Femtosecond Infrared Transient Absorption Dynamics of
Benzimidazole-Based Ruthenium Complexes on TiO
2
Films for
Dye-Sensitized Solar Cells
Hung-Yu Hsu, Chi-Wen Cheng, Wei-Kai Huang, Yuan-Pern Lee,* and Eric Wei-Guang Diau*
Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsinchu 30010, Taiwan
*
S Supporting InformationABSTRACT: By means of femtosecond infrared transient absorption spectra, we measured the interfacial electron-transfer dynamics for benzimidazole-based heteroleptic ruthenium dyes (RD5, RD12, RD15−RD18) sensitized on TiO2thin films. For all measurements, the first singlet metal-to-ligand charge-transfer states (1MLCT) of the ruthenium complexes were excited at 519 nm and the injected
electrons in the conduction band of TiO2 were probed at 4.3 μm. All transient
signals featured two rising components on a femtosecond−picosecond scale due to a two-step electron injection and an offset (N719, RD16−RD18) or a slow-decay (RD5, RD12, and RD15) component on a nanosecond−microsecond scale due to a back electron transfer. A complicated two-step kinetic model was derived analytically to interpret the observed two rising components for which the rapid (τ1< 300 fs) and slow (τ2= 10−20 ps) electron injections arose from the singlet 1MLCT and
triplet 3MLCT states, respectively. The amplitudes of the two electron-injection
components (A1and A2) were controlled by the rate coefficient of the1MLCT→3MLCT intersystem crossing; the variations of
A1and A2are consistent with the trend of the corresponding Stokes shifts rationalized with a conventional energy-gap law for nonradiative transitions. Compared with the kinetics observed for the N719 dye, the involvement of a benzimidazole ligand in RD dyes had the effect of accelerating the two electron injections, thus improving the short circuit current of the device. RD dyes substituted withfluorine atoms and/or thiophene units in the benzimidazole ligands showed a retardation of3MLCT electron injection relative to that of the nonsubstituted RD5 dye. Acceleration of the BET process was observed for the RD5 dye (9 ns), and both fluoro-substituted dyes (14 ns for RD12 and 21 ns for RD15) and thiophene-substituted dyes (nonobservable for RD16−RD18) had significantly retarded BET kinetics. The observed kinetics of the3MLCT electron injection for all RD dyes is
satisfactorily simulated with the Marcus theory.
■
INTRODUCTIONDye-sensitized solar cells (DSSC) attract much attention because of their promising features as cost-effective devices for efficient solar energy conversion.1−4Over the last 20 years, many highly efficient light-harvesting photosensitizers, such as ruthenium complexes,5,6 zinc porphyrins,7−9 and metal-free organic dyes,10,11 have been designed and characterized for DSSC. For example, the devices made of homoleptic Ru complexes, such as N3 and N719 dyes, attained remarkable photovoltaic performance, η ∼ 11%, of power conversion efficiency (PCE) under 1 sun illumination;12,13the device made of a push−pull zinc porphyrin dye (YD2-oC8) cosensitized with an organic dye (Y123) using cobalt-based electrolyte further attainedη = 12.3%.9However, the enduring stability of these devices has become a major challenge when commerci-alization of DSSC is considered. In our previous study, we designed a series of hetroleptic Ru complexes containing bidentate pyridine-benzimidazole ligands with fluoro-substitu-tion to retard the electron transfer between semiconductor and electrolyte.14In spite of the fact that the performance of the device using RD12 as a photosensitizer is superior to that using N719, the molar absorption coefficients of these dyes are
smaller than that of N719, which limits the light-harvesting ability to further enhance the device performances. For this reason, we put thiophene derivatives on the pyridine part of the ancillary ligands in to increase the molar absorption coefficients of these dyes in a study of the second phase.15 The device performance made of the champion dye, RD18, achieved PCE exceeding 10% with the enduring performance of the RD18 device being superior to that of the N719 device.
