行政院國家科學委員會專題研究計畫 成果報告
登革病毒感染產生自體抗體對內皮細胞的影響及疫苗研發
的考量
計畫類別: 個別型計畫 計畫編號: NSC91-3112-B-006-002-執行期間: 91 年 05 月 01 日至 92 年 04 月 30 日 執行單位: 國立成功大學微生物免疫學研究所 計畫主持人: 林以行 共同主持人: 劉校生 計畫參與人員: 林秋烽,鄭憲仁,蕭玉翎,萬書文 報告類型: 完整報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中
華
民
國 92 年 7 月 31 日
行政院國家科學委員會補助專題研究計畫
登革病毒感染產生自體抗體對內皮細胞的影響及疫苗研發的考
量
Anti-endothelial cell autoantibody production in dengue virus
infection and its concern in vaccine development
計畫類別:■ 個別型計畫
□ 整合型計畫
計畫編號:NSC 91-3112-B006-002
執行期間:九十一年五月一日至九十二年四月三十日
計畫主持人:林以行 (成大醫學院微生物及免疫學研究所)
共同主持人:劉校生 (成大醫學院微生物及免疫學研究所)
莊偉哲 (成大醫學院生物化學研究所)
計畫參與人員:
博士後研究員:林秋烽 (成大醫學院微生物及免疫學研究所)
博士班研究生:鄭獻仁 (成大醫學院基礎醫學研究所)
碩 士 班 研 究 生 :
蕭
玉
翎
、
(成大醫學院微生物及免疫學研
萬
書
究所)
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整
報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
■
成果報告
□
期中進度報告
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究
計畫、列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公
開查詢
執行單位:國立成功大學醫學院微生物及免疫學研究所
中 華 民 國 九十二 年 七 月 三十一 日
摘要
感染登革病毒 (dengue virus; DV) 會出現輕微的登革熱 (dengue fever; DF) 症狀甚或演變至嚴重的登革出血熱和登革休克症候群 (dengue hemorrhagic fever and dengue shock syndrome; DHF/DSS)。血管病變及出血性症狀是 DHF/DSS 的主 要病徵,即使有許多致病假說來解釋病理的發生,然而導致 DHF/DSS 的真正機 制仍未清楚。我們認為 DV 係利用”分子模擬”的特性引發與宿主相似蛋白的交叉 反應性抗體,除了病毒體會直接傷害宿主細胞外,病毒引發宿主產生自體免疫反 應也可能參與登革病症的發生。實驗證明抗 DV NS1 抗體 (anti-DV NS1) 與內皮 細胞發生結合作用亦會導致內皮細胞進行細胞凋亡。實驗發現 anti-DV NS1 抗體 會誘發 NO 的表現量增加並參與內皮細胞凋亡。針對 anti-DV NS1 抗體刺激內皮 細胞所造成訊息傳遞之研究,結果顯示抗體可引起內皮細胞內蛋白質的磷酸化現 象以及 NF-κB 蛋白的活化作用。我們也觀察到內皮細胞的細胞激素 (IL-6)、化 學趨化激素 (IL-8, MCP-1) 但不包括 RANTES,均會受 anti-DV NS1 抗體的刺激 而增加其表現量。在病人血清中亦偵測出大量 MCP-1 的存在。另外,anti-DV NS1 抗體刺激生成細胞黏著分子 (ICAM-1) 的表現,同時也促進週邊血液的單核球 附著於受刺激的內皮細胞表面。而這樣的現象則可被預先處理中和抗體包括 anti-MCP-1 及 anti-ICAM-1 所競爭抑制,顯示了 MCP-1 調控 ICAM-1 引發細胞 黏著的可能機制。未來的研究方向之一將探討 NF-κB 之於調控細胞激素、化學 趨化激素及細胞黏著分子表現的分子機制,以瞭解 anti-DV NS1 抗體造成內皮細 胞活化的作用。而 NO 的生成與 NF-κB 的活化亦將是研究的重點。我們遂以小 鼠動物模式探討 anti-DV NS1 抗體的病理效應,發現不管是主動免疫小鼠病毒 NS1 蛋白亦或是被動給予 anti-DV NS1 抗體都會造成小鼠體內 AST 及 ALT 的表 現增加。肝組織的切片病理觀察顯示部分區域有細胞壞死及巨噬細胞浸潤的發 生。根據本計畫的研究結果,我們認為內皮細胞自體抗體的產生會造成細胞失去 正常機能,這樣的效應可能在 DV 感染所引起的致病機制上扮演了一個重要的角 色。而這些發現將有助於提供重要的訊息做為疫苗研發的考量。 關鍵詞:登革病毒、登革出血熱/登革休克症候群、內皮細胞自體抗體、NS1 抗 體、細胞凋亡、細胞激素、化學趨化激素、細胞黏著分子
Abstr act
Dengue virus (DV) infection causes dengue fever or severe life-threatening dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS). Vascular leakage is a major hallmark associated with DHF/DSS in DV infection. The puzzle regarding the pathogenesis of hemorrhagic syndrome is poorly understood, although several hypotheses have been suggested. We have proposed a mechanism of molecular mimicry in which antibodies (Abs) directed against DV nonstructural protein 1 (NS1) cross-react with endothelial cells and cause damage. Anti-DV NS1 Abs induced endothelial cells to undergo apoptosis via production of NO. Further studies showed protein tyrosine phosphorylation and NF-κB activation after anti-DV NS1 stimulation. The cytokine and chemokine production, including IL-6, IL-8 and MCP-1, but not RANTES, in endothelial cells increased after treatment with anti-DV NS1 Abs. High levels of MCP-1 production was also observed in dengue patient sera. The increase in both ICAM-1 expression and the adhesive ability of peripheral blood mononuclear cells to endothelial cells was observed, and these effects were inhibited by pretreatment with anti-MCP-1 or anti-ICAM-1 Abs. The regulatory role of NF-κB on the expression of IL-6, IL-8, MCP-1, and ICAM-1 during anti-DV NS1-induced endothelial cell activation is currently under investigation. The relationship between NO production and NF-κB activation also remains to be clarified. Moreover, the effects of anti-NS1 Abs were further examined in animal model. Results showed that the levels of AST and ALT in mouse sera increased after either active immunization of recombinant DV NS1 to generate anti-NS1 Abs or passive administration with anti-NS1 Abs. Gross and histological examination revealed tissue damage and macrophage infiltration in mouse livers. Taken together, immune activation and apoptosis occur in endothelial cells after stimulation by anti-DV NS1 Abs, which may be involved in the pathogenesis of DV infection. The findings in the present study may provide valuable information for dengue vaccine development.
