國立臺灣大學生命科學院動物學研究所 博士論文
Graduate Institute of Zoology College of Life Science
National Taiwan University Doctoral dissertation
p62在蛋白質堆疊物的降解作用之調控機制 The role of the autophagic cargo receptor p62 in the
clearance of aggregation-prone proteins
董盈岑
Ying-Tsen Tung
指導教授﹕黃偉邦 博士、廖永豐 博士
Wei-Pang Huang, Ph.D. and Yung-Feng Liao, Ph.D.
中華民國 102 年 1 月
January 2013
國立臺灣大學博士學位論文 口試委員會審定書
p62 在蛋白質堆疊物的降解作用之調控機制
The role of the autophagic cargo receptor p62 in the clearance of aggregation-prone proteins
本論文條董盈本君(仿 3b41019) 在國立臺灣大學動物 學研究所完成之博士學位論文,於民國 101 年 12
月13
日承下列考試委員審查通過及口試及格,特此證明
口試委員:
(指導教授)
谷之立\
;有 r;(~
戶頭芝在埔
同長已
動物學研究所所長
為三衷方家
L 蓋孟之
i
中文摘要
神經退化性疾病的共同特徵,是在神經內或周圍有不正常的蛋白質堆疊,
許多研究指出,這些異常堆疊的蛋白質,是導致神經退化的主因。目前已知選擇 性的細胞自噬作用(selective autophagy),可以特定清除這些堆疊物,做為一種保 護 神 經 的 機 制 。 有 證 據 顯 示 , 在 哺 乳 動 物 細 胞 中 , 這 樣 的 選 擇 性 是 經 由 p62/sequestosome1 做為受體:它可以跟自噬體上的 LC3 結合,且被觀察到會共同 沉積含特定致病蛋白的堆疊物中,例如亨丁頓舞蹈症(Huntington’s disease, HD) 的突變型 Huntingtin (Htt),以及阿茲海默氏症(Alzheimer’s disease, AD)的 Tau。
本論文的第一部分,延續了之前在酵母菌模式生物的發現,找出在 LC3 上 負責與 p62 結合的胺基酸位點,顯示從酵母菌到哺乳動物都利用相同的分子機 制,將受體連結到自噬體中。而我們的實驗也證實,當 LC3 與 p62 無法形成連 結時,突變 Htt 型蛋白堆疊便無法經由選擇性細胞自噬作用所清除,進一步支持 p62 可作為細胞自噬作用降解特定蛋白質堆疊的受體。
p62 在神經退化性疾病的重要性,促使我們去研究它基因表現上的調控,與 其蛋白質功能的相關性。本論文的第二部份,陳述我們發現 presenilin-1 (PS1)可 經由活化 Akt/AP-1 訊息傳導途徑,來促進 p62 基因的轉錄表現。我們也測試了 幾個在家族遺傳性 AD 病人中特有的 PS1 基因突變型,發現當細胞表現這些突變 的 PS1 時,卻無法維持其 p62 基因的正常表現,導致細胞沒有足夠的 p62 來幫 助清除 Tau 蛋白質。此研究找到了 PS1 調控 p62 表現進而影響 Tau 降解的新功能,
也更加確認 p62 在神經退化性疾病中,作為清除蛋白質堆疊的重要分子。
本論文的研究成果,證實 LC3-p62 的結合,在選擇性細胞自噬作用中擔任 舉足輕重的角色,負責將誘發神經退化性疾病的蛋白質堆疊送至自噬體中。另一 方面,也找到調控 p62 表現的新途徑,幫助我們更進一步了解 AD 及其他神經退 化性疾病的致病機轉。
關鍵字: 細胞自噬作用;p62/sequestosome-1;神經退化性疾病;presenilin-1
ii
Summary
The accumulation of certain misfolded protein aggregates in the brain is a common feature in various neurodegenerative diseases, and is accepted as a major causative factor of neurodegeneration. Aggrephagy, the process by which protein aggregates are selectively degraded through macroautophagy, plays an essential role in protecting neurons from aggregate-induced neurotoxicity. Recent findings have identified p62/sequestosome1 as a cargo receptor that interacts with the autophagosomal membrane associated protein LC3, and recruits ubiquitin-positive protein aggregates into autophagosomes. The finding that p62 is co-localized with inclusion bodies in the brains of patients with Huntington’s disease (HD) and Alzheimer’s disease (AD) suggests a critical role for p62 in neurodegeneration.
Previous findings have identified residues in a yeast LC3 homologue, Atg8, that are essential for interaction of Atg8 with the cargo receptor Atg19 in selective autophagic processes. In the first part of my thesis, I describe our attempts to determine whether such interaction is evolutionally conserved from yeast to mammals.
By using an amino acid replacement approach, we determined that three residues in LC3 corresponding to those in Atg8 were essential for p62 binding. Furthermore, while disruption of the LC3-p62 complex formation did not alter overall autophagic activity, it was sufficient to impede the autophagy-mediated clearance of aggregation–prone mutant Huntingtin (Htt), the cytotoxic protein which induces the
iii
pathological phenotypes of HD.
The protective role of p62 in the clearance of aggregation-prone proteins prompted us to investigate how p62 expression is regulated under pathological conditions. In the second part of my thesis, I describe our discovery that p62 expression is transcriptionally regulated by presenilin 1 (PS1), a protein which is mutated in the majority of patients with early-onset familial Alzheimer’s disease (FAD). The PI3K/Akt/AP-1 pathway was found to be required for PS1-mediated regulation of p62 expression. Moreover, down-regulation of p62 by either PS1 deficiency or over-expression of FAD-linked PS1 mutants compromised clearance of aggregation-prone Tau, which forms intracellular neurofibrillary tangles in the AD brain; these findings thus confirm the essential role of p62 in the clearance of neurotoxic protein aggregates.
Together, our studies emphasize the importance of the LC3-p62 interaction in selective autophagy, and the requirement of p62 for the removal of neurodegeneration-associated protein aggregates. Furthermore, the identification of PS1-dependent transcriptional regulation of p62 expression uncovers a novel PS1/p62-mediated molecular mechanism underlying the pathogenesis of AD and related neurodegenerative diseases.
Key words: autophagy;p62/sequestosome-1;neurodegenerative diseases;
presenilin-1
iv
Table of Contents
中文摘要... i
Summary ... ii
Table of Content ... iv
List of figures ... vi
Chapter 1. Introduction ... 1
1.1 Neurodegenerative disease ... 2
1.2 Huntington’s disease ... 4
1.3 Alzheimer’s disease ... 8
1.4 Proteasome ... 16
1.5 Autophagy ... 20
1.6 p62/sequestosome 1 ... 31
Chapter 2. The evolutionarily conserved cargo-receptor binding site of LC3 interacts with p62 to mediate autophagy-dependent degradation of mutant Huntingtin ... 41
2.1 Abstract... 42
2.2 Introduction ... 43
2.3 Materials and Methods ... 45
2.4 Results ... 53
2.5 Discussion... 69
Chapter 3. Presenilin-1 regulates the expression of sequestosome-1/p62 and governs p62-dependent degradation of Tau independent of γ-secretase activity ... 73
3.1 Abstract... 74
3.2 Introduction ... 75
3.3 Materials and Methods ... 78
3.4 Results ... 89
v
3.5 Discussion... 114
Chapter 4. Concluding remarks and future perspectives ... 121
References ... 127
Appendix: List of publications ... 147
vi
List of Figures
Fig 1.1 Schematic depiction of macroautophagy, lipidation process and aggrephagy. .... .24 Fig 1.2 Schematic layout of the domain organization of p62 ... 32 Fig 2.1 Mutations in the ubiquitin core of LC3 attenuate its interaction with endogenous p62. ... 56 Fig 2.2 Mutation within the ubiquitin core of LC3 results in reduced lipidation of LC3. . 59 Fig 2.3 Autophagosomal recruitment and degradation of p62 are impaired in cells expressing mutant LC3 ... 63 Fig 2.4 Overexpression of mutant LC3 abolishes p62-dependent autophagic clearance of Htt. ... 67 Fig 3.1 PS1 deficiency reduces p62 protein levels. ... 90 Fig 3.2 PS1 deficiency reduces p62 mRNA levels. ... 94 Fig 3.3 PS1 transcriptionally regulates p62 expression independent of γ-secretase activity..
... 97 Fig 3.4 The PS1-dependent regulation of p62 expression is mediated by an Akt/AP1 pathway ... 102 Fig 3.5 Downregulation of PS1 enhances Tau accumulation through impairment of p62-dependent Tau degradation.. ... 107 Fig 3.6 Overexpression of PS1 containing FAD-linked mutations fails to restore the impaired p62 expression and p62-dependent Tau degradation in PS1-deficient cells.. ... 111 Fig 3.7 Overexpression of secretase-inactive PS1-D257A mutant rescued the pAkt level, p62 expression and p62-dependent Tau degradation in PS1-deficient cells…....113
1
Chapter 1
Introduction
2
1.1 Neurodegenerative disease
Neurodegenerative disease is a general term for a number of disorders that are characterized by gradual loss of central or peripheral structures of the nervous system.
As neurons are highly differentiated, non-proliferating cell types, recovery from their degeneration is difficult. Aging is a major risk factor for neurodegenerative diseases, as it makes patients more prone to neuronal atrophy, as well as interfering with the replacement of lost neurons. It is therefore important to uncover the mechanisms underlying neurodegenerative processes, which may pave the way for the prevention or treatment of detrimental neurodegenerative diseases.
Common features
A common feature of age-related neurodegenerative diseases is the accumulation of misfolded proteins within or around neurons. Different neurodegenerative diseases are characterized by aggregates composed of distinct protein components. For example, aggregates of α-synucleins in Parkinson’s disease
(PD); prions in Bovine spongiform encephalopathy (BSE); SOD1 in amyotrophic lateral sclerosis (ALS); huntingtin (Htt) in Huntington’s disease (HD); and amyloid-β (Aβ) and Tau in Alzheimer’s disease (AD) [1]. While the constituents of inclusions
are disease specific, they share strikingly similar structural characteristics.
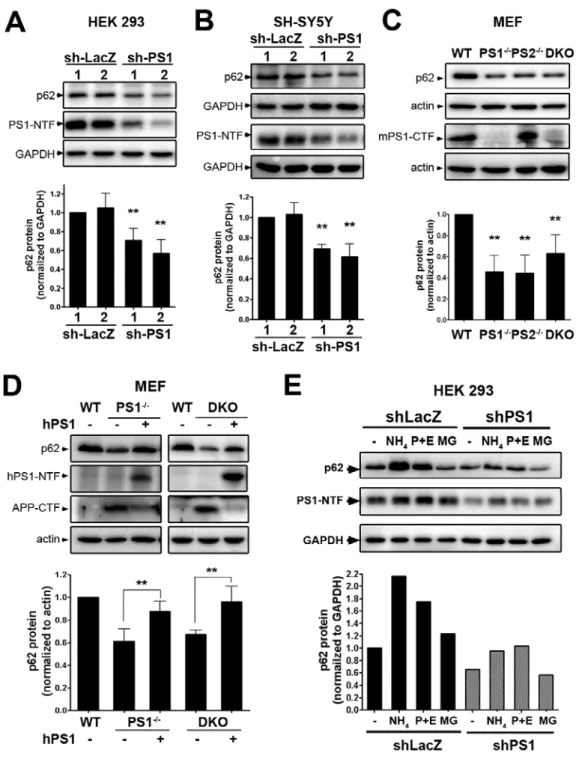
3
These disease-related proteins behave normally under physiological conditions, but may become aggregation-prone due to mutations that alter amino acid residues or translational modifications that alter secondary structures. Such mutations or modifications result in improper folding of the protein and exposure of hydrophobic patches, thereby enhancing self oligomerization via hydrophobic interaction. After formation of oligomeric (globular) intermediates, protofibrillar structures are assembled. The protofibrillar intermediates further elongate first into filaments and then fibrils, finally resulting in a helical assemblage with a cross β structure, visible under a light microscope [2]. In most cases these large inclusion bodies are ubiquitin-positive; however, they are usually resistant to degradation [3]
The accumulation of neurodegenerative disease-related protein aggregates has been shown to impair basic cellular functions. For example, aggregate formation can induce oxidative stress and mitochondrial dysfunction, which lead to neuronal death [4]. Synaptic activity and membrane integrity of neurons are also disrupted by aggregates [5]. Furthermore, proteasomal degradative activity is decreased when abnormal protein aggregates form inside cells [3, 6], thereby further promoting accumulation of inclusion bodies.
Interestingly, recent findings argue that oligomeric soluble species exert greater neurotoxic effects than large protein aggregates. This hypothesis emerged from the
4
observation that application of oligomers composed of 10~20 molecules induced cell death, while application of large aggregates (> 200mers) did not cause significant cytotoxicity [7, 8]. Animal models that express aggregation-prone proteins were treated with specific antibodies to remove soluble proteins, resulting in amelioration of behavior deficits, while the aggregates remained intact [9]. Hence it is possible that the formation of aggregates is a protective mechanism to sequester the toxic oligomeric species [10, 11]. Despite these findings, further investigations are required to elucidate which polymeric stage exerts the most detrimental effects on cellular functions; in addition, it remains widely accepted that degradative systems involved in the clearance of these aggregate-prone proteins play a key role in protecting against neurotoxicity.
In the following sections, I will first discuss specific protein aggregates implicated in two neurodegenerative diseases: HD and AD. I will subsequently introduce the degradative systems involved in these diseases, and describe the current understanding of the regulatory mechanisms involved in the degradation of aggregation-prone proteins.
1.2 Huntington’s disease
Huntington’s disease (HD) is named after Dr. George Huntington, who first
5
accurately described the symptoms of patients with this disease [12, 13]. It has been estimated by a systematic analysis that the world-wide prevalence of HD is about 3 per 100,000 people [14].HD is an adult onset disease, the pathological changes of which start with neurodegeneration in the striatum of the basal ganglia, a brain region controlling the voluntary motor functions and the cognitive and emotional functions.
The most common symptom of this disease is involuntary movement of head and limbs, which resembles dance-like movements. As such, HD is also called Huntington's chorea; chorea is derived from the Greek word choreia, meaning a type of dance. Patients also develop cognitive dysfunction and psychiatric problems during aging, which correspond with progressive neurodegeneration from the striatum to other brain regions, such as the cerebral cortex, the hippocampus, the hypothalamus and parts of the thalamus [15]. Patients usually present with severe dementia, mental illness and other complications around 15 to 20 years after the onset of HD, which finally leads to death. Treatments are now available to alleviate the symptoms, but no therapies can alter the course of this disease.
Huntintin (Htt)
It is now clear that HD is an autosomal dominant disease which occurs in patients carrying mutations in the Htt gene. This gene is located on human
6
chromosome 4 and contains poly(CAG) repeats within the exon 1 region. In healthy subjects, the number of CAG repeats in the Htt gene range from 3 to 25. Once the number of CAG repeats exceed 36, the resulting long polyQ-containing Htt proteins tend to aggregate inside neurons [16]. Direct intermolecular interactions between
polyQ extensions of mutant Htt proteins, or intramolecular interactions between polyQ and the N terminus cause a dramatic conformational change from the α-helical
structure of normal Htt to a misfolded parallel β-sheet form [17]. The misfolded confomation is more prone to form aggregates.
The length of the polyQ repeat has been shown to inversely correlate with the onset age of HD [18]. However, the number of neurons containing Htt inclusions do not positively correlate with neurodegeneration [19], suggesting that soluble monomers or, more likely, oligomers, contribute to neurotoxicity, while insoluble Htt
deposition may be a protective response. In support of this view, microinjection of soluble β-sheet monomers and β-sheet oligomers of an expanded poly-Q protein into
cell cultures caused severe cytotoxicity, which was rescued by a poly-Q binding peptide that prevents β-sheet conformation transition [20].
The cytotoxic effects of mutant Htt have been extensively studied. BDNF transportation is crucial for striatum neurons to maintain neuronal functions, but can be impaired by mutant Htt [21]. Htt can translocate between the cytosol and the
7
nucleus, and nuclear mutant Htt has been reported to attenuate expression of several genes [22]. Furthermore, mutant Htt was found to transfer between cells in culture [23], indicating that, like prions, mutant Htt is able to spread to neighboring cells, inducing formation of neurotoxic aggregates and contributing to progressive neuronal death [24].
The above evidence suggests that polyQ extension in Htt causes a gain-of-toxic function that promotes HD pathogenesis. On the other hand, genetic depletion of Htt expression in a mouse model caused early developmental lethality [25], which could be reversed by breeding knock-in mice that expressed Htt containing 50 polyQ repeats [26]. Therefore, Htt plays a role during development regardless of poly Q expansion.
In addition, this protein is required for suppression of caspase-mediated apoptosis [27], and may also be involved in maintaining pro-survival Akt pathways [28]. Loss of the normal protective Htt function may act synergistically with mutant Htt to cause neurodegeneration in HD patients.
Htt can be modified via phosphorylation [29-31], ubiquitination [32, 33], acetylation [34] and sumoylation [35]. These post-translational modifications can regulate the stability of Htt or contribute to the toxicity of the mutant form. Enzymes involved in the above modifications are potential therapeutic targets for HD. On the other hand, Htt with a poly-Q extension has been reported to impede both proteasomal
8
and autophagy systems [36-38], leading to greater accumulation of toxic Htt species.
Strategies to enhance cellular degradative systems may prove to be potent therapies.
1.3 Alzheimer’s disease
Alzheimer’s disease (AD) is named after Dr. Alois Alzheimer, who first reported the symptoms and pathologies of a patient with the disease [39]. As of the time of writing, more than 35 million people worldwide are affected by this disease.
In Taiwan, the prevalence of AD in elderly people is around 2~4%, which means that more than 25,000 people suffer from AD [40]. This prevalence is estimated to increase dramatically in the next decade due to the ageing population. The enormous medical costs to patients’ families and society will inevitably become an important social issue.
Progression of AD is accompanied by gradual neuronal loss [41]. The first region to undergo degeneration is the entorhinal cortex-II (EC-II), which is connected to the hippocampus, the main area in the brain for memory storage [41-43]. Other brain regions connected to EC-II subsequently shrink. Severe loss of the cortical regions becomes apparent within 7 to 10 years following disease onset, and accounts for cognitive and emotional deficits. Hence, patients in the late stage of AD not only lose their short-term memory, but also suffer from cognitive dysfunction and
9
psychiatric problems that dramatically disrupt their quality of life and reduce life expectancy.
Alois Alzheimer used a silver staining method to uncover two abnormal structures in the brain of the first patient identified with AD -- protein deposits scattered over the cortex (amyloid plaques) and intraneuronal bundles of fibrils (neurofibrillary tangles, NFTs) [39]. Both pathological hallmarks are now widely regarded as the primary causes of neurodegeneration in AD.
Amyloid-β (Aβ) and Presenilins (PSs)
Amyloid plaques are composed of fibrillar aggregates of amyloid-β peptide (Aβ). These 39-43 amino acid polypeptides are derived from an integral membrane protein called amyloid-β precursor protein (APP) [44]. Aβ deposits are closely
associated with dystrophic neurites, active microglia, and reactive astrocytes [45]. The neurotoxicity of Aβ fibrils, which has been demonstrated in various in vitro systems, is closely correlated with the pathogenesis of AD, and blockage of Aβ fibril formation prevents Aβ toxicity [46]. A recent report showed that soluble Aβ oligomers exert
neurotoxic effects by inhibiting hippocampal long-term potentiation (LTP) in rats in vivo [47], strongly suggesting Aβ-induced neurodegeneration in AD pathogenesis.
Thus, understanding how Aβ is generated and how this pathogenetic process can be
10
regulated is extremely crucial.
Aβ is generated through sequential cleavages by two proteases: β- and γ-secretase [48]. First, APP is cleaved by β-secretase at a site 28 residues N-terminal to the transmembrane domain [49], releasing a large soluble fragment (sAPPβ) into
the lumen/extracellular space, and retaining a 99-residue C-terminal fragment (C99-CTF) in the membrane. This membrane-tethered C99 can then be further
processed by γ-secretase through an unusual cleavage that apparently occurs within the transmembrane domain of C99, thereby producing Aβ peptides.
γ-Secretase has been regarded as a prime therapeutic target, because it catalyzes the final proteolytic step in the generation of Aβ. The enzyme is a large membrane
protein complex composed of presenilin (PS), nicasatrin (NCT), Aph1 and Pen2.
Evidence compiled from pharmacological studies, mutagenesis, affinity labeling, and biochemical isolation strongly suggests that γ-secretase is an aspartyl protease with an
active site located at the interface of the PS heterodimer [50], which is thought to be the active form of PS [51, 52]. Significant accumulation of γ-secretase substrates is found
in ES cells derived from mice deficient in both PS homologues (PS1-/-, PS2-/-), concordant with the lack of detectable Aβ in these animals [53, 54]. Two highly
conserved aspartate residues residing within the sixth and seventh transmembrane domains (TM6 and TM7) of presenilins are required for γ-secretase activity [55].
11
Research on familial AD (FAD) has identified several mutations in the genes
encoding APP, PS1, and PS2. Approximately 90% of FAD mutations are found in PS1 and PS2, and the majority of these mutations alter the proteolytic activity of γ-secretase, leading to an increase in relative production of aggregation-prone Aβ42 peptides [56].
However, emerging evidence implies both secretase-dependent and -independent roles of PS1 in diverse cellular activities, which may also contribute to neurodegeneration in AD. The secretase-independent function of PS1 will be further discussed in chapter 3.
Tau
The NFTs are composed of Tau proteins, which are encoded by the human gene MAPT (microtubule-associated protein Tau). As suggested by the name of the encoding gene, Tau proteins were first co-purified with microtubules in 1975 [57], and later studies revealed a major role of this protein in stabilization of microtubules [58]. The region required for such interaction is located in the C-terminus of the Tau protein, and consists of either three or four 18 amino acid repeats that result from alternative splicing [59, 60]. The positively charged repeats allow Tau to bind directly to the negatively charged microtubule surface. The highly acidic N-terminal region contains zero to two negatively charged inserts (29 amino acids of each) which are
12
also derived from alternative splicing. Therefore, six Tau isoforms exist in the adult human nervous system. Between the C-terminal repeat regions and the N-terminal acidic domain is a proline-rich region with PPXXP or PXXP motifs; these motifs are believed to bind to plasma membrane-associated proteins, including the Fyn kinase [61, 62]. Association between Tau and Fyn kinase contributes to dendritic localization
of Fyn and promotes Fyn-meditated phosphorylation of NMDA receptors, leading to exaggerated excitotoxicity in the presence of toxic Aβ species.
Tau proteins have been found to undergo several post-translational modifications. Phosphorylation of Tau is the most prominent modification in AD, and is thus a subject of extensive study [63]. There are 79 postulated sites of phosphorylation in the longest Tau isoform. Phospho-serine or -threonine surrounding repeat domains have been shown to disrupt the interaction between Tau and
microtubules. Under normal conditions, the phospho-state of Tau is tightly regulated by kinases, which include glycogen synthase kinase 3β (GSK3 β), Cdk5, MAP kinase
(p38), JNK, cyclic AMP-dependent kinase (PKA), Ca2+/calmodulin-dependent kinase (CaMKII), and protein kinase C (PKC), and by phosphatases, including protein phosphatase (PP) 1, PP2A, PP2B (calcineurin), and PP2C [63]. However, the brains of most AD patients exhibit abnormal accumulation of hyper-phosphorylated Tau (eight or more phosphates per protein, as compared to only 2~3 phosphates in a
13
normal Tau protein), and the extent of phosphorylation correlates with the progression of the disease [64].
More than 45 phospho-residues have been identified in AD patients [65], and a subset of antibodies recognizing different AD-associated phospho-residues on Tau have been developed for diagnostic purposes [63]. Although the phosphorylation sites that underlie pathogenesis remain unclear, phospho-Thr231, -Ser235 and -Ser262, all of which are observed at early pre-tangle stages, were shown to be critical for the dissociation of Tau from microtubules, leading to destabilization of the latter [66, 67].
Therefore, intracellular trafficking of neurotrophins and proteins essential for metabolism is blocked in the presence of hyper-phosphorylated Tau. Furthermore, formation of Tau aggregates through hyper-phosphorylation results in attenuation of proteasomal activity [68], supporting the pivotal role of hyper-phosphorylated Tau in neurodegeneration. It should be noted that spatial and temporal phosphorylation of Tau by diverse kinases contributes to the formation of hyper-phosphoryated Tau in AD, and promotes self-assembly of Tau into insoluble filament structures, thus complicating the development of stratagies targeting Tau phosphorylation through
inhibition of protein kinases [69]. For example, PKA-mediated phosphorylation of Tau facilitates subsequent phosphorylation by GSK3β, which is accompanied by loss
of memory in a rat model [70].
14
Conversely, there is also evidence suggesting protective roles for hyper-phosphorylated Tau, as there are reports that it renders neurons more resistant to apoptosis and oxidative stress [71]. This is consistent with the observation that Tau proteins in neurons from fetal brains possess more phosphorylated residues [72].
Whether these protective phosphorylated sites are the same as those detected in AD remains unaddressed. Nevertheless, the levels of total Tau and phophorylated Tau in cerebrospinal fluid is tightly correlated with cognitive deficits in AD, suggesting that Tau is an important factor in the disease.
Aβ versus Tau hypothesis
The formation of two pathological hallmarks, amyloid plaques and neurofibrillary tangles, in AD patients strongly implies that Aβ and Tau contribute to
pathogenesis of the disease. However, which of these is the primary cause of AD has
been debated among researchers for a long time. In the past decade, the amyloid hypothesis claiming that Aβ acts upstream of Tau to exert neurotoxicity and facilitate
hyper-phosphorylation of Tau is predominant in the field of research [56]. This concept is substantially supported by the genetic discovery of FAD-linked mutations in APP, PS1, and PS2, but not in Tau. As mentioned earlier, the common feature of these mutations is to increase the relative production of Aβ42, which accelerates the
15
aggregation of amyloid peptides. Several transgenic mouse models expressing either a FAD-linked human APP mutant alone or in combination with a human PS1 mutant
exhibit neurodegeneration and intraneuronal NFT accumulation, supporting the role of Aβ in driving AD pathogenesis.
Interestingly, mounting evidence suggests that Tau is indispensible for Aβ-induced neurotoxicity. Neurodegeneration was observed in Aβ42-treated primary
neuron cultures derived from wild-type mice, but no toxic effects were detected in those from Tau KO mice that underwent the same treatment with Aβ42 [73]. Another study reported that mutant APP-expressing J20 mice exhibit Aβ plaque pathology
accompanied by premature lethality and memory defects. Crossing J20 mice with Tau-null mice can ameliorate these pathological phenotypes [74]. Later, Ittner, L.
M. et al. demonstrated that the interaction between Tau and Fyn leads to postsynaptic localization of Fyn and induces phosphorylation of NMDA receptors by Fyn [61].
Phospho-NMDA receptors tend to associate with postsynaptic density protein 95 (PSD95), forming a complex required for Aβ-induced excitotoxicity. According to the
above evidence, Itner and Gotz proposed the “Tau axis hypothesis” [75], which suggests that formation of Aβ oligomers in dendrites results in synaptic dysfunction at the early stage of AD. As Aβ also induces accumulation of hyper-phosphorylated Tau
(possibly through activation of NMDA receptors), neurons are rendered more
16
vulnerable to Aβ toxicity, contributing to neuronal loss during the progression of the
disease.
Taken together, these results suggest that although Aβ is the principle causative factor for AD, excessive amounts of Tau are also required to mediate Aβ-driven
toxicity. Strategies to prevent Tau accumulation are therefore potential therapeutic approaches to block neurodegeneration. In the following sections, I summarize the two main cellular degradative systems, both of which are involved in clearance of neurodegeneration-related proteins and are thus therapeutic targets for these diseases.
1.4 Proteasome
Both the ubiquitin-proteasomes and the autophagosome/lysosome systems are indispensible degradative complexes in our cells. Functional defects in these complexes can severely impair cellular protein quality control. While each is composed of different cellular elements, the two systems also share common elements, and have been recently shown to undergo cross-talk with each other [76, 77].
The 26S proteasome is a complex that resembles a tube with upper and lower covers composed of several subunits [78]. The tubule structure is the 20S core particle
of the proteasome, consisting of two outer α-rings and two inner β-rings, each of which is formed by seven subunits. Catalytic activity is conferred by the β-ring.
17
Different β-subunits possess either trypsin-like, chymotrypsin-like, or peptidyl-glutamyl peptide-hydrolyzing activities, all locating to the inner face of the
tube. Exchange between these subunits contributes to catalytic specificities of the proteasome. The α-rings connect with the 19S regulatory particles that constitute the
upper and lower covers. The precisely-organized 19S regular particles control the opening of the α-rings, and are involved in substrate recognition, unfolding and translocation.
The ubiquitin-proteasome system is tightly controlled to enable degradation of specific proteins. Such selectivity is achieved by the ubiquitin conjugation process [79, 80]. Proteins that fail to fold properly are tagged with 8 kDa ubiquitin molecules through covalent binding at Lys residues. Three enzymes are required for the ubiquitin conjugation process—first, ubiquitin is linked to an ubiquitin-activating E1 enzyme, which is accompanied by ATP hydrolysis to activate the ubiquitin through adenylylation. Second, the ubiquitin is transferred to an ubiquitin-conjugating E2 enzyme. Finally, an ubiquitin ligase E3 enzyme binds to both the E2 conjugase and the specific substrate, allowing the ubiquitin to form a covalent bond with a lysine residue on the substrate. While only two E1 and around one hundred E2 enzymes have been identified, it has been shown that more than one thousand E3 ligases are present in eukaryotic cells, which enables restricted and precise degradation of target
18
proteins.
Ubiquitin contains seven lysine residues, each of which can be conjugated with another ubiquitin, resulting in the formation of linear chain linkages or branched chain linkages on the substrate (polyubiquitination). K48 linkages, consisting of at least four ubiquitin molecules that are all linked through the lysine residues at position 48, are the most abundant form of polyubiquitin chains. It has been shown that proteins targeted with K48 linkages are the most rapidly degraded substrates of proteasomes.
The K11 linkage was also demonstrated to be a tag for proteasomal degradation in some cases [81, 82]. Other types of lysine linkages are specifically responsible for different cellular activities. For example, the K33 linkage is involved in kinase modification [83], while K29 and K63 linkages are associated with endocytosis trafficking and autophagic degradation (to be discussed in later sections) [84, 85].
After polyubiqutination, most target proteins can directly bind to the ubiquitin receptors on the 19S regulatory particles of proteasomes. In some cases, they require specific shuttle factors such as Rad23, ubiquilin 1/2 and p62 for transportation to proteasomes [86]. Common features of these shuttle proteins are the presence of at least one ubiquitin associated (UBA) domain to interact with ubiquitin chains on the substrates, and a ubiquitin-like (UBL) domain that binds to proteasomal Ub receptors.
Once the substrate protein docks to a proteasome, it undergoes deubiquitination
19
and unfolding, as mediated by certain subunits of 19S regulatory particles. For example, Rpn11 of the lid subcomplex is required for removal of entire ubiquitinated chains [87]. Both deubiquitination and the unfolding reaction involve high energy demands, and therefore a subgroup of 19S regulatory particles possess ATPase activity to provide sufficient driving force [86]. Finally, the unfolded proteins without ubiquitin enter the catalytic tube for digestion. Short peptides (around 8 amino acids) are generated and released back into the cytosol for further use.
Proteasome dysfunction has been observed in aging brains and in the affected brain areas of a number of neurodegenerative diseases [88]. Frame shift mutations of ubiquitin were identified in AD [89], indicating a link between proteasome impairment and neurodegeneration. In support of this link, treatment of cell models with chemical inhibitors targeting S20 core particles induced formation of ubiquitin positive-inclusion bodies and caused cell death [90]. Furthermore, transgenic mice that lacked functional 26S proteasomes in the central nervous system developed intraneuronal inclusions and presented with neurodegeneration [91]. Overall, loss of quality control in protein turnover by proteasome damage can contribute to neuropathogenesis, and the proteasomal system is thus a potential therapeutic target for neurodegenerative diseases.
20
1.5 Autophagy
While the proteasome is required for degradation of short-lived and small soluble proteins, the relatively large sized membrane compartments of the autophagy/lysosome machinery allows the digestion of long-lived macromolecules and even organelles. Autophagy/lysosomal processes are highly conserved from yeast to mammals. Three types of autophagy have been identified, namely microautophagy, chaperone-mediated autophagy and macroautophagy.
Microautophagy
Microautophagy is the direct engulfment of molecules surrounding lysosomes by the dynamic activity of the lysosomal membrane. Although it was discovered over 40 years ago [92], the mechanisms are not yet completely understood. Nevertheless, the process of microautophagy can be divided into five steps, according to the involvement of distinct molecules and morphological changes of the lysosome [93].
The first step is the invagination of the lysosomal membrane and the resulting formation of autophagic tubes towards the interior of lysosome. This initial step can be induced by starvation or by inhibition of the mammalian target of rapamycin (mTor). Certain macroautophagy-related proteins (Atgs), including Atg3, Atg4, Atg5, Atg7, Atg8, Atg10 and Atg12, are required for organization of the autophagic tube
21
during the process of microautophagy (please refer to the “Macroautophagy” section for descriptions of Atgs). Additionally, vacuolar transporter chaperone (VTC) complexes were found localized to the invagination site in yeast, where they serve as scaffolds that interact with cytoskeleton proteins, membrane proteins and regulatory molecules [94]. Changes in the protein/lipid composition of the autophagic tube then leads to formation of a vesicle at the tip (vesicle formation step) with a lollipop shape.
This vesicle undergoes expansion (expansion step) and finally pinches off into the
lumen from the tube (scission step), which is driven by GTPase. At the final stage, the
released vesicles are degraded by lysosomal hydrolases, and the digested products are
transported back to the cytosol for recycling through the permease Atg22p.
Since microautophagy ends with consumption of the lysosomal membrane, and its regulatory machineries overlap with those of macroautophagy, it has been proposed that microautophagy is a compensatory mechanism for macroautophagy (which contributes the membrane source of lysosomes) to balance the size of lysosomes. However, microautophagy not only engulfs substrates non-selectively by invagination, but also specifically entraps damaged peroxosomes (micropexophagy), non-essential portions of the nucleus (piecemeal microautophagy of the nucleus, PMN) or impaired mitochondria (micromitophagy) in yeast models [93, 95], implying an active role of microautophagy in maintaining cellular physiological functions.
22
Chaperone-mediated autophagy (CMA)
Chaperone-mediated autophagy (CMA) requires specific chaperone proteins to transport cytosolic molecules directly into lysosomes. Proteins containing a pentapeptide (KFERQ) motif are recognized and bound by heat shock cognate protein of 70 kDa (hsc70) [96]. This interaction allows substrates to dock to a receptor, lysosomal associated membrane protein type 2A (LAMP2A), which is located on the lysosomal membrane. On the cytosolic side, several co-chaperones assist in the unfolding of the substrate [97], while a subgroup of hsc70 molecules on the luminal side provide a pulling force to facilitate translocation of the substrate through LAMP2A into the lysosome [98]. In contrast to microautophagy and macroautophagy, CMA is restricted to degrading soluble proteins (not large organelles), as substrates must be able to pass directly through transporters in the lysosomal membrane.
CMA can be induced by stress, including nutrient deprivation and oxidative damage [99]. During these conditions, nonessential proteins and oxidized proteins are removed partly through CMA. It has also been shown that CMA is required for presentation of endogenous antigens by MHC-II [100], suggesting diverse roles of CMA in physiological functions. Interestingly, the KFERQ-like motif is also present in proteins related to neurodegenerative diseases, such as α-synuclein, Htt and APP, mutations in all of which can cause conformational changes that may result in toxic
23
polymers. Among these potential substrates, wild-type α-synuclein has been proved to
undergo CMA-mediated degradation, while mutant forms do not translocate into the lysosome through LAMP2A [101]. In addition, CMA has been shown to degrade endogenous wild-type Htt [102], and expansion of the polyQ tract in Htt may disrupt its transportation across lysosomal membranes.
Macroautophagy
Macroautophagy is characterized by the formation of a double-membraned autophagosome that fuses with a lysosome to degrade its contents. It is the most extensively investigated type of autophagy, and hereafter use of the term “autophagy”
will refer to macroautophagy, if not otherwise indicated.
Macroautophagy is initiated by the formation of the phagophore assembly site (PAS); this encloses a portion of the cytoplasm, which becomes a double-membrane-limited autophagosome [103]. The degradation of contents within autophagosomes is subject to fusion with lysosomes [104] (Fig.1.1a). The process of autophagy is tightly governed by a series of autophagy-related proteins (Atgs), ranging from Atg1 to Atg36, that were first identified in yeast and are thought to be functionally conserved from yeast to human [105].
24
Fig.1.1 Schematic depiction of macroautophagy, lipidation process and aggrephagy.
(a) Non-selective autophagy: cytosolic material (bulk cytoplasm, protein aggregates, organelles or pathogens) is sequestered by an expanding membrane sac, resulting in the formation of a double-membrane vesicle, an autophagosome. The outer membrane of the autophagosome subsequently fuses with a lysosome, the cargo-containing membrane compartment is then lysed, and the contents are degraded. (b) Covalent binding of LC3-I/Atg8 to phosphatidylethanolamine (PE), mediated by Atg7, Atg3 and Atg5-Atg12-Atg16L complex, is essential for the translocation to autophagosome, which in turn mediates autophagosome formation. (c) p62-mediated aggrephagy: ubiquitinated proteins initially interact with the UBA domain within p62, and are selectively entrapped into autophagosomes through interaction between LC3 and p62 for their degradation.
One group of Atg proteins essential for the initial stages of autophagy is comprised of Atg1 and Atg13. The Atg1-Atg13 complex acts downstream of Target of Rapamycin (TOR), AMPK and the Ras/PKA pathway to regulate autophagy [106, 107].
25
Two mammalian homologs of Atg1, uncoordinated 51-like kinase 1 (ULK1) and ULK2, have also been shown to coordinate with mammalian Atg13 to induce autophagy [108, 109]. Upon activation, the ULK complex translocates to the PAS forming site (adjacent to the ER), where it may contribute to the relocalization of the phosphatidylinositol 3-OH kinase (PI3K) complex, BECLIN 1-VPS34 [110]. This promotes the generation of phosphatidylinositol 3-phosphate (PI3P) at the PAS, which is crucial for activating certain Atg complexes (namely Atg12-Atg5-Atg16L1).
The core Atg machinery, including Atg3, Atg4, Atg5, Atg7, Atg8, Atg10, Atg12, and Atg16, functions as a ubiquitin-like conjugation system during the early stage of autophagy [111]. The ubiquitin-like protein Atg12 is conjugated to Atg5 through the sequential action of Atg7 (ubiquitin-activating enzyme) and Atg10 (ubiquitin-conjugating enzyme) [112]. The Atg5-Atg12 conjugate then forms a large complex with Atg16-L.
The other ubiquitin-like protein, Atg8, is first synthesized as a precursor form, and its C-terminus is subsequently processed by the Atg4 cysteine protease to expose a glycine residue. The processed Atg8 is then transferred to phosphatidylethanolamine (PE) by Atg7 (the same ubiquitin-activating enzyme required by Atg12), Atg3 (a ubiquitin-conjugating enzyme) and the Atg5-Atg12-Atg16L complex (a ubiquitin E3 ligase) (Fig 1.1b). The PAS localization of the Atg5-Atg12-Atg16L complex therefore
26
determines the site at which Atg8 is converted from an unlipidated species (form-I) to a lipidated one (form-II), allowing the attachment of lipidated Atg8 to both sides of the forming autophagosome membrane. As Atg8 is the only molecule known to persist on autophagosomes throughout autophagy, it has been commonly used as a marker to identify autophagosomes [113].
At least six homologues of Atg8 have been identified in mammals, including
three microtubule-associated protein 1 light chain 3 proteins (LC3A, B and C), γ-aminobutyrate receptor-associated protein (GABARAP), and two GABARAP-like
proteins (GABARAPL1 and 2) [114, 115]. LC3B has been reported to be the major homologue involved in starvation-induced autophagy. However, cell type-specific distribution of LC3 isoforms, and the observation that all these homologues can be processed to conjugate with PE, suggest a complicated involvement of these proteins in the regulation of autophagy. It has recently been shown that the N-terminal α−
helices of LC3 and GABARAPL2 are crucial for membrane fusion during autophagosome biogenesis [116]. Interestingly, overexpression of LC3 resulted in accumulation of larger Atg16-positive phagophores, while overexpression of GABARAPL2 reduced the number of phagophores, indicating that LC3s are required for expansion of phagophore membranes, and the GABARAP subfamily is involved in autophagosome maturation [117].
27
In order to catabolize the confined materials, the outer membranes of the autophagosome must first fuse with lysosomes or late endosomes, a process that requires the small GTPase Rab7A and LAMP1/2 [118]. By recycling the degraded products for sources of energy production and macromolecule synthesis, cells are able to survive under stress conditions. Interestingly, the released molecules can reactivate mTOR activity, which impedes autophagy and induces formation of proto-lysosomal tubules and vesicles; these vesicles form autolysosomes that finally mature into functional lysosomes. Hence, mTOR also participates in negative feedback regulation to maintain the homeostasis of lysosomal membranes [119].
The essential roles of autophagy in controlling the balance of protein metabolism have been intensively studied in various biological contexts. An increase in autophagic activity has been shown to expand the lifespan of various animal models [120-122]. Conditional deletion of Atg5 ot Atg7 in the central nervous system of mice results in loss of autophagic activity and concomitant accumulation of ubiquitinated proteins, leading to significant neurodegeneration [123, 124]. These findings clearly demonstrate that autophagy-mediated clearance of unwanted aggregates is crucial for neuronal survival, consistent with the notion that a decline in autophagy is tightly associated with normal aging [125]. Moreover, disruption of autophagy is also linked with a number of neurodegenerative diseases.
28
For HD, genetic blockage of autophagic activity hampered degradation of mutant Htt [126]. Activation of autophagy by the insulin signaling pathway promoted clearance of poly-Q-expanded Htt [127]. Rapamycin treatment, which inhibits mTor and in turn induces autophagy, has been reported to reduce Htt aggregates in cells overexpressing mutant Htt and in a mouse model of HD, and alleviate neurodegeneration [128, 129], strengthening the protective role of autophagy in HD.
Abnormal accumulation of autophagosomes and other pre-lysosomal autophagic vacuoles are found in dystrophic neocortical and hippocampal pyramidal neurons from AD patients [130]. This pathological phenotype can be simulated by inhibition of lyososomal proteolysis in neuronal cultures [131]. Autophagosomal-lysosomal dysfunction and memory deficits were observed in an APP transgenic mouse model, and these defects were rescued by enhancement of lysosomal activity [132]. These lines of evidence suggest that failures in the later stages of autophagy, rather than dysregulation of autophagic induction, account for the autophagosome/lysosome pathology in AD.
Accumulating evidence suggests that autophagy participates in the removal of detrimental Tau and Aβ peptides, and the APP-CTF precursor. For example, Aβ can
be internalized and transported into the autophagic machinery for degradation via the Aβ-binding receptor α7nAChR [133], implying that enhancement of this degradative
29
pathway should exert neuroprotective effects. However, unlike HD, the role of
autophagy is more complicated in AD. Convincing data reveal an unfavorable function of autophagy in facilitating the production of intracellular Aβ [134]. The two faces of autophagy in homeostasis of Aβ potentially place it in a unique and intriguing
position for AD pathogenesis (refer to [135] for detailed information).
Selective autophagy
Although the autophagic pathway appears to non-selectively engulf cytosolic molecules, it can also trap certain cargos for selective turnover, which is pivotal for quality control of specific proteins and for removal of damaged organelles. Several types of selective autophagy have been identified and named for their distinct substrates, including mitophagy (turnover of mitochondria), ERphagy (turnover of ER), pexophagy (turnover of peroxisomes), nucleophagy (turnover of nucleus), lipophagy (turnover of lipid droplets), aggrephagy (turnover of aggregation-prone proteins), and xenophagy (turnover of pathogens) [136]. These forms of selective autophagy have been shown to have profound roles in the pathogenesis of diverse diseases.
These distinct forms of selective autophagy require precise regulatory systems, and emerging studies are making efforts to elucidate the molecules involved. It has
30
been reported that a cargo receptor is necessary to bring the substrates into autophagosomes. An adaptor may also be required to make a connection between the cargo-receptor complex and the Atg proteins on the forming phagophores.
The best characterized form of selective autophagy is the yeast cytosol-to-vacuole targeting (Cvt) pathway, a process specifically required for the maturation and vacuole transportation of the vacuolar aminopetidase 1 (Ape1), aminopetidase 4 (Ape4), α-mannosidase 1 (Ans1) and Ty1 transposon [137]. These substrates first form a Cvt complex to interact with the autophagy receptor Atg19. The adaptor protein Atg11 then binds to Atg19 and recruits the whole complex to the phagophore; Atg1/ULK1 at this site then regulates formation of the autophagic membrane. The cargo receptor Atg19 also interacts with Atg8, the only protein known to be retained on the autophagosome membrane, thereby allowing transportation of the Cvt complex to lysosomes via entrapment within autophagosomes.
In mammals, no Atg19 homologues have been thus far identified, but a few potential cargo receptors required for selective autophagy have been discovered. A common feature of these receptors is the presence of at least one consensus
“LC3-interacting region (LIR)” [138, 139]. The protein p62/sequestosome1 was the first cargo receptor identified in mammals, and is currently the best characterized [140-142]. It is required not only for selective autophagy, and especially aggrephagy
31
(Fig 1.1c), but also for diverse cellular functions. Mammalian adaptor proteins are less understood; to date, only the ALFY adaptor is known to recruit the Atg5-Atg12 complex to phagophores and directly interact with the cargo-receptor complex (reviewed in [143]).
Interestingly, the cargo receptor p62 has been shown to be present in inclusion
bodies within degenerating brain neurons of patients with various neurodegenerative diseases; these include aggregations of α-synuclein in PD, Htt in HD, TDP-43 in ALS,
and Tau in AD [144-146]. In the next section, I will discuss in detail the role of p62 in diverse physiological functions and neurodegeneration-related pathological conditions.
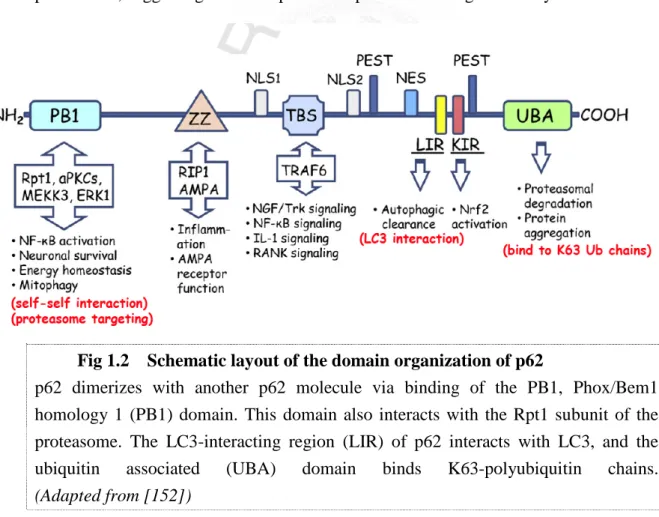
1.6 p62/sequestosome 1 The structure of p62
The complementary (c)DNA of p62 was cloned in 1996 [147], and the encoding protein was subsequently named sequestosome 1, on account of its ability to form cytosolic inclusion bodies with ubiquitinated proteins, and to ”sequester” these proteins in specialized compartments [148]. p62 messenger (m)RNA is ubiquitously expressed in a variety of tissues [147], and the protein is conserved throughout metazoa. Multiple protein-protein interaction motifs have been characterized in the
32
p62 protein [149] (Fig 1.2). At the N-terminus of p62, there is a PB1 domain which
mediates self interaction. As such, p62 can bind to other proteins containing PB1;
these proteins are usually signaling molecules, including PKCζ, MEKK3, MEK5 and
ERK, and p62 acts as a scaffold protein to regulate the various signaling pathways [150, 151]. In addition, p62 can assemble into homopolymers via the PB1 domain, which is crucial for the formation of inclusion bodies or sequestosome structures. It is interesting to note that this domain also interacts with the Rpt1 subunit of the proteasome, suggesting a role for p62 in the proteasomal degradative system.
Fig 1.2 Schematic layout of the domain organization of p62
p62 dimerizes with another p62 molecule via binding of the PB1, Phox/Bem1 homology 1 (PB1) domain. This domain also interacts with the Rpt1 subunit of the proteasome. The LC3-interacting region (LIR) of p62 interacts with LC3, and the
ubiquitin associated (UBA) domain binds K63-polyubiquitin chains.
(Adapted from [152])
33
The C-terminus of p62 contains an ubiquitin-associated (UBA) domain which allows it to bind to ubiquitinated proteins, and in particular K63-polyubiquitinated proteins, with high affinity [153]. It has been reported that this domain enables p62 to co-localize with certain neurodegeneration-related polyubiquitinated protein aggregates, such as α-synuclein, Tau, and Huntingtin (Htt), in brain autopsies from patients [145, 146]. Interestingly, overexpression of p62 mutated in either the PB1 domain or the UBA motif significantly reduced the number of sequestosomes, as compared to those observed in cells expressing wild type p62 [154]. This finding indicates that both p62 self interaction and association of ubiquitin-positive proteins with p62 are required for the formation of inclusion bodies. Of note, the UBA domain is also required to recognize cargo for sorting to the autophagy or proteasome, which will be discussed in later sections.
Other motifs for protein interaction in p62 include a ZZ domain for AMP receptor binding [155], a TBS domain for association with an E3 ligase, TRAF6 [156], and a KIR domain for association with Keap1, a negative regulator of Nrf2 [157]. A recent study also identified two nuclear localization signal domains (NLS1 and NLS2) and one nuclear export motif (NES) in p62 [158]. It also showed that nuclear p62 contributes to nuclear protein aggregate formation in frontotemporal lobar degeneration and spinocerebellar ataxia.
34
p62 as a shuttle factor for proteasomal degradation
The presence of a UBA domain, a TBS domain (for TRAF6 binding) and a PB1 domain (for direct targeting to the proteasome subunit Rpt1) strongly suggest involvement of p62 in proteasome degradation. In support of this hypothesis, depletion of p62 expression in a cell model system resulted in accumulation of polyubiquitinated proteins [153]. In addition, two substrates, TrkA and Tau, have been reported to be shuttled to the proteasome in a p62-dependent manner. Upon NGF stimulation, the NGF receptor, TrkA, is subject to K63-linked polyubiquitination by TRAF6, and subsequently interacts with the UBA domain of p62. This association targets TrkA to Rpt1 and facilitates deubiquitination of TrkA, which is essential for its turnover by the proteasome [159]. Therefore, depletion of p62 results in stabilization of TrkA in PC12 cells. On the other hand, Tau- and ubiquitin-positive inclusion bodies in AD brains were found to co-localize with the E3 ligase TRAF6 and p62. Depletion of p62 blocked the Tau-proteasome (Rpt1) interaction and Tau degradation [160].
Furthermore, neurofibrillary tangle-like structures containing hyperphosphorylated Tau were observed in the brains of p62–/– mice [161]. It remains possible that homeostasis of other substrates may also be regulated through the TRAF6/p62/proteasome axis, pending further investigation.
35
p62 as a cargo receptor in selective autophagy
In addition to its role as a shuttling factor for proteasome degradation, Bjorkoy et al. (2005) demonstrated that some p62 proteins are constrained inside LC3-positive
autophagosomes. Blockage of autophagy increases the size and number of p62-containing inclusion bodies [154], suggesting a role for p62 in autophagic degradation. More recently, an LC3-interacting region (LIR) was identified within p62, N-terminally proximal to its UBA domain. Biochemical analyses showed that the LIR domain is required for p62-LC3 binding and the subsequent entrapment of p62 into autophagosomes for degradation [140, 141]. In cultured cells, overexpression of p62 mutated in either the LIR or UBA domain results in a significant increase in cytosolic ubiquitin-positive inclusion bodies [141], similar to those observed in neurodegenerative diseases. In the first part of my PhD project (described in chapter 2), I identified some of the residues in LC3 essential for LC3-p62 complex formation, and demonstrated the importance of this conserved p62-LC3 interaction in the turnover of cytotoxic Huntingtin aggregates.
It has become clear that the p62-LC3 complex not only mediates aggrephagy, but is also required for selective autophagic degradation of post-mitotic midbody rings, damaged mitochondria (mitophagy), excessive peroxisomes (pexophagy) and intracellular pathogens such as S. enteric (xenophagy) [138]. A recent study found that
36
during Mycobacterium tuberculosis infection, p62 recruits antibacterial proteins (rather than bacteria) into autolysosomes, which results in bacterial death when the bacteria-containing phagosomes fuse with autolysosomes [162]. In all of the selective autophagy processes described above, ubiquitination is a prerequisite for p62 to recognize specific substrates.
It should be noted that autophagic receptors other than p62 have been recently identified. These proteins, which include NBR1, Nix and NDP52, contain at least one LIR domain that interacts with LC3 [163-165]. These cargo receptors are believed to coordinately regulate substrate selectivity under different conditions, although elucidation of the underlying mechanisms awaits further investigation.
Regulation of p62 expression
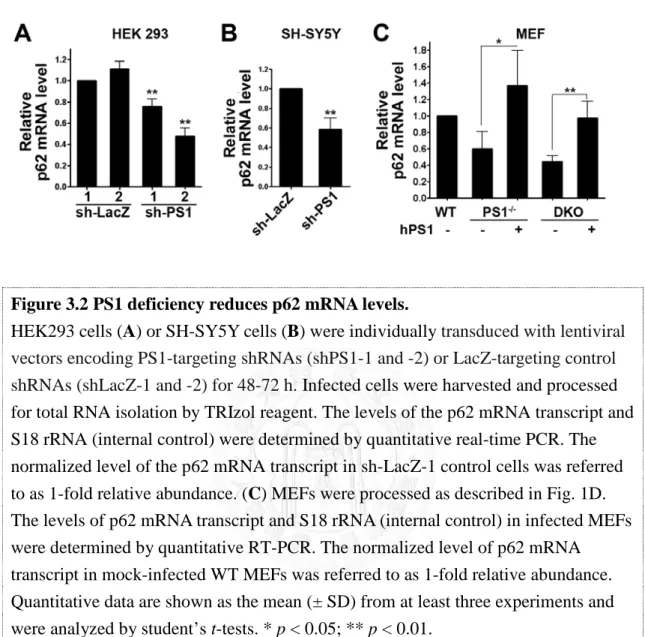
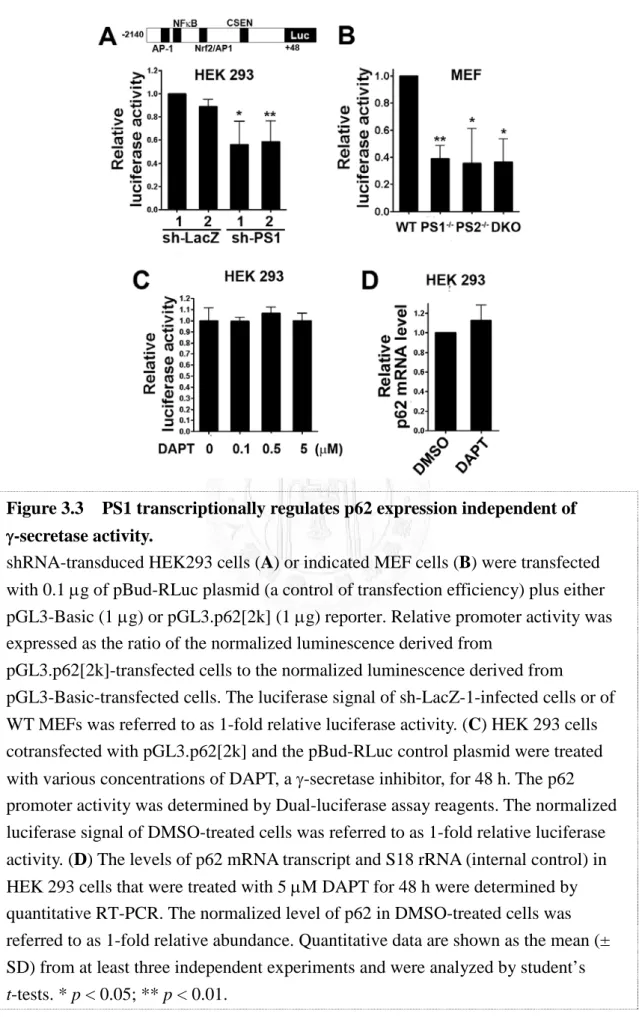
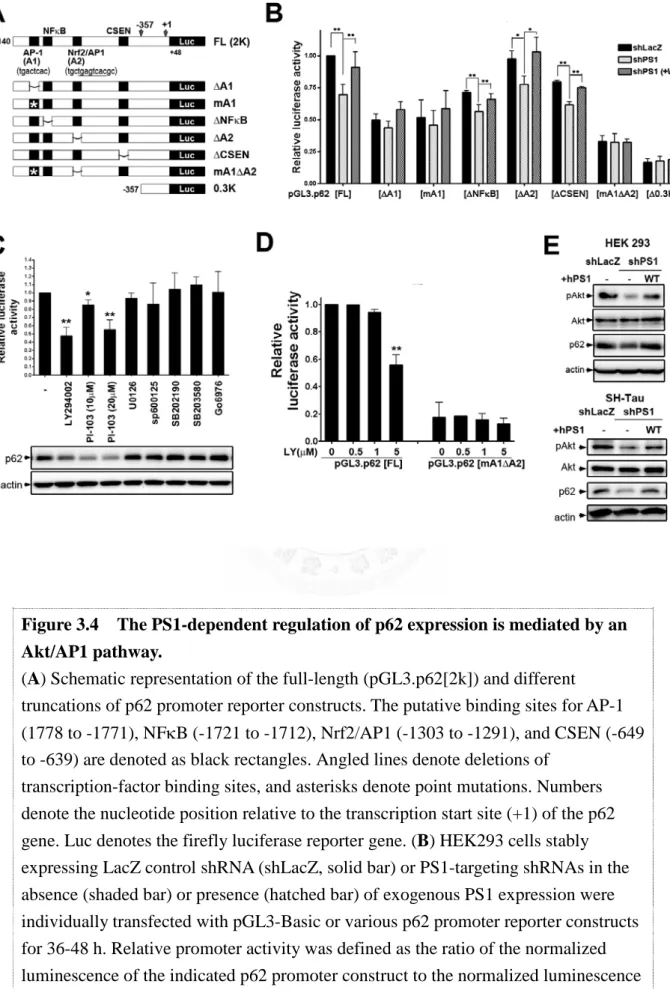
Expression of the multifunctional p62 protein requires extensive regulation.
Transcription of p62 has been shown to be rapidly activated by a variety of extracellular signals, including phorbol 12-myristate 13-acetate (PMA) and calcium ionomycin in peripheral blood mononuclear cells, serum and platelet-derived growth factor (PDGF) in serum-starved NIH3T3 cells [147], and prostaglandin J2 (PGJ2) in neuroblastoma SK-N-SH cells [166]. Analysis of the 5’-flanking region of the p62 gene revealed the presence of putative binding sites for the transcription factors AP-1,
37
Sp1, NF-κB, c-myc, Ets-1 family proteins, MyoD, and C/EBP [167], which may coordinately contribute to the early p62 induction response. It has been reported that the oncogene Ras induces binding of the activator protein (AP)-1 to the p62 promoter, thereby enhancing the production of p62 in human cancer cells [168]. In addition, several of the aforementioned putative transcription factor binding sites are embedded in CpG islands, which are sensitive to oxidative damage [169, 170]. Therefore, the expression of p62 can also be attenuated under conditions of chronic oxidative stress that commonly occur in aging. On the other hand, acute induction of oxidative stress by electrophile treatment also simulates p62 transcription, by activating nuclear factor (NF)-E2-related factor 2 (Nrf2) [171], a redox-sensitive transcription factor, indicating a pivotal role of p62 in response to oxidative and electrophilic stress.
The pathogenic functions of p62 in disease
Altered expression of p62 has been linked to various diseases. An abundance of p62 proteins is linked to breast tumors, liver cancers and liver cirrhosis [172, 173]. A mouse model with reduced levels of a key autophagic molecule, beclin 1, developed spontaneous liver and lung tumors with accumulation of p62 protein [174].
Furthermore, liver tumors developed spontaneously in a liver-specific ATG7-deficient mouse model; this was suppressed by depletion of p62 protein, suggesting that
38
increased levels of p62 contribute to tumorgenesis [175]. Additional studies have shown that p62 is an important mediator of Ras-induced lung adenocarcinomas. The
interaction between p62 and TRAF6 triggers polyubiquitination of IκB kinase (IKK), which in turn activates NFκB signaling [176], a major signaling pathway downstream
of Ras. Therefore, Ras induces excessive expression of p62, thereby activating the NFκB pathway in cancer cells; this in turn decreases the number of reactive oxygen
species (ROS) and enhances tumor survival [168]. In addition, the KIR sequence of p62 also allows it to chelate Keap1, a negative regulator of the stress responsive transcription factor Nrf2 [157]. Up-regulation of p62 in tumor cells may therefore also alleviate oxidative stress by promoting Nrf2 activity, which in turn induces the expression of key ROS scavengers [177]. As mentioned in previous section, Nrf2 enhances p62 transcription, resulting in a positive feedback loop that prolongs the antioxidative response under conditions of stress.
Mutations of p62 are also implicated in Paget’s disease of bone (PDB), a disease characterized by abnormal bone destruction and regrowth. Most of the mutations are within or surrounding the UBA region, indicating that impaired binding of p62 to ubiquitin contributes to the pathogenesis of PDB [178]. However, the most prevalent PDB mutation, P392L, does not alter the binding affinity of p62 to ubiquitin, and nor does it affect the formation of the sequestosome [179, 180]. Instead, the
39
P392L mutation reduces interaction between p62 and the tumor suppressor Cyclindromatosis (CYLD), a deubiquitinase enzyme that negatively regulates osteoclastogenesis [181]. The p62-CYLD complex may be the key for osteoclast development/bone resorption activity in PDB.
The presence of p62 in neuronal inclusions of a variety of neurodegenerative diseases implies a crucial role of this protein in neuropathy. The ability of p62 to serve as a scaffold and act as a shuttle factor in the proteasomal and selective autophagic systems supports the hypothesis that its protective function involves sequestering misfolded proteins and mediating their turnover. The case for this hypothesis was strengthened by a recent study demonstrating that p62 immunostaining co-localized with pathological inclusions in PD brains, but was homogenously distributed in healthy neurons of both control and PD brains [182].
Several studies have provided evidence that PD-associated PINK1 and parkin mutations might work together to impair mitophagy and promote the pathogenesis of PD [183, 184]. Depolarization of the mitochondrial membrane triggers accumulation of the mitochondrial kinase PINK1 at the outer mitochondrial membrane, which in turn induces translocation of the E3 ligase parkin to mitochondria. Parkin catalyses K27 and K63-linked ubiquitination of voltage-dependent anion channel 1 (VDAC1) on depolarized mitochondria, allowing binding of p62 and subsequent recruitment of
40
LC3 to accomplish mitophagy [183, 184]. Therefore, down-regulation of p62 may also contribute to defects in mitochondrial turnover in PD.
The role of p62 in AD has recently come under increased scrutiny. Du et. al.
observed an age-dependent increase in oxidative damage to the p62 promoter in the brains of patients with sporadic AD, and this was inversely correlated with the level of p62 protein [169]. Overall, it seems that p62 functions as a neuroprotective protein in AD, although the regulatory mechanisms of this protein remain to be determined.
The relationship between p62 and AD is discussed in detail in the second part of my PhD project (described in chapter 3). I also address the role of a novel transcriptional control pathway in regulating p62 expression in familial AD.
In summary, we are intrigued by the multifunctional and protective role of p62 in the clearance of aggregation-prone proteins. In the following chapters, we focus on the role of the evolutionarily conserved LC3-p62 interaction in the autophagic removal of neurodegeneration-associated Htt aggregates, and FAD-associated regulation of p62 expression.
41
Chapter 2
The evolutionarily conserved cargo-receptor binding site of LC3 interacts with p62 to mediate autophagy-dependent degradation of mutant
Huntingtin
42
2.1 Abstract
Autophagy has been found to play a critical role in the clearance of various aggregated proteins that contribute to the pathogenesis of human diseases. In mammalian cells, p62/sequestosome-1 protein binds to both LC3 and polyubiquitinated cargo proteins destined to autophagy-mediated degradation. LC3, the mammalian homologue of yeast Atg8, is translocated to autophagosomes upon induction of autophagy and regulates autophagosome formation in cultured mammalian cells. Our previous findings have identified several hydrophobic or positively charged residues in Atg8 that are essential for its interaction with the cargo receptor Atg19 in selective autophagic processes of yeast. We thus sought to determine whether such interaction is evolutionally conserved from yeast to mammals.
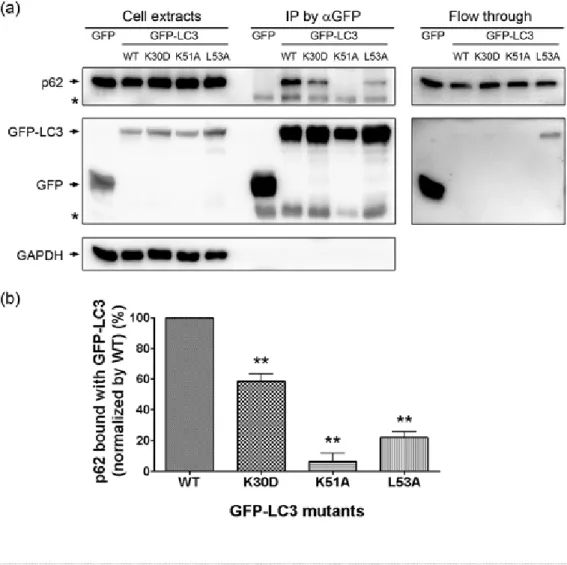
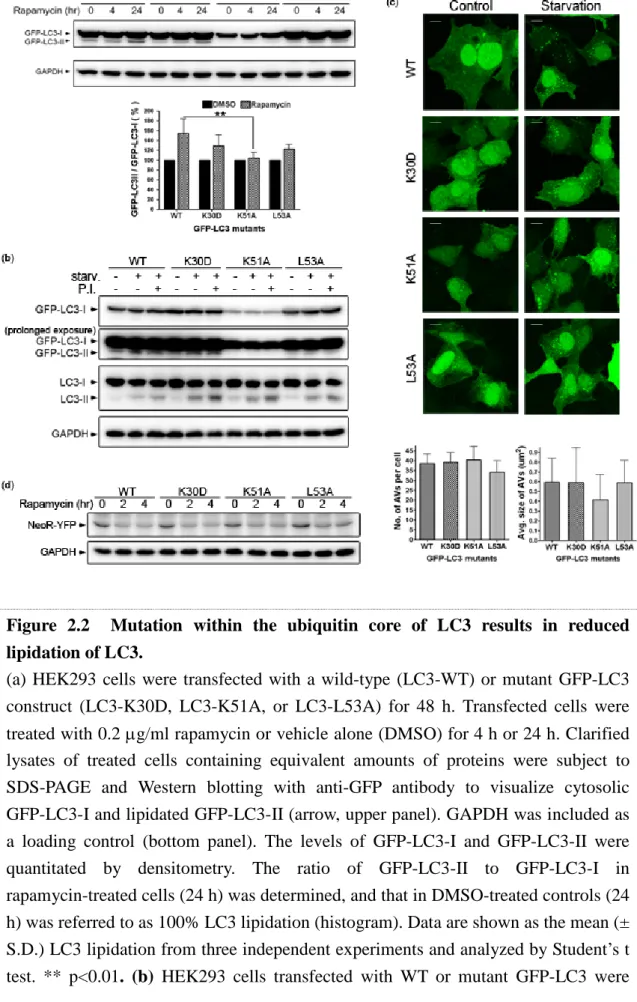
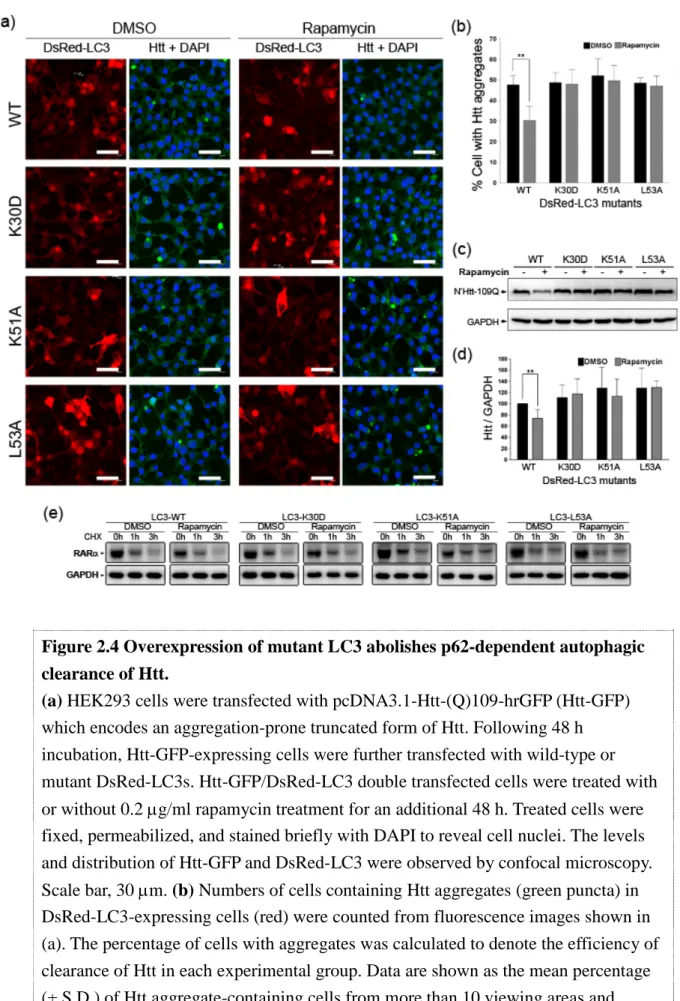
Using amino acid replacement approach, we demonstrated that cells expressing mutant LC3 (LC3-K30D, LC3-K51A, or LC3-L53A) all exhibit disrupted LC3-p62 interaction and impaired autophagic degradation of p62, suggesting that the p62-binding site of LC3 is localized within an evolutionarily conserved domain.
While cells overexpressing LC3 mutants did not exhibit normal autophagic activity, the autophagy-mediated clearance of aggregation–prone mutant Huntingtin, the cytotoxic protein inducing pathological phenotypes of Huntintung’s disease, was significantly defective in cells expressing LC3 mutants. These results suggest that p62