 主 持 人 : 黃憲達 計畫參與人員: 碩士班研究生-兼任助理人員:梁超 碩士班研究生-兼任助理人員:陳冠州 碩士班研究生-兼任助理人員:蔡文婷 碩士班研究生-兼任助理人員:蘇晟漢 博士班研究生-兼任助理人員:黃熙淵 博士班研究生-兼任助理人員:許勝達 博士班研究生-兼任助理人員:簡佳宏 公 開 資 訊 : 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢中文摘要: 本跨領域整合計畫將延續上期研究成果,首要目的在利用已建 立生物分子序列-結構-功能-生化路徑間的關係,深入研究 分子間交互作用機制、演化關係並建立基因調控網路模型。基 因序列、蛋白質及其生物功能間的複雜關係與疾病、藥物、生 醫研究密不可分,生化反應網絡由生物分子的交互作用組成, 這是蛋白質體學中亟待解決的問題。本計畫的研究成果,有助 於高精確度地分析、預測蛋白質交互作用及生化網路的構成方 式,以及未來藥物的發展。簡言之,即是以序列、結構資訊與 演算法為基礎,解析生物系統(biological systems),並以實驗驗 證之。 本計畫包括四個子計畫,分別是以生化網路演化關係研究分子 交互作用與生化路徑(子計畫一)、智慧型最佳化方法用於基因 網路的重建與分析(子計畫二)、建立大型分子交互作用資料庫 系統與資料探索(子計畫三),以及蛋白質於細胞位置之預測與 蛋白質間的交互作用:由基因體序列到蛋白質功能(子計畫 四)。四者的緊密結合,可以涵蓋由基因、蛋白質、訊息傳遞路 徑(signaling pathway)、代謝路徑(metabolic pathway)到基因調控 網路(gene regulatory networks)間各層次的完整研究。
玆列出子計畫三(即本精簡報告之子計畫)研究目標條列如下: 一. 建置超大型生物分子交互作用(molecular interactions)及生化 路徑(pathways)資料庫(database),涵蓋基因、蛋白質、生物分 子交互作用、生化路徑、基因調控網路,進行收集及整合,並 自動更新。這個資料庫系統將具備完整性、支援物種完整以及 不重複等特性(子計畫三、二、一及四)。 二. 以演化式最佳化方法等機器學習(machine learning)方法重建 基因調控網路,同時對實驗資料不足之情況,提出有效之改善 方法。並利用子計畫一及二提出的分子交互作用模型與子計畫 三資料庫得到之知識,使重建的網路模型符合真實之生物系 統。 三. 針對疾病(disease)、癌症治療(cancer therapy)、環境微生物 及生質能源等相關議題中的特定生物系統機制,以多角度探討 蛋白質-蛋白質、蛋白質-DNA 及蛋白質-RNA 之間的交互作 用,以及這些交互作用在生化路徑及調控網路中扮演的角色, 並以實驗驗證之(子計畫三、二、一及四)。
英文摘要: The primary theme of our integrated project is to investigate
molecular interactions and evolution relationships, and build various pathways (i.e. signaling pathways and metabolic pathways) and networks (e.g. gene regulatory networks). Disease, drug and biomedical researches correlated closely with the complications among gene sequence, protein and biological functions. The fruitful outcomes of our project will be in accurately analyzing and
predicting molecular interactions and biochemical network components, moreover, drug developments. Furthermore, our predictions will be immediately confirmed by cellular and viral experiments.
The project is composed of four subprojects: Network evolution for studying molecular interactions and pathways (subproject 1); intelligent optimization methods for reconstruction and analysis of gene networks (subproject 2); data management and exploration of molecular interactions and pathways (subproject 3), and Protein subcelluar localization prediction and protein-protein interactions: from genomic sequences to protein functions (subproject 4). This integrated project studies molecular interactions and networks on genes, proteins, signaling pathways, metabolic pathways, and gene regulatory networks.
The specific aims of subproject 3 (for this report) are listed as follows:
1. To establish an integrated database system, including sequence, structure, molecular interaction, biochemical pathways, and gene regulatory networks. This large database can update and integrate information w automatically. The database is designed as a comprehensive, broad species coverage and non-redundant molecular interaction repository.
2. To reconstruct the gene regulatory networks by optimization methods, such as machine learning methods, and to revise
deficiency of experimental data. We will fit the reconstructed gene regulatory networks to the natural biological systems based on the molecular interactions models (subprojects 1 and 2) and the biological databases (subproject 3).
3. To cooperate with biological experiments to evaluate our computational results on some important biochemical interactions and networks, such as the mechanisms in diseases, cancer therapy and production of biomass energy. We will use multiple strategies to explore protein-protein, protein-DNA and protein-RNA
interactions, and find out the roles of these interactions in these networks.