Photoinduced interfacial electron transfer (ET) in dye-sensitized wide band gap semiconductors has been a subject of intense research interest in recent years.16−20 The rates of electron injection from the molecular excited state to the conduction band (CB) of the semiconductor thin films and subsequent charge recombination play key roles in determining the device performance.21−23 One of the highly efficient sensitizers used for DSSC is the N3 dye, which is known to Special Issue: Michael Grätzel Festschrift
Received: January 14, 2014
Revised: April 4, 2014
Published: April 8, 2014
inject electrons into nanocrystalline TiO2film with practically near unit quantum efficiency.24The kinetics of ET from N3 to TiO2has been intensively studied in the past decade.25−29The electron injection has been found to occur over a wide time scale, from <100 fs to several tens of picoseconds. This behavior was rationalized in terms of a two-state mechanism: the fast and slow components being contributed to the injection from the singlet metal-to-ligand charge-transfer (1MLCT) and triplet metal-to-ligand charge-transfer (3MLCT) excited states of the ruthenium complex, respec-tively.25−27The electron injection dynamics are very similar in the two dyes (either the protonated N3 or the deprotonated N719 dye), and dye aggregation has only moderate effects on the electron-transfer dynamics.30
In this study, we present dynamics of interfacial ET of six Ru dyes, RD5, RD12, RD15, RD16, RD17, and RD18 (corre-sponding molecular structures are shown in Chart 1) sensitized on TiO2 films using the technique of femtosecond transient
absorption spectroscopy (fs-TAS) with visible-pump at 519 nm and infrared-probe at 4.3μm. The transients of these sensitized films all exhibited a rising feature described by a biexponential function; the fast and slow rise components are assigned to the electron injections from the 1MLCT and 3MLCT states,
respectively. The transients showed a decay feature on the time scale of 10 ns only for thefilms of RD5, RD12, and RD15, but such a decay feature was nonobservable for the N719 and the RD16−RD18 films. We conclude that the fluorine substitution in the benzimidazole ligands retards the back electron transfer (BET) as observed for the RD12 and RD15films, whereas the thiophene substitution in the ligands retards the electron injection as observed for the RD16−RD18 films. The observed interfacial ET dynamics on the sensitized films is consistent with the photovoltaic performance of the corresponding devices.
■
EXPERIMENTSThe dyes of the RD series were synthesized according to the procedure reported elsewhere,14,15 and the N719 dye was purchased from Everlight Chemical, Taiwan. The TiO2 films were prepared by screen printing with TiO2nanoparticles (NP)
and nanorods (NR) according to a conventional synthetic procedure reported elsewhere.31,32 For the working electrode, the TiO2NP and NR were coated on a TiCl4pretreated and
antireflective coated FTO glass substrate (TEC 7, Hartford) to obtain the required film thickness. The TiO2 films were
immersed in solutions containing the target Ru complexes (RD5, RD12, RD15, or RD16−RD18, 3 × 10−4 M) and chenodeoxycholic acid (CDCA, 3 × 10−4 M) in anhydrous CH3CN/t-BuOH (v/v, 1:1) for 3 h. The counter electrode was
made on spin-coating the H2PtCl6/isopropyl alcohol solution onto a FTO glass substrate with a typical size 1.0 × 1.5 cm2
through thermal decomposition. The device was assembled with two electrodes and sealed with a hot-meltfilm (SX1170, thickness∼25 μm). The electrolyte solution contained I2(0.03 M), LiI (0.1 M), PMII (1 M), guanidinium thiocyanate (GuSCN, 0.1 M), and 4-tert-butylpyridine (0.5 M) in a mixture of acetonitrile and valeronitrile (volume ratio×85:15).
The photovoltaic characteristics were determined with a digital source meter (Model 2400, Keithley Instruments) under 1 sun AM1.5G irradiation from a solar simulator (XES-40S1, SAN-EI). The spectra of incident photon-to-current conversion efficiency (IPCE) were recorded with a system comprising a Xe lamp (Model A-1010, PTi, 150W), monochromator (PTi, 1200 g mm−1blazed at 500 nm), and Keithley 2400 source meter.
