行政院國家科學委員會專題研究計畫 成果報告
實驗及理論計算小分子醇類於氣相及異相催化分解及氣化
反應(3/3)
研究成果報告(完整版)
計 畫 類 別 : 個別型 計 畫 編 號 : NSC 99-2113-M-009-002- 執 行 期 間 : 99 年 07 月 01 日至 100 年 06 月 30 日 執 行 單 位 : 國立交通大學應用化學系(所) 計 畫 主 持 人 : 林明璋 計畫參與人員: 學士級-專任助理人員:于秀琴 碩士班研究生-兼任助理人員:李鎮全 博士班研究生-兼任助理人員:王載德 博士班研究生-兼任助理人員:黃雯妃 博士後研究:徐振峰 處 理 方 式 : 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢中 華 民 國 100 年 09 月 09 日
I 中文摘要
交通大學於97 年 1 月正式成立綠色能源科技中心(CGET, Center for Green
Energy Technology),並委以林明璋院士主持,以全力發展本校再生能源之研究。 中心的主要研究目標將朝跨領域整合型研究的方向來發展。專注的跨領域研究, 將利用最先進的研發技術來從事新型矽薄膜多層光電池、量子光電池、有機光電 池及乙醇燃料電池等研究,並配合高階的理論計算來更進一步瞭解這些重要體系 的光化學及燃料系統的化學反應及物理特性。 本三年期國科會個人計畫(97 年 7 月 1 日至 100 年 6 月 30 日),配合 CGET 發展跨領域研究的方向為主軸,和各研究領域的專家學者密切合作,以進行下列 重 要 課 題 的 基 礎 研 究 :(1)利用交通大學的衝擊波管做小醇類分子(CH3OH, C2H5OH 及 i-C3H7OH)熱分解及燃燒反應機制確立及有關基礎反應化學動力學的 研究;(2)小醇分子在固態氧化物表面熱分解及氧化的高階量子計算以供給固態 氧化物燃料電池( SOFC)大型模擬的應用。 此報告包含99 年 7 月 1 日至 100 年 6 月 30 日之研究成果。 關鍵詞:再生小醇分子、熱分解及氧化反應、氧化物燃料電池( SOFC)電極反應
II 英文摘要
At National Chiao Tung University (NCTU), an interdisciplinary renewable energy research center, CGET (Center for Green Energy Technology), was established in January 2008 with emphasis on studies of a new generation of silicon thin-film solar cells, organic solar cells and ethanol fuel cells. Prof. M. C. Lin of Emory University has assumed the Center’s directorship to coordinate the interdisciplinary research program funded by the Ministry of Education through the MOE-ATU Project. He is also in charge of the coordination of the ethanol fuel cell sub-project.
The proposed research support from NSC covers the period of July 1, 2008 – June 30, 2011 for the administration of CGET by the PI as well as for collaborative studies with faculty members at NCTU on high temperature kinetics and mechanisms for small alcohol decomposition and oxidation reactions using a shock tube currently available at NCTU. The measured kinetics for elementary processes related to their decomposition and oxidation at high temperatures such as H + ROH and OH + ROH
(R = CH3, C2H5 and i-C3H7), as well as the decomposition of RO radicals (CH3O,
C2H5O, i-C3H7O and their structural isomers, CH2OH, CH3CHOH, CH3CHOCH3,
CH3COHCH3 and CH3CHOHCH2), will be interpreted with high-level ab initio
molecular orbital and statistical theory calculations.
In addition, the decomposition and oxidation of the alcohols and alkoxyl radicals on solid oxide fuel cell electrode surfaces will be investigated by large-scale quantum chemical calculations to elucidate their decomposition mechanisms and predict their decomposition kinetics based on computed potential energy surfaces.
關鍵詞:Decomposition and oxidation, Small alcohols, Ethanol SOFC, Quantum calculations
III 目 錄 中文摘要...I 英文摘要...II 目 錄...III 報告內容...1 附 錄...11
1 報告內容 一、前言
This report covers the period Jul. 1, 2010 – Jun 30, 2011 centering on: 1), Determination of the kinetics and mechanisms for thermal decomposition and oxidation of small alcohols at high temperatures using a shock tube currently available at NCTU. The measured kinetics for elementary processes have been interpreted with high-level ab initio molecular orbital and statistical theory calculations; 2), Reactions related to the operation and optimization of ethanol solid oxide fuel cell (SOFC); 3), Management of the Center for Green Energy Technology and the Center for Interdisciplinary Molecular Sciences at NCTU. In the proposed collaborative research, 15 full papers have been completed and submitted for publications with additional 11 papers credited to NSC for works carried out at Emory University but written or edited by the PI during the past year visiting NCTU. Representative cases studied are discussed briefly below. More detailed results can be found in the list of publications covering the reporting period.