行政院國家科學委員會補助專題研究計畫
■成果報告
□期中進度報告
生物系統內分子交互作用及生化路徑之大規模分析--(子計畫三)建立大
型分子交互作用資料庫系統與資料探索(3/3)
Data management and exploration of molecular interactions and
pathways (3/3)
計畫類別:□ 個別型計畫 ■ 整合型計畫
計畫編號: NSC 99-2627-B-009-003
執行期間:
2010 年 08 月 01 日至 2011 年 07 月 31 日
計畫主持人: 黃憲達 教授
共同主持人:
計畫參與人員: (請 Sherry 更新)
博士班研究助理 林豐茂、許博凱、許勝達
碩士班研究助理 黃至昶、邱致閔、吳維芸
碩士班研究助理 李佳融
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整報
告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年■二年後可公開查詢
執行單位:國立交通大學 生物科技系、生物資訊所
行政院國家科學委員會專題研究計畫成果報告
生物系統內分子交互作用及生化路徑之大規模分析--(子計畫三)建立大
型分子交互作用資料庫系統與資料探索(3/3)
Data management and exploration of molecular interactions and
pathways (3/3)
計畫編號:
NSC 99-2627-B-009-003
執行期限:99 年 8 月 1 日至 100 年 7 月 31 日
主持人:黃憲達 教授/主任 國立交通大學 生物科技系暨生物
資訊所
計畫參與人員:
(請 Sherry 更新)
博士班研究助理 林豐茂、許博凱、許勝達
碩士班研究助理 黃至昶、邱致閔、吳維芸
碩士班研究助理 李佳融
目
錄
中文摘要... iii 英文摘要... iv 一、研究計畫簡述 ... 5 二、相關研究成果... 13 參考文獻... 18中文摘要
本跨領域整合計畫將延續上期研究成果,首要目的在利用已建立生物分子序列- 結構-功能-生化路徑間的關係,深入研究分子間交互作用機制、演化關係並建立基 因調控網路模型。基因序列、蛋白質及其生物功能間的複雜關係與疾病、藥物、生醫 研究密不可分,生化反應網絡由生物分子的交互作用組成,這是蛋白質體學中亟待解 決的問題。本計畫的研究成果,有助於高精確度地分析、預測蛋白質交互作用及生化 網路的構成方式,以及未來藥物的發展。簡言之,即是以序列、結構資訊與演算法為 基礎,解析生物系統(biological systems),並以實驗驗證之。 本計畫包括四個子計畫,分別是以生化網路演化關係研究分子交互作用與生化路 徑(子計畫一)、智慧型最佳化方法用於基因網路的重建與分析(子計畫二)、建立大型 分子交互作用資料庫系統與資料探索(子計畫三),以及蛋白質於細胞位置之預測與蛋 白質間的交互作用:由基因體序列到蛋白質功能(子計畫四)。四者的緊密結合,可以 涵蓋由基因、蛋白質、訊息傳遞路徑(signaling pathway)、代謝路徑(metabolic pathway) 到基因調控網路(gene regulatory networks)間各層次的完整研究。玆列出子計畫三(即本精簡報告之子計畫)研究目標條列如下: 一. 建置超大型生物分子交互作用(molecular interactions)及生化路徑(pathways)資 料庫(database),涵蓋基因、蛋白質、生物分子交互作用、生化路徑、基因調控網 路,進行收集及整合,並自動更新。這個資料庫系統將具備完整性、支援物種完 整以及不重複等特性(子計畫三、二、一及四)。 二. 以演化式最佳化方法等機器學習(machine learning)方法重建基因調控網路,同時 對實驗資料不足之情況,提出有效之改善方法。並利用子計畫一及二提出的分子 交互作用模型與子計畫三資料庫得到之知識,使重建的網路模型符合真實之生物 系統。 三. 針對疾病(disease)、癌症治療(cancer therapy)、環境微生物及生質能源等相關議 題中的特定生物系統機制,以多角度探討蛋白質-蛋白質、蛋白質-DNA 及蛋白質 -RNA 之間的交互作用,以及這些交互作用在生化路徑及調控網路中扮演的角色, 並以實驗驗證之(子計畫三、二、一及四)。
英文摘要
The primary theme of our integrated project is to investigate molecular interactions and evolution relationships, and build various pathways (i.e. signaling pathways and metabolic pathways) and networks (e.g. gene regulatory networks). Disease, drug and biomedical researches correlated closely with the complications among gene sequence, protein and biological functions. The fruitful outcomes of our project will be in accurately analyzing and predicting molecular interactions and biochemical network components, moreover, drug developments. Furthermore, our predictions will be immediately confirmed by cellular and viral experiments.
The project is composed of four subprojects: Network evolution for studying molecular interactions and pathways (subproject 1); intelligent optimization methods for reconstruction and analysis of gene networks (subproject 2); data management and exploration of molecular interactions and pathways (subproject 3), and Protein subcelluar localization prediction and protein-protein interactions: from genomic sequences to protein functions (subproject 4). This integrated project studies molecular interactions and networks on genes, proteins, signaling pathways, metabolic pathways, and gene regulatory networks.
The specific aims of subproject 3 (for this report) are listed as follows:
1. To establish an integrated database system, including sequence, structure, molecular interaction, biochemical pathways, and gene regulatory networks. This large database can update and integrate information w automatically. The database is designed as a comprehensive, broad species coverage and non-redundant molecular interaction repository.
2. To reconstruct the gene regulatory networks by optimization methods, such as machine learning methods, and to revise deficiency of experimental data. We will fit the reconstructed gene regulatory networks to the natural biological systems based on the molecular interactions models (subprojects 1 and 2) and the biological databases (subproject 3).
3. To cooperate with biological experiments to evaluate our computational results on some important biochemical interactions and networks, such as the mechanisms in diseases, cancer therapy and production of biomass energy. We will use multiple strategies to explore protein-protein, protein-DNA and protein-RNA interactions, and find out the roles of these interactions in these networks.
一、研究計畫簡述
Motivation and the aims: The availability of molecular experiment data such as
sequence data, structural data, functional information and molecular interactions makes it possible to computationally explore the sequence-structural-function relationship in biological systems. However, comprehensive data collection and efficient data management method are crucial when doing such a biological data-driven analysis. In this proposed sub-project, in order to support the investigation of the other sub-projects in this proposed overall project, we plan to establish a biological data warehouse to integrate a variety of biological databases containing molecular interaction data, such as protein-protein interaction, protein-DNA interaction and metabolic pathways. The molecular interaction data warehouse, namely BioNet, can provide effective both the experimentally validated molecular interactions and the computationally predicted molecular interacted relationships among proteins and nucleotides. The data warehouse will be comprehensive, broad species coverage and non-redundant molecular interaction repository.
Data Warehousing System: Effective analysis of genome sequences and associated functional data requires access to many different kinds of biological information. A data warehouse plays an important role for storage and analysis for genome sequence and functional data. For example, when analyzing gene expression data, it may be useful to have access to the sequences upstream of the genes, or to the cellular location of their protein products. Such information is currently stored in different formats at different sites in a way that does not readily allow integrated analyses.