The fs-TAS measurements were carried out using a typical pump−probe method with a tunable visible or IR probed system described elsewhere;33 Figure 1 shows the optical layout, and the function of the system is described below. The femtosecond pulses were generated with a regenerative amplifier (SPTF-100F-1K-XP, Spectra Physics) seeded with a mode-locked Ti:sapphire laser system (Mai Tai Sp, Spectra Physics) and pumped with a Nd:YLF laser (Empower 30, Spectra Physics, 1 kHz). The laser pulse centered at 800 nm (2.5 mJ pulse−1, full width at half-maximum (fwhm)∼100 fs) was equally split into two parts to pump two optical parametric amplifiers (TOPAS-C, Light Conversion) to provide tunable femtosecond pulses. For the present study, the pump beam at 519 nm was generated by an optical parametric amplifier (TOPAS-C-1). The pump pulse (∼200 nJ) was focused onto a sample cell containing the sample plate, target RD dye Chart 1. Molecular Structures of the Heteroleptic Ruthenium Complexes RD5, RD12, RD15, RD16, RD17, and RD18
adsorbed on a TiO2 thin film (∼3 μm) immersed in
acetonitrile, and two sapphire windows sealed with a Teflon spacer of thickness∼0.2 mm.
Another optical parametric amplifier (TOPAS-C-2) was used to generate the mid-IR probe beam at 4.3μm. This IR probe pulse was split into two equal parts for use as sample and reference beams; the sample probe beam spatially overlaps with the pump beam. The optical delay between the pump and the probe beams was achieved with a stepping translational stage (SGSP(MS)26-200(X), Sigma Koki). The sample cell was rotated with a speed of∼10 degree s−1so that each pump pulse excited a fresh sample region. After passing through the sample, the two IR probe beams were collimated and dispersed with a spectrograph (Micro HR, Jobin-Yvon Horiba) and detected separately with a 2 × 32-element Hg/Cd/Te array detector (IR-6416, Infrared Systems Development Corp.) to yield spectra of resolution 3−4 cm−1. Every other pump pulse was
blocked with a synchronized chopper (MC1000A, Thorlabs) operated at 500 Hz. The instrument response function (IRF) was characterized with a Gaussian function of fwhm∼300 fs. By varying the temporal delay between the excitation and the probe pulses via a stepping translational stage (delay line D), we obtained the transient absorption profile (ΔA vs t). The polarization between pump and probe pulses wasfixed at the magic angle 54.7° with a Berek compensator.
■
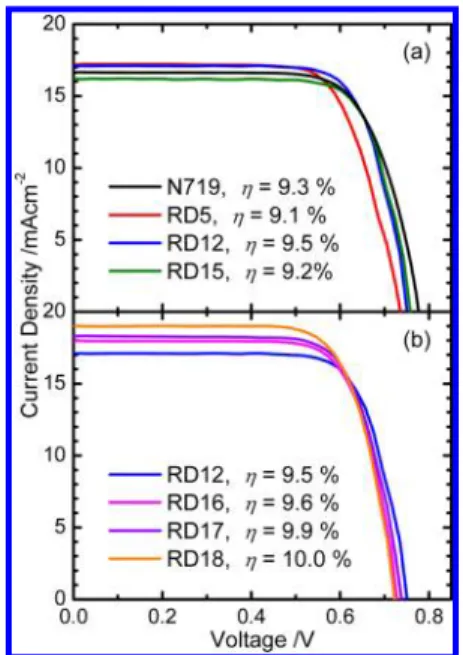
RESULTS AND DISCUSSIONThe devices of the Ru dyes were fabricated with the TiO2film of thickness∼22 μm (17 μm of the active layer and 5 μm of the scattering layer) for photovoltaic measurements; Figure 2 presents the current−voltage characteristics for those devices under AM1.5G 1 sun illumination; the resulting photovoltaic parameters are summarized in Table S1 of the Supporting Information. As shown in Figure 2a, the device performances of thefluoro-substituted Ru dyes showed a systematic trend on JSC
with the order RD5∼ RD12 > RD15 but an opposite trend on VOC, i.e., more fluorine atoms in the ligands led to lower
photocurrents but higher photovoltages for the devices. In our preceding work,14we concluded that this trend of JSCmight be
due to (i) the dyes with lower levels of lowest unoccupied molecular orbital (LUMO) and (ii) small amount of dye loading for the ligands containing morefluorine atoms, whereas the trend of VOC was due to the retardation of charge
recombination with more fluorine atoms.14 For the devices made of RD16−RD18, as shown in Figure 2b, the JSC values
increased with increasing number of thiophene units, showing an order RD18 > RD17 > RD16 > RD12, but the thiophene substitutes also (i) down-shifted the edge of the potential band of TiO2and (ii) accelerated the charge recombination so that a
VOCof the RD18 device smaller than that for the RD12 device was observed.15The VOCvalues of the devices made with dyes