Keywor ds: dengue virus; dengue hemorrhagic fever/dengue shock syndrome;
anti-endothelial cell autoantibody; anti-NS1; apoptosis; cytokine; chemokine; adhesion molecule
目錄
中 文 摘 要 I 英 文 摘 要 I I 目 錄 I I I 前 言 1 研 究 目 的 3 結 果 與 討 論 4 計 畫 成 果 自 評 7 參 考 文 獻 8 圖 表 1 2前言
Dengue, an anthropod-borne disease caused by dengue virus (DV), is a major public health problem throughout subtropical and tropical regions of the world (49, 54). DV belongs to the genus Flavivirus of the family Flaviviridae, and is subgrouped into four serotypes. The genome of DV consists of a single-stranded RNA nearly 11 kb in length which is plus-stranded and infectious. DV genome codes for three structural proteins, capsid (C), preM, which is a precursor to membrane (M), and envelope (E), as well as seven nonstructural proteins, NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5. Infection with DV is associated with a wide range of clinical severity, from mild febrile illness to life-threatening diseases including dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) (19, 25, 54). To date, there are no effective strategies to prevent the progression of DHF/DSS, because its pathogenic mechanisms are not fully understood (13, 14, 23, 36, 37, 56). Antibody-dependent enhancement (ADE) of infection has been suggested as an explanation of the high risk for hemorrhage and shock syndrome in the secondary heterologous DV infection (20, 21, 22, 50). Virus variation and virus load may also be responsible for the severity of dengue disease. In addition to a direct effect of DV, abnormal immune responses to DV infection, such as the production of cytokines or chemokines and the activation of complement or immune cells, may also underlie the pathogenesis of disease (5, 11, 18, 30, 37, 47, 61). Immune deviations are also involved. In addition to the host factors, the virus variation, virulence and dynamics may account for severe dengue disease (53, 62). Accumulated evidence has shown that host cell apoptosis induced by DV infection may also be involved (34, 48). It is likely that several mechanisms are involved in the pathogenesis of DV infection, although their relative importance remains undefined.
In dengue pathology, the onset of vascular leakage and hemorrhage is suggested to be a result of multiple factors, including thrombocytopenia, coagulopathy, and vasculopathy (4, 24, 51). The pathogenesis of the endothelial dysfunction related to vascular leakage syndrome is not well understood. The antibody may play a role that involved in the pathogenesis of DHF. This is explained by the theory of antibody dependent enhancement, whereby cross reactive but non-neutralizing antibodies from a previous infection bind to new infecting serotype and facilitate virus entry into FcR-bearing cells resulting in higher peak viral titers (20, 21, 22, 44, 46). Antibody-enhanced infection of DV in peripheral blood monocytes could modulate endothelial cell function by an indirect route (1). Previous studies have shown that dengue virus can infect endothelial cells, leading to chemokine production, complement activation, and apoptosis (2, 3). In dengue infection, higher viral titers are associated with more severe disease. Higher titers may result in an amplified cascade of cytokines and complement, platelet destruction, and consumption of coagulation factors, which result in plasma leakage and hemorrhagic manifestations (15, 31). Mouse anti-DV NS1 Abs have also been shown to cross-react with human endothelial cells. Endothelial cell death may play a role in the fulminant but short-lived vascular leakage of DHF pathogenesis, as has been suggested previously (9). The clinical features indicate that vascular endothelia in serosal tissues are preferentially affected by dengue pathogenic mechanism, resulting in ascites and pleural effusion for most of the plasma leakage in DSS.
For vaccine development, NS1 has been the target of many research interests (7, 63, 64). Both passive transfer of anti-NS1 monoclonal antibodies and active immunization of purified NS1 conferred protection in mice (17, 26, 57). Protection by antibodies to NS1 is thought to involve immune recognition of NS1 at the infected-cell surface, followed by complement-mediated lysis. DV NS1 is expressed in a GPI-linked form that results in signal transduction, as evidenced by tyrosine phosphorylation of cellular proteins (33). Since immunization of NS1 does not elicit anti-virion antibodies, the ADE of virus replication in Fc receptor-bearing cells of a subsequent DV infection would be unlikely. Therefore, NS1 has been thought to
possess the potential to confer immunity without the risk of causing life-threatening DHF/DSS (32). Although plasmid encoding E and purified E or fusion proteins have been evaluated as potential DV vaccines, the ADE effect mediated by anti-E still needs to be taken into consideration (58, 59). Furthermore, anti-E monoclonal antibodies enhance binding of DV to human platelets via non-Fc receptors. Antibodies against E protein peptide bind to human plasminogen and inhibit plasmin activity. Therefore, anti-E antibodies may also be involved in dengue pathogenesis (29, 66). Nevertheless, it should be noted that anti-NS1 antibodies generated in mice have recently been shown to cross-react with human blood clotting, integrin/adhesin proteins, and bind to human endothelial cells (16).
Autoimmune responses characterized by the production of cross-reactive antibodies to platelets and/or endothelial cells have been shown in several viral infections such as cytomegalovirus (CMV), Epstein-Barr virus (EBV) and human immunodeficiency virus (HIV) (35, 55). The anti-endothelial cell autoantibodies from systemic sclerosis patients induced endothelial cell damage (6, 8, 10). Thus, endothelial cell apoptosis may be related to the disruption of the endothelial barrier and then lead to the transient leakage syndrome. Recently, it was shown that the autoantibodies that bound to and induced endothelial cell apoptosis recognized human CMV late protein UL94 (45). Studies of mouse antibodies generated to dengue NS1 showed the cross-reactivity with human fibrinogen, thrombocytes and endothelial cells (16). We have also reported the presence of anti-platelet autoantibodies in dengue patient sera (39) and in dengue virus-infected mice developing thrombocytopenia (28). The possible effects of anti-DV NS1 Abs on the coagulopathy, vasculopathy or other pathological manifestations were studied. We demonstrated that endothelial cells underwent apoptosis after being bound by anti-NS1 Abs generated in mice (41). The signaling pathways were investigated and showed the expression of inducible NO synthase (iNOS) and the production of NO after induction by anti-NS1. Addition of the NOS inhibitor protected cells from anti-NS1-induced apoptosis. The possible relations of NO production with the changes in Bcl-2, Bcl-xL, Bax, and p53 expression and cytochrome c release were also demonstrated (41).
The incidence and geographic distribution of dengue, a mosquito-borne disease, have greatly increased in recent years (19, 37, 54). In Taiwan, this disease has become an emerging public health problem, and an increasing number of cases are being reported. So far, there are no dengue vaccines available. The major reason for this is that the pathogenesis of DHF/DSS is not well understood. We propose a mechanism of molecular mimicry in which antibodies directed against DV NS1 can cross-react with endothelial cells and induce these cells to undergo apoptosis. When together with previous findings that anti-NS1 also cross-reacts with platelets (39) and that transient thrombocytopenia in DV-infected mice is associated with the generation of anti-platelet antibodies (28), we suggest that the onset of autoimmune responses in DV infection may have implications in DHF pathogenesis. Gene expression profiling in endothelial cells, including cytokines, chemokines, adhesion molecules, and the molecules involved in apoptotic signaling, after anti-NS1 stimulation will gain insight into the vessel endothelium disruption. The immunogenicity and safety of using modified NS1 as the vaccine candidate will be assessed at both protein and DNA levels. The results obtained from this study will provide important information about dengue therapy and vaccine development.