A data warehouse system is a repository of integrated information, which can be provided for query or analysis. Data warehouse systems can collect and maintain information form multiple distributed, autonomous, or heterogeneous information sources, related data retrieved from the information source can be processed and transformed into internal types available for data warehouse systems. The related data may be integrated with other information already existing in a data warehouse system, for part of results responded to user has been calculated and restored in a data warehouse system.
The main functions of data warehouse systems are retrieving, filtering, integrated related information required by complex queries. In contradiction to on-demand approach (extract data only when processing queries) of traditional databases, data warehouse system provides an in-advance approach (interested data are retrieved from information source in advance). It is not necessary to re-calculate the whole query result, because some parts of the result has been calculated and restored in repository (data warehouse), and these parts can be used directly by data warehouse system. Therefore, required time can be reduced by handing complex queries to data warehouse system. Because processed information has been stored in data warehouse, there exists inconsistency between data warehouse and underlying information sources.
The information sources are usually database systems, but may be non-traditional data such as general files, HTML and SGML documents, news wires, knowledge base, and legacy systems, etc. Each information source connects to a wrapper/monitor. The major tasks of the wrapper/monitor are the translation and change detection. The wrapper is responsible for translating the schema of the information source it concerns to the schema which is used by the data warehouse system. The monitor module is in charge of
detecting any change from the information source it connects to, and reporting those changes to the component above, the integrator. Any change from information sources will be propagated to the integrator. The integrator is responsible for bringing source data into the data warehouse, propagating changes in the source relations to the data warehouse, and maintaining the data extracted in the data warehouse, which may include merging, filtering and summarizing information from different information sources. When storing integrated data, it may need to obtain further information from the same or different information sources. Then it would send requests to the appropriate wrapper modules below it.
Methods: We plan to design and implement the architecture of the molecular
interaction data warehouse. The data sources, which will be integrated into the molecular interaction data warehouse, are divided into two categories. (1) The
experimentally validated molecular interaction databases that will be integrated in the
first year are listed as follows: MINT, BIND, DIP, TRNSFAC, RegulonDB, EcoCyc and KEGG. The curated databases containing the experimentally validated molecular interaction data in public domain are obtained and integrated into the warehouse. (2) The
computationally predicted molecular interactions databases that will be integrated in
the second year are lists as follows: STRING, InterDom and Predictome. These databases will be integrated to enhance the molecular interaction data warehouse to provide more molecular interactions. We plan to integrate a variety of putative molecular interaction databases into the data warehouse.
We plan to refine the molecular interaction data in the warehouse to make non-redundant molecular interactions. To provide effective interface for end users and research developers in the other sub-projects, we plan to design and implement the application interface and graphical interface to the molecular data warehouse. The application interface can provide data extraction functionalities from the data warehouse for researchers. The graphical interface can facilitate the representation of biological networks or pathways reconstructed by the molecular interaction data.
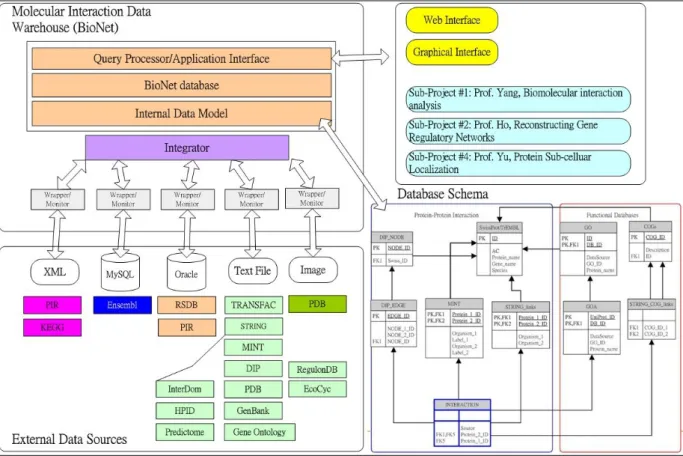
Figure 1. The proposed system architecture.
System Architecture: In this section, we will briefly describe the proposed methods to
collect and integrate multiple molecular interaction data sources. The proposed system architecture is shown in Fig. 1. The molecular interaction data warehouse, namely
BioNet, comprises the external data sources, the internal data model, the unify query
interface, the wrapper and monitor, and the integrator. The external data sources in different formats such as MINT, STRING and BIND and so son. The relational data model will be selected as the internal data model. Especially, the system will provide web interface and graphical interface to facilitate the access of the molecular interaction data in the BioNet warehouse. In order to support the requirements of the molecular interaction data for the investigation of the other sub-projects of the overall project, application interface is provided to effectively communicate with other analyzing programs developed in other sub-projects.
Data Collection ~ To collect molecular interaction data sources
Data Sources: Experimentally Validated Molecular Interaction Databases
DIP: Database of Interacting Proteins (1) documents experimentally determined
protein-protein interactions. Up to March 2005, there are 44,349 interactions among 17,048 proteins. Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) is a database of known and predicted protein-protein interactions. STRING (2) currently contains 444,238 genes in 110 species. Molecular Interaction Network Database (BIND) (3) is an archive information about interactions, molecular complexes and pathways occur among protein, RNA, DNA, gene. Up to March 2005, there are 77,732 interactions among 32,551 proteins. A Molecular INTeractions database (MINT) (4) is a relational database designed to store interactions between biological molecules. Presently MINT contains 18,115 interactions among 42,481 proteins.
KEGG: The Kyoto Encylopedia of Genes and Genomes (KEGG) (5,6) database consists of the
PATHWAY database for the computerized knowledge on molecular interaction networks such as pathways and complexes, the GENES database for the information about genes and proteins generated by genome sequencing projects, and the LIGAND database for the information about chemical compounds and chemical reactions that are relevant to cellular processes. Pathway data is provided by the KEGG database based on the KEGG Markup Language (KGML) which is currently available only for metabolic pathway.