in the RD series were smaller than that made with the N719 dye, whereas the JSCvalues for the former were all superior to
those for the latter. To understand the above-mentioned observed trend of photovoltaic performances, we measured the fs-TAS for the thin-film samples sensitized with these Ru dyes. Panels a and b of Figure 3 show the fs-TAS signals (normalizedΔA vs time) for TiO2films sensitized with N719/ RD5/RD12/RD15 dyes and RD12/RD16/RD17/RD18 dyes, respectively. For the measurements, thefilms were immersed in acetonitrile to avoid heat accumulation in the excited dye molecules; the excitation wavelength wasfixed at 519 nm, and the probe wavelength at 4.3 μm, which corresponds to the injected electron signals on TiO2.34As shown in Figure 3, all transient signals show kinetics that may be described by a biphasic rising character shown in the insets in the sub-picosecond to 10 ps time scale, together with an offset (N719 or RD16−RD18) or a slow-decay feature (RD5, RD12, and RD15). Accordingly, the transient signals, S(t), werefitted by a kinetic model with three components convoluted with the IRF (fwhm∼300 fs):35
= − − τ + − − τ − τ
S t( ) [(1 A1)e t/1 (1 A2)e t/2]e t/d (1) in whichτ1andτ2represent two time coefficients with a rising
character andτd a time coefficient with a decay character; A1 and A2 are two corresponding pre-exponential factors.
According to eq 1, the fitted results are represented as solid curves shown in Figure 3 with the corresponding fitted parameters summarized in Table 1. The transient of the N719 film served as a reference, which gives a pulse-limited rapid rise (τ1< 0.3 ps), a well-determined slow rise (τ2= 26 ps), and an offset signal with a slow-decay coefficient (τd) that cannot be
determined within the experimental time scale. Our N719 results are consistent with those of the other Ru systems Figure 1. Optical layout of the femtosecond visible pump/infrared
probe transient absorption spectrometer. M1 and M2 are gold mirrors; M3−M6 are silver mirrors; L1−L4 are lenses; BS1 and BS2 are beam splitters; B is the Berek compensator; C is the chopper; D is the delay stage; S is the sample.
Figure 2.Current−voltage characteristics of DSSC sensitized with (a) N719, RD5, RD12, and RD15 dyes and (b) RD12, RD16, RD17, and RD18 dyes under AM1.5G 1 sun illuminations.
reported elsewhere.16,36−39 We now discuss the observed formation and relaxation dynamics of these thin-film samples based on the kinetic model applied herein.
For the two rising parts of the transients, a two-state injection model has been proposed to explain the complex injection kinetics caused by the competition between the electron injection and the excited-state relaxation pathways.36−39 As is shown in the schematic kinetic model in Figure 4, electrons were injected from the1MLCT and the3MLCT excited states
to the CB of TiO2with rate coefficients kSand kT, respectively.
The rate coefficient for the thermal relaxation of the hot electrons in the CB of TiO2 is represented by kHR. The rate
coefficient of intersystem crossing kISC incorporates the electronic relaxation from the 1MLCT to the 3MLCT states
as well as any vibrational energy relaxation and solvation stabilization occurring in the 3MLCT state. According to this
kinetic model, we are able to derive the expressions for the injected hot and cold electrons in the CB of TiO2:
** = + − − − − + N t k N k k k ( ) [e k t e k k t] e S 0 ISC S HR ( ) HR ISC S (2) * = + + − − + − + − − ⎡ ⎣ ⎢ ⎤ ⎦ ⎥ N t N B k B k k k N B N B C ( ) ( ) e e e k k t k t k t e 0 1 HR 2 T ISC S ( ) 0 1 0 2 ISC S HR T (3)
in which Ne**(t) and Ne*(t) are the populations of unrelaxed
(hot) and relaxed (cold) electrons, respectively, in the semiconductor. N0 is the initial population of the excited
state of the dye. The coefficients B1, B2, and C can be expressed
as (kS)/(kISC + kS − kHR), (kISC)/(kISC + kS − kT), and
−(N0(B1kHR+ B2kT))/(kS+ kISC) + B1N0+ B2N0, respectively.