研究目的
The onset of vascular leakage and hemorrhagic diathesis is one of the life-threatening complications that occurs in dengue patients, yet the pathogenic mechanisms are not well understood. We have proposed a mechanism of molecular mimicry in which antibodies directed against DV NS1 can cross-react with endothelial cells and induce these cells to undergo apoptosis. Immune responses to infectious agents are the critical defense mechanisms in a host, yet abnormal immunity may also be involved in the pathogenesis of disease. We hypothesize that the host-virus interactions that induce autoimmune responses may have implications in DHF/DSS pathogenesis. Gene expression profiling after anti-DV NS1 stimulation in endothelial cells will be studied which may have implications in dengue disease pathogenesis. For safety in vaccine development, the epitopes in NS1 that may elicit autoimmune responses should be avoided. The genes encoding endothelial cell surface molecules which share epitopes with DV NS1 and are recognized by cross-reactive antibodies will be identified. The modified NS1 in both protein and DNA levels without the detrimental epitopes will be prepared for vaccine development.
Specific Aims:
1. The mechanisms involved in the effects of anti -NS1 on endothelial cells
after binding will be examined. Using microarray technique, gene expression profiling after anti-DV NS1 stimulation will be dissected. The expression of proteins will be confirmed by immunohistochemistry. The expression of cytokines, chemokines, adhesion molecules, and the molecules involved in apoptotic signaling will be investigated.
2. Endothelial cell surface molecules recognized by anti-DV NS1 will be
screened using cDNA library from endothelial cells. The epitopes shared between NS1 and speci fic surface molecules will be identified. The recombinant NS1 with modifications to remove epitopes that may elicit the pathogenic effects will be prepared and analyzed for: (a) their immunogenicity, (b) protective efficacy in the animal model, and (c) lack of pathogenic effects both in vitro and in vivo.
3. We propose to develop DNA vaccine by construction of modified NS1 genes
in a mammalian expression system and testing the protective efficacy and potential pathogenic effects based on the knowledge obtaine d from Specific Aims 1 and 2.
結果與討論
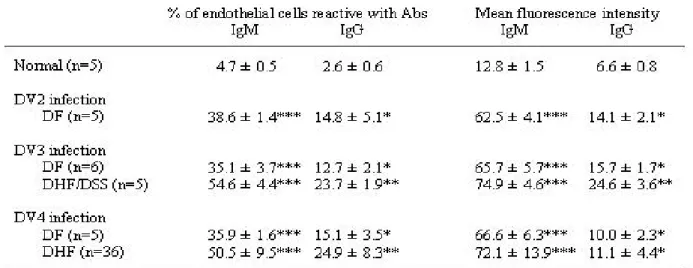
Vascular leakage and hemorrhagic syndrome are the clinical features associated with DHF/DSS, yet the mechanisms remain unclear (4, 24). Studies using patient sera collected from an outbreak in southern Taiwan showed that dengue patients produced Abs which cross-reacted with endothelial cells and induced damage (40). The percentages of endothelial cells reactive with DHF/DSS patient sera were higher than those with dengue fever patient sera. To further examine the relationship between the levels of autoantibodies and dengue serotypes or disease severity, sera obtained from the Vietnamese collaborators and Dr. C. C. Liu from National Cheng Kung University Hospital were tested. The anti -endothelial cell Abs derived from p atients infected by different dengue serotypes and at different stages and different grades were detected. Results showed that the levels of anti-endothelial cell Abs were similar in patients infected with DV2, 3 or 4 (Table 1). Consistent with previous fi ndings, the levels of anti-endothelial cell Abs were higher in DHF/DSS than in DF patient sera (Tables 1 and 2), and were higher in acute phase than in convalescent or later phases (Table 2). Preliminary studies did not show any correlation of anti-endothelial cell Abs levels with DHF grades (Table 3). Sample size of patient sera needs to be further increased to gain an insight into the role of anti-endothelial cell Abs in DHF pathogenesis. The effects of these autoantibodies on endothelial cells were further demonstrated by their abilities in induction of endothelial cell apoptosis (Table 4 and Fig. 5). Further studies showed that the cross-reactive Abs were, at least in part, due to the Abs against DV NS1 as demonstrated by NS1 preabsorption experiments (40). The cross-reactivity of anti-DV NS1 with endothelial cells had also been demonstrated using mouse Abs directed against NS1. The signaling pathways of anti-NS1-induced apoptosis were investigated and suggested the involvement of NO during this process. Production of NO caused upregulation of p53 and Bax and downregulation of Bcl -2 and Bcl-xL, which resulted in cytochrome c release and caspase-3 activation (41).
Immune responses to infectious agents are the critical defense mechanisms in a host, yet abnormal immunity may also be involved in the pathogenesis of disease (12). We demonstrated in the present study that the generation of anti-DV NS1 Abs can cross -react with endothelial cells and induce cell damage. When taken together with previous findings th at anti-NS1 present in dengue patient sera also cross -reacted with platelets and that transient thrombocytopenia in DV-infected mice was associated with the generation of anti-platelet Abs, this finding suggests that the onset of autoimmune responses in DV infection may have implications in DHF pathogenesis. NO-modulated endothelial cell apoptosis may play a role in the disruption of vessel endothelium and contribute to the pathogenesis of vascular leakage in DHF. Several questions remain: first, whether th ese in vitro findings are involved in the in vivo mechanism; second, whether a mechanism similar to the one found in DV -2 studies would occur in DV -1, -3, and -4. Our unpublished data indicate that the sera from dengue patients infected with DV-2, -3, or -4 (we have no DV-1 patient serum) show endothelial cell binding activity and induced apoptosis. Studies are required to identify the endothelial cell surface molecules that can be recognized by anti-NS1 Abs. Whether the epitopes shared between endothelial cell surface molecules and NS1 are conserved in all four dengue viruses remains to be assessed.
Further studies directed toward Specific Aim 1 concerning the mechanisms involved in the effects of anti -NS1 Abs on endothelial cells
after binding showed protein tyrosine phosphorylation (Fig. 1) and NF -κB activation (Fig. 2) after anti-NS1 stimulation. In dengue pathology, various cytokines and chemokines including TNF -α, IL-6, IL-8, and RANTES have been detected in patient sera with DHF/DSS and in DV-infected endothelial cell culture supernatants (27, 52). We are also interested in investigating whether anti-NS1 Abs can stimulate cytokine and chemokine production. Studies showed the production of IL-6, IL-8 and MCP-1, but not RANTES, by endothelial cells after treatment with anti-NS1 Abs. Production of IL-6, IL-8 and MCP-1 could be observed at as early as 6 h after stimulation, whereas RANTES could not be observed up to 24 h (Fig. 3, Table 5). The MCP-1 production had also been detected by RT-PCR (Fig. 4). We were also interested in investigating the possible role of adhesion molecules that may lead to adhesion of leukocytes, which may cause more damage to endothelial cells and contribute to the pathogenicity of dengue disease. The increase in both ICAM-1 expression and the adhesive ability of peripheral blood mononuclear cells to endothelial cells were observed (Fig. 5). Anti-JEV NS1 did not show a similar effect. Taken together, both immune activation and apoptosis occur in endothelial cells after stimulation by anti-NS1 Abs.