DIP (1) (Database of Interacting Proteins) database documents experimentally determined
protein-protein interactions. The database integrates the diverse body of experimental evidence on protein-protein interactions into a single, easily accessible online database. The DIP was initially developed to store and organize information on binary protein-protein interactions that was retrieved from individual research articles (1). Up to June 2004, there are 44,349 interactions among 17,048 proteins.
BIND (Molecular Interaction Network Database) (3) is an archive information about
interactions, molecular complexes and pathways occur among protein, RNA, DNA, gene. The BIND records come from published literature and submissions. BIND developed a graphical analysis tool that provides users with a view of the domain composition of proteins in interaction and complex records to help relate functional domains to protein interactions. An interaction network clustering tool has also been developed to help focus on regions of interest. Up to June 2004, there are 77,732 interactions among 32,551 proteins.
MINT ex (4) is a relational database designed to store interactions between biological
molecules. Beyond cataloguing the formation of binary complexes, MINT was conceived to store other type of functional interactions namely enzymatic modifications of one of the partners. Presently MINT focuses on experimentally verified protein-protein interactions with special emphasis on mammalian organisms. Both direct and indirect relationships are considered. MINT consists of entries extracted from the scientific literature by curators. The interaction data can be easily extracted and viewed graphically through the 'MINT Viewer'. Presently MINT contains 18,115 interactions among 42,481 proteins.
consensus, however, at present most consensus descriptions are unreliable in the sense that they tend to give many false positives when compared against the genome sequences of even modest length”. Despite of these limitations, this study describes the binding sites using consensus patterns. The latest version of TRANSFAC (release 8.4) contains 5,919 transcription factors and 14,782 transcriptional factor binding sites.
We also integrate gene and protein function databases that offers more relationships among those unknown proteins. Therefore, Gene Ontology (GO) (10) database is used and briefly described in the following. GO provides structured, controlled vocabularies and classifications that cover several domains of molecular and cellular biology and are freely available for community use in the annotation of genes, gene products and sequences. Gene Ontology Annotation (GOA1) (11) is an integrated resource of GO annotations to the UniProt2
(12) Knowledgebase. The GOA database uses the GO vocabulary to provide high quality electronic and manual annotations to gene products contained in UniProt (Swiss-Prot, TrEMBL, and PIR-PSD).
Data Sources: Computational Predicted Molecular Interaction Databases
STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) (2) is a
database of known and predicted protein-protein interactions. The interactions include direct (physical) and indirect (functional) associations; there are derived from four sources: genomic context, high throughput experiments, co-expression, and previous knowledge. Functional links between proteins can often be inferred from genomic associations between the genes that encode them: groups of genes that are required for the same function tend to show similar species coverage, are often located in close proximity on the genome (in prokaryotes), and tend to be involved in gene-fusion events. STRING is a pre-computed global resource for the exploration and analysis of these associations. STRING contains a unique scoring-framework based on benchmarks of the different types of associations against a common reference set, integrated in a single confidence score per prediction. STRING currently contains 444,238 genes in 110 species.
Predictome (13) is a database of predicted links between the proteins of 44 genomes based on the implementation of three computationalmethods—chromosomal proximity, phylogenetic profiling and domain fusion—and large-scale experimental screeningsof protein–protein interaction data. The combination ofdata from various predictive methods in one database allowsfor their comparison with each other, as well as visualizationof their correlation with known pathway information. As a repositoryfor such data, Predictome is an ongoing resource for the community,providing functional relationships among proteins as new genomicdata emerges.
HPID (The Human Protein Interaction Database) (14) is designed to provide human
protein interaction information precomputed from existing structural and experimental data, to predict potential interactions between proteins submitted by users, and to deposit new human protein interaction data directly from users through the web server. It aims to serve comprehensive protein interaction information using both bioinformatic and experimental data. Two types of interactions are available from the precomputed data and from online prediction: human protein interactions at the protein superfamily levels, and human protein interactions transferred from yeast protein interactions. ____________________________________
1
GOA http://www.ebi.ac.uk/GOA
2
Data Sources: Other Related Biological Databases (Gene Annotations and Protein Annotations)
GenBank (15) is a database of nucleotide sequences from over 14,000 organisms obtained primarily through submissions from individual laboratories using Third Party Annotation (TPA), BankIt, or Sequin program and batch submissions from large-scale sequencing project of Expressed Sequence Tags (EST), Sequence-Tag Sites (STS), Genome Survey Sequence (GSS), High-Throughput Genomic (HTG) and High-Throughput cDNA (HTC). UniGene (16) is a largely automated analytical system for producing an organized view of the transcriptome. HomoloGene (16) is a resource for exploring putative homology relationships among genes, bringing together curated homology information and results from automated sequence comparisons. UniGene clusters have been used as a source of gene sequences for automated comparisons.
UTRdb (17) is a specialized database of 5’ and 3’ untranslated regions of eukaryotic mRNA. The 5’ and 3’ untranslated regions of eukaryotic mRNAs may play a crucial role in the regulation of gene expression controlling mRNA localization, stability and translational efficiency. ARED (AU-Rich Element Database) [28] is the database whose aim is to search for ARE-mRNA (cDNA) using 3’UTR-specific ARE consensus sequences. The search strategy used in this database is a comprehensive approach for extracting mRNA (cDNA) with full-length coding regions while dealing with inconsistencies in GenBank in regard to nucleic acid molecule type.
Data Integration ~ To establish the molecular interaction data warehouse
Effective analysis of molecular interaction data requires access to many different sources of biological information. A data warehouse plays an important role for the storage and the analysis for molecular interaction data. A data warehouse is a repository of information collected from multiple sources, stored under a unified schema, and which usually resides at a single site.