Therefore, Ne**(t) is described by the sum of a
single-exponential rising function and a single-single-exponential decay function: the rising term is determined by the combined rate coefficients (kISC+ kS), and the decay term is determined by
kHR; Ne*(t) is described by a sum of three single-exponential
rising functions characterized by the rate coefficients (kISC +
kS), kHR, and kT, respectively. Our IR-probe monitored both hot
and cold injected electrons in the CB of TiO2, and the
corresponding TAS signal Se(t) is expressed as σ υ σ υ
= ** ** + * *
S te( ) Ne ( )t ( ) N te( ) ( ) (4) where σ**(υ) and σ*(υ) are cross sections of hot and cold injected electrons, respectively. Assuming that σ**(υ) and σ*(υ) are similar within the experimental uncertainties, the kHR
terms involved in eqs 2 and 3 are canceled in eq 4, which can be expressed by two single-exponential rising functions charac-terized by only (kISC+ kS) and kT.
Equation 4 is the expression that represents the two-step injection kinetic model without consideration of the slow-decay component characterized by the decay time coefficient τd
Figure 3.Normalized infrared transient absorbance (ΔA) of the TiO2 films sensitized with (a) N719, RD5, RD12, and RD15 dyes and (b) RD12, RD16, RD17, and RD18 dyes. The excitation wavelength was 519 nm; the probe wavelength was 4.3μm. The thin lines present the experimental data, and the thick lines arefitted curves. The insets show magnified portions of the transients at shorter time delay for clarity.
Table 1. Time Coefficients and Relative Amplitudes of the Temporal Profiles for Sensitized TiO2Films Obtained from a
Nonlinear Curve Fit According to Eq 1a
dye A1 τ1(ps) A2 τ2(ps) τd(ns) N719 0.53± 0.01 <0.3 0.47± 0.01 25.7± 1.8 n.a. RD5 0.61± 0.01 <0.3 0.39± 0.01 11.8± 1.0 8.9± 0.8 RD12 0.60± 0.02 <0.3 0.40± 0.01 10.4± 1.1 14.4± 2.5 RD15 0.56± 0.01 <0.3 0.44± 0.01 14.3± 1.3 21.3± 6.1 RD16 0.48± 0.01 <0.3 0.52± 0.01 11.8± 0.6 n. a. RD17 0.46± 0.01 <0.3 0.54± 0.01 13.9± 0.6 n. a. RD18 0.57± 0.01 <0.3 0.43± 0.01 16.0± 0.8 n. a.
aThe transients were obtained with excitation at 519 nm and probe at 4.3μm; each uncertainty represents one standard deviation.
Figure 4. Schematic kinetic model representing the two electron injections (with kSand kT), intersystem crossing from the3MLCT to the 1MLCT states (kISC), hot electron relaxation in the conduction band (CB) of TiO2(kHR), and back electron-transfer (kBET) process occurring in the dye/TiO2interface of a DSSC device.
shown in eq 1. Accordingly, we assigned the observed rapid (τ1) and slow (τ2) rising time coefficients as
τ1−1=kISC+kS (5)
τ2−1=kT (6) This two-step kinetic model was initially proposed by Asbury et al. for the N3 dye,36for whichτ1< 100 fs andτ2∼ 50 ps, on excitation at 530 nm, comparable to our results for the N719 dye excited at 519 nm. Asbury et al. observed also a decay time coefficient ∼90 ps upon excitation at 400 nm. This 90 ps decay component might be attributed to the hot electron relaxation (kHR) when the excitation was performed at 400 nm where
σ**(υ) of the hot electrons became significantly greater than σ*(υ) of the cold electrons.
As summarized in Table 1, the injection kinetics of all RD dyes consist of a distinct ultrafast component which is within the instrument response time of ∼300 fs (τ1) and a slow component of ∼10 ps (τ2). For the fluoro-substituted
benzimidazole-based Ru dyes (Figure 3a), the fast-injection amplitude (A1) showed the trend RD5 ∼ RD12 > RD15 >
N719. This phenomenon indicates that the benzimidazole ligand in RD5 had the effect of enhancing the contribution of the fast-injection because kS is the dominant parameter determining the value of A1indicated in eq 2. The involvement
of strong electron-pulling species like fluorine atoms could reduce the ability for the fast-injection process so as to decrease the corresponding amplitude A1 in a systematic manor. This also led to the variation of the slow-injection amplitude A2,
which shows an opposite trend (A1+ A2= 1).
For the thiophene-substituted benzimidazole-based Ru dyes (Figure 3b), the fast-injection amplitude (A1) showed the order RD12 > RD18 > RD16∼ RD17. Because of the heavy atom effect (sulfide in the thiophene unit),40thiophene substitution in RD dyes could lead to the enhancement of the ISC rate coefficient (kISC) and thus increase the amplitude A2 responsible for the slow injection as indicated in eq 3. The variation of A1is opposite to that of A2; therefore, the above observation can be rationalized with the heavy-atom effect only for RD16 and RD17 but not for RD18, which contains one more thiophene unit than the other two dyes. To explain this anomalous phenomenon, a conventional energy-gap law for nonradiative transition like ISC may be applied.
The energy gap between the1MLCT and the3MLCT states
can be determined by the Stokes shift between the absorption peakλabsof the1MLCT state and the photoluminescence peak
λPL of the 3MLCT state. The absorption spectra and the
corresponding PL spectra of the RD dyes are shown in Figure S1 of the Supporting Information, and the resulting Stokes shifts of these RD dyes are summarized in Table S2 of the Supporting Information. We observed that the Stokes shifts have the trend RD12 > RD18 > RD16 > RD17 because thiophene substitution in the benzimidazole ligand stabilizes the triplet state more effectively than the singlet state. Therefore, we may predict the values of kISC with the order
RD17 > RD16 > RD18 > RD12, which is consistent with the variation of the slow-injection amplitude A2, which shows the
same order. Note that the energy-gap law is also applied for the fluoro-substituted RD dyes, for which the trend of the A2values
is consistent with the inverse trend of the Stokes shifts showing the order RD5 > RD12 > RD15.
The insets of Figure 3a,b magnify the initial region of the transients for the two series of RD dyes, showing the feature of
the slow-injection component (time coefficient τ2) with the order N719 (25.7 ps) > RD15 (14.3 ps) > RD12 (10.4 ps)∼ RD5 (11.8 ps) and the order RD18 (16.0 ps) > RD17 (13.9 ps) > RD16 (11.8 ps) > RD12 (10.4 ps). These results indicate that the electron injection from the3MLCT state to the CB of TiO2
(rate coefficient kT) slows when morefluorine atoms and/or
more thiophene units were substituted. First, the triplet-state injection rate coefficients of all the RD dyes are faster than that of the N719 dye, indicating the superior electron-injection feature for this series of dyes containing benzimidazole ligands. Second, thefluoro-substitution retards the triplet-state injection because of the electron-pulling properties of thefluorine atoms. Third, the thiophene-substitution also slows down the triplet-state injection for the same reason. However, increasing the number of thiophene units increases the absorption coefficients of the dye that enhanced the JSCof the device significantly.15
For the offset or the slow-decay component observed in the TAS signals, we assigned this process to the electron transfer from the CB of TiO2back to the oxidized dye (BET) with the
corresponding decay rate coefficient (kBET) equal to the inverse
ofτdexpressed in eq 1. This assignment is consistent with the
ns-TAS results of Tachibana et al. showing that the BET process in the N3/TiO2 film was on the microsecond−
millisecond scale.24Katoh et al. studied the BET quantum yield of a black dye system and concluded that the fast BET process (<100 ps) results from the charge recombination at the surface state of TiO2.