二、結果與討論
1. Ab initio chemical kinetics for the O(3P) + CH3 reaction: The effect of
roaming transition states on the CHO + H2 formation
The O(3P) + CH3 reaction is one of the most influential processes in the
combustion of hydrocarbon and bio-fuels. It produces H atom and CH2O as well as
CHO + H2. The latter product pairs were first detected by Leone and coworkers by
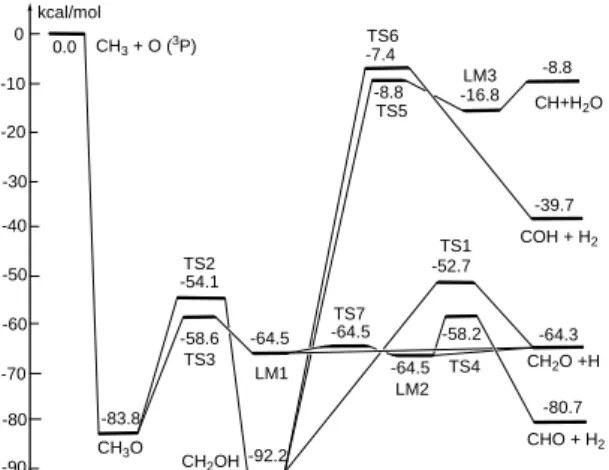
time-resolved CO emission with FTIR [1]. The surprising result was reported by Harding and Klippenstein through on-the-flight CPMD calculations to be a reaction which takes place without a transition state [2]. We investigated the kinetics and mechanism of this peculiar reaction at the CCSD/aug-cc-pVTZ, CCSD/aug-cc-pVDZ and B3LYP/6-311+G(3df,2p) levels of theory. The energies of all stationary points have been refined by single-point energy calculations at the CCSD(T)/aug-cc-pVTZ//CCSD/aug-cc-pVTZ level of theory. The predicted PES
presented in Fig. 1 shows that both CH2O + H and CHO + H2 can be produced by
isomerization/decomposition of the excited CH3O, from which the TS3 → LM1
step is favorable for the CH2O + H formation and the consecutive well-defined
path TS3 → LM1 → TS7 → LM2 → TS4 step leads to the CHO + H2 products,
contrary to the conclusion reached earlier by Harding and Klippenstein that the formation of the latter products occurred without a transition state. The rate constants and the individual product branching ratios predicted with the micro-canonical VTST/RRKM theory according to the CCSD(T) potential energy surface are in good agreement with experimental data as illustrated in Fig. 2 and Fig. 3, respectively.
TS7 and TS4 shown in Fig. 1 are effectively roaming transition states for the
formation of CHO + H2 which are competitive only with the presence of a large
2 0 -50 -10 kcal/mol -40 -20 -30 -52.7 -92.2 -54.1 -83.8 -58.6 CH2O +H CH3O CHO + H2 TS2 TS1 CH2OH TS3 -80.7 -58.2 TS4 -80 -70 -60 -8.8 -16.8 -8.8 TS5 LM3 CH+H2O 0.0 CH3 + O (3P) TS6 -7.4 COH + H2 -39.7 -64.3 -90 TS7 LM1 LM2 -64.5 -64.5 -64.5
Fig. 1. Schematic energy diagram of the CH3+O (3P) predicted on the ground
electronic doublet state potential energy surface at the CCSD(T)/aug-cc-pVTZ//CCSD/aug-cc-pVTZ level of theory.
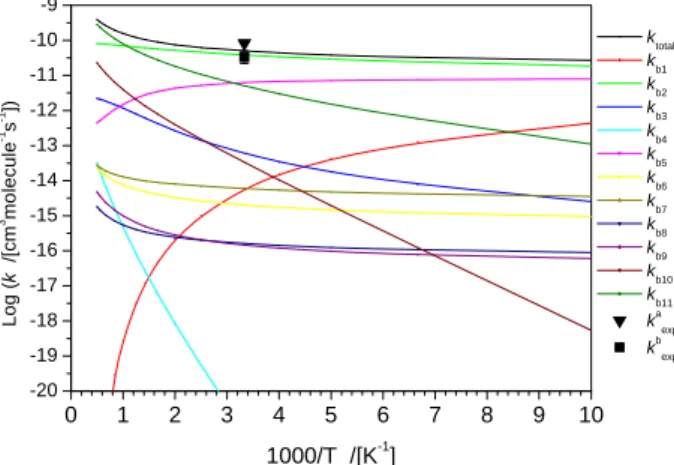
0 1 2 3 4 5 6.0x10-11 8.0x10-11 1.0x10-10 1.2x10-10 1.4x10-10 1.6x10-10 1.8x10-10 2.0x10-10 2.2x10-10 k /[cm 3molecule -1s -1] 1000/T /[K-1] B3LYP CCSD CASPT2 a b c d e f g h i j k l m n o
Fig. 2. Predicted total rate constant in the CH3 + O reaction in the temperature range
from 200 to 2600 K, computed with different Morse potentials for the CH3 + O
association process comparing with available data. a. Washida, 1973; b. Washida, 1980; c. Biordi, 1975; d. Bhaskaran, 1980; e. Plumb; 1982; f. Slagle, 1987; g. Zellner, 1988; h. Oser, 199; i. Lim, 1992; j. Fockenberg, 1999; k. Fockenberg, 2002; l. Hack, 2005; m. Dean, 1987; n. Yagi, 2004; o Harding, 2005. 0 500 1000 1500 2000 2500 3000 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0 HCO+H2: this work
Seakins and Leone, JPC, 1992 Fockenberg, et al., JPC, 1999, 2000, 2002 Hack, et al., PCCP, 2005, Branchiong ratio T /[K] CH2O+H: this work
Seakins and Leone, JPC, 1992 Fockenberg, et al., JPC, 1999, 2000, 2002 Hack, et al., PCCP, 2005,
Fig. 3. Individual product rate constants and total rate constant in the CH3 + O
3
2. Kinetics for CH3O and CH2OH isomerization/decomposition reactions
Both CH3O and CH2OH isomeric radicals play a key role in the
decomposition and oxidation of CH3OH. They are also important intermediates in
the combustion of hydrocarbons at the end of the oxidation chain processes. They
can be directly formed by reactions such as CH3 + OxH and CH3 + O2. We have
studied the ground electronic doublet-state potential energy surface of the C1H3O1
system at the CCSD(T)/aug-cc-pVTZ and G2M levels of theory (see Fig. 1). The stationary points were optimized by using the CCSD/aug-cc-pVTZ, CCSD/aug-cc-pVDZ and B3LYP/6-311+G(3df, 2p) methods. The result shows
that there are three low energy barrier processes including CH2O + H → CHO +
H2, CH2O + H → CH2OH, and CH2O + H → CH3O. For these three reactions
computed at the CCSD(T)/aug-cc-pVTZ// CCSD/aug-cc-pVTZ level, the forward potential barriers are predicted to be 6.1, 11.6, and 5.7 kcal/mol, with the corresponding reverse barriers of 22.5, 39.5, and 25.2 kcal/mol, respectively. The
heats of formation of CH2OH and CH3O are predicted to be -1.2 ± 0.4 and 7.6 ±
0.3 kcal/mol, respectively, using isodesmic reactions. The rate constant for the hydrogen abstraction reaction has been calculated by the canonical variational transition state theory with quantum tunneling and small-curvature corrections to
be k(CH2O + H → CHO + H2) = 2.28 ×10-19 T2.65 exp(-766.5/T) cm3molecule–1s–1
for the temperaturerange 200–3000 K. The rate constants for the addition and
decomposition reactions have been calculated by the microcanonical RRKM theory with the time-dependent master equation solution of the multiple quantum well system in the temperature range 200 – 3000 K at 1 Torr – 100 atm pressures. The predicted rate constants are in good agreement with most of available data
(see Figs. 4 and 5). At 1 atm Ar pressure, the rate constants for the CH2OH and
CH3O formation can be expressed in units of cm3molecule-1s-1 as:
ka1atm(CH2OH) = 7.00 × 10-10 T-1.40 exp(-2612.5/T) (200 – 1000 K)
= 3.41 × 107 T-6.23 exp(-7720.3/T) (1000 – 3000 K)
ka1atm(CH3O) = 2.32 × 10-10 T-1.22 exp(-1813.2/T) (200 – 800 K)
= 3.10 × 108 T-6.79 exp(-5573.9/T) (800 – 3000 K)
and those for the CH2OH and CH3O decomposition reactions can be expressed in
unit of s-1 as:
kd1atm(CH2OH) = 4.52 × 1034 T-7.11 exp(-22176.3/T) (500 – 3000 K)
4 0 1 2 3 4 5 -17 -16 -15 -14 -13 -12 -11 -10 -9 CVT TST CVT/ZCT CVT/SCT L og ( k /[c m 3molecu le -1s -1]) 1000/T /[K-1 ] a b c d e f g h i j k l m n
Fig. 4. Rate constants, k(CH2O+H), of the CH2O + H → CHO + H2.
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 -28 -26 -24 -22 -20 -18 -16 -14 -12 -10 kd(CH3O, low-P) kd(CH2OH, low-P) Log ( k /[ c m 3 mol e cu le -1 s -1 ]) 1000/T /[K-1] Ar N2 He a b c d e f
Fig.5. Low pressure rate constants, kd0(CH2OH) and kd0(CH3O), for the decomposition
reactions of CH2OH and CH3O, comparing with available data in the literature..
3. Thermal decomposition of C2H5OH in shock waves under highly diluted
conditions
The thermal decomposition of C2H5OH highly diluted in Ar (1 and 3 ppm)
has been studied by monitoring H atoms using the atomic resonance absorption spectrometry (ARAS) technique behind reflected shock waves over the temperature range 1450 − 1760 K at fixed pressure; 1, 1.45 and 2 atm. The rate constant and the product branching fractions have been determined by analyzing temporal profiles of H atoms; the effect of the secondary reactions on the results has been examined by using a detailed reaction mechanism composed of 103 elementary reactions. The apparent rate constant of ethanol decomposition can be
expressed as k1/s−1 = (5.28 ± 0.14) × 1010 exp[−(23530 ± 980)/T] (T = 1450 –
1670 K, P = 1 − 2 atm.) without a detectable pressure dependence within the tested pressure range of this study. Branching fractions for producing
CH3+CH2OH (1a) and H2O+C2H4 (1b) have been examined by a quantitative
measurement of H atoms produced in the successive decompositions of the
products CH2OH (1a): the pressure dependence of the branching fraction for
channel (1a) is obtained by a linear least-squares analysis of the experimental data
and can be expressed as 1a = (0.71±0.07) − (826±116)/T, (0.92±0.04) −
(1108±70)/T, (1.02±0.10) − (1229±168)/T for T = 1450 − 1760 K, at P = 0.99, 1.45 and 2.0 atm., respectively. The rate constant obtained in this study is found to
5
be consistent with previous theoretical and experimental results; however, the pressure dependence of the branching fraction obtained in this study is smaller than those of previous theoretical works. Modification of the parameters for the decomposition rate in the fall-off region is suggested to be important to improve the practical modeling of the pyrolysis and combustion of ethanol. These results have been published in J. Phys. Chem. A, 2011, 115, 8086 [3].
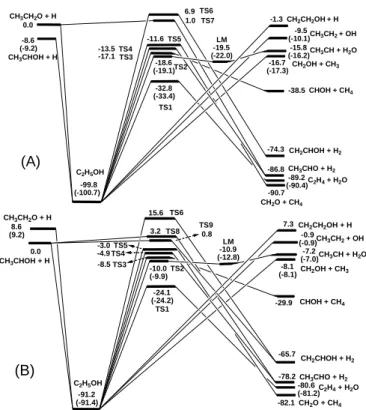
4. Ab initio kinetics for the C2H5O1 + H reactions
Both CH3CH2O and CH3CHOH are stable but highly reactive intermediates
of ethanol decomposition and oxidation reactions. Their reactions with H atoms, key chain carriers in these high temperature processes, generate a number of even
more reactive small radicals such as CH3, CH3O, CH2OH and OH. In this project,
we have investigated the chemical activation reactions of these two C2-containing radicals with H atoms can be mechanistically presented below for the association/decomposition and direct abstraction processes:
(a) CH3CH2O + H → CH3CH2OH* → CH3CH2OH (+M) (a1)
→ CH2OH + CH3 (a2) → CH3CH2 + OH (a3) → CH3CHOH + H (a4) →(TS1)→ CH2CH2 + H2O (a5) → (TS2)→LM→ CH3CH + H2O (a6) → (TS3) → CHOH + CH4 (a7) → (TS4) → CH3CHO + H2 (a8) → (TS5) → CH2O + CH4 (a9) → (TS7) → CH3CHO + H2 (a10) (b) CH3CHOH + H → CH3CH2OH* → CH3CH2OH (+M) (b1) → CH2OH + CH3 (b2) → CH3CH2 + OH (b3) → CH3CH2O + H (b4) →(TS1)→ CH2CH2 + H2O (b5) → (TS2)→LM→ CH3CH + H2O (b6) → (TS3) → CHOH + CH (b7) → (TS4) → CH3CHO + H2 (b8) → (TS5) → CH2 + CH4 (b9) → (TS8) → CH3CHO + H2
(b10) → (TS9) → CH2CHOH + H2
(b11)
The various transition states (TSn) and variational transition states for radical-radical production channels are summarized in Fig. 6:
6 CH3CH2O + H CH3CHOH + H C2H5OH TS1 C2H4 + H2O TS2 TS3 TS4 CH3CH + H2O CHOH + CH4 TS5 CH2O + CH4 -99.8 (-100.7) CH2OH + CH3 -16.7 (-17.3) -32.8 (-33.4) -89.2 (-90.4) -18.6 (-19.1) -15.8 (-16.2) -17.1 -38.5 -13.5 -86.8 -11.6 -90.7 6.9 CH2CHOH + H2 -74.3 CH3CHO + H2 -9.5 (-10.1)CH3CH2 + OH 1.0 TS6 TS7 -8.6 (-9.2) 0.0 -19.5 (-22.0) LM (A) -1.3 CH2CH2OH+ H CH3CH2O + H CH3CHOH + H C2H5OH TS1 C2H4 + H2O TS2 TS3 TS4 CH3CH + H2O CHOH + CH4 TS5 CH2O + CH4 -91.2 (-91.4) CH2OH + CH3 -8.1 (-8.1) -24.1 (-24.2) -80.6 (-81.2) -10.0 (-9.9) -7.2 (-7.0) -8.5 -29.9 -4.9 -78.2 -3.0 -82.1 15.6 CH2CHOH + H2 -65.7 CH3CHO + H2 -0.9 (-0.9)CH3CH2 + OH 3.2 TS6 TS8 0.0 8.6 (9.2) -10.9 (-12.8) LM TS9 0.8 (B) 7.3 CH2CH2OH + H
Fig. 6. Schematic diagrams of the potential energy surfaces predicted at the CCSD(T)/6-311+G(3df, 2p)//BH&HLYP /6-311+G(3df, 2p) level of theory
with zero-point vibrational energy corrections. (a) CH3CH2O + H and (b)
CH3CHOH + H. [E(CH3CH2O) = -154.1134354 a.u.; E(CH3CHOH) =
-154.129074 a.u and E(H) = -0.499810 a.u.; E(CH3CH2OH) = -154.788661
a.u]. The numbers in parentheses are predicted at the CCSD(T)/6-311+G(3df, 2p)//CCSD/6-311+G(3df, 2p) level of theory with ZPE correction.
[E(CH3CH2O) = -154.113875 a.u.; E(CH3CHOH) = -154.1296977 a.u and E(H)
= -0.499810 a.u.; E(CH3CH2OH) = -154.789264 a.u].
The potential energy surfaces of the two processes have been evaluated at the CCSD(T)/6-311+G(3df, 2p) level of theory with geometric optimization carried out at the BH&HLYP/6-311+G(3df, 2p) level. The direct hydrogen abstraction channels and the indirect association/decomposition channels from the chemically activated ethanol molecule have been considered for both reactions. The rate
constants for both reactions have been calculated at 100-3000 K and 10-4 Torr-103
atm Ar pressure by micro-canonical VTST/RRKM theory with master equation solution for all accessible product channels. The results show that the major
product channel of the CH3CH2O + H reaction is CH3 + CH2OH (a2) under the
atmospheric pressure conditions. Only at high pressure and low temperature, the
rate constants for CH3CH2OH formation by collisional deactivation (a1) become
dominant. For CH3CHOH + H, there are three major product channels; at high
temperatures, CH3+CH2OH production (b2) predominates at low pressures (P <
100 Torr), while the formation of CH3CH2OH by collisional deactivation (b1)
becomes competitive at high pressures and low temperatures (T < 500K). At
high temperatures, the direct hydrogen abstraction reaction producing CH2CHOH
+ H2 becomes dominant (b11). Rate constants for all accessible product channels
in both systems have been predicted and tabulated for modeling applications. The
predicted value for CH3CHOH + H at 295 K and 1 Torr pressure agrees closely
7
the rate constants for the thermal unimolecular decomposition of ethanol giving key accessible products have been predicted; those for the two major product channels taking place by dehydration and C-C breaking agree closely with available literature data (see Fig. 8); this work has been published in ref. [4]..
0 1 2 3 4 5 6 7 8 9 10 -20 -19 -18 -17 -16 -15 -14 -13 -12 -11 -10 -9 Log ( k /[c m 3 m o lecule -1 s -1 ]) 1000/T /[K-1] ktotal kb1 1 k b2 A k b3 a k b4 kb5 kb6 kb7 kb8 kb9 kb10 kb11 kaexpt kb expt
Fig. 7. Predicted braching ratios for the CH3CHOH + H reaction at 1 Torr, 1 atm, and
100 atm. 0.5 1.0 1.5 -10 -5 0 5 10 (B) L og ( k /[ s -1 ]) 1000/T /[K-1] this work a b c d 0.5 1.0 1.5 -10 -5 0 5 10 (A) Log ( k /[ s -1 ]) 1000/T /[K-1] this work a b c d
Fig. 8. Comparison of the predicted constants with those in the literature for the
decomposition reactions of (A) CH3CH2OH → CH3 + CH2OH and (B)
8
5. Unimolecular decomposition of the C3H7O1 isomeric radicals
The C3H7O1 radicals, (CH3)2COH, (CH3)2C(H)O and CH2CH(OH)CH3, are
intermediates of n- and iso- propanols, propane and larger hydrocarbon combustion reactions. The kinetics and mechanisms of their unimolecular decomposition and isomerization reactions have not been well characterized. In this work, the mechanisms for the isomerization and decomposition of these radicals have been studied at the CCSD(T) /6-311+G(3df,2p)//B3LYP/6-311+G (3df, 2p) level. The following product channels for these radical isomers are identified as shown in Fig. 9:
CH2C(H)OHCH3 → H + CH2C(OH)CH3 (1) → CH3 + CH2C(H)OH (2) → OH + CH2C(H)CH3 (3) → H + Propylene oxide (4) (CH3)2C(H)O → CH3 + CH3C(H)O (5) → H + (CH3)2CO (6) (CH3)2COH → H + (CH3)2CO (7) → CH4 + CH3CO (8) → H2O + CH3CCH2 (9) (CH3)2C(H)O ⇔ CH2C(H)OHCH3 (10, 11) (CH3)2C(H)O ⇔ (CH3)2COH (12, 13) CH2C(H)OHCH3 ⇔ (CH3)2COH (14, 15)
The rate constants for the low-lying energy channels are evaluated by using The transition-state-theory (TST) and the Rice-Ramsperger-Kassel-Marcus
(RRKM) theory. Among these radicals, (CH3)2COH is the most stable one,
CH2CH(OH)CH3 and (CH3)2COH lie above it by 7.8 and 11.3 kcal/mol,
respectively. For the (CH3)2CHO decomposition, the formation of CH3C(H)O +
CH3 is the dominant channel with 14.9 kcal/mol barrier; formation of (CH3)2CO +
H has an 18.4 kcal/mol barrier. Rate constant results show that at 1 atm (N2),
branching ratio of CH3C(H)O + CH3 is larger than 99% in the temperature range
of 298 ~ 3000 K. For the CH2CH(OH)CH3 decomposition, formation of
CH2=C(H)CH3 + OH, CH2=C(H)OH + CH3 needs to overcome 27.0, 30.9
kcal/mol barriers, respectively. For the (CH3)2COH decomposition, the lowest
energy channel is the formation of (CH3)2CO + H with 31.5 kcal/mol barrier.
The calculated rates for the formation of CH3C(H)O + CH3, (CH3)2CO + H and
CH2=C(H)CH3 + OH are in good agreement with available experimental values.
The results by coupling all the channels show that comparing with the low-lying energy processes, the isomerization rates among the isomers can be ignored due to their higher isomerization barriers.
9
Fig.9. Schematic energy diagram for the dissociation of IPA radicals computed at the CCSD(T)/6-311+G(3df,2p)//B3LYP/6-311+G(3df,2p) level.
6. Photo-fragmentation of C6H5CHO at 266, 248 and 193 nm
We had reported the preliminary result of this extensive study in our last report; the work resulted from a collaboration with IAMS (C. K. Ni and Y. T. Lee) and NCTU’s reaction dynamics laboratory of Y-P Lee employing the multi-mass ion imaging photo-fragmentation machine and step-scan tr-FTIR emission technique, respectively aid by our high-level ab initio MO calculations [5]. We also characterized the potential energies with the CCSD(T)/6-311+G(3df,2p) method and predicted the branching ratios for various channels of dissociation. Upon photolysis at 248 and 266 nm, two major channels for formation of HCO and CO, with relative branching of 0.37: 0.63 and 0.20 : 0.80, respectively, were
observed The C6H5 + HCO channel has two components with large and small
recoil velocities; the rapid component with average translational energy of ~25 kJ
mol−1 dominates. The C6H6 + CO channel has a similar distribution of
translational energy for these two components. Secondary dissociation channels
C6H5CO (from H elimination) → C6H5 + CO and C6H6 (from CO elimination) →
C6H5 + H or HCO (from HCO elimination) → CO + H, might also occur. IR
emission from internally excited C6H5CHO, ν3 (v = 1) of HCO, and levels v ≤ 2, J
≤ 43 of CO was observed; the latter has average rotational energy ~13 kJ mol−1
and vibrational energy ~6 kJ mol−1. Upon photolysis at 193 nm, similar
distributions of energy were observed, except that the C6H5 + HCO channel
becomes the only major channel with a branching ratio of 0.82 ± 0.10 and an
increased proportion of the slow component; IR emission from levels ν1 (v = 1)
and ν3 (v = 1 and 2) of HCO and v ≤ 2, J ≤ 43 of CO were observed; the latter has
average energy similar to that observed in photolysis at 248 nm. The observed product yields at different dissociation energies are consistent with statistical-theory predicted results based on the computed singlet and triplet potential energy surfaces given below:
CH2CH(OH)CH3 (CH3)2C(H)O (CH3)2COH 0.00 TS12 29.1 38.0 23.7 12.7 (CH3)2CO + H 3.5 21.9 18.4 8.0 CH2=C(H)OH + CH3 59.7 42.7 Propylene oxide + H -7.8 CH3C(H)O + CH3 19.3 28.1 -7.5 TS1 32.6 CH2=C(OH)CH3+ H 25.7 CH 3CC H2 + H2O 18.0 65.1 70.5 TS8 TS9 TS5 TS7 TS6 TS4 TS2, 30.9 TS10 CH3CO + CH4 TS11 31.6 25.5 TS3 27.0 CH2=C(H)CH3+ OH
10 (A) 0.0 2.5 367.8 TS1 CO+C6H6(1A1g) kJ/mol 365.3 463.6 TS7 + H 466.9 413.0 C6H5 + HCO S0 481.5 (248 nm) 618.6 (193 nm) C6H5 + TS8 479.9 397.1 TS2 286.3 C6H4CO + H2 C6H5CHO (1A') 588.5 C6H5 + COH t-C6H5COH (1A') 211.9 TS3 332.3 306.4TS4 230.3 c-C6H5COH (1A) 442.2 TS5 C6H5C + OH 683.0 247.9 C6H4CHOH (1A) 429.3 TS6 C6H5+CO+H C6H4CH+OH 610.3 C6H5CO+H 449.0 (266 nm) 436.0 TS 9 S1 S2 S3 S4 S5 S6 S7 S8 S9 S10 0.0001 338.2 353.2 0.0195 263.3 453.9 0.2424 240.2 497.0 0.0000 225.0 531.3 0.1433 199.7 598.2 0.0016 191.2 623.6 0.2305 190.5 627.4 0.0003 188.0 635.8 0.0037 187.5 637.5 0.2052 187.1 638.7 Oscillator
strengthsExcitation energiesnm kJ/mol
TS 10 363.7 C<C6H4 + H2O 428.9 518.3 TS 11 (B) 0.0 kJ/mol 365.3 463.6 466.9 S0 481.5 (248 nm) 618.6 (193 nm) 400.0 TS12 306.8 C6H5CHO (1A') C6H5CHO (3A") 405.5 TS13 C6H6CO (3A) 298.5 362.4 TS14 CO+C6H6( 3B 1u) 361.6 588.5 457.7 TS16 C6H5COH (3A) 315.2 683.0 596.5 413.0 C6H5+HCO C6H5CO+H 421.3 TS15 479.9 C6H5+TS8 C6H5C+OH C6H5+COH C6H5+CO+H TS17 TS7+H 449.0 (266 nm) 581.0 TS18 535.4 TS19 TS20 610.5 C6H4CH+OH t-C6H4CHOH (3A") 330.5 718.5 426.8 TS21 S5 S6 S7 S8 S9 S10 0.0001 338.2 353.2 S1 0.0195 263.3 453.9 S2 0.2424 240.2 497.0 S3 0.0000 225.0 531.3 S4 0.1433 199.7 598.2 0.0016 191.2 623.6 0.2305 190.5 627.4 0.0003 188.0 635.8 0.0037 187.5 637.5 0.2052 187.1 638.7 Oscillator
strengthsExcitation energiesnm kJ/mol
410.5 324.0 c-C6H4CHOH (3A") TS22 683.0 TS23 661.3 TS 24 558.03 672.6 C<C6H4 (3B1) + H2O C-C6H4 (3A")+ H2O
Fig. 10. Singlet (A) and triplet (B) potential energy surfaces of C6H5CHO [5].
7. References:
[1]. P. W. Seakins and S. R. Leone, J. Phys. Chem., 1992, 96, 4478
[2]. T. P. Marcy, R. R. Diaz, D. Heard, S. R. Leone, L. B. Harding and S. J. Klippenstein, J. Phys. Chem. A, 2001, 105, 8361.
11
[3]. Chih-Wei Wu, Hiroyuki Matsui, Niann-Shiah Wang and M. C. Lin, “Shock Tube Study on the Thermal Decomposition of Ethanol”, J. Phys. Chem. A, 2011, 115, 8086-8092.
[4]. Z. F. Xu, Kun Xu and M. C. Lin, “Thermal Decomposition of Ethanol. IV. Ab
Initio Chemical Kinetics for Reactions of H Atoms with CH3CH2O and
CH3CHOH Radicals”, J. Phys. Chem. A, 2011, 115(15), 3509-3522.
[5]. Arnab Bagchi, Yu-Hsuan Huang, Z. F. Xu, P. Raghunath, Yuan T. Lee, Chi-Kung Ni, M. C. Lin and Yuan-Pern Lee, "Photodissociation dynamics of
benzaldehyde (C6H5CHO) at 266, 248, and 193 nm", Chemistry--An Asian
Journal, accepted.
8. Publications for 2010-2011:
A. Research results on reaction kinetics and dynamics:
[1]. S. Y. Wu, P. Raghunath, J. S. Wu and M. C. Lin*, “Ab initio Chemical Kinetic
Study for Reactions of H Atoms with SiH4 and Si2H6: Comparison of Theory
and Experiment”, J. Phys. Chem. A, 114(1), 633-639 (2010).
[2]. D. H. Varma, P. Raghunath and M. C. Lin*, “Ab Initio Chemical Kinetics for
the Reaction of H atom with Si3H8”, J. Phys. Chem. A, 114, 3642-48 (2010).
[3]. Ku-We Lu, Hiroyuki Matsui*, Ching-Liang Huang, P. Raghunath, Niann-Shiah Wang and M. C. Lin, A Shock Tube Study on the Thermal
Decomposition of CH3OH”, J. Phys. Chem. A, 114, 5493-5502 (2010).
[4]. P. Raghunath and M. C. Lin*, “Ab Initio Chemical Kinetics for SiH3
Reactions with SixH2x+2 (x=1-4)”, J. Phys. Chem. A, 114, 13353–13361
(2010).
[5]. Keith Freel, J. Park, M. C. Lin and M. C. Heaven*, “Cavity ring-down spectroscopy of the phenyl radical in a pulsed discharge supersonic jet expansion”, Chem. Phys. Lett., 507(4-6), 216-220 (2011).
[6]. Z. F. Xu, Kun Xu and M. C. Lin*, “Thermal Decomposition of Ethanol. IV.
Ab Initio Chemical Kinetics for Reactions of H Atoms with CH3CH2O and
CH3CHOH Radicals”, J. Phys. Chem. A, 115(15), 3509-3522 (2011).
[7]. Chih-Wei Wu, Hiroyuki Matsui*, Niann-Shiah Wang and M. C. Lin*, “Shock Tube Study on the Thermal Decomposition of Ethanol”, J. Phys. Chem. A, 115, 8086-8092 (2011).
[8]. Arnab Bagchi, Yu-Hsuan Huang, Z. F. Xu, P. Raghunath, Yuan T. Lee, Chi-Kung Ni*, M. C. Lin* and Yuan-Pern Lee*, "Photodissociation dynamics
of benzaldehyde (C6H5CHO) at 266, 248, and 193 nm", Chemistry--An Asian
Journal, accepted.
B. Research results on studies relevant to solar cells and fuel cells:
[1]. YongMan Choi, M. C. Lin and Meilin Liu*, “Rational Design of Novel Cathode Materials in Solid Oxide Fuel Cells using First-Principles Simulations”, J. Power Sources, 195, 1441-45 (2010)
[2]. Wei-Ta Chen, Kuei-Bo Chen, Ming-Fang Wang, Sheng-Feng Weng, Chi-Shen
Lee* and M. C. Lin, “Enhanced Catalytic Activity of Ce1-xMxO2 (M = Ti, Zr,
and Hf) Solid Solution with Controlled Morphologies”, Chem. Comm., 46, 3286-3288 (2010).
[3]. Chih-Wei Wu, Chih-Wei Lu, Y-P Lee, Yu-Jong Wu*, Bing-Ming Cheng and
M. C. Lin*, “Blue/Near UV light emission from hybrid InN/TiO2
12
[4]. Hsin-Tsung Chen*, P. Raghunath and M. C. Lin*, “Computational
Investigation of O2 Reduction and Diffusion on 25% Sr-doped LaMnO3
Cathodes in Solid Oxide Fuel Cells", Langmuir, 27(11), 6787-93 (2011). [5]. W. F. Huang, P. Raghunath, and M. C. Lin*, “Computational Study on the
Reactions of H2O2 on TiO2 Anatase (101) and Rutile (110) Surfaces”, J.
Comp. Chem., 32(6), 1065-1081 (2011).
[6]. Tsai-Te Wang, P. Raghunath, Yun-Fang Lu, Yu-Chang Liu, Chwei-Huawn Chiou and M. C. Lin*, “Observation of Significant Enhancement in the
efficiency of a DSSC by InN Nanoparticles over TiO2-Nanoparticle Films”,
Chem. Phys. Lett., 510(1-3), 126-130 (2011).
[7]. Zhe Cheng, Jeng-Han Wang, YongMan Choi, Lei Yang, M. C. Lin and Meilin Liu*, “From Ni-YSZ to Sulfur-Tolerant Anode Materials for SOFCs: Electrochemical Behavior, in situ Characterization, Modeling, and Future Perspectives”, Energy and Eviron. Sci., in press.
C. Other publications credited to NSC supports are listed below:
[1]. S. C. Xu and M. C. Lin*, “Ab initio Chemical Kinetics for the NH2 + HNOx
Reactions: III. Kinetics and Mechanism for NH2 + HONO2”, Int. J. Chem.
Kinet., 42(2), 69-78 (2010).
[2]. R. S. Zhu and M. C. Lin*, “An Ab Initio Chemical Kinetic Study on the
Reactions of H, OH and Cl with HOClO3”, Int. J. Chem. Kinet., 42(4),
253-261 (2010).
[3]. Z. F. Xu and M. C. Lin*, “Computational Studies on Metathetical and Redox
Processes of HOCl in the Gas Phase: (II) Reactions with ClOx (x = 1−4)”,
Phys. Chem. A, 114(2), 833-838 (2010)
[4]. J. Park, Sonya Cates and M. C. Lin*, “Photolytically and Thermally Initiated
Reactions of NH3 with NOx (x=1,2)”, Combust. Sci. Technol., 182, 365-79
(2010).
[5]. S. C. Xu, S. Irle and M. C. Lin*, “Quantum Chemical Prediction of Reaction
Pathways and Rate Constants for Reactions of NO and NO2 with
Monovacancy Defects on Grapite (0001) Surfaces”, J. Phys. Chem. C, 114, 8375-8382 (2010).
[6]. Z. F. Xu and M. C. Lin*, “Computational Studies on Metathetical and Redox Processes of HOCl in the Gas Phase: (III) Its Self-Reaction and Interactions
with HNOx (x = 1-3)”, J. Phys. Chem. A, 114, 5320-26 (2010).
[7]. Z. F. Xu and M. C. Lin*, "Ab Initio Chemical Kinetic Study on Cl + ClO and Related Reverse Processes", J. Phys. Chem. A, 114, 11477-82 (2010).
[8]. R. S. Zhu and M. C. Lin*, “Ab Initio Chemical Kinetic Study on the
Reactions of ClO with C2H2 and C2H4”, J. Phys. Chem. A, 114(51),
13395-13401 (2010).
[9]. R. S. Zhu and M. C. Lin*, “Ab Initio Chemical Kinetics for Reactions of ClO
with Cl2O2 Isomers”, J. Chem. Phys., 134(5), 054307/1-054307/6 (2011).
[10]. R. S. Zhu and M. C. Lin*, “Ab initio chemical kinetics for ClO reactions with
HOx, ClOx and NOx (x=1, 2): A Review”, Comput. Theoret. Chem., 965(2-3),
328-339, (2011).
[11]. R. S. Zhu and M. C. Lin*, “First-Principles Study of Water Effect on the Sublimation of Ammonium Perchlorate”, Int. J. Energ. Mater. Chem. Propul., in press.
國科會補助計畫衍生研發成果推廣資料表
日期:2011/09/09國科會補助計畫
計畫名稱: 實驗及理論計算小分子醇類於氣相及異相催化分解及氣化反應(3/3) 計畫主持人: 林明璋 計畫編號: 99-2113-M-009-002- 學門領域: 物理化學無研發成果推廣資料
99 年度專題研究計畫研究成果彙整表
計畫主持人:林明璋 計畫編號: 99-2113-M-009-002-計畫名稱:實驗及理論計算小分子醇類於氣相及異相催化分解及氣化反應(3/3) 量化 成果項目 實際已達成 數(被接受 或已發表) 預期總達成 數(含實際已 達成數) 本計畫實 際貢獻百 分比 單位 備 註 ( 質 化 說 明:如 數 個 計 畫 共 同 成 果、成 果 列 為 該 期 刊 之 封 面 故 事 ... 等) 期刊論文 0 0 0% 研究報告/技術報告 0 0 0% 研討會論文 0 0 0% 篇 論文著作 專書 0 0 0% 申請中件數 0 0 0% 專利 已獲得件數 0 0 0% 件 件數 0 0 0% 件 技術移轉 權利金 0 0 0% 千元 碩士生 1 1 100% 博士生 2 2 100% 博士後研究員 1 1 100% 國內 參與計畫人力 (本國籍) 專任助理 1 1 100% 人次 期刊論文 15 5 100% 研究報告/技術報告 0 0 0% 研討會論文 0 0 0% 篇 論文著作 專書 0 0 0% 章/本 申請中件數 0 0 0% 專利 已獲得件數 0 0 0% 件 件數 0 0 0% 件 技術移轉 權利金 0 0 0% 千元 碩士生 0 0 0% 博士生 0 0 0% 博士後研究員 0 0 0% 國外 參與計畫人力 (外國籍) 專任助理 0 0 0% 人次其他成果
![Fig. 4. Rate constants, k(CH 2 O+H), of the CH 2 O + H → CHO + H 2 . 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5-28-26-24-22-20-18-16-14-12-10kd(CH3O, low-P)kd(CH2OH, low-P)Log (k /[cm3molecule-1s-1]) 1000/T /[K -1 ] Ar N2 He a b c d e f](https://thumb-ap.123doks.com/thumbv2/9libinfo/8421104.180523/8.892.295.580.124.341/fig-rate-constants-cho-ch-log-molecule-ar.webp)
![Fig. 10. Singlet (A) and triplet (B) potential energy surfaces of C 6 H 5 CHO [5]. 7](https://thumb-ap.123doks.com/thumbv2/9libinfo/8421104.180523/14.892.189.724.107.1014/fig-singlet-triplet-b-potential-energy-surfaces-cho.webp)