In order to efficiently manage the information from multiple biological databases to facilitate the implementation of the proposed system, we incorporate the concept of data warehousing system to construct a biological data warehouse, which maintains, updates and integrates all the required biological databases in the proposed system.
We design and implement a data warehouse to integrate multiple heterogeneous biological data sources in the data types of text-file, XML, image, MySQL database model, or Oracle database model. The relational database model is incorporated in the internal database model of the biological data warehouse. Wrappers and monitors are designed for each type of biological database, which the wrappers are capable of converting the external data into the internal data model and the monitors monitor and update the states of the external data sources. The proposed data warehousing system also
database model, and Oracle database model are relational database model, which is the same as the internal database model in the data warehousing system. Especially, some external data sources contain more than one data types, e.g., the protein structures in the protein data bank (PDB) is in text-file as well as structural images. Generally, most of the external data sources provide the data files which can be downloaded freely and directly.
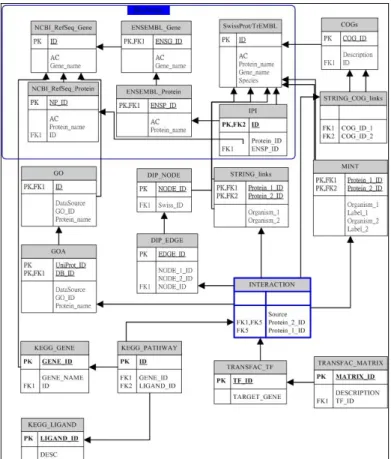
Figure 2. Database schema of the molecular interaction data warehouse.
The Internal Data Model: The proposed biological data warehouse can convert the
molecular interaction data in a variety of data formats into the coherent data model and store the data into the warehouse. We plan to select the relational data model as the internal data model. The internal database schema based on relational database model is designed to maintain the required molecular interaction information from different external data sources. The draft of the database schema is given in Fig. 2. For instance, the table (relation) INTERACTION stores the molecular interaction between protein and protein or protein and nucleotide.
The Integrator and the Wrapper/Monitor: In order to integrate the external data sources into the internal database in the molecular interaction data warehouse system, the integrator is responsible for bringing source data into the data warehouse, propagating changes in the source relations to the data warehouse, and maintaining the data extracted in the data warehouse, which may include merging, filtering and summarizing information from different information sources. When storing integrated data, it may need to obtain further information from the same or different information sources. Then it would send requests to the appropriate wrapper modules below it.
The wrapper/monitor module for each biological database are designed and implemented. The major tasks of the wrapper/monitor are the translation and change detection. The wrapper is responsible for translating the schema of the information source it concerns to the schema which is used by the data warehouse system. The monitor module is in charge of detecting any change from the information source it connects to, and reporting those changes to the component above, the integrator. Any change from information sources were propagated to the integrator.
Query interface, web interface and application interface: As to the interface of the
proposed molecular interaction data warehouse, we plan to provide a unify query interface for the development of web interface, graphical interface (visualization tool) and other applications or analyses in other sub-projects. Web interface was provided to facilitate end users or biologists to access the molecular interaction data in the BioNet warehouse. The application interface was provided for the development of the analyses of molecular interaction and molecular network.
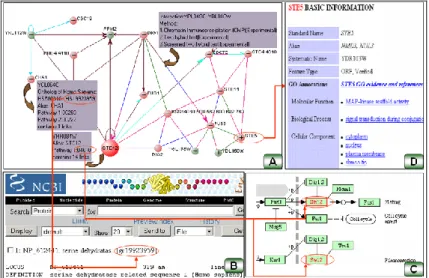
Figure 3. An example of user interface from VisANT (18).
Visualization of the molecular interaction network: The protein-protein interactions, protein-DNA interaction, protein-RNA interactions or gene functional associations can be denoted and viewed as an undirected graph G = ( V , E ) , where x , y V and server E. Let x and y represent proteins and ( x , y ) E represents an interaction or association between proteins/genes x and y (19). A variety of network visualization tools, such as Cytoscape (20), Osprey (21) and VisANT (18), are able to display varying aspects of the physical interaction, expression profiles and the related protein annotations. An
