R E V I E W
Open Access
Dysregulated transcriptional and post-translational
control of DNA methyltransferases in cancer
Ruo-Kai Lin
1,3,4and Yi-Ching Wang
2*Abstract
Cancer is a leading cause of death worldwide. Aberrant promoter hypermethylation of CpG islands associated with
tumor suppressor genes can lead to transcriptional silencing and result in tumorigenesis. DNA methyltransferases
(DNMTs) are the enzymes responsible for DNA methylation and have been reported to be over-expressed in various
cancers. This review highlights the current status of transcriptional and post-translational regulation of the DNMT
expression and activity with a focus on dysregulation involved in tumorigenesis. The transcriptional up-regulation of
DNMT gene expression can be induced by Ras-c-Jun signaling pathway, Sp1 and Sp3 zinc finger proteins and virus
oncoproteins. Transcriptional repression on DNMT genes has also been reported for p53, RB and FOXO3a transcriptional
regulators and corepressors. In addition, the low expressions of microRNAs 29 family, 143, 148a and 152 are associated
with DNMTs overexpression in various cancers. Several important post-translational modifications including acetylation
and phosphorylation have been reported to mediate protein stability and activity of the DNMTs especially DNMT1. In
this review, we also discuss drugs targeting DNMT protein expression and activation for therapeutic strategy against
cancer.
Keywords: DNA methyltransferase, Cancer, Tumor suppressor gene, Transcription, Post-translational modifications
Introduction
Cancer is a leading cause of death worldwide, accounting
for 8.2 million deaths in 2012 [1]. The process of
tumori-genesis needs to be initiated and promoted by molecular
abnormalities including oncogenes activation and tumor
suppressor genes (TSGs) inactivation [2]. Methylation
of CpG islands is one of the epigenetic modifications
in mammalian genome that modulates gene expression
without changes in the DNA sequence [2]. Aberrant
promoter hypermethylation of CpG islands associated
with TSGs can lead to transcriptional silencing and result
in tumorigenesis. DNA methylation is frequently not
restricted to a single CpG island but affects multiple
independent loci, reflective of a widespread deregulation
of DNA methylation pattern in different types of tumors
[3,4]. Development of genome-wide high-throughput
technologies has facilitated the identification of global
DNA methylation pattern [5,6]. For example, genomic
screening of 98 different primary human tumors has
revealed that on an average there exist about 600 aberrantly
methylated CpG islands in each tumor [7]. In addition,
an increase of methylation variability may contribute
to tumor heterogeneity [8]. Collectively, dysregulation
of DNA methylation is apparently one of the major
barriers to effective cancer diagnosis and treatment in
different types of cancer.
Epigenetic disorders give rise to several significant
human diseases including various cancers, neuron disorder,
psychosis, and cardiovascular diseases, many of which are
associated with altered expression and activity of DNA
methyltransferases (DNMTs) [9-13]. DNMTs are the
en-zymes responsible for DNA methylation through transfer
of methyl group to cytosine residue of CpGs [2]. Five types
of DNMTs have been identified, viz. DNMT1, 2, 3A, 3B,
and 3L. DNMT1 comprises a large N-terminal domain
with regulatory function and a smaller C-terminal catalytic
domain [14]. The regulatory domain harbors different
motifs and is involved in the intracellular delivery and
regulation of catalytic activity of DNMT1. DNMT1 has
been shown to prefer hemimethylated over
unmethy-lated DNA 30- to 40-fold in vitro [15-17]. It is referred to
as a
“maintenance” methyltransferase and is the primary
* Correspondence:[email protected]
2
Department of Pharmacology and Institute of Basic Medical Sciences, National Cheng Kung University, No.1, University Road, Tainan 70101, Taiwan Full list of author information is available at the end of the article
© 2014 Lin and Wang; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
enzyme responsible for copying the methylation patterns
after DNA replication. DNMT1 localizes to replication
foci and interacts with PCNA, a processivity factor for
DNA replication complex [18]. However, evidences show
that DNMT1 may also work together with DNMT3A
and DNMT3B in de novo methyltransferase activity in
certain genome in both embryonic cells and differentiated
somatic cells [19,20]. Many interacting proteins have been
reported to bind to their N-terminal region by
biochem-ical interaction assay [14]. For example, DNMT1 directly
interacts with histone modifying enzymes such as histone
H3K9 methyltransferase SUV39H1, histone H3K27
meth-yltransferase EZH2, and histone deacytelase HDAC1 and
HDAC2 [14,21]. DNMT1 also interacts with
methyl-CpG-binding proteins such as MBD2, MBD3 and MeCP2 and
with the heterochromatin binding protein HP1 [14].
Notably, DNMT1, DNMT3A, and DNMT3B are
over-expressed in a coordinate manner in most tumor tissues
and at a significantly higher level in cancer than in
non-tumorous tissues [22-24]. The mechanism underlying
DNMTs overexpression is worthy of comprehensive
discus-sion. Delineating mechanisms of DNMTs overexpression
will provide more information and strategies to remedy the
altered epigenetic states. It will offer more exciting
oppor-tunities that can reactivate epigenetically silenced TSGs and
critical anti-cancer pathways [25].
Transcriptional regulation of
DNMT gene expression
The earlier study on transcriptional regulation of DNMT
mediated by Ras-c-Jun signaling pathway provided a
mo-lecular explanation for the role of DNMT1 to
carcinogen-esis [26,27]. The expressions of DNMT1, DNMT3A and
DNMT3B genes are also controlled by Sp1 and Sp3 zinc
finger proteins [28,29]. Wilms' tumour 1 protein has been
shown to directly transactivate DNMT3A expression [30].
Homeobox B3 can bind to and activate DNMT3B gene
[31]. In addition to transcription factors, several important
transcriptional repressors have been reported to suppress
the DNMT1, DNMT3A and DNMT3B gene expression,
including p53, RB and FOXO3a (Table 1 and Figure 1).
The major findings are described below.
The p53-mediated regulation of
DNMT genes
The tumor suppressor gene p53 encodes a transcription
factor that mediates many downstream effects such as
growth arrest and apoptosis through activation or
repres-sion of its target genes [46]. However, the p53 gene is a
frequent target of missense mutation rendering it unable
to recognize the p53 consensus binding sites therefore loss
of transcriptional function of p53 in cancers [47].
Se-quencing analyses reveal that point mutation and small
intragenic deletion/insertion of p53 gene are significantly
associated with DNMT1 protein overexpression [32]. A
study shows that deletion of p53 in the HCT116 human
colon carcinoma cell line results in increase of DNMT1
mRNA and protein [48]. Wild-type p53 decreases DNMT1
promoter activity and expression level through the exon 1
region (−19 to +317) of DNMT1 promoter, which contains
p53 putative binding sites, in lung cancer cells [32,48].