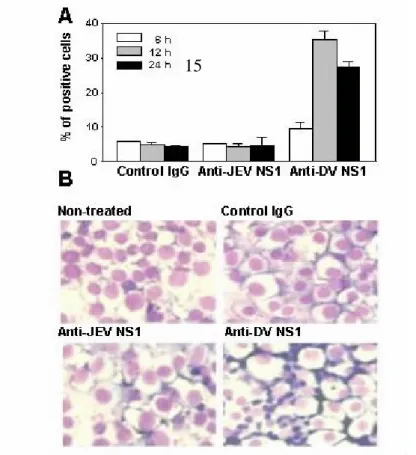
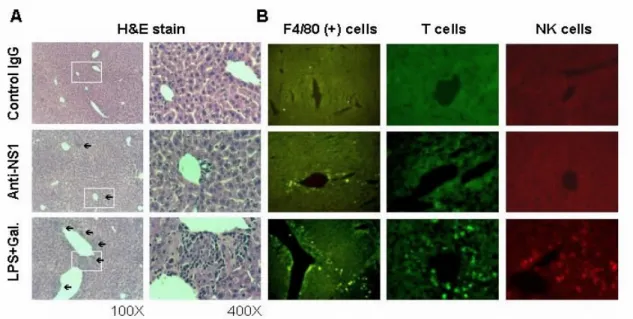
Passive administration of anti-NS1 Abs had been shown to confer protection in mice when challenged with lethal doses of dengue virus. However, these previous studies only monitored the survival rates of mice and did not examine the potential histopathologic effects. The effects of anti-NS1 Abs were thus examined in animal models. Studies indicated that either active immunization of recombinant DV NS1 to generate anti -NS1 Abs (Fig. 6) or passive administration with anti-NS1 Abs (Table 6) ca used increased concentrations of AST and ALT, but not BUN, in mouse sera, suggesting a pathogenic effect of anti-NS1 Abs on the liver. Gross and histological examinations revealed tissue damage (Fig. 7 and Fig. 8A) and macrophage infiltration in mouse livers (Fig. 8B). Since dengue patient sera could not cross-react with liver cell lines (Fig. 9), an interaction between autoantibodies and liver cells was therefore less likely involved. Liver damage is one of the clinical manifestations of DHF (65). Dengue virus had been reported to infect liver cells and induce apoptotic cell death (42, 43). In this study, we found that dengue patient sera could not cross-react with liver cell lines, suggesting that an interaction between autoantibodies and liver cells was not involved. The possibilities such as the reaction of autoantibodies with liver vessel endothelial cells and the deposition of immune-complexes leading to complement activation are currently under investigation. Based on the established animal model and the studies using endothelial cell line as described above, whether the modified/truncated NS1 may elicit the pathogenic effects as for the studies directed toward Specific Aim 2 can therefore be examined both in vivo and in vitro.
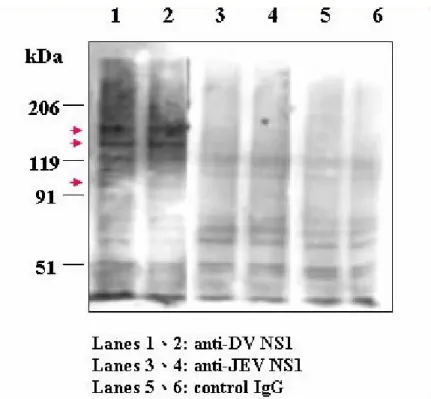
For Specific Aim 2, the surface molecules of endothelial cells recognized by anti-NS1 are targeted for identification. Endothelial cell membrane extracts were run on SDS -PAGE, followed by Western blot analysis using anti-NS1 Abs for detection. Results showed the presence of prote in bands with molecular weights of approximately 95 -, 130- and 150-kDa (Fig. 10). Alternatively, we will purify endothelial cell membrane proteins after biotin-labeling, and then detect the possible candidate membrane proteins by using anti-NS1 and streptavidin-conjugated anti-mouse IgG to confirm the former results. These protein bands are to be sequenced. In addition, the cDNA library from endothelial cells will be either constructed and clonally selected or used for in vitro transcription and translation of endothelial cell proteins, and the surface molecules will be screened through their binding activity with anti-DV NS1 Abs. The
epitopes shared between NS1 and specific surface molecules on endothelial cells will be deciphered. Based on the results, various truncated or modified DV NS1 gene in E. coli expression system will be constructed.
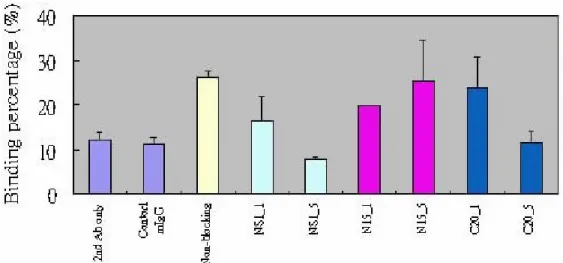
For alternative approach, the inhibition of endothelial cell binding activity have been tested by pretreatment of anti-NS1 with synthetic peptides. Results showed that the amino acids 1-15 of DV NS1 (N15) partially inhibited platelet-binding activity of patient sera (Fig. 11), but not endothelial cell binding (40). This suggest that the surface molecules on endothelial cells recognized by anti-NS1 may not be necessarily the same as those on platelets. Furthermore, the NS1 homologous proteins corresponding to the endothelial cell surface molecules were searched from the protein databases. Preliminary data show that mucin-like protein sequences are partially homologous to C-terminal peptide of NS1 protein. Pretreatment with the synthetic peptide C20 designed from NS1 (314-333 a.a.) can inhibit the cross-reaction between anti-NS1 Abs and endothelial cells (Fig. 12). In order to confirm the importance of speculated epitopes, we will prepare Abs against N15 and C20 to test their reactivity with endothelial cells. Constructions of three truncated NS1 proteins (e.g. d_N15, d_C20, and d_N15/C20 NS1) are currently in progress. The different modified NS1 sequences are obtained from constructed plasmid pCRII-TOPO-NS2L by using PCR method with designed primers. These modified NS1 sequences are ready now to be cloned into plasmid pRSET of E. coli expression system. Sequence confirmation of these modified NS1 clones remains to be proceeded.
It has recently been reported that DV NS1 is expressed in a GPI-linked form that results in signal transduction, as evidenced by tyrosine phosphorylation of cellular proteins (40). Our preliminary results showed tyrosine phosphorylation following anti-NS1 stimulation in endothelial cells (Fig. 1). The signal transduction pathways from tyrosine phosphorylation to iNOS expression leading to the changes in p53 and Bcl-2 family proteins require further delineation. Endothelial cells have been shown to become apoptotic in response to antagonists of integrin αvβ3 that cause p53 activation and p21WAF1/CIP1 expression (60). The surface molecules of endothelial cells, GPI-linked or not, recognized by anti-NS1 still need to be identified.
In this project, we hypothesize that the host-virus interplay leading to the autoimmune responses may have implications in DHF pathogenesis. The percentages of endothelial cells reactive with DHF/DSS patient sera were higher than those of dengue fever sera. In addition to the endothelial cell damage mediated by cross-reactive antibodies, a combination of factors including platelet binding activity of autoantibodies, degree of coagulation and fibrinolysis activation, as well as host immune deviation during dengue virus infection may also be associated with the onset of DHF. For the safety concern of vaccine development, the epitopes that may elicit the pathogenic effects such as autoimmunity should be avoided. The epitopes which are shared between dengue virus proteins (e.g., NS1) and specific surface molecules are to be deciphered. The endothelial cell surface molecules recognized by autoantibodies that are common or distinct from those of platelets also remain to be elucidated.
計畫成果自評
登革病毒 (DV) 感染造成致病的發生機制是多因的,我們的研究顯示 DV 感 染產生自體免疫反應生成抗內皮細胞自體抗體,係源自於病毒利用其 NS1 蛋白 所引發的分子模擬自體免疫反應,進而產生與宿主細胞有交叉結合作用的病理效 應。本計畫初始的目標為發展以登革病毒非結構性蛋白 1 (NS1) 做為基礎抗原蛋 白,研發中和性登革疫苗抗體。先前的研究發現,NS1 在登革病毒感染時參與了 自體免疫抗體生成的機制。而根據 DV 感染引起種種的病理反應都顯示除了病毒 的直接傷害外,愈來愈多的證據顯示宿主因子可能參與 DHF/DSS 的病程發展與 機制。我們的研究顯示 DV NS1 抗體 (anti-DV NS1) 就是一種自體免疫抗體,對 血管內皮細胞均有結合作用並造成對細胞功能上的影響。我們認為來自病毒的直 接感染和抗體的合併效應,亦或是抗體直接的影響都有可能是造成內皮細胞死亡 進而導致血管內皮系統失去正常機能進而參與出血性病症的發生。抗體與內皮細 胞的結合作用會造成細胞死亡的結果,除此之外,相關激素的生成如 IL-6、IL-8 及 MCP-1 亦會受到抗體的作用。換句話說,除了病毒感染的細胞活化反應,抗 DV NS1 抗體似乎也可刺激內皮細胞造成細胞不正當的活化反應生成激素,而是 否與 DV 的致病機制有關是必須再進一步釐清。小鼠動物模式的實驗顯示了 anti-DV NS1 的綜合病理影響,而肝組織的局部傷害符合了登革病理臨床上的觀 察。對於抗體如何造成肝組織病變的發生機轉則有待進一步的研究。基於這些結 果的發現,警示我們在研發疫苗之前,針對抗體所可能參與的自體免疫反應,有 必要去釐清它所隱藏的潛在性致病角色。我們以 NS1 抗體為研究對象,希冀瞭 解抗體其自體免疫反應可能會對宿主細胞造成的影響,並研究自體免疫反應在登 革病毒引發嚴重出血性及休克併發症 DHF/DSS 之致病機制中可能扮演的角色。 研究結果希望能提供我們重要的訊息,特別是找出 NS1 蛋白引發自體免疫反應 的抗原決定位。計劃將此決定位去除或予以修飾的方式,排除 NS1 所引發的 cross-reactive 自 體 免 疫 抗 體 。 如 此 一 來 ,才 能 研 發 具 保 護 性 而無致 病 性 的 candidate vaccines。本計劃的內容依據原先 propose 的計劃目標執行,部分研究 成果業已期刊發表 (39, 40, 41)。我們仍持續研究 NS1 找出會引發自體免疫抗體 的抗原決定位。而根據本計畫執行所得的這些結果都將為我們未來研發登革疫苗 提供重要的考量因素。參考文獻
1. Anderson, R., S. Wang, C. Osiowy, and A. C. Issekutz. 1997. Activation of
endothelial cells via antibody-enhanced dengue virus infection of peripherial blood monocytes. J. Virol. 71:4226.
2. Andrews, B. S., A. N. Theofilopoulos, C. J. Loskutoff, W. E. Brandt, and F. J.
Dixon. 1978. Replication of dengue and Junin viruses in cultured rabbit and human endothelial cells. Infect. Immun. 20:776.
3. Avirutnan, P., P. Malasit, B. Seliger, S. Bhakdi, and M. Husmann. 1998. Dengue
virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J. Immunol. 161:6338.
4. Bhamarapravati, N. 1989. Hemostatic defects in dengue hemorrhagic fever. Rev.
Infect. Dis. 11:S826.
5. Bokisch, V. A., F. H. Top, Jr., P. K. Russell, F. J. Dixon, and H. J. Muller-Eberhard.
1973. The potential pathogenic role of complement in dengue hemorrhagic shock syndrome. N. Engl. J. Med. 289:996.
6. Bordron, A, M. Dueymes, Y. Levy, C. Jamin, J. P. Leroy, J. C. Piette, Y. Shoenfeld,
and P. Y. Youlnou. 1998. The binding of some human antiendothelial cell antibodies induces endothelial cell apoptosis. J. Clin. Invest. 101:2029.
7. Brandt WE. 1988. Current approaches to the development of dengue vaccines and
related aspects of the molecular biology of flaviviruses. J. Infect. Dis. 157:1105.
8. Brasile, L., J. M. Kremer, and J. L. Clarke. 1989. Identification of an autoantibody
to vascular endothelial cell-specific antigens in patients with systemic vasculitis. Am. J. Med. 87:74.
9. Butthep, P., A. B. Bunyaratvej, and N. Bhamarapravati. 1993. Dengue virus and
endothelial cell: a related phenomenon to thrombocytopenia and granulocytopenia in dengue hemorrhagic fever. Southeast Asian J. Trop. Med. Public Health 24:246.
10. Carvalho, D, C. O. S. Savage, C. M. Black, and J. D. Pearson. 1996. IgG
antiendothelial cell autoantibodies from scleroderma patients induce leukocyte adhesion to human vascular endothelial cells in vitro. Induction of adhesion molecule expression and involvement of endothelium-derived cytokines. J. Clin. Invest. 97:111.
11. Chaturvedi, U. C., E. A. Elbishbishi, R. Agarwal, R. Raghupathy, R. Nagar, R.
Tandon, A. S. Pacsa, O. I. Younis, and F. Azizieh. 1999. Sequential production of cytokines by dengue virus-infected human peripheral blood leukocyte cultures. J. Med. Virol. 59:335.
12. Chaturvedi, U. C., E. A. Elbishbishi, R. Agarwal, and A. S. Mustafa. 2001.
Cytotoxic factor-autoantibodies: posibile role in the pathogenesis of dengue haemorrhagic fever. FEMS Immunol. Med. Microbiol. 30:181.
13. Chen, Y., T. Maguire, and R. M. Marks. 1996. Demonstration of binding of
dengue virus envelope protein to target cells. J. Virol. 70:8765.
14. Chen, Y., T. Maguire, R. E. Hileman, J. R. Fromm, J. D. Esko, R. J. Linhardt, and
R. M. Marks. 1997. Dengue virus infectivity depends on envelope protein binding to target cell heparin sulfate. Nature Med. 3:866.
15. Despres, P., M. P. Frenkiel, P. E. Ceccaldi, C. Duarte dos Santos, and V. Deubel.
1998. Apoptosis in the mouse central nervous system in response to infection with mouse-neurovirulent dengue viruses. J. Virol. 72:823.
16. Falconar, A. K. I. 1997. The dengue virus nonstructural-1 protein (NS1) generates
antibodies to common epitopes on human blood clotting, integrin/adhesin proteins and binds to human endothelial cells: potential implications in haemorrhagic fever pathogenesis. Arch. Virol. 142:897.
17. Falgout, B., M. Bray, J. J. Schlesinger, and C. J. Lai. 1990. Immunization of mice
with recombinant vaccinia virus expressing authentic dengue virus NS1 protect against lethal dengue virus encephalitis. J. Virol. 64:4356.
18. Green, S., D. W. Vaughn, S. Kalayanarooj, S. Nimmannitya, S. Suntayakorn, A.
Nisalak, R. Lew, B. L. Innis, I. Kurane, A. L. Rothman, and F. A. Ennis. 1999. Early immune activation in acute dengue illness is related to development of plasma leakage and disease severity. J. Infect. Dis. 179:755.
19. Gubler, D. J. 1998. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev.
11:480.
20. Halstead, S. B., and E. J. O’Rourke. 1977. Antibody-enhanced dengue virus
infection in primate leukocytes. Nature 265:739.
21. Halstead, S. B. 1979. In vivo enhancement of dengue virus infection in rhesus
monkeys by passively transferred antibody. J. Infect. Dis. 140:527.
22. Halstead, S. B., C. N. Venkateshan, M. K. Gentry, and L. K. Larsen. 1984.
Heterogeneity of infection enhancement of dengue 2 strains by monoclonal antibodies. J. Immunol. 132:1529.
23. Halstead, S. B. 1988. Pathogenesis of dengue: challenges to molecular biology.
Science 239:476.
24. Hathirat P., P. Isarangkura, T. Srichaikul, C. Survatte, and C. Mitrakul. 1993.
Abnormal hemostasis in dengue hemorrhagic fever. Southeast Asian J. Trop. Med. Public Health 24:80.
25. Henchal, E. A., and J. R. Putnak. 1990. The dengue viruses. Clin. Microbiol. Rev.
3:376.
26. Henchal, E. A., L. S. Henchal, and J. J. Schlesinger. 1988. Synergistic interactions
of anti-NS1 monoclonal antibodies protect passively immunized mice from lethal challenge with dengue-2 virus. J. Gen. Virol. 209:2101.
27. Hober, D., L. Poli, B. Roblin, P. Gestas, E. Chungue, G. Granic, P. Imbert, J. L.
Pecarere, R. Vergez-Pascal, and P. Wattre. 1993. Serum levels of tumor necrosis factor-alpha (TNF-alpha), interleukin-6 (IL-6), and interleukin-1 beta (IL-1 beta) in dengue-infected patients. Am. J. Trop. Med. Hyg. 48:324.
28. Huang, K. J., S. Y. J. Li, S. C. Chen, H. S. Liu, Y. S. Lin, T. M. Yeh, C. C. Liu,
and H. Y. Lei. 2000. Manifestation of thrombocytopenia in dengue-2-virus-infected mice. J. Gen. Virol. 81:2177.
29. Huang, Y. H., B. I. Chang, Y. H. Lei, H. S. Liu, C. C. Liu, H. L. Wu, and T. M.
Yeh. 1997. Antibodies against dengue virus E protein peptide bind to human plasminogen and inhibit plasmin activity. Clin. Exp. Immunol. 110:35.
30. Huang, Y. H., H. Y. Lei, H. S. Liu, Y. S. Lin, C. C. Liu, and T. M. Yeh. 2000.
Dengue virus infects human endothelial cells and induces IL-6 and IL-8 production. Am. J. Trop. Med. Hyg. 63:71.
31. Huang, Y. H., C. C. Liu, S. T. Wang, H. Y. Lei, H. S. Liu, Y. S. Lin, H. L. Wu, and
T. M. Yeh. 2001. Activation of coagulation and fibrinolysis during dengue virus infection. J. Med. Virol. 63:247.
32. Huang, J. H., J. J. Wey, Y. C. Sun, C. Chin, L. J. Chien, and Y. C. Wu. 1999.
Antibody responses to an immunodominant nonstructural 1 synthetic peptide in patients with dengue fever and dengue hemorrhagic fever. J. Med. Virol. 57:1.
33. Jacobs, M. G., P. J. Robinson, C. Bletchly, J. M. Mackenzie, and P. R. Young.
2000. Dengue virus nonstructural protein 1 is expressed in a glycosyl-phosphatidylinositol-linked form that is capable of signal transduction. FASEB J. 14:1603.
34. Jan, J. T., B. H. Chen, S. H. Ma, C. I. Liu, H. P. Tsai, H. C. Wu, S. Y. Jiang, K. D.
Yang, and M. F. Shaio. 2000. Potential dengue virus-triggered apoptotic pathway in human neuroblastoma cells: arachidonic acid, superoxide anion, and NF-κB are sequentially involved. J. Virol. 74:8680.
35. Kaplan, C., F. Morinet, and J. Cartron. 1992. Virus-induced autoimmune
thrombocytopenia and neutropenia. Semin. Hematol. 29:34.
36. Kurane, I., A. L. Rothman, P. G. Livingston, S. Green, S. J. Gagnon, J. Janus, B. L.
Innis, S. Nimmannitya, A. Nisalak, and F. A. Ennis. 1994. Immunopathologic mechanisms of dengue hemorrhagic fever and shock syndrome. Arch. Virol. Suppl. 9:59.
37. Lei, H. Y., T. M. Yeh, H. S. Liu, Y. S. Lin, S. H. Chen, and C. C. Liu. 2001.
Immunopathogenesis of dengue virus infection. J. Biomed. Sci. 8:377.
38. Li, J., C. A. Bombeck, S. Yang, Y. M. Kim, and T. R. Billiar. 1999. Nitric oxide
suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J. Biol. Chem. 274:17325.
39. Lin, C. F., H. Y. Lei, C. C. Liu, H. S. Liu, T. M. Yeh, S. T. Wang, T. I. Yang, F. C.
91 8
Sheu, C. F. Kuo, and Y. S. Lin. 2001. Generation of IgM anti-platelet autoantibody in dengue patients. J. Med. Virol. 63:143.
40. Lin, C. F., H. Y. Lei, A. L. Shiau, C. C. Liu, H. S. Liu, T. M. Yeh, S. H. Chen, and
Y. S. Lin. 2003. Antibodies from dengue patient sera cross-react with endothelial cells and induce damage. J. Med. Virol. 69:82.
41. Lin, C. F., H. Y. Lei, A. L. Shiau, H. S. Liu, T. M. Yeh, S. H. Chen, C. C. Liu, S. C.
Chiu, S. C. Chiu, and Y. S. Lin. 2002. Endothelial cell apoptosis induced by antibodies against dengue virus nonstructural protein 1 via production of nitric oxide. J. Immunol. 169:657.
42. Lin, Y. L., C. C. Liu, H. Y. Lei, T. M. Yeh, Y. S. Lin, R. M. Y. Chen, and H. S. Liu.
2000a. Infection of five human liver cell lines by dengue-2 virus. J. Med. Virol. 60:425.
43. Lin, Y. L., C. C. Liu, J. I. Chuang, H. Y. Lei, T. M. Yeh, Y. S. Lin, Y. H. Huang,
and H. S. Liu. 2000b. Involvement of oxidative stress, NF-IL-6, and RANTES expression in dengue-2-virus-infected human liver cells. Virology 276:114.
44. Littaua, R., I. Kurane, and F. A. Ennis. 1990. Human IgG receptor II mediates
antibody-dependent enhancement of dengue virus infection. J. Immunol. 144:3183.
45. Lunardi, C., C. Bason, R. Navone, E. Millo, G. Damonte, R. Corrocher, and A.
Puccetti. 2000. Systemic sclerosis immunoglobulin G autoantibodies bind the human cytomegalovirus late protein UL94 and induce apoptosis in human endothelial cells. Nature Med. 6:1183.
46. Mady, B. J., D. V. Erbe, I. Kurane, M. W. Fanger, and F. A. Ennis. 1991.
Antibody-dependent enhancement of dengue virus infection mediated by bispecific antibodies against cell surface molecules other than Fcγ receptors. J. Immunol. 147:3139.
47. Malasit, P. 1987. Complement and dengue hemorrhagic fever/shock syndrome.
Southeast Asian J. Trop. Med. Pub. Hlth. 18:316.
48. Marianneau, P., A. Cardona, L. Edelman, V. Deubel, and P. Despres. 1997.
Dengue virus replication in human hepatoma cells activates NF-κB which in turn induces apoptotic cell death. J. Virol. 71:3244.
49. Monath, T. P. 1994. Dengue: the risk to developed and developing countries. Proc.
Natl. Acad. Sci. USA 91:2395.
50. Morens, D. M. 1994. Antibody-dependent enhancement of infection and the
pathogenesis of viral disease. Clin. Infect. Dis. 19:500.
51. Nimmannitya, S. 1987. Clinical spectrum and management of dengue
haemorrhagic fever. Southeast Asian J. Trop. Med. Pub. Hlth. 18:392.
52. Raghupathy, R., U. C. Chaturvedi, H. Al-Sayer, E. A. Elbishbishi, R. Agarwal, R.
Nagar, S. Kapoor, A. Misra, A. Mathur, H. Nusrat, F. Azizieh, M. A. Y. Khan, and A. S. Mustafa. 1998. Elevated levels of IL-8 in dengue hemorrhagic fever. J. Med. Virol. 56:280.
53. Rico-Hesse, R., L. M. Harrison, R. A. Salas, D. Tovar, A. Nisalak, C. Ramos, J.
Boshell, M. T. R. de Mesa, R. M. R. Nogueira, and A. Travassos da Rosa. 1997. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 230:244.
54. Rigau-Perez, J. G., Clark, G. G., Gubler, D. J., Reiter, P., Sanders, E. J., and
Vorndam, A. V. 1998. Dengue and dengue haemorrhagic fever. Lancet 352:971.
55. Rose, N. R. 1998. The role of infection in the pathogenesis of autoimmune disease.
Semin. Immunol. 10:5.
56. Rothman, A. L., and F. A. Ennis. 1999. Immunopathogenesis of dengue
hemorrhagic fever. Virology 257:1.
57. Schlesinger, J. J., M. W. Brandriss, and E. E. Walsh. 1987. Protection of mice
against dengue 2 virus encephalitis by immunization with the dengue 2 virus non-structural glycoprotein NS1. J. Gen. Virol. 68:853.
58. Srivastava, A. K., J. R. Putnak, R. L. Warren, and C. H. Jr. Hoke. 1995. Mice
immunized with a dengue type 2 virus E and NS1 fusion protein made in Escherichia coli are protected against lethal dengue virus infection. Vaccine 13:1251.
59. Staropoli, I., M. P. Frenkiel, F. Megret, and V. Deubel. 1997. Affinity-purified
dengue-2 virus envelope glycoprotein induces neutralizing antibodies and protective immunity in mice. Vaccine 15:1946.
60. Stromblad, S., J. C. Becker, M. Yebra, P. C. Brooks, and D. A. Cheresh. 1996.
Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin
αvβ3 during angiogenesis. J. Clin. Invest. 98:426.
61. Suvatte, V., D. Pongpipat, S. Tuchinda, A. Patanawongs, P. Tuchinda, and S.
Bukkavesa. 1973. Studies on serum complement C3 and fibrin degradation products in Thai hemorrhagic fever. J. Med. Assoc. Thai. 56:24.
62. Vaughn, D. W., S. Green, S. Kalayanarooj, B. L. Innis, S. Nimmannitya, S.
Suntayakorn, T. P. Endy, B. Raengsakulrach, A. L. Rothman, F. A. Ennis, and A. Nisalak. 2000. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 181:2.
63. Velzing, J., J. Groen, M. T. Drouet, G. van Amerongen, C. Copra, A. D. M. E.
Osterhaus, and V. Deubel. 1999. Induction of protective immunity against dengue virus type 2: comparison of candidate live attenuated and recombinant vaccines. Vaccine 17:1312.
64. Venugopal, K., and E. A. Gould. 1994. Towards a new generation of flavivirus
vaccines. Vaccine 12:966.
65. Wang, L. Y., W. Y. Chang, S. N. Lu, and T. P. Chen. 1990. Sequential changes of
serum transaminase and abnormal sonography in patients with suspected dengue fever. Kao Hsiung I Hsueh Ko Hsueh Tsa Chih 6:483.
66. Wang, S., R. He, J. Patarapotikul, B. L. Innis, and R. Anderson. 1995.
Antibody-enhanced binding of dengue-2 virus to human platelets. Virology 213:254.
Table 1. Anti-endothelial cell IgM/IgG levels in dengue patients infected with
different serotypes.
Table 2. Anti-endothelial cell IgM/IgG levels in different disease stages.
Table 3. Anti-endothelial cell IgM/IgG levels in DHF patients with different disease
Table 4. Endothelial cell apoptosis induced by sera of dengue patients infected with
different serotypes.
Table 5. Production of IL-6, IL-8 and MCP-1 by endothelial cells after treatment with
anti-NS1 Abs for various time periods.
Table 6. AST, ALT, and BUN levels in the sera of mice after passive administration
with anti-NS1 Abs.
Fig. 1. Tyrosine phosphorylation of cellular proteins after treatment with anti-NS1
Abs in endothelial cells. After serum-free starvation for 1 h, HMEC-1 cells were treated with 5 µg of anti-DV NS1, anti-JEV NS1 or control IgG for 30 min. The tyrosine phosphorylated protein levels were analyzed by Western blot and quantified by the optical density (A), followed by staining with FITC-conjugated tyrosine phosphorylated IgG (PY-20) and flow cytometry analysis (B). EGM (endothelial cell growth medium) is used as positive control in cell signal transduction.
Fig. 2. NF-κB activation induced by anti-NS1 Abs in endothelial cells. (A) HMEC-1 cells were treated with 5µg of anti-DV NS1 or anti-JEV NS1 IgG for 3 or 24 h, and the expressions of NF-κB in cytoplasm and nucleus were determined by Western blot. C, cytoplasm; N, nucleus; P, NF-κB p65 fragment as a positive control. (B) The translocation of cytosolic NF-κB to nucleus in HMEC-1 cells after treatment with
13
anti-DV NS1 or control IgG for 3 h was detected by immunohistochemical staining using NF-κB specific Abs.
Fig. 3. Production of IL-6, IL-8 and MCP-1, but not RANTES, by endothelial cells
after treatment with anti-NS1 Abs. HMEC-1 cells were treated with 5 µg of anti-DV NS1, anti-JEV NS1, or control IgG for 24 h. The expression of IL-6, IL-8, MCP-1, and RANTES were detected by immunohistochemical staining (A) and flow cytometry analysis (B).
Fig. 4. MCP-1 but not RANTES mRNA expression after anti-DV NS1 treatment.
Total cellular mRNA were extracted at 24 h from HMEC-1 cells after treated with anti-DV NS1 (5 µg/105 cells) or with 100 m.o.i. of dengue-2 virus, and detected by RT-PCR. The β-actin was used as an internal control.
Fig. 5. A. The expression of ICAM-1 in endothelial cells after treatment with
anti-NS1 Abs. HMEC-1 cells were treated with 5 µg of anti-DV NS1, anti-JEV NS1, or control IgG for 6, 12, or 24 h. Cells were labeled with mouse anti-human ICAM-1 Abs followed by FITC-conjugated goat anti-mouse IgG. The ICAM-1 expression was detected by flow cytometry. B. The adhesive ability of peripheral blood mononuclear cells (PBMC) to anti-DV NS1-treated endothelial cells. HMEC-1 cells were cultured in 8-well glass chamber slides and treated with 5 µg of anti-DV NS1, anti-JEV NS1, or control IgG for 1 h. Cells were washed with sterile PBS, and then cocultured with fresh human PBMC for 2 h. Cell adhesion was observed by Liu’s stain.
Fig. 6. AST, ALT, and BUN levels in the sera of NS1-immunized mice. Sera were
collected from C3H/NeN mice which were immunized with PBS, recombinant DV NS1 protein or JEV NS1 protein in adjuvant for five times. The concentrations of AST, ALT, and BUN were measured using an automatic analyzer.
Fig. 7. Histopathological changes in the livers of C3H/HeN mice immunized with
NS1 protein. Mice were immunized with PBS (B), JEV NS1 (C), or DV NS1 (D-H) in adjuvants for five times. The normal mouse control is shown in (A). The concentrations of AST and ALT in each of the mice are also shown. (HE staining; magnification: left panels, 100X; right panels, 400X)
12 16
Fig. 8. Histopathological changes (A) and macrophage infiltration (B) in the livers of
mice after passive administration with anti-DV NS1 Abs. C3H/NeN mice were intravenously injected with 500 µg of anti-DV NS1, anti-JEV NS1, or control IgG. (A) Arrows indicate localization of infiltrated cells. (HE staining, magnification: left panels, 100X; right panels, 400X). (B) The frozen sections were detected by FITC-conjugated F4/80, FITC-conjugated anti-CD3, and PE-congugated anti-CD16 Abs. LPS + galactosamine was used as positive control.
Fig. 9. The cross-reactivity of dengue patient sera is not observed in liver cell lines
tested. Chang liver, HA22T and Huh7 cell lines were incubated with 1:25 dilution of various patient or normal control sera, followed by FITC-conjugated anti-human IgM or IgG and analyzed by flow cytometry. The percentages of HUVEC that reacted with patient or control sera are shown. *P < 0.05 vs. Normal.
Fig. 10. Analysis of membrane fractions of endothelial cells recognized by anti-DV
NS1. HMEC-1 cell membrane proteins were prepared and separated by 12.5% SDS-PAGE, detected by Western blot analysis with anti-DV NS1, anti-JEV NS1, and control IgG. The figures to the left indicate, in kDa, the migration profile of prestained markers.
Fig. 11. Platelet binding activity of patient sera or mouse anti-NS1 Abs can be
inhibited by preabsorption with NS1 amino acids 1-15 peptides. Human platelets were incubated with 1:25 dilution of dengue fever patient sera (A) or 5 µg of mouse anti-NS1 IgG (B) which were preabsorbed with 5 or 25 µg of DV2 NS1 protein or NS1 amino acids 1-15 peptides of DV2, DV3 or JEV, followed by FITC-conjugated anti-human IgM or anti-mouse IgG and analyzed by flow cytometry. The percentages of platelets that reacted with non- or preabsorbed Abs are shown as mean ± SD.
Fig. 12. Endothelial binding activity of mouse anti-NS1 Abs can be inhibited by
preabsorption with NS1 C20 peptides. HMEC-1 cells were incubated with 5 µg of mouse anti-DV NS1 IgG which were preabsorbed with 1 or 5 µg of DV2 NS1 protein, NS1 peptides N15 (1-15 a.a.), or C20 (314-333 a.a.), followed by FITC-conjugated anti-mouse IgG and analyzed by flow cytometry. The percentages of HMEC-1 cells that reacted with non- or preabsorbed Abs are shown as mean ± SD.