二、相關研究成果
在研究的過程中,各個子計畫密切合作,共建立了八個與生物資訊及系統生物 相關網站,以提供國內外專家學者使用,使成果可以共享與廣泛測試,並達到 各子計畫的整合。我們也從使用者的回饋,修正我們的方法,讓我們的方法更 符合生物學家的需求。 1. sRNAMap: http://srnamap.mbc.nctu.edu.tw/ 2. FMM: http://FMM.mbc.nctu.edu.tw/ 3. RiboSW: http://ribosw.mbc.nctu.edu.tw/ 4. N-Ace: http://n-ace.mbc.nctu.edu.tw/ 5. MASA: http://masa.mbc.nctu.edu.tw/ 6. miRExpress: http://mirexpress.mbc.nctu.edu.tw/7. miRTarBase: http://mirtarbase.mbc.nctu.edu.tw/ (第三年), Nucleic Acids Research, 2011
8. miRTar: http://mirtar.mbc.nctu.edu.tw (第三年), BMC Bioinformatics, 2011
9. miRStart: http://miRstart.mbc.nctu.edu.tw ( 第 三 年 ), Nucleic Acids Research, 2011
10. RegPhos: http://RegPhos.mbc.nctu.edu.tw ( 第 三 年 ), Nucleic Acids Research, 2011
總計畫、子計畫二與子計畫三共同合作建立的 sRNAMap 資料庫 (Nucleic Acids Research, 2009),為全球第一個收集實驗驗證的微生物非編碼小片段核醣核酸 (Small non-coding RNAs, 簡稱 sRNAs)及其轉錄因子與標的基因之資料庫。資 料來源主要透過文獻整理及整合相關資料庫,提供使用者非編碼小片段核醣核 酸的詳細資訊,如 RNA 結構、Transcription Start Site 資訊、基因表現等,進 一步提供實驗驗證的相對應轉錄因子與標的基因。此研究成果,對於 sRNAs 的相關研究,有很大的助益。本計畫總計畫共同主持人曾慶平教授與子計畫三 主持人黃憲達、子計畫二主持人何信瑩,正進行 sRNA 實驗驗證及探討數個延 伸性的議題。(sRNAs 可直接調控菌體內的基因表現、DNA 複製與影響蛋白質 活性,除了直接參與代謝途徑外,與菌體吸附、致病機制、毒素釋放、抗藥性 等等現象亦息息相關) 總計畫、子計畫二與子計畫三共同合作發展二個智慧型最佳化方法用於基因網 路的重建與分析。一個是基於 S-System model 的基因調控網路方法 iTEAP, 可以得出各基因間調控的量化模型,目前已受邀發表在演化式計算重要應用專 書中。另一個基因網路重建的方法 GRNet,除了使用有時間序列關係的基因表
現量的資料,更透過使用文獻與各種物種公開的基因調控資料庫中已知的調控 關係(子計畫二與子計畫三共同合作),來建立有效且能夠不受實驗雜訊影響的 網路重建方法,並藉由生物實驗來驗證建立的網路的正確性(總計畫與子計畫 二共同合作)。目前已完成模擬實驗分析,將規劃實驗來進一步作用在微生物 非編碼小片段核醣核酸及其轉錄因子與標的基因(總計畫與子計畫三共同合 作)。 推行此跨領域整合型研究計畫,在人才培育、儀器設備、研究能量提昇、研究 團隊知識交流、論文發表經費等貢獻良多。推行此跨領域整合型研究計畫,本 研究團隊證實以生物資訊之策略,進行生物學上的可能性之探討,大量減少逐一 測試之時間及資源,再以實驗直接驗證、探討生物學及計算生物上之差異,可 有效建立系統生物之流程。 本團隊的計畫執行成果與績效良好,本“生物資訊跨領域計畫”對於國內發展生 物資訊貢獻卓著。惟系統生物學正快速發展中,因此可考慮以”系統生物與生 物資訊跨領域” 取代“生物資訊跨領域”。 建立分子交互作用資料庫。 子計畫三與總計畫共同主持人曾慶平教授及子計 畫二主持人何信瑩教授,進行細菌之 small RNA 相關研究。相關合作成果已共 同發表 sRNAMap 資料庫 (Nucleic Acids Research, 2009),為全球第一個收集實 驗驗證的微生物非編碼小片段核醣核酸(Small non-coding RNAs, 簡稱 sRNAs) 及其轉錄因子與標的基因之資料庫。基於這個研究成果,可以證明本計畫各子 計畫間合作密切,進行生物資訊跨領域合作計畫。目前本團隊正進行數個與 small RNA 相關的數個延伸性的議題。
miRNA 與 mRNA 交互作用研究。 我們與陽明大學鄒安平教授及中央研究院 基因體研究中心蕭宏昇研究員,進行 HCC (Hepatocellular carcinoma)與 miR-122 之相關研究。本團隊運用生物資訊分析方法預測出 45 個 miR-122 target genes 做為 candidates,鄒安平教授進行實驗驗證,其中 35 target genes 被證實為 miR-122 之 target genes,合作之成果發表於 Hepatology (SCI IF=11.355),為生 物資訊、分子生物與實驗動物三個領域合作的成功範例。
Regulatory RNA 偵測。 我們發展了一個截至目前最為準確的預測方法,用以 偵測 mRNA 上之 riboswitches,相關成果發表於 RNA (SCI IF=5.018)。此研究 成果對於 mRNA 上 riboswitches 的註解,以及探討 riboswitches 在生物體中的 調控功能很有幫助 (Riboswitches 為生物體參與重要代謝功能的 Regulatory RNA。其中牽涉了 Gene transcriptional regulation、post-transcriptional regulation、 RNA stability 等調控)。
Tools for post-translational modifications。我們針對蛋白質後轉譯修飾中的 Acetylation 及 methylation 發展電腦分析工具,相關研究成果已分別發表於 Journal of Computational Chemistry (2009 and 2010)。
評量上述成果,本計畫執行進度符合原預定目標。
發表期刊論文
1. M. McDonald, W.C. Wang, H.D. Huang*, and J.Y. Leu* (2011) "Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences", PLoS Biology, Vol. 9, No. 6, e1000622. (IF=12.916, Rank=8/283, Biochemistry and Molecular Biology)
2. T.Y. Lee, J. B.K. Hsu, W.C. Chang* and H.D. Huang* (2011) "RegPhos: a system to explore the protein kinase-substrate phosphorylation network in humans" Nucleic
Acids Research, Vol.39, pp. D777-87.
3. S.D. Hsu, F.M. Lin, W.Y. Wu, C. Liang, C.J. Lee, C.Y. Huang, A.P. Tsou and H.D.
Huang* (2011) "miRTarBase: a database curates experimentally validated
microRNA-target interactions"Nucleic Acids Research, Vol.39, pp. D163-9.
4. C.H. Chien, Y.M. Sun, W.C. Chang, P.Y. Chiang-Hsieh, T.Y. Lee, W.C. Tsai, J.T. Horng, A.P. Tsou*, H.D. Huang* (2011) "Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data" Nucleic Acids Research (accepted)
5. X. Zhang, H. Zhao, S. Gao, W.C. Wang, S. Katiyar-Agarwal, H.D. Huang, N. Raikhel and H. Jin* (2011) " Arabidopsis Argonaute 2 Regulates Innate Immunity via miRNA393*-Mediated Silencing of a Golgi-Localized SNARE Gene, MEMB12"
Molecular Cell, Vo. 42, No. 3, pp. 356-366. (IF=14.608, Rank=7/283, Biochemistry and Molecular Biology)
6. J.G. Chang, D.M. Yang, W.H. Chang, L.P. Chow, W.L. Chan, H.H. Lin, H.D. Huang, Y.S. Chang, C.H. Hung, and W.K. Yang (2011) "Small molecule amiloride modulates oncogenic RNA alternative splicing to devitalize human cancer cells" PLoS One, Vol. 6, No. 6, e18643.