39
In our case, the TAS signals of N719 show no observable decay contribution but the transients of RD5−RD15 do exhibit a slow-decay feature with decay coefficients in the 10−20 ns range showing the order RD15 > RD12 > RD5 (Figure 3a). Therefore, the BET process slows when more fluorine atoms are substituted in the benzimidazole ligands, an effect similar to the aforementioned triplet-state electron-injection kinetics. The decay coefficients of BET exhibit the order N719 > RD15 > RD12 > RD5, which is consistent with the trend of VOC of the corresponding devices. For the thiophene-substituted dyes RD16−RD18, the corresponding BET kinetics cannot be determined within the observed time scale, indicating that the thiophene units in the bezimidazole ligands play an important role in retarding the BET process. The smaller VOC values of the thiophene-substituted dyes are
due to the down shifts of the potential band edge of TiO2upon
sensitization by these RD dyes.15
We discuss the kinetics of slow electron injection (kT)
according to a formulation based on Marcus theory:41
π πλ λ λ Δ = ℏ | ̂ Δ | − + Δ k G H G k T G k T ( ) 2 ( ) 4 exp[ ( ) /4 ] ET 2 B 2 B (7) in whichΔG denotes the energy difference between donor and acceptor, Ĥ the average electronic coupling between donor and acceptor, and λ the total reorganization energy. The driving force of the system is related to ΔG, which is evaluated according to the energy difference between the LUMO level of the dye and the conduction-band edge of TiO2.
14,15
Because the acceptor is a semiconductor, kT is determined on
convolution of kETwith the density of states of TiO2treated
as a continuous distribution:42
∫
ρΔ = − Δ
kT( G) (E G k) ET( ) dE E
Here we neglected the contribution from surface and defect states below the edge of the conduction band of TiO2; the
density of states of TiO2is described with this expression:43
ρ π = * ℏ ⎜ ⎟ ⎛ ⎝ ⎞ ⎠ E E m E E ( ) d 1 2 2 d 2 2 3/2 (9) in which m* is the effective mass of an electron in the conduction band of the semiconductor. According to eqs 7−9, energy-dependent kT(ΔG) is predicted theoretically by fixing the parameters to reported values: 7.5mefor m*, 560 cm−1for
Ĥ, and 0.21 eV for λ.41,44−47 Figure 5 presents a simulated
semilog plot of kT versus −ΔG as a solid curve. The experimental data of the RD dyes obtained from the slow injection are shown also as open circles in the figure for comparison.
In general, the experimental data follow satisfactorily a trend predicted with the Marcus theory (kT vs −ΔG), but some
deviations exist because of simulated results based on the same electronic coupling parameter (560 cm−1) and the same reorganization energy (0.21 eV) for all RD dyes. As the anchoring groups of the dyes are all the same (dicarboxylic bipyridine, dcbpy), we reasonably assume that all RD systems have the same electronic coupling effect between the anchoring ligand and the semiconductor. The observed deviations between experiment and theory might accordingly be rationalized by the reorganization energies λ that differ upon substitution of fluorine atoms or thiophene groups in the ancillary ligands of the Ru complexes.
■
CONCLUSIONWe studied interfacial electron-transfer dynamics for a series of heteroleptic ruthenium complexes containing varied fluoro-substituted and thiophene-fluoro-substituted benzimidazole ligands sensitized on TiO2 films using femtosecond infrared transient absorption spectral method excited at 519 nm and probed at 4.3 μm. The observed transients featured two rising components that can be described by a two-state electron-injection kinetic model: the rapid rise has a pulse-limited time coefficient (τ1<300 fs) corresponding to the injection from the
singlet 1MLCT state, whereas the slow rise has a time
coefficient in the tens of picoseconds scale (τ2 = 10−20 ps)
corresponding to the injection from the triplet 3MLCT state.
The transients also featured an offset or a slow-decay component in the tens of nanoseconds scale corresponding to the back electron-transfer process. We found that the
benzimidazole ligands had the effect of accelerating the two electron-injection processes in comparison with the injection kinetics observed for the N719 dye. Substitution withfluorine atoms and/or thiophene units in the ligand led, however, to retardation of the injection kinetics, which is consistent with the electron-pulling nature of the substituents. The relative amplitudes of the two electron-injection components were controlled by the rate coefficient of the 1MLCT → 3MLCT intersystem crossing process, and the variations of A1and A2in all Ru dyes are consistent with the trend of the corresponding Stokes shifts rationalized by a conventional energy-gap law for nonradiative transitions. The Marcus theory accounts for the kinetics of the slow electron injection. Our kinetic results show the superior character of electron injection for RD dyes in this series and are consistent with the JSC values in the corresponding dye-sensitized solar cells.
■
ASSOCIATED CONTENT*
S Supporting InformationPhotovoltaic parameters of DSSC with photosensitizers N719, RD5, RD12, RD15, RD16, RD17, and RD18 under simulated AM1.5G illumination (Table S1); maximum peaks of the absorption and PL spectra and the corresponding Stokes shifts for the heteroleptic ruthenium complexes N719, RD5, RD12, RD15, RD16, RD17, and RD18 obtained from the results shown in Figure S1 (Table S2); absorption and PL spectra of RD5, RD12, RD15, RD16, RD17, and RD18 in DMF (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
■
AUTHOR INFORMATION Corresponding Authors *E-mail: [email protected]. *E-mail: [email protected]. NotesThe authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSMinistry of Science and Technology of Taiwan and Ministry of Education of Taiwan, under the ATU program, provided financial support of this project.
■
REFERENCES(1) Kamat, P. V.; Tvrdy, K.; Baker, D. R.; Radich, J. G. Beyond Photovoltaics: Semiconductor Nanoarchitectures for Liquid-Junction Solar Cells. Chem. Rev. (Washington, DC, U.S.) 2010, 110, 6664−6688. (2) Ning, Z.; Fu, Y.; Tian, H. Improvement of Dye-Sensitized Solar Cells: What We Know and What We Need to Know. Energy Environ. Sci. 2010, 3, 1170−1181.
(3) Vougioukalakis, G. C.; Philippopoulos, A. I.; Stergiopoulos, T.; Falaras, P. Contributions to the Development of Ruthenium-Based Sensitizers for Dye-Sensitized Solar Cells. Coord. Chem. Rev. 2011, 255, 2602−2621.
(4) Li, L. L.; Diau, E. W. G. Porphyrin-Sensitized Solar Cells. Chem. Soc. Rev. 2013, 42, 291−304.
(5) Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. (Washington, DC, U.S.) 2010, 110, 6595−6663.
(6) Yu, Q.; Wang, Y.; Yi, Z.; Zu, N.; Zhang, J.; Zhang, M.; Wang, P. High-Efficiency Dye-Sensitized Solar Cells: The Influence of Lithium Ions on Exciton Dissociation, Charge Recombination, and Surface States. ACS Nano 2010, 4, 6032−6038.
Figure 5.kTversus−ΔG for the slow electron injection predicted with the Marcus theory. The open circles denote experimental data; the solid curve is the simulated result according to eqs 7−9.
(7) Imahori, H.; Umeyama, T.; Ito, S. Largeπ-Aromatic Molecules as Potential Sensitizers for Highly Efficient Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1809−1818.
(8) Martínez-Díaz, M. V.; de la Torre, G.; Torres, T. Lighting Porphyrins and Phthalocyanines for Molecular Photovoltaics. Chem. Commun. (Cambridge, U.K.) 2010, 46, 7090−7108.
(9) Yella, A.; Lee, H. W.; Tsao, H. N.; Yi, C.; Chandiran, A. K.; Nazeeruddin, M. K.; Diau, E. W. G.; Yeh, C. Y.; Zakeeruddin, S. M.; Grätzel, M. Porphyrin-Sensitized Solar Cells with Cobalt (II/III)-Based Redox Electrolyte Exceed 12% Efficiency. Science 2011, 334, 629−634.
(10) Ning, Z.; Tian, H. Triarylamine: A Promising Core Unit for Efficient Photovoltaic Materials. Chem. Commun. 2009, 5483−5495.
(11) Zeng, W.; Cao, Y.; Bai, Y.; Wang, Y.; Shi, Y.; Zhang, