A R T I C L E
Journal of
Materials Chemistry
www .rsc .or g/mat erials
Synthesis and characterization of nano-sized LiFePO 4 cathode materials prepared by a citric acid-based sol–gel route
Kuei-Feng Hsu,
aSun-Yuan Tsay
aand Bing-Joe Hwang*
ba
Department of Chemical Engineering, National Cheng Kung University,
No.1 Ta-Hsueh Road, Tainan 701, Taiwan, ROC. E-mail: [email protected]
b
Department of Chemical Engineering, National Taiwan University of Science &
Technology, 43 Keelung Road, Sec. 4, Taipei 106, Taiwan, ROC.
E-mail: [email protected]
Received 5th May 2004, Accepted 26th May 2004
First published as an Advance Article on the web 1st July 2004
LiFePO
4/carbon composite cathode materials were synthesized by a sol–gel process. The citric acid in the developed sol–gel process plays the role not only as a complexing agent but also as a carbon source, which improves the conductivity of the composites and hinders the growth of LiFePO
4particles. Nano-sized LiFePO
4particles without the impurity phase were successfully synthesized. The grain size of LiFePO
4particles in the range of 20–30 nm is obtained at calcined temperatures from 450 to 850 uC. Increasing the calcination temperature leads to a decrease in the carbon content but an increase in the conductivity of the composites in the range of 400–850 uC. However, the conductivity slightly decreases if the calcination temperature further increases to 950 uC. The LiFePO
4/carbon composite synthesized at 850 uC shows the highest conductivity (10
23S cm
21), the highest specific capacity, and the best rate capability among the synthesized materials. It is worthy to note that the cell performance of the LiFePO
4depends on the electrochemical cycling procedure employed.
Introduction
Lithium secondary batteries have been considered as an attrac- tive power source for a wide variety of applications, such as cellular phones, notebook computers, camcorders, electric vehicles, etc. Recently, LiFePO
4has been extensively studied for use as cathode in rechargeable lithium ion batteries due to its low cost, high reversibility and safety. Orthorhombic LiFePO
4has an ordered olivine structure. This material has a relatively larger theoretical capacity of 170 mAh g
21com- pared with other iron-based compounds. This material is environmentally friendly and thermally stable, and shows very good electrochemical performance. LiFePO
4crystallizes in the Pnmb space group, and consists of distorted LiO
6, FeO
6, and PO
4units. The cation arrangement in LiFePO
4differs significantly from that in layered or spinel structures. There is no continuous network of FeO
6edge-shared octahedral that might contribute to the electronic conductivity. Instead, the divalent Fe
21ions occupy the corner-shared octahedra. The P
51is located in tetrahedral sites, and Li
1resides in chains of edge-shared octahedra.
1The strong covalent bonding between the oxygen and P
51to form the (PO
4)
32unit allows for greater stabilization in such structures compared to layered oxides.
2Among the characteristics of cathode materials, their electri- cal conductivity and lithium ion diffusion coefficient are two of the most important issues responsible for the rate capability of batteries. LiFePO
4has inherently low electronic conductivity, which results in its poor rate capability
3and therefore many approaches have been considered to overcome its poor con- ductivity, such as metal doping, carbon coating and co- synthesizing with carbon by solid-state methods.
4–7However, large particle size and poor particle size distribution (PSD) of LiFePO
4powder is usually obtained in a solid-state method.
Moreover, increasing the grain size of LiFePO
4leads to a decrease in its lithium ion diffusion coefficient and results in its poor rate capability. Therefore, it is of great importance to synthesize LiFePO
4powder with small grain size and high electronic conductivity for the improvement of its rate
capability. In this work, LiFePO
4/carbon composites are synthesized by a sol–gel process, in which citric acid plays the role of both complexing agent and carbon source. The effect of sintering temperature on carbon content, the form of the residual carbon, the structure of LiFePO
4, the electronic conductivity and electrochemical properties of the synthesized materials were investigated and discussed in detail.
Experimental
A sol–gel method was developed to synthesize LiFePO
4/carbon composite cathode materials in this study. Citric acid is employed as a chelating agent
8and a carbon source in the developed process. Traditionally, the chelating agent provides the mixing of cations at the molecular level in a sol–gel process.
Here citric acid is also a carbon source, which can prevent the oxidation of ferrous ions and afford the network structure of carbon for electron conduction. Stoichiometric amounts of ferrous oxalate dihydrate (17.99 g) and lithium nitrate (6.895 g) were dissolved in 1 M nitric acid solution (60 ml) into which 20 ml citric acid solution (66 wt%) was added dropwise with continuous stirring. To this solution, a saturated solution of ammonium dihydrogen phosphate (10 ml, 53 wt%) was then added. In these experiments, the molar ratio of chelating agent to the total metal ion was maintained at unity. The mixtures were heated gently with continuous stirring for 4 h to remove the excess water of about 50 ml. The resulting gel precursor was dried in a circulation oven for a week at 60 uC. The precursors were further calcined at 400–950 uC in 99.99% nitrogen atmo- sphere for 2 h. The heating rate of the furnace was 10 uC min
21. The thermal decomposition behavior of the gel precursors was examined with a thermo-gravimetric analyzer (TGA, Perkin Elmer, TAC 7/DX) under N
2flow. The TGA spectra were acquired in the temperature range from 30 to 850 uC at a heating rate of 5 uC min
21. To determine the carbon content, elemental analysis was performed (EA, Heraeus CHN–O Rapid Analyzer). Phase purity was verified from powder X-ray
DOI:10.1039/b406774f
diffraction (XRD, Rigaku, RINT2000) using Cu Ka radiation with 2h in the range from 10 to 60 u at a scan rate of 4 u min
21. The conductivity of the LiFePO
4was measured on the pressed pellet using Autolab PGSTAT 30 equipment (Euo Chemie B. V., Netherlands) with Frequency Response Analysis (FRA) software under an oscillation potential of 10 mV from 0.85 MHz to 0.01 Hz. The nanoscale microstructure was examined using a transmission electron microscope (TEM, JEOL, JEM 1010). The samples were dispersed into water and the suspension solution was dropped onto a standard copper TEM grid. The disordering of the formed carbon was examined by Raman spectroscopy in the range of 500–3000 cm
21(Dilor XY with Ar laser of 20 mW at 514 nm). Electrochemical characterization was carried out with coin-type cells. The slurry consisting of 77.5% LiFePO
4/carbon active material, 10%
Super P carbon black, and 12.5% poly(vinylidene fluoride) (PVdF) dissolved in N-methyl-2-pyrrolidinone (NMP) solvent was prepared. The obtained slurry was then cast on to the Al current collector and dried for 2 h in an oven at 120 uC. The resulting electrode film was subsequently pressed and punched into the circular disc. The electrode films are preserved in an argon-filled glove box (Unilab, Mbruan). The coin cell was fabricated using the lithium metal as a counter electrode. The electrolyte used consisted of a 1 M solution of LiPF
6in a mixture 1:1 by volume of ethylene carbonate (EC) and diethyl carbonate (DEC). The separator (Celgard 2400, Hoechst Celenese Corp) was soaked in an electrolyte for 24 h prior to use. All the weighing procedures and coin cell assembly were performed in the argon-filled glove box by keeping both the oxygen and moisture levels at less than 1 ppm. The charge–
discharge measurements were performed on the coin cell using the programmable battery tester (Maccor 2300) at different c-rates in a potential range of 3.0–4.0 V.
Results
Thermal analysis
The TGA plot for the gel precursor obtained from the developed process is shown in Fig. 1. A 70% weight-loss was observed during the temperature sweep to 600 uC. The change of weight-loss becomes insignificant when the temperature is further increased to 850 uC. Three discrete regions of weight- loss were found in the regions 100–200, 200–400 and 400–
600 uC. The first weight-loss region, i.e. between 100 and 200 uC is mainly due to the desorption of water (ca. 20 wt%). The second weight-loss region, i.e. between 200 and 400 uC, arises from the pyrolysis of citric acid, oxalate and phosphate (ca.
40 wt%). Subsequently, pyrolysis of the remaining organic compounds occurs in the temperature range from 400 to 600 uC (ca. 10 wt%). The final product remains at about 30 wt%
LiFePO
4/carbon, which is consistent with the theoretical yield of LiFePO
4(Fe: 7.5 wt%; Li: 0.9 wt%; PO
4: 12.7 wt%) and the amount of the residual carbon (12 wt%) in the composite material measured by elementary analysis, as shown in Table 1.
Structure
Fig. 2 shows the X-ray diffraction patterns of the LiFePO
4/ carbon synthesized at various sintering temperatures from 400 to 950 uC. The material formed at 400 uC still remains amor- phous, as shown in Fig. 2. However, a crystalline material without the impurity phase was obtained when the sintering temperature was higher than 450 uC. The phase of the synthesized material was confirmed with the JCPDS file (no. 40-1499). The grain size of the LiFePO
4powders calcu- lated from Scherrer’s equation,
9as shown in Table 1, is in the range of 20–30 nm which is much smaller than that reported in the literature.
6,7,10–12The grain growth of the LiFePO
4is
Fig. 1 Thermo-gravimetric analysis for the gel precursor in N2at theheating rate of 5uC min21.
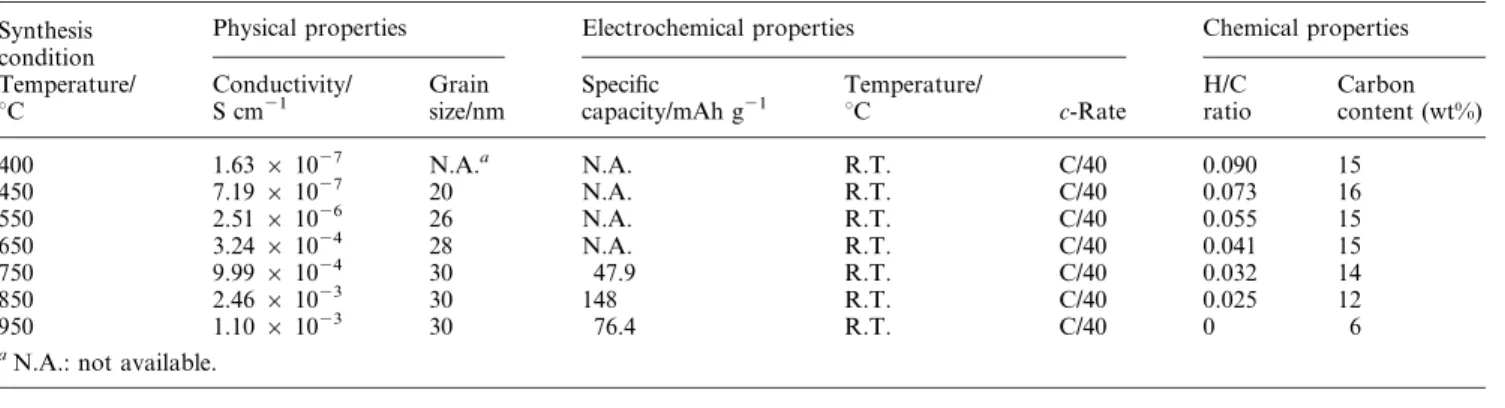
Table 1 Conductivity, carbon content and average capacity at different sintering temperatures Synthesis
condition
Physical properties Electrochemical properties Chemical properties
Temperature/
uC Conductivity/
S cm21
Grain size/nm
Specific
capacity/mAh g21
Temperature/
uC c-Rate
H/C ratio
Carbon content (wt%)
400 1.63 6 1027 N.A.a N.A. R.T. C/40 0.090 15
450 7.19 6 1027 20 N.A. R.T. C/40 0.073 16
550 2.51 6 1026 26 N.A. R.T. C/40 0.055 15
650 3.24 6 1024 28 N.A. R.T. C/40 0.041 15
750 9.99 6 1024 30 47.9 R.T. C/40 0.032 14
850 2.46 6 1023 30 148 R.T. C/40 0.025 12
950 1.10 6 1023 30 76.4 R.T. C/40 0 6
aN.A.: not available.
Fig. 2 X-Ray diffraction patterns (Cu Ka) of LiFePO4/carbon formed at various temperatures by the sol–gel process.
insignificant as the sintering temperature increases from 450 to 950 uC, indicating that the formed carbon can provide a network structure to impede the grain growth of the LiFePO
4. An experiment without the addition of citric acid to synthesize LiFePO
4particles via the similar procedure proposed here was performed. As shown in Fig. 3, the LiFePO
4powders with an impurity phase of Fe
2O
3were observed, indicating that the citric acid plays an important role in the proposed process.
Raman spectra
The Raman spectra of the synthesized powders depend on the sintering temperatures. At low temperatures (below 400 uC), the Raman spectra of the powders were similar to those of the precursor. At temperatures higher than 400 uC, thermal pyrolysis of the organic compounds in the precursor took place. The soft amorphous hydrocarbon may form in the range of 400–600 uC but transforms to hard carbon at temperatures higher than 600 uC
13during the pyrolysis process. Fig. 4 shows the Raman spectra of the LiFePO
4/carbon samples sintered at different temperatures. Two peaks were observed, at 1600 and 1358 cm
21. The strong Raman band at 1600 cm
21is one of the E
2gmodes (or mode G), which has been assigned to the vibrational mode corresponding to the movement in opposite directions of two neighboring carbon atoms in a graphene sheet.
14The 1358 cm
21band is assigned to the D mode which originally is not a Raman-active mode of the graphene sheet.
This mode is generally associated with the defects in the curved graphene sheet and staging disorder. The peak intensity ratio (I
D/I
G) between 1358 and 1600 cm
21in Raman shifts was calculated to be an index of the degree of disordering. An increase in the I
D/I
Gratio reflects a greater degree of disorder.
The I
D/I
Gratio of the synthesized powders decreases from 1.32 to 0.95 as the sintering temperature increases from 600 to 900 uC, indicating that citric acid was pyrolyzed to form graphitized carbons with a lower degree of disordering at higher sintering temperatures.
15Morphology
As mentioned in the Introduction, the particle size and morphology of the samples have a great influence on the electrochemical performance and conductivity of the electrode materials used in rechargeable lithium batteries. TEM investigations were conducted to examine the effect of sintering temperature on the particle size and morphology of the synthesized powders, as shown in Fig. 5. It was found that the LiFePO
4powders with a particle size of less than 100 nm are uniformly and completely covered with carbon. In the TEM image, the dark region is LiFePO
4and the light grey region is carbon, indicating that the LiFePO
4is surrounded by carbon.
The particles of LiFePO
4are embedded in a network structure of carbon, which can prevent its grain growth. It is in good agreement with the XRD observations. The network structure of the formed carbon can provide good electronic contact between the synthesized LiFePO
4particles. To investigate the effect of sintering temperature on the conductivity of the composite powders, the amount of the formed carbon and the conductivity of the samples were measured by EA and AC impedance, respectively, and shown in Table 1. As the sintering temperature is increased from 400 to 850 uC, the amount of carbon and the value of the H/C ratio decrease from 15 to 12%
and from 9 to 2.5%, respectively, but the conductivity increases from 1.63 6 10
27to 2.46 6 10
23S cm
21. However, the con- ductivity of the sample sintered at 950 uC decreases slightly to 1.10 6 10
23S cm
21although the amount of carbon and
Fig. 3 X-Ray diffraction patterns (Cu Ka) of LiFePO4/carbonprepared by the sol–gel process without citric acid.
Fig. 4 Raman spectra of LiFePO4/carbon powders prepared at different temperatures by the sol–gel method. From top to bottom:
950, 850, 750, and 650uC, respectively.
Fig. 5 TEM images of LiFePO4/carbon prepared at different temperatures by the sol–gel method: (a) 450, (b) 550, (c) 650, (d) 750, (e) 850, and (f) 950uC.
the value of the H/C ratio decrease further to 6 and 0%, respectively.
Electrochemistry
The cell performance of the synthesized LiFePO
4depended strongly on the sintering temperature, as shown in Table 1.
There is no electrochemical activity for the samples sintered at 450, 550 and 650 uC at 1/40 C although a crystalline material without the impurity phase was obtained, indicating that the conductivity of the carbon in the composite powders is still not good enough. The material synthesized at 850 uC showed the best cell performance, as is shown in Fig. 6. The reversible
capacity obtained for the material synthesized at 850 uC is 148 mAh g
21. A flat discharge profile was observed over a wide potential range at 3.4 V, indicating that the two-phase redox reaction proceeds via a first-order transition between FePO
4and LiFePO
4.
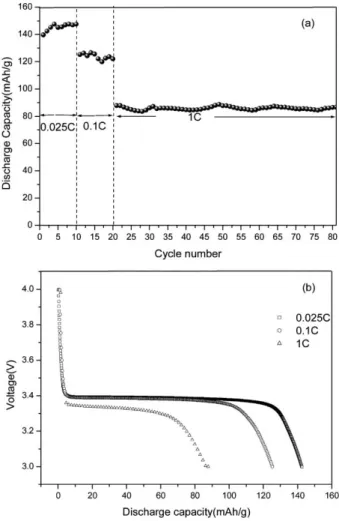
1No capacity fading but a slight increase in capacity was observed after several cycles. Two cycling pro- cedures were employed in this work. One starts from a low to a high c-rate. The other one is in its reverse. Fig. 7(a) and Fig. 8(a) show the charge capacity observed in the continuous cycling at rates varying from 0.025 to 1C and 0.2 to 0.05C, respectively, in the potential range of 3.0–4.0 V. Fig. 7(b) and Fig. 8(b) show discharge profiles of the LiFePO
4/carbon at various c-rates. When an extremely low c-rate (0.025C) is used, at first, the discharge plateau is 3.4 V and the discharge plateau still remains as 3.4 V at 0.1 C, as shown in Fig. 7(a).
The discharge capacity decreases with an increase in c-rate because of the limitations of lithium diffusion and electronic conduction.
When a high c-rate is firstly applied, the discharge plateau drops to 3.2 V and remains as 3.2 V even when the c-rate is switched back to 0.05C, as shown in Fig. 8(b). The capacity with the cycling procedure from a low to high c-rate is higher than that from a high to low c-rate. From these results, it was found that the cell performance of the LiFePO
4/carbon cathode was strongly influenced by the cycling procedures.
Ceder and co-workers have reported that Li motion in the olivine crystal structure occurs through one-dimensional channels.
16The channel for Li ion diffusion is probably obstructed by Li
117,18or Fe
21ions and becomes unavailable if a high c-rate is applied at the first cycle. However, this phenomenon can be avoided if a low c-rate is employed at the first cycle.
Fig. 6 Discharge profiles of LiFePO4/carbon sintered at 850uC for 2 h.
Fig. 7 Cyclability of the LiFePO4/carbon cells at various c-rates (from low to high c-rate): (a) discharge capacity vs. cycle number; (b) voltage vs. discharge capacity.
Fig. 8 Cyclability of the LiFePO4/carbon cells at various c-rates (from high to low c-rate): (a) discharge capacity vs. cycle number; (b) voltage vs. discharge capacity.
Discussion
From the XRD observations, the LiFePO
4samples with an olivine structure were successfully synthesized by a sol–gel method using citric acid as a chelating agent. The chelating agent provides the mixing of cations at the molecular level in a sol–gel process as well as a carbon source, which can prevent the oxidation of ferrous ions and afford the network structure of carbon for electron conduction. Meanwhile, nano-sized LiFePO
4particles can be obtained because their grain growth is impeded by the formation of the network structure of carbon.
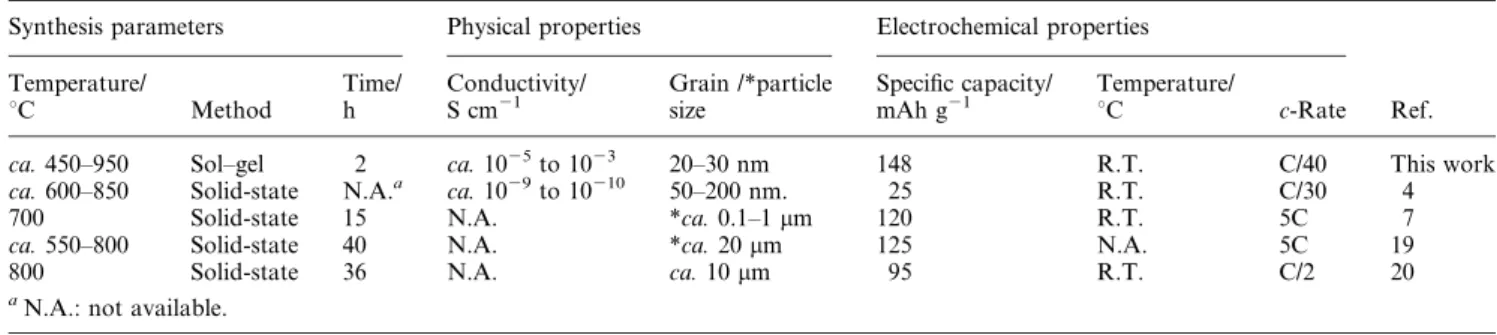
These advantages are essential for providing good rate capability of cathode materials but cannot be produced from a traditional solid-state process. The sintering time (2 h) for the developed process is much less than that (w15 h) for the solid- state processes,
7,19,20as shown in Table 2. It is of paramount importance for mass production.
The grain size (v50 nm) of the LiFePO
4powders syn- thesized by the developed sol–gel process is smaller than that (w50 nm) prepared by the solid-state methods. It is suggested that the formed carbon in the developed process can prevent the grain growth of LiFePO
4during the sintering process.
Consequently, the LiFePO
4powders with small grain size and uniform particle size distribution can be obtained by the developed sol–gel process. The rate capability would be improved because the lithium ion diffusion is enhanced due to the reduction of grain size for the synthesized materials. The conductivity of the LiFePO
4/carbon composite depends on the intrinsic conductivity of the formed carbon and LiFePO
4powders as well as the contact resistance between the carbon and LiFePO
4powders. Since the grain size of the synthesized LiFePO
4powders with pure olivine structure is similar at various sintering temperatures, the variation of their con- ductivity is mainly due to the change of intrinsic conductivity of carbon and the contact resistance between the LiFePO
4particles. Increasing the sintering temperature leads to a decrease in carbon content, H/C ratio and the degree of dis- ordering (I
D/I
G). The lower the H/C ratio and degree of disordering (I
D/I
G), the higher the intrinsic conductivity of the formed carbon. The contact resistance between the LiFePO
4particles is ascribed to the network structure of the formed carbon. A higher carbon content would provide a better network structure of carbon for electron conduction and lower contact resistance between the LiFePO
4particles.
Consequently, the conductivity of the synthesized materials results from the compensation between the contact resistance and the intrinsic conductivity of the formed carbon. It was found that the conductivity (2.46 6 10
23S cm
21) of the LiFePO
4/carbon composite synthesized at 850 uC is the best among the synthesized materials, as shown in Table 1. The electrochemical performance of the cathode materials is strongly influenced by their structure, grain size and con- ductivity. Since the LiFePO
4materials with pure phase and similar grain size can be obtained at various temperatures, the cell performance is mainly influenced by their conductivity. The cell performance of the LiFePO
4/carbon composite material
synthesized at 850 uC is the best among the synthesized materials, implying that conductivity plays the most important role in the cell performance of the LiFePO
4/carbon composite materials.
Conclusions
The nano-sized LiFePO
4particles were successfully synthesized by a sol–gel method at a moderate sintering temperature (850 uC) and the LiFePO
4particles were well covered with a network structure of carbon. The carbon can suppress the growth of the LiFePO
4particle during the sintering process and enhance the electronic conductivity of the composite powders.
The electronic conductivity of the LiFePO
4/carbon composites is greatly improved, reaching a value of 2.46 6 10
23S cm
21at room temperature. It was found that the rate capability of the synthesized LiFePO
4/carbon composite is much improved because of its small grain size and good electronic conductivity.
The electrochemical properties of the LiFePO
4/carbon cathode were strongly influenced by the cycling procedure. The electrochemical performance of the LiFePO
4/carbon cathode is better if the cell is initially charged at a lower c-rate.
Acknowledgements
Financial support received from Ministry of Education (EX-91-E-FA09-5-4) and Ultrafine company and also basic support from National Cheng-Kung University and National Taiwan University of Science & Technology are gratefully acknowledged.
References
1 K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188.
2 O. Garcia-Moreno, M. Alvarez-Vega, F. Garcia-Alvarado, J. Garcia-Jaca, J. M. Gallardo-Amores, M. L. Sanjuan and U. Amador, Chem. Mater., 2001, 13, 1570.
3 M. Thackeray, Nature Mater., 2001, 1, 81.
4 S-T Chung, J. T. Bloking and Y-M Chiang, Nature Mater., 2002, 1, 123.
5 S. Yang, P. Y. Zavalij and M. S. Whittingham, Electrochem.
Commun., 2001, 3, 505.
6 N. Ravet, Y. Chouinard, J. F. Magnan, S. Besner, M. Gauthier and M. Armand, J. Power Sources, 2001, 97, 503.
7 H. Huang, S.-C. Yin and L. F. Nazar, Electrochem. Solid State Lett., 2001, 4, A170.
8 B. J. Hwang, R. Santhanam and D. G. Liu, J. Power Sources, 2001, 97, 443.
9 B. D. Cullity and S. R. Stock, Elements of X-Ray Diffraction, Prentice Hall Publishers, New Jersey, USA, 2001, 3rd edn., ch. 5.2.
10 G. Li, H. Azuma and M. Tohda, J. Electrochem. Soc., 2002, 149, A743.
11 F. Croce, A. D. Epifanio, J. Hassoun, A. Deptula and T. Olczac, Electrochem. Solid State Lett., 2002, 5, A47.
12 G. Arnold, J. Garche, R. Hemmer, S. Stro¨bele, C. Vogler and M. Wohlfahrt-Mehrens, J. Power Sources, 2003, 119, 247.
13 Z. Sun, X. Shi, X. Wang and Y. Sun, Diamond Relat. Mater., 1999, 8, 1107.
Table 2 Physical and electrochemical properties of LiFePO4synthesized by different methods
Synthesis parameters Physical properties Electrochemical properties
Ref.
Temperature/
uC Method
Time/
h
Conductivity/
S cm21
Grain /*particle size
Specific capacity/
mAh g21
Temperature/
uC c-Rate
ca. 450–950 Sol–gel 2 ca. 1025to 1023 20–30 nm 148 R.T. C/40 This work
ca. 600–850 Solid-state N.A.a ca. 1029to 10210 50–200 nm. 25 R.T. C/30 4
700 Solid-state 15 N.A. *ca. 0.1–1 mm 120 R.T. 5C 7
ca. 550–800 Solid-state 40 N.A. *ca. 20 mm 125 N.A. 5C 19
800 Solid-state 36 N.A. ca. 10 mm 95 R.T. C/2 20
aN.A.: not available.
14 H. Hiura, T. W. Ebbesen, K. Tanigaki and H. Takahashi, Chem.
Phys. Lett., 1993, 202, 509.
15 M. Doeff Marca, Yaoqin Hu, Frank McLarnon and Robert Kostecki, Electrochem. Solid State Lett., 2003, 6, A207.
16 D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid State Lett., 2004, 7, A30.
17 A. S. Andersson and J. O. Thomas, J. Power Sources, 2001, 97, 498.
18 P. P. Prosini, L. Cianchi, G. Spina, M. Lisi, S. Scaccia, M. Carewska, C. Minarini and M. Pasquali, J. Electrochem.
Soc., 2001, 148, A125.
19 Z. Chen and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1184.
20 P. P. Prosini, D. Zane and M. Pasquli, Electrochim. Acta, 2001, 46, 3517.