7. W. Wu, H. Xiao, A. Laguna-Fernandez, G. Villarreal, K.C. Wang, G.G. Geary, W.C. Wang, H.D. Huang, J. Zhou, Y.S. Li, S. Chien, G. Garcia-Cardena, J.Y-J. Shyy* (2011) "Flow-regulation of Krüppel-like Factor 2 Is Mediated by MicroRNA-92a"
Circulation (accepted) (IF=14.816, Rank=1/95, Cardiac & Cardiovascular Systems) 8. T.Z. Lee, Yi-Ju Chen, T.C. Lu, H.D. Huang, and Y.J. Chen* (2011) "SNOSite: exploiting
maximal dependence decomposition to identify cysteine S-nitrosylation with substrate site specificity" PLoS One (accepted)
9. J.B.K. Hsu, C.M. Chiu, S.D. Hsu, W.Y. Huang, C.H. Chien, T.Y. Lee and H.D. Huang* "miRTar: an integrated system for identifying miRNA-target interactions in Human"
BMC Bioinformatics (accepted)
10. T.H. Chang, L.C. Wu, Y.T. Chen, H.D. Huang, B.J. Liu, K.F. Cheng, J.T. Horng (2011) "Characterization and prediction of mRNA polyadenylation sites in human genes",
Medical & Biological Engineering & Computing, Vol. 49, No. 4, pp. 463-72.
11. W. Sun, Y.S.J. Li, H.D. Huang, J. Y-J. Shyy*, S. Chien* (2010) "MicroRNA: a master regulator of cellular processes for bioengineering systems" Annual Review of
Biomedical Engineering, Vol. 12, pp. 1-27. (IF=11.235, Rank=1/59, Engineering, Biomedical)
12. T.Y. Lee, J.B.K. Hsu, F.M. Lin, W.C. Chang*, P.C. Hsu, and H.D. Huang* (2010) "N-Ace:
using solvent accessibility and physicochemical properties to identify protein N-Acetylation sites" Journal of Computational Chemistry, Vol. 31, No. 15, pp. 2759-71.
13. K.Y. Lin, C.P. Cheng , B. Chang , W.C. Wang , Y.W. Huang , Y.S. Lee, H.D. Huang, Y.H. Hsu, N.S. Lin* (2010) "Global analyses of small interfering RNAs derived from Bamboo mosaic virus and its associated satellite RNAs in different plants" PLoS One, Vol. 5, No. 8, e11928.
14. S.C. Peng, Y.T. Lai, H.Y. Huang, H.D. Huang, and Y.S. Huang* (2010) "A novel role of CPEB3 in regulating EGFR gene transcription via association with Stat5b in neurons" Nucleic Acids Research, Vol. 38, No. 21, pp. 7446-57.
15. L.C. Wu, J.L. Huang, J.T. Horng*, H.D. Huang (2010) “An expert system to identify co-regulated gene groups from time-lagged gene clusters using cell cycle expression data” Expert Systems with Applications, Vol. 37, No. 3, pp. 2202-2213.
16. T.H. Chang, L.C. Wu, J.H. Lin, H.D. Huang, B.J. Liu, K.F. Cheng, J.T. Horng* (2010) "Prediction of small non-coding RNA in bacterial genomes using support vector machines" Expert Systems with Applications, Vol. 37, No. 8, pp. 5549-5557.
17. C.H. Chou†,, W.C. Chang†, C.M. Chiu, C.C. Huang, and H.D. Huang* (2009) “FMM: a
web server for metabolic pathway reconstruction and comparative analysis”,
Nucleic Acids Research, Vol. 37, Vol. 37, W129-34.
18. T.H. Chang, H.D. Huang*, L.C. Wu, C.T. Yeh, B.J. Liu, J.T. Horng* (2009) “Computational Identification of riboswitches based on RNA conserved functional sequences and conformations”, RNA, 15(7), pp.1426-30.
19. W.C. Tsai†,, Paul W.C. Hsu†,, T.C. Lai†, († joint first authorship), G.Y. Chau, C.W. Lin,
C.M. Chen, C.D. Lin, Y.L. Liao, J.L. Wang, Y.P. Chau, M.T. Hsu, M. Hsiao*, H.D. Huang*, A.P. Tsou*. (2009) “MicoRNA-122, a tumor suppressor microRNA that regulates
21. W.C. Chang, T.Y. Lee, D.M. Shien, J. B.K. Hsu, P.C. Hsu, T.Y. Wang, J.T. Horng, H.D.
Huang* and R.L. Pan* (2009) “Incorporating support vector machine for identifying
protein tyrosine sulfation sites” Journal of Computational Chemistry, Vol. 30, No. 9, pp. 2526-2537.
22. H.Y. Huang, H.Y. Chang, C.H. Chou, C.P. Tseng, S.Y. Ho, and H.D. Huang*. (2009) “sRNAMap: Genomic maps for small non-coding RNAs, their regulators and their targets in microbial genomes” Nucleic Acids Research, Vol.l 37, D150-D154.
23. W.C. Wang, F.M. Lin, W.C. Chang, K.Y. Lin, H.D. Huang*, and N.S. Lin* (2009) “miRExpress: Analyzing high-throughput sequencing data for profiling microRNA expression,” BMC Bioinformatics, 10, 328.
24. T.Y. Lee, J.B.K. Hsu, W.C. Chang, T.Y. Wang, P.C. Hsu, and H.D. Huang*. (2009) “A Comprehensive Resource for Integrating and Displaying Protein Post-Translational Modifications”, BMC Research Notes, 2(1):111.
25. L.C. Wu, H.D. Huang, Y.C. Chang, Y.C. Lee, J.T. Horng*. (2009) “Detecting LTR structures in human genomic sequences using profile hidden Markov models”
Expert Systems with Applications, Vol. 36, No. 1, pp. 668-674.
26. L.C. Wu, J.X. Lee, H.D. Huang, B.J. Liu, J.T. Horng*. (2009) “An expert system to predict protein thermostability using decision tree” Expert Systems with
參考文獻
1. Xenarios, I., Salwinski, L., Duan, X.J., Higney, P., Kim, S.M. and Eisenberg, D. (2002) DIP, the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res, 30, 303-305.
2. Christian von Mering, M.H., Daniel Jaeggi, Steffen Schmidt, Peer Bork, and Berend Snel. (2003) STRING: a database of pridicted functional associations between proteins. Nucleic Acids Res, 31, 258-261.
3. Bader, G.D., Betel, D. and Hogue, C.W. (2003) BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res, 31, 248-250.
4. Zanzoni, A., Montecchi-Palazzi, L., Quondam, M., Ausiello, G., Helmer-Citterich, M. and Cesareni, G. (2002) MINT: a Molecular INTeraction database. FEBS Lett, 513, 135-140.
5. Kanehisa, M., Goto, S., Kawashima, S. and Nakaya, A. (2002) The KEGG databases at GenomeNet. Nucleic Acids Res, 30, 42-46.
6. Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. and Hattori, M. (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res, 32, D277-280.
7. Matys, V., Fricke, E., Geffers, R., Gossling, E., Haubrock, M., Hehl, R., Hornischer, K., Karas, D., Kel, A.E., Kel-Margoulis, O.V. et al. (2003) TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res, 31, 374-378.
8. Wingender, E. (2004) TRANSFAC, TRANSPATH and CYTOMER as starting points for an ontology of regulatory networks. In Silico Biol, 4, 55-61.
9. Brazma, A., Jonassen, I., Vilo, J. and Ukkonen, E. (1998) Predicting gene regulatory elements in silico on a genomic scale. Genome Res, 8, 1202-1215.
10. Harris, M.A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C. et al. (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res, 32 Database issue, D258-261.
11. Camon, E., Barrell, D., Lee, V., Dimmer, E. and Apweiler, R. (2004) The Gene Ontology Annotation (GOA) Database - An integrated resource of GO annotations to the UniProt Knowledgebase. In Silico Biol, 4, 5-6.
12. Bairoch, A., Apweiler, R., Wu, C.H., Barker, W.C., Boeckmann, B., Ferro, S., Gasteiger, E., Huang, H., Lopez, R., Magrane, M. et al. (2005) The Universal Protein Resource (UniProt). Nucleic Acids Res, 33 Database Issue, D154-159.
13. Mellor, J.C., Yanai, I., Clodfelter, K.H., Mintseris, J. and DeLisi, C. (2002) Predictome: a database of putative functional links between proteins. Nucleic Acids Res, 30, 306-309.
14. Han, K., Park, B., Kim, H., Hong, J. and Park, J. (2004) HPID: the Human Protein Interaction Database. Bioinformatics, 20, 2466-2470.
15. Benson, D.A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J. and Wheeler, D.L. (2003) GenBank. Nucleic Acids Res, 31, 23-27.
16. Wheeler, D.L., Church, D.M., Edgar, R., Federhen, S., Helmberg, W., Madden, T.L., Pontius, J.U., Schuler, G.D., Schriml, L.M., Sequeira, E. et al. (2004) Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res, 32 Database issue, D35-40.
17. Pesole, G., Liuni, S., Grillo, G., Licciulli, F., Mignone, F., Gissi, C. and Saccone, C. (2002) UTRdb and UTRsite: specialized databases of sequences and functional elements of 5' and 3' untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res, 30, 335-340.
18. Hu, Z., Mellor, J., Wu, J. and DeLisi, C. (2004) VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics, 5, 17.
19. Han, K. and Ju, B.H. (2003) A fast layout algorithm for protein interaction networks. Bioinformatics, 19, 1882-1888.
國科會補助計畫衍生研發成果推廣資料表
日期:2011/10/31國科會補助計畫
計畫名稱: (子計畫三)建立大型分子交互作用資料庫系統與資料探索(3/3) 計畫主持人: 黃憲達 計畫編號: 99-2627-B-009-003- 學門領域: 生物資訊跨領域研究無研發成果推廣資料
99 年度專題研究計畫研究成果彙整表
計畫主持人:黃憲達 計畫編號: 99-2627-B-009-003-計畫名稱:生物系統內分子交互作用及生化路徑之大規模分析--(子計畫三)建立大型分子交互作用資 料庫系統與資料探索(3/3) 量化 成果項目 實際已達成 數(被接受 或已發表) 預期總達成 數(含實際已 達成數) 本計畫實 際貢獻百 分比 單位 備 註 ( 質 化 說 明:如 數 個 計 畫 共 同 成 果、成 果 列 為 該 期 刊 之 封 面 故 事 ... 等) 期刊論文 0 0 100% 研究報告/技術報告 0 0 100% 研討會論文 0 0 100% 篇 論文著作 專書 0 0 100% 申請中件數 0 0 100% 專利 已獲得件數 0 0 100% 件 件數 0 0 100% 件 技術移轉 權利金 0 0 100% 千元 碩士生 0 0 100% 博士生 0 0 100% 博士後研究員 0 0 100% 國內 參與計畫人力 (本國籍) 專任助理 0 0 100% 人次 期刊論文 5 10 60% 研究報告/技術報告 0 0 100% 研討會論文 1 1 100% 篇 論文著作 專書 0 0 100% 章/本 申請中件數 0 0 100% 專利 已獲得件數 0 0 100% 件 件數 0 0 100% 件 技術移轉 權利金 0 0 100% 千元 碩士生 4 4 100% 博士生 3 3 100% 博士後研究員 0 0 100% 國外 參與計畫人力 (外國籍) 人次其他成果