In addition, wild-type p53 protein has been shown to
negatively regulate DNMT1 expression by forming a
complex with Sp1 protein and chromatin modifiers on
the DNMT1 promoter [32]. Low level of exogenous Sp1
expression enhances the repressive activity of endogenous
p53 on the DNMT1 promoter, whereas a high level of Sp1
expression upregulates DNMT1 gene expression level in
A549 (p53 wild-type) cells. In H1299 (p53 null) cells,
exogenous Sp1 induces DNMT1 expression in a
dose-dependent manner. A high level of Sp1, via its
COOH-terminal domain, induces interaction between p53 and
MDM2, resulting in degradation of p53 by
MDM2-mediated ubiquitination [32]. Clinical data from 102 lung
cancer patients indicated that overexpression of DNMT1
is significantly associated with p53 mutation and high
expression of Sp1 protein. In addition, patients with
over-expression of both DNMT1 and Sp1 proteins show poor
prognosis [32]. Cell and clinical data provide evidence
that deregulation of DNMT1 is associated with gain of
transcriptional activation of Sp1 and/or loss of repression
of p53. DNMT1 overexpression is involved in epigenetic
alterations of multiple TSGs that ultimately leads to lung
tumorigenesis and poor prognosis [32].
Some reports have also shown that Sp1 and Sp3 increase
the activity of DNMT1, DNMT3A, and DNMT3B
pro-moters by physical binding to their propro-moters in mouse
NIH3T3 cells or human embryonic kidney 293T cells
[28,29]. p53 is shown to suppress the promoter activity
and mRNA/protein expression of DNMT3A through
binding to its promoter and the suppression can be
atten-uated by knockdown of p53 [33]. Whether overexpression
of DNMT3A and DNMT3B resulted from the loss of
transcriptional regulation of p53/Sp1 warrants further
in-vestigation. It is justifiable to propose that overexpression
of DNMTs is associated with the gain of transcriptional
activation of Sp1 and/or the loss of repression of p53
(Figure 1A).
The RB-mediated regulation of
DNMT genes
The RB (retinoblastoma) protein is a tumor suppressor,
which plays a pivotal role in the negative control of the
cell cycle and in tumor progression [49]. The RB protein
represses gene transcription, required for transition from
G1 to S phase, by directly binding to the transactivation
domain of E2F and by binding to the promoter of the
target genes as a complex with E2F [50]. RB also represses
transcription by remodeling chromatin structure through
interaction with proteins such as HP1, SWI/SNF, HDAC1
and SUV39H1, which are involved in DNA methylation,
nucleosome remodeling, histone deacetylation and histone
methylation, respectively [51-54].
The mouse and human DNMT1 promoters are found to
contain E2F binding sites that are required for RB/E2F
regulation in wtPrE (wild-type prostate epithelial cell line)
[34]. DNMT1 is negatively regulated by E2F-RB-HDAC
pathway in mouse NIH3T3 embryonic fibroblast, monkey
COS-7 kidney cell, and saos-2 human osteosarcoma cell
lines [35]. In addition, DNMT1 mRNA can be diminished
by overexpression of RB protein in saos-2 cells and are
induced by deletion of RB gene in wtPrE cells [34,35].
RB also suppresses DNMT3A promoter activity and
mRNA/protein expression through binding with E2F1
protein to the DNMT3A promoter [36]. Repression of
DNMT3A by RB leads to the decrease of methylation
level globally and TSG specifically, such as RARβ, FHIT,
and RASSF1A genes [36]. Together, these data suggest that
RB is a transcriptional repressor of DNMT1 and DNMT3A
genes (Figure 1B).
The FOXO3a-mediated regulation of
DNMT genes
Forkhead O transcription factor 3a (FOXO3a) belongs to
a large protein family of transcriptional regulators
charac-terized by a conserved DNA-binding domain termed the
“forkhead-box” [55]. To date, many reports indicate a
tumor suppressor role for FOXO3a. For example, ectopic
overexpression of FOXO3a significantly impairs tumor
growth in cell and xenograft models in breast cancer and
promotes apoptosis in leukemia and prostate cancer cells
[56,57]. In addition, restrained transcriptional activity of
FOXO3a in cancer cells results in promoting angiogenesis
and tumor progression [58-60]. FOXO3a has been shown
to transcriptionally up-regulate apoptotic-related gene
such as p27kip [61], Bim [62], and Fas ligand [63]. In
contrast, FOXO3a could transcriptionally repress
micro-RNA21, which suppresses the expression of Fas ligand
[64]. Of note, the gene deletion of FOXO3a is found in
early-stage lung adenocarcinoma in smokers and tobacco
carcinogen-initiated lung tumors in mice [37,65].
Restor-ation of FOXO3a in FOXO3a-deficient lung cancer cells
increases the cell apoptosis response to nicotine-derived
nitrosamino ketone-mediated DNA damage [66]. The
last-mentioned two studies implicate that loss of FOXO3a
may contribute to lung cancer pathogenesis.
We recently showed that FOXO3a negatively regulates
DNMT3B promoter activity by interacting with the binding
element FOXO3a (+166 ~ +173) of DNMT3B promoter
[67]. Ectopically overexpressed FOXO3a or combined
treatment with doxorubicin to induce FOXO3a nuclear
accumulation leads to further binding at the distal FOXO3a
Table 1 Transcriptional regulation of
DNMT promoter activity and/or mRNA expression
Pathways Mechanisms DNMTs mRNA/promoters References
Down regulation
p53 p53/Sp1 transcriptional repression DNMT1/3A/3B [32]
RB/E2F RB/E2F transcriptional repression DNMT1/3A [33-36]
FOXO3a Transcriptional repression DNMT3B [37]
Up regulation
Ras/AP-1 AP1 transcriptional activation DNMT1 [26,27,38]
Sp1 Transcriptional activation DNMT1/3A/3B [28,29,32]
Sp3 Transcriptional activation DNMT1/3A/3B [28,29]
E2F Transcriptional activation DNMT1 [34,39]
ERK Unknow DNMT1/3A [40]
17β-estradiol ER-dependent transcription activation DNMT3B [41]
Homeobox B3 Promoter binding DNMT3B [31]
Wilms' tumour 1 Transcriptional activation DNMT3A [30]
Viruse induction
LMP1 Activation of JNK/AP-1 pathway DNMT1 [42]
BKV Tag and E1a pRB/E2F pathway DNMT1 [43]
HBx Promoter transcriptional activator DNMT1/3A [44]
HBx Promoter transcriptional repression DNMT3b [44]
HIV-1 Through transcription factor AP-1 DNMT1 [45]
LMP1: latent membrane protein 1.
BKV Tag and E1a: Human polyomavirus BKV large T antigen and adenovirus E1a. HBx: Hepatitis B virus X protein.
site (−249 ~ −242). Abundant FOXO3a represses DNMT3B
promoter by establishing a repressed chromatin structure,
while knockdown of FOXO3a results in an open chromatin
structure and high DNMT3B mRNA and protein
expres-sion. Importantly, enforced abundant nuclear accumulation
of FOXO3a could decrease expression of DNMT3B with
synergistic inhibition of tumor growth and decrease in
methylation status on TSGs in human lung tumor
xeno-graft specimens [67]. It is plausible that FOXO3a binds to
the FOXO3a DNA element of the DNMT3B promoter to
repress DNMT3B expression (Figure 1C).
Transcriptional deregulation of
DNMT genes by MDM2
overexpression
p53 protein is known to be degraded in cytoplasm by
ubiquitin-mediated proteasomal degradation pathway
modulated by MDM2 [68]. MDM2, an E3 ubiquitin ligase,
also physically interacts with RB and FOXO3a
result-ing in degradation of RB and FOXO3a proteins [69,70].
Overexpression of MDM2 has been demonstrated in
many human cancers [36,71]. In addition, oncogenic
ERK phosphorylates FOXO3a at Ser
294, Ser
344, and
Ser
425thereby enhancing the interaction with MDM2
and results in promoting degradation of FOXO3a [69].
Therefore, we hypothesized that MDM2 plays a critical
role in regulating the DNMT genes by synergistically
destabilizing p53, RB and FOXO3a proteins. To test
this hypothesis we analyzed the relationship of MDM2
protein with p53, RB, FOXO3a and DNMT proteins in
lung cancer cell, xenograft and patient models.
Dra-matic induction of DNMT3A and DNMT3B expression
by ectopic overexpression MDM2 suggests a negative
A
D
B
C
Figure 1 Transcriptional regulation onDNMT gene expression. (A) p53 transcriptionally suppresses DNMTs through binding with Sp1 protein to the DNMT1, 3A and 3B promoters. (B) RB transcriptionally suppresses DNMT1/3A through binding with E2F1 protein to the DNMT1 and 3A promoters. (C) FOXO3a binds to the FOXO3a DNA element of the DNMT3B promoter to repress DNMT3B transcription. (D) Clinically, overexpressed MDM2 dramatically induces DNMT1, DNMT3A, and DNMT3B expression by negative control over p53, RB and FOXO3a leading to methylation of multiple TSGs and tumorigenesis.
control of MDM2 over RB and FOXO3a [36,67]. Note
that treatment with the MDM2 inhibitor, Nutlin-3,
sig-nificantly reduces DNMT3A and DNMT3B expression
and methylation of TSGs, as well as tumor growth in vivo
[36,67]. Clinically, MDM2 overexpression inversely
corre-lates with expression of p53, RB and FOXO3a proteins in
tumor tissues from lung cancer patients. Importantly, a
sub-group of patient with gene expression signature of
DNMTs high, p53/RB/FOXO3a low, and MDM2 high
ex-pression profile correlating with poor survival [33,36,67].
This defined signature may serve as a prognostic marker
in lung cancer patients whose genomic DNA may exert
promoter hypermethylation in multiple TSGs (Figure 1D).
The microRNA-mediated regulation of DNMTs
MicroRNAs (miRs) are small, noncoding RNAs that
regulate expression of many genes. Recent studies
sug-gest that abnormal expressions of miRs are involved in
pathogenesis of different types of human cancers [72].
Previous reports have shown that expression profiles
of miRs in lung cancer are different from normal lung.
The miR-29 family (29a, 29b, and 29c) has intriguing
complementarities to the 3'-UTRs of DNM3A and
DNMT3B [73]. The expression of miR-29s is inversely
correlated to DNMT3A and DNMT3B in lung cancer
tissues, and miR-29s directly target the 3'-UTRs of
both DNMT3A and DNMT3B. The enforced expression
of miR-29s in lung cancer cell lines restores normal
pat-terns of DNA methylation. The miR-29s further induces
re-expression of methylation-silenced TSGs, such as FHIT
and WWOX, and inhibits tumorigenicity in vitro and
in vivo [73]. Enforced miR-29b expression in acute
mye-loid leukemia cells also results in marked reduction in the
expression of DNMT1, DNMT3A, and DNMT3B and
ultimately to re-expression of p15
INK4band ESR1 via
pro-moter DNA hypomethylation [74]. Of note, an inverse
correlation between miR-29c expression and DNMT3A
and DNMT3B protein expression has been reported in
melanomas [75].
In addition to miR-29s, ectopic expression of
miRNA-148a in lung cancer cell lines also results in a significant
reduction in the expression of DNMT1 [76]. Using
lucifer-ase reporter assay, DNMT1 mRNA was found to be a
tar-get of miR-148b and miR-152 [77]. Antagomir-mediated
knock-down and re-expression of miRs assays support
that miR-148b, miR-29c, and miR-26b down-regulate
DNMT3B gene in breast cancer cells [78]. Furthermore,
overexpression of miR-148b and -152 in pancreatic cancer
cell lines decreases DNMT1 expression, restores normal
DNA methylation patterns and induces re-expression of
TSGs, like BNIP3 and SPARC [77]. It is to be noted that
miR-143 was reported to directly target DNMT3A. In
colorectal cancer tissues, the miR-143 expression was
observed to be inversely correlated with DNMT3A mRNA
and protein expression [79]. Specifically, miR-1741,
miR-16c, miR-222 and miR-1632 are found to influence
expression of DNMT3A or DNMT3B, possibly through
their 3′-UTR post-transcriptional regulation [80]. Table 2
summarizes the regulation of DNMTs by miRs.
Post-translational modification of DNMT proteins
Several important post-translational modification including
acetylation and phosphorylation have been reported to
mediate protein stability and activity of the DNMTs
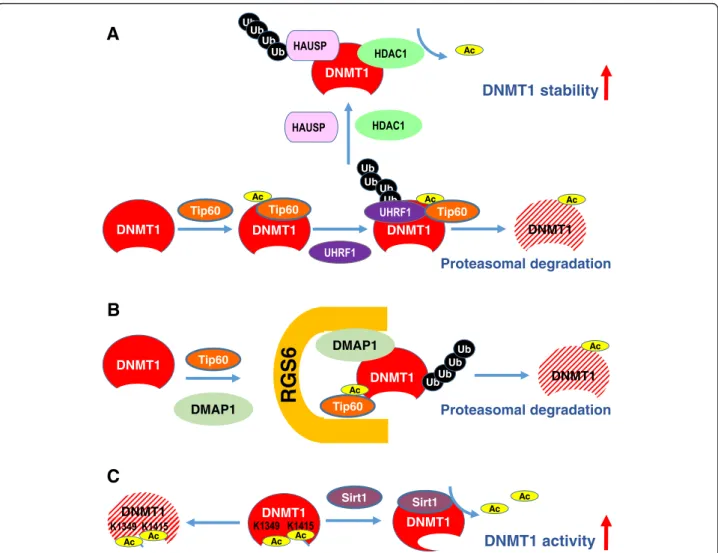
es-pecially DNMT1 (Figures 2 and 3). The major findings
are described below.
Acetylation-mediated DNMT stability and activity
DNMT1 has been shown to be destabilized by
acetylation-mediated ubiquitination. DNMT1 physically interacts with
acetyltransferase Tip60, herpesvirus-associated ubiquitin
specific protease (HAUSP), ubiquitin-like containing PHD
and RING finger domains 1 (UHRF1), HDAC1 and PCNA
on chromatin [81]. Tip60 promotes acetylation of DNMT1,
which triggers ubiquitination by the E3 ligase UHRF1,
thereby targeting DNMT1 for proteasomal degradation
during late S phase [82]. Conversely, HAUSP and
HDAC1 protect DNMT1 from degradation through
deubiquitination and deacetylation, respectively [81]
(Figure 2A). In addition, the pleiotropic regulator of G
protein signaling (RGS) family member RGS6 facilitates
Tip60-mediated degradation of DNMT1 [83]. RGS6 may
serve as a scaffold of Tip60, DNMT1 and
Dnmt1-associated protein (DMAP1) to facilitate Tip60 acetylation
of DNMT1 and subsequent DNMT1 polyubiquitylation
and degradation [83] (Figure 2B). In contrast, the histone
deacetylase SIRT1 physically associates with DNMT1 and
deacetylates acetylated DNMT1 in vitro and in vivo [84].
Using mass spectrometry analysis, 12 new acetylated lysine
sites are identified in DNMT1 [85]. Deacetylation of
dif-ferent lysines on DNMT1 by SIRT1 has difdif-ferent effects
on the functions of DNMT1. For example, deacetylation
of Lys1349 and Lys1415 by SIRT1 in the catalytic domain
of DNMT1 enhances the methyltransferase activity of
DNMT1 (Figure 2C). Collectively, these findings suggest
that deacetylation of the identified acetylated lysine sites
in DNMT1 may be involved in the impaired activity of
DNMT1.
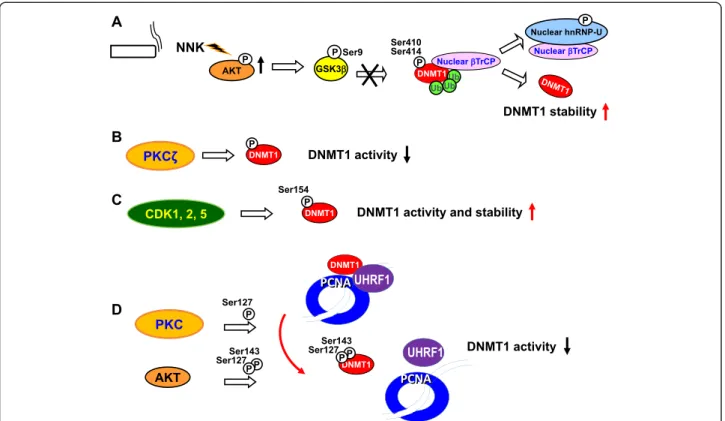
Phosphorylation-mediated DNMT stability and activity
Lin et al. found that the tobacco-specific nitrosamine
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)
in-creases DNMT1 protein expression and activity [86].
Strong nuclear staining of DNMT1 protein in lung
tumor tissues is significantly associated with smoking
status of lung cancer patients [22,86]. Cigarette smoking
is a dominant risk factor for lung cancer. Among the
multiple components of tobacco smoke, 20 carcinogens
Table 2 Regulation of DNMT expression by miRNAs
Pathway Regulated regions DNMTs Cancer types References
miR-16c 3'-UTRs DNMT3B in vitro [80]
miR-26b ND DNMT3B breast cancer [78]
miR-29a 3'-UTRs DNMT3A/3B lung cancer [73]
miR-29b 3'-UTRs DNMT1/3A/3B lung, ALL and melanomas [73,74]
miR-29c 3'-UTRs DNMT3A/3B breast, lung and melanomas [73,75,78]
miR-143 3'-UTRs DNMT3A colorectal [79]
miR-148a ND DNMT1 lung and pancreas [76,77]
miR-148b ND DNMT3B breast cancer [78]
miR-152 ND DNMT1 pancreas [77]
miR-222 3'-UTRs DNMT3B in vitro [80]
miR-1632 3'-UTRs DNMT3B in vitro [80]
miR-1741 3'-UTRs DNMT3A in vitro [80]
ND: non-determined.
ALL: Acute lymphoblastic leukemia.
Figure 2 Acetylation-mediated DNMT stability and activity. (A) Tip60 promotes acetylation of DNMT1, which triggers ubiquitination by the E3 ligase UHRF1, thereby targeting DNMT1 for proteasomal degradation. (B) RGS6 serves as a scaffold to facilitate Tip60 acetylation of DNMT1 and subsequent DNMT1 degradation. (C) Deacetylation of Lys1349 and Lys1415 by SIRT1 in the catalytic domain of DNMT1 enhances the methyltransferase activity of DNMT1.
convincingly cause lung tumors in laboratory animals or
humans. Of these, NNK is likely to play a major role
because NNK has been shown to induce lung tumor in
rats, mice and hamsters [87]. In addition, exposure of
NNK not only causes gene mutation, but also stimulates
the promoter hypermethylation on multiple TSGs in
bladder, liver, and lung cancers, including FHIT,
RASSF1A, and p16
INK4A, DAPK1, RUNX3, RARβ and
SFRP genes [88-93]. Notably, NNK stimulates the AKT,
NF-κB, EGFR, and ERK1/2 signal pathways resulting in
increased cell proliferation and survival [94-96]. It has
previously been shown that AKT inactivates GSK3β
Ser/Thr kinase, which phosphorylates its substrate protein
and recruits the E3-ubiqutin ligase
βTrCP, leading to
substrate degradation [97,98]. In addition, stabilization
of DNMT1 protein is regulated by inhibiting
GSK3β-mediated phosphorylation and proteasomal degradation
of DNMT1 upon activation of T cell receptor signaling
[99]. Lin et al. showed that GSK3β interacts with
DNMT1 to phosphorylate DNMT1 at Ser410 and Ser414
and promotes binding of DNMT1 by
βTrCP leading to
proteasomal degradation of DNMT1. They also
demon-strated that DNMT1 contains a domain, ESGXXS, similar
to the conserved binding motif DSGXXS of
βTrCP [86].
This study suggests that NNK activates AKT, then inhibits
GSK3β/βTrCP–mediated protein degradation, leading to
DNMT1 protein accumulation [86]. In addition, DNMT1/
βTrCP interaction could be interrupted by treating cells
with NNK. NNK also induces
βTrCP translocation to the
cytoplasm by interacting with phosphorylated
heteroge-neous nuclear ribonucleoprotein U (hnRNP-U) shuttling
protein mediated by AKT. Therefore, NNK exposure
re-sults in DNMT1 nuclear accumulation and
hypermethyla-tion of the promoters of multiple TSGs [86]. Figure 3A
shows the phosphorylation-mediated DNMT stability
con-trol induced by the cigarette carcinogen NNK.
In addition to Ser410 and Ser414 of DNMT1, recent
studies have suggested that the enzymatic activity of
DNMT1 is possibly modulated by phosphorylation of
Ser/Thr residues located in the N-terminal domain of
the enzyme [100-102]. Protein kinase C (PKC)
α, βI,
βII, δ, γ, η, ζ and μ preferentially phosphorylate the
N-terminal domain of human DNMT1 [102].
Phosphoryl-ation of DNMT1 by PKCζ reduces its methyltransferase
activity in vitro [102] (Figure 3B). In addition,
phosphoryl-ation of DNMT1 at Ser154 by CDKs, including CDK1, 2
and 5, is important to enhance enzymatic activity and
pro-tein stability of DNMT1 [100] (Figure 3C). AKT and PKC
Figure 3 Phosphorylation-mediated DNMT stability and activity. (A) Cigarette carcinogen NNK activates AKT, inhibits GSK3β-mediated DNMT1 phosphorylation at Ser410 and Ser414, thereby leading to DNMT1 protein accumulation. (B) Phosphorylation of DNMT1 by PKCζ reduces its methyltransferase activity. (C) Phosphorylation of DNMT1 at Ser154 by CDK1, 2 and 5 enhances enzymatic activity and protein stability of DNMT1. (D) Phosphorylation of DNMT1 at Ser127 and/or Ser143 decreases its interaction with UHRF1 ubiquitin-like protein and renders DNMT1 less efficient to catalyze the DNA methyltransferase activity at the replication fork.
are capable of phosphorylating DNMT1 at the residues
Ser127/143 and Ser127, respectively [101].
Phosphoryl-ation of the DNMT1 at Ser127 and/or Ser143 decreases
the capacity of the protein to interact with PCNA and
UHRF1 proteins and renders DNMT1 less efficient to
catalyze methylation [101] (Figure 3D). Interestingly,
phosphorylation of DNMT1 at Ser143 by AKT1 interferes
with the methylation of Lys142 by SET7, a known histone
methyltransferase involved in proteasome-mediated
deg-radation of DNMT1 [103].
The impact of viruses on the regulation of
DNMT
genes
Several viruses have been reported to increase DNMTs
expression (Table 1). Epstein-Barr virus (EBV) is closely
associated with human malignancies, including
nasopha-ryngeal carcinoma, Burkitt's lymphoma, T-cell lymphoma,
gastric carcinoma [104,105]. Epigenetic regulation of EBV
plays a central role in viral latency and viral-associated
carcinogenesis [105]. EBV latent membrane protein 1
(LMP1) activates cellular DNMTs, resulting in
hyperme-thylation and silencing of E-cadherin. LMP1-mediated
DNMT1 activation involves JNK but not NF-κB and p38
mitogen-activated protein kinases [42]. The EBV
onco-gene product LMP1, induces promoter hypermethylation
of RARβ2 via up-regulation of DNMT1, DNMT3A, and
DNMT3B proteins, leading to decrease in RARβ2
expres-sion in nasopharyngeal carcinoma cell lines [43]. Human
polyomavirus BKV large T antigen and adenovirus E1a
also strongly increase DNMT1 expression. Mutation of
the E2F sites within the DNMT1 promoter dramatically
abrogates transcriptional activation, suggesting that BKV
viral induction of DNMT1 may be through modulation of
pRB/E2F pathway [39].
The hepatitis B virus (HBV) X protein (HBx) plays a
key role in the molecular pathogenesis of HBV-related
hepatocellular carcinoma. HBx expression increases total
DNMT activities and selectively promotes regional
hyper-methylation of specific TSGs, including RASSF1A, GSTP1,
and CDKN2B, in pHBx-transfected cells [44]. Another
study shows that enforced HBx suppresses RASSF1A
possibly via induction of DNMT1 and DNMT3B
ex-pression [106].
Human immunodeficiency virus type 1 (HIV-1) also has
been reported to induce DNMT1 through the responsive
element residing in the
−1634 to +71 of DNMT1
pro-moter [45]. The increase in expression of DNMT1 and
overall genomic methylation as well as hypermethylation
of the p16
INK4Agene are found when infected with HIV-1
in Hut 78 lymphoid cells [107]. HIV infection of human
regulatory T cells down-regulates FOXP3 expression
me-diated by increasing DNMT3B levels and DNA
methyla-tion in the FOXP3 gene [108]. Therefore, the ability of
increased DNMT activity to downregulate the expression
of critical genes may be one of the mechanisms for
dys-function of T cells in HIV-1-infected individuals.
Concluding remark
DNMTs are the enzymes which catalyze the CpG DNA
methylation and have been reported to be over-expressed
in various cancers. The mechanisms of DNMT
over-expression are worthy of investigation. The transcriptional
up-regulation on DNMT gene expression can be induced
by Ras-c-Jun signaling pathway, Sp1 and Sp3 zinc finger
proteins, wilms' tumour 1, homeobox B3 and various
hu-man viruses. Loss of transcriptional repression control on
DNMT genes has also been reported. For example, p53
transcriptionally suppresses DNMTs through binding with
Sp1 protein to the DNMT promoters. RB transcriptionally
suppresses DNMT1/3A through binding with E2F1
pro-tein to the DNMT1 and 3A promoters. FOXO3a binds to
the FOXO3a DNA element of the DNMT3B promoter to
repress DNMT3B transcription. In addition, overexpressed
MDM2 may induce DNMT1, DNMT3A, and DNMT3B
expression by negative control over p53, RB and FOXO3a.
Low expressions of some miRs such as miR-29s, miR-143,
miR-148a and miR-152 are associated with DNMT
overexpression in various cancers. Several important
post-translational modification including acetylation
and phosphorylation have been reported to affect
pro-tein stability and activity of the DNMTs especially
DNMT1. Therefore, drugs targeting DNMT protein
in-activation and depletion, such as MDM2, AKT and
CDKs inhibitors may prove to be a good therapeutic
strategy for cancer treatment. Combined treatment with
the known DNMT inhibitors such as decitabine could
be a potential therapeutic strategy through epigenetic
modulation warranting further investigation in cancer
treatment.
Abbreviations
DNMT:DNA methyltransferase; FOXO3a: forkhead O transcription factor 3a; HBx: hepatitis B virus X protein; HAUSP: herpesvirus-associated ubiquitin specific protease; hnRNP-U: heterogeneous nuclear ribonucleoprotein U; LMP1: latent membrane protein 1; miR: microRNA; NNK: nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; RB: retinoblastoma; RGS: regulator of G protein signaling; TSG: tumor suppressor gene; UHRF1: ubiquitin-like containing PHD and RING finger domains 1.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RKL and YCW wrote the review. Both authors read and approved the final manuscript.
Acknowledgements
This work was supported by Taiwan National Science Council grant 100-2320-B-038-002 to RKL; and Taiwan National Science Council grant 102-2627-B-006-010 and Taiwan Ministry of Health and Welfare grant 103-TDU-PB-211-133005 to YCW.
Author details
1
Graduate Institute of Pharmacognosy, Taipei Medical University, 250 Wu-Hsing Street, Taipei 110, Taiwan.2Department of Pharmacology and
Institute of Basic Medical Sciences, National Cheng Kung University, No.1, University Road, Tainan 70101, Taiwan.3Program for the Clinical Drug
Discovery from Botanical Herbs, Taipei Medical University, 250 Wu-Hsing Street, Taipei 110, Taiwan.4Program for Clinical Pharmacogenomics and
Pharmacoproteomics, Taipei Medical University, 250 Wu-Hsing Street, Taipei 110, Taiwan.
Received: 21 May 2014 Accepted: 1 July 2014 Published: 19 August 2014
References
1. International Agency for Research on Cancer: GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. 2013. 2. Taby R, Issa JP: Cancer epigenetics. CA Cancer J Clin 2010, 60:376–392. 3. Belinsky SA: Gene-promoter hypermethylation as a biomarker in lung
cancer. Nat Rev Cancer 2004, 4:707–717.
4. Momparler RL: Cancer epigenetics. Oncogene 2003, 22:6479–6483. 5. Lee EJ, Luo J, Wilson JM, Shi H: Analyzing the cancer methylome through
targeted bisulfite sequencing. Cancer Lett 2013, 340:171–178. 6. Ma X, Wang YW, Zhang MQ, Gazdar AF: DNA methylation data analysis
and its application to cancer research. Epigenomics 2013, 5:301–316. 7. Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X,
Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O'Dorisio MS, Held WA, Cavenee WK, Plass C: Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet 2000, 24:132–138.
8. Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP: Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011, 43:768–775.
9. Jamaluddin MS, Yang X, Wang H: Hyperhomocysteinemia, DNA methylation and vascular disease. Clin Chem Lab Med 2007, 45:1660–1666. 10. Costa E, Dong E, Grayson DR, Guidotti A, Ruzicka W, Veldic M: Reviewing
the role of DNA (cytosine-5) methyltransferase overexpression in the cortical GABAergic dysfunction associated with psychosis vulnerability. Epigenetics 2007, 2:29–36.
11. Kanai Y: Alterations of DNA methylation and clinicopathological diversity of human cancers. Pathol Int 2008, 58:544–558.
12. Egger G, Liang G, Aparicio A, Jones PA: Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429:457–463.
13. Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B: DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002, 416:552–556.
14. Hermann A, Gowher H, Jeltsch A: Biochemistry and biology of
mammalian DNA methyltransferases. Cell Mol Life Sci 2004, 61:2571–2587. 15. Pradhan S, Talbot D, Sha M, Benner J, Hornstra L, Li E, Jaenisch R, Roberts
RJ: Baculovirus-mediated expression and characterization of the full-length murine DNA methyltransferase. Nucleic Acids Res 1997, 25:4666–4673.
16. Fatemi M, Hermann A, Pradhan S, Jeltsch A: The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J Mol Biol 2001, 309:1189–1199.
17. Goyal R, Reinhardt R, Jeltsch A: Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res 2006, 34:1182–1188.
18. Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF: Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 1997, 277:1996–2000.
19. Ko YG, Nishino K, Hattori N, Arai Y, Tanaka S, Shiota K: Stage-by-stage change in DNA methylation status of Dnmt1 locus during mouse early development. J Biol Chem 2005, 280:9627–9634.
20. Ratnam S, Mertineit C, Ding F, Howell CY, Clarke HJ, Bestor TH, Chaillet JR, Trasler JM: Dynamics of Dnmt1 methyltransferase expression and intracellular localization during oogenesis and preimplantation development. Dev Biol 2002, 245:304–314.
21. Hernandez-Munoz I, Taghavi P, Kuijl C, Neefjes J, van Lohuizen M: Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol Cell Biol 2005, 25:11047–11058.
22. Lin RK, Hsu HS, Chang JW, Chen CY, Chen JT, Wang YC: Alteration of DNA methyltransferases contributes to 5'CpG methylation and poor prognosis in lung cancer. Lung Cancer 2007, 55:205–213.
23. Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, Sasaki H: Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97:1172–1179.
24. Qu Y, Mu G, Wu Y, Dai X, Zhou F, Xu X, Wang Y, Wei F: Overexpression of DNA methyltransferases 1, 3a, and 3b significantly correlates with retinoblastoma tumorigenesis. Am J Clin Pathol 2010, 134:826–834. 25. Esteller M: DNA methylation and cancer therapy: new developments and
expectations. Curr Opin Oncol 2005, 17:55–60.
26. Bakin AV, Curran T: Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science 1999, 283:387–390.
27. Bigey P, Ramchandani S, Theberge J, Araujo FD, Szyf M: Transcriptional regulation of the human DNA Methyltransferase (dnmt1) gene. Gene 2000, 242:407–418.
28. Jinawath A, Miyake S, Yanagisawa Y, Akiyama Y, Yuasa Y: Transcriptional regulation of the human DNA methyltransferase 3A and 3B genes by Sp3 and Sp1 zinc finger proteins. Biochem J 2005, 385:557–564. 29. Kishikawa S, Murata T, Kimura H, Shiota K, Yokoyama KK: Regulation of
transcription of the Dnmt1 gene by Sp1 and Sp3 zinc finger proteins. Eur J Biochem 2002, 269:2961–2970.
30. Szemes M, Dallosso AR, Melegh Z, Curry T, Li Y, Rivers C, Uney J, Magdefrau AS, Schwiderski K, Park JH, Brown KW, Shandilya J, Roberts SG, Malik K: Control of epigenetic states by WT1 via regulation of de novo DNA methyltransferase 3A. Human Mol Genet 2013, 22:74–83.
31. Palakurthy RK, Wajapeyee N, Santra MK, Gazin C, Lin L, Gobeil S, Green MR: Epigenetic silencing of the RASSF1A tumor suppressor gene through HOXB3-mediated induction of DNMT3B expression. Mol Cell 2009, 36:219–230.
32. Lin RK, Wu CY, Chang JW, Juan LJ, Hsu HS, Chen CY, Lu YY, Tang YA, Yang YC, Yang PC, Wang YC: Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res 2010, 70:5807–5817.
33. Tang YA, Tsai YT, Lin RK, Hsu HS, Chen CY, Wang YC: Deregulation of p53 and RB Transcriptional Control of DNA Methyltransferases in Lung Cancer. J Cancer Res Pract 2014, 30:88–101.
34. McCabe MT, Davis JN, Day ML: Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res2005, 65:3624–3632.
35. Kimura H, Nakamura T, Ogawa T, Tanaka S, Shiota K: Transcription of mouse DNA methyltransferase 1 (Dnmt1) is regulated by both E2F-Rb-HDAC-dependent and -independent pathways. Nucleic Acids Res 2003, 31:3101–3113.
36. Tang YA, Lin RK, Tsai YT, Hsu HS, Yang YC, Chen CY, Wang YC: MDM2 overexpression deregulates the transcriptional control of RB/E2F leading to DNA methyltransferase 3A overexpression in lung cancer. Clin Cancer Res 2012, 18:4325–4333.
37. Herzog CR, Blake DC Jr, Mikse OR, Grigoryeva LS, Gundermann EL: FoxO3a gene is a target of deletion in mouse lung adenocarcinoma. Oncol Rep 2009, 22:837–843.
38. Rouleau J, MacLeod AR, Szyf M: Regulation of the DNA methyltransferase by the Ras-AP-1 signaling pathway. J Biol Chem 1995, 270:1595–1601. 39. McCabe MT, Low JA, Imperiale MJ, Day ML: Human polyomavirus BKV
transcriptionally activates DNA methyltransferase 1 through the pRb/E2F pathway. Oncogene 2006, 25:2727–2735.
40. Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, Richardson B: Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum 2003, 48:746–756.
41. Cui M, Wen Z, Yang Z, Chen J, Wang F: Estrogen regulates DNA methyltransferase 3B expression in Ishikawa endometrial adenocarcinoma cells. Mol Biol Rep 2009, 36:2201–2207.
42. Tsai CL, Li HP, Lu YJ, Hsueh C, Liang Y, Chen CL, Tsao SW, Tse KP, Yu JS, Chang YS: Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res 2006, 66:11668–11676.
43. Seo SY, Kim EO, Jang KL: Epstein-Barr virus latent membrane protein 1 suppresses the growth-inhibitory effect of retinoic acid by inhibiting retinoic acid receptor-beta2 expression via DNA methylation. Cancer Lett 2008, 270:66–76.
44. Park IY, Sohn BH, Yu E, Suh DJ, Chung YH, Lee JH, Surzycki SJ, Lee YI: Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology 2007, 132:1476–1494. 45. Youngblood B, Reich NO: The early expressed HIV-1 genes regulate
DNMT1 expression. Epigenetics 2008, 3:149–156.
46. Selivanova G: Wild type p53 reactivation: From lab bench to clinic. FEBS Lett 2014, 588:2628-2638
47. Sigal A, Rotter V: Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res 2000, 60:6788–6793. 48. Peterson EJ, Bogler O, Taylor SM: p53-mediated repression of DNA
methyltransferase 1 expression by specific DNA binding. Cancer Res 2003, 63:6579–6582.
49. Giacinti C, Giordano A: RB and cell cycle progression. Oncogene 2006, 25:5220–5227.
50. Dyson N: The regulation of E2F by pRB-family proteins. Genes Dev 1998, 12:2245–2262.
51. Siddiqui H, Fox SR, Gunawardena RW, Knudsen ES: Loss of RB compromises specific heterochromatin modifications and modulates HP1alpha dynamics. J Cell Physiol 2007, 211:131–137.
52. Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T: Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. Embo J 2001, 20:2536–2544.
53. Zhang HS, Dean DC: Rb-mediated chromatin structure regulation and transcriptional repression. Oncogene 2001, 20:3134–3138.
54. Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T: Rb targets histone H3 methylation and HP1 to promoters. Nature 2001, 412:561–565. 55. Tsai KL, Sun YJ, Huang CY, Yang JY, Hung MC, Hsiao CD: Crystal structure
of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res 2007, 35:6984–6994. 56. Medema RH, Kops GJ, Bos JL, Burgering BM: AFX-like Forkhead
transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404:782–787.
57. Modur V, Nagarajan R, Evers BM, Milbrandt J: FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem 2002, 277:47928–47937. 58. Arden KC: Multiple roles of FOXO transcription factors in mammalian
cells point to multiple roles in cancer. Exp Gerontol 2006, 41:709–717. 59. Greer EL, Brunet A: FOXO transcription factors at the interface between
longevity and tumor suppression. Oncogene 2005, 24:7410–7425. 60. Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara
R, DePinho RA, Zeiher AM, Dimmeler S: Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest 2005, 115:2382–2392.
61. Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ: Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol 2000, 20:9138–9148.
62. Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH: The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol 2002, 168:5024–5031. 63. Yang JY, Xia W, Hu MC: Ionizing radiation activates expression of
FOXO3a, Fas ligand, and Bim, and induces cell apoptosis. Int J Oncol 2006, 29:643–648.
64. Wang K, Li PF: Foxo3a regulates apoptosis by negatively targeting miR-21. J Biol Chem 2010, 285:16958–16966.
65. Mikse OR, Blake DC Jr, Jones NR, Sun YW, Amin S, Gallagher CJ, Lazarus P, Weisz J, Herzog CR: FOXO3 encodes a carcinogen-activated transcription factor frequently deleted in early-stage lung adenocarcinoma. Cancer Res 2010, 70:6205–6215.
66. Blake DC Jr, Mikse OR, Freeman WM, Herzog CR: FOXO3a elicits a pro-apoptotic transcription program and cellular response to human lung carcinogen nicotine-derived nitrosaminoketone (NNK). Lung Cancer 2010, 67:37–47. 67. Yang YC, Tang YA, Shieh JM, Lin RK, Hsu HS, Wang YC: DNMT3B
overexpression by deregulation of FOXO3a-mediated transcription repression and MDM2 overexpression in lung cancer. J Thorac Oncol 2014, 9:1305-1315.
68. Khoo KH, Verma CS, Lane DP: Drugging the p53 pathway: understanding the route to clinical efficacy. Nat rev Drug discov 2014, 13:217–236. 69. Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC,
Chang CJ, Huang WC, Huang H, Kuo HP, Lee DF, Li LY, Lien HC, Cheng X, Chang KJ, Hsiao CD, Tsai FJ, Tsai CH, Sahin AA, Muller WJ, Mills GB, Yu D, Hortobagyi GN, Hung MC: ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol 2008, 10:138–148.
70. Bhattacharya S, Ghosh MK: HAUSP, a novel deubiquitinase for Rb - MDM2 the critical regulator. FEBS J 2014, 281:3061-3078.
71. Landers JE, Cassel SL, George DL: Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild-type p53 protein. Cancer Res 1997, 57:3562–3568.
72. Iorio MV, Croce CM: microRNA involvement in human cancer. Carcinogenesis 2012, 33:1126–1133.
73. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM: MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A 2007, 104:15805–15810.
74. Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, Havelange V, Volinia S, Blum W, Rush LJ, Perrotti D, Andreeff M, Bloomfield CD, Byrd JC, Chan K, Wu LC, Croce CM, Marcucci G: MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113:6411–6418.
75. Nguyen T, Kuo C, Nicholl MB, Sim MS, Turner RR, Morton DL, Hoon DS: Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics 2011, 6:388–394.
76. Chen Y, Min L, Zhang X, Hu S, Wang B, Liu W, Wang R, Gu X, Shen W, Lv H, Zou J, Xu X, Chen L: Decreased miRNA-148a is associated with lymph node metastasis and poor clinical outcomes and functions as a suppressor of tumor metastasis in non-small cell lung cancer. Oncol Rep 2013, 30:1832–1840.
77. Azizi M, Teimoori-Toolabi L, Arzanani MK, Azadmanesh K, Fard-Esfahani P, Zeinali S: MicroRNA-148b and microRNA-152 reactivate tumor suppressor genes through suppression of DNA methyltransferase-1 gene in pancreatic cancer cell lines. Cancer Biol Ther 2014, 15(4):419–427. 78. Sandhu R, Rivenbark AG, Coleman WB: Loss of post-transcriptional
regulation of DNMT3b by microRNAs: a possible molecular mechanism for the hypermethylation defect observed in a subset of breast cancer cell lines. Int J Oncol 2012, 41:721–732. 79. Ng EK, Tsang WP, Ng SS, Jin HC, Yu J, Li JJ, Rocken C, Ebert MP, Kwok TT,
Sung JJ: MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br J Cancer 2009, 101:699–706.
80. Lee JY, Jeong W, Lim W, Lim CH, Bae SM, Kim J, Bazer FW, Song G: Hypermethylation and post-transcriptional regulation of DNA methyltransferases in the ovarian carcinomas of the laying hen. PloS one 2013, 8:e61658.
81. Hong Q, Shao ZM: Ubiquitination/deubiquitination and acetylation/ deacetylation: making DNMT1 stability more coordinated. Acta Pharmacol Sin 2011, 32:139–140.
82. Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S, Kao HY, Xu Y, Willis J, Markowitz SD, Sedwick D, Ewing RM, Wang Z: DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci Signal 2010, 3:ra80.
83. Huang J, Stewart A, Maity B, Hagen J, Fagan RL, Yang J, Quelle DE, Brenner C, Fisher RA: RGS6 suppresses Ras-induced cellular transformation by facilitating Tip60-mediated Dnmt1 degradation and promoting apoptosis. Oncogene 2013, doi: 10.1038/onc.2013.324.
84. Peng L, Yuan Z, Ling H, Fukasawa K, Robertson K, Olashaw N, Koomen J, Chen J, Lane WS, Seto E: SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol Cell Biol 2011, 31:4720–4734.
85. Kar S, Deb M, Sengupta D, Shilpi A, Parbin S, Torrisani J, Pradhan S, Patra S: An insight into the various regulatory mechanisms modulating human DNA methyltransferase 1 stability and function. Epigenetics 2012, 7:994–1007.
86. Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF, Wang YC: The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest 2010, 120:521–532.
87. Hecht SS: Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 1999, 91:1194–1210.
88. Kim DH, Nelson HH, Wiencke JK, Zheng S, Christiani DC, Wain JC, Mark EJ, Kelsey KT: p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res 2001, 61:3419–3424.
89. Kim DH, Kim JS, Ji YI, Shim YM, Kim H, Han J, Park J: Hypermethylation of RASSF1A promoter is associated with the age at starting smoking and a poor prognosis in primary non-small cell lung cancer. Cancer Res 2003, 63:3743–3746.
90. Kim JS, Kim H, Shim YM, Han J, Park J, Kim DH: Aberrant methylation of the FHIT gene in chronic smokers with early stage squamous cell carcinoma of the lung. Carcinogenesis 2004, 25:2165–2171.
91. Hutt JA, Vuillemenot BR, Barr EB, Grimes MJ, Hahn FF, Hobbs CH, March TH, Gigliotti AP, Seilkop SK, Finch GL, Mauderly JL, Belinsky SA: Life-span inhalation exposure to mainstream cigarette smoke induces lung cancer in B6C3F1 mice through genetic and epigenetic pathways. Carcinogenesis 2005, 26:1999–2009.
92. Vuillemenot BR, Hutt JA, Belinsky SA: Gene promoter hypermethylation in mouse lung tumors. Mol Cancer Res 2006, 4:267–273.
93. Pulling LC, Vuillemenot BR, Hutt JA, Devereux TR, Belinsky SA: Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res 2004, 64:3844–3848.
94. Askari MD, Tsao MS, Schuller HM: The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol 2005, 131:639–648. 95. Laag E, Majidi M, Cekanova M, Masi T, Takahashi T, Schuller HM: NNK
activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int J Cancer 2006, 119:1547–1552.
96. Tsurutani J, Castillo SS, Brognard J, Granville CA, Zhang C, Gills JJ, Sayyah J, Dennis PA: Tobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cells. Carcinogenesis 2005, 26:1182–1195.
97. Sharma M, Chuang WW, Sun Z: Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J Biol Chem 2002, 277:30935–30941.
98. Taketo MM: Shutting down Wnt signal-activated cancer. Nat Genet 2004, 36:320–322.
99. Li C, Ebert PJ, Li QJ: T cell receptor (TCR) and transforming growth factor beta (TGF-beta) signaling converge on DNA
(cytosine-5)-methyltransferase to control forkhead box protein 3 (foxp3) locus methylation and inducible regulatory T cell differentiation. J Biol Chem 2013, 288:19127–19139.
100. Lavoie G, St-Pierre Y: Phosphorylation of human DNMT1: implication of cyclin-dependent kinases. Biochem Biophys Res Commun 2011, 409:187–192.
101. Hervouet E, Lalier L, Debien E, Cheray M, Geairon A, Rogniaux H, Loussouarn D, Martin SA, Vallette FM, Cartron PF: Disruption of Dnmt1/PCNA/UHRF1 interactions promotes tumorigenesis from human and mice glial cells. PloS one 2010, 5:e11333.
102. Lavoie G, Esteve PO, Laulan NB, Pradhan S, St-Pierre Y: PKC isoforms interact with and phosphorylate DNMT1. BMC Biol 2011, 9:31.
103. Esteve PO, Chang Y, Samaranayake M, Upadhyay AK, Horton JR, Feehery GR, Cheng X, Pradhan S: A methylation and phosphorylation switch between an adjacent lysine and serine determines human DNMT1 stability. Nat Struct Mol Biol 2011, 18:42–48.
104. Matsusaka K, Funata S, Fukayama M, Kaneda A: DNA methylation in gastric cancer, related to and Epstein-Barr virus. World J Gastroenterol 2014, 20:3916–3926.
105. Tempera I, Lieberman PM: Epigenetic regulation of EBV persistence and oncogenesis. Semin Cancer Biol 2014, 26C:22–29.
106. Qiu X, Zhang L, Lu S, Song Y, Lao Y, Hu J, Fan H: Upregulation of DNMT1 mediated by HBx suppresses RASSF1A expression independent of DNA methylation. Oncol Rep 2014, 31:202–208.
107. Fang JY, Mikovits JA, Bagni R, Petrow-Sadowski CL, Ruscetti FW: Infection of lymphoid cells by integration-defective human immunodeficiency virus type 1 increases de novo methylation. J Virol2001, 75:9753–9761. 108. Pion M, Jaramillo-Ruiz D, Martinez A, Munoz-Fernandez MA, Correa-Rocha R:
HIV infection of human regulatory T cells downregulates Foxp3 expression by increasing DNMT3b levels and DNA methylation in the FOXP3 gene. AIDS 2013, 27:2019–2029.
doi:10.1186/2045-3701-4-46
Cite this article as: Lin and Wang: Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell & Bioscience 2014 4:46.
Submit your next manuscript to BioMed Central
and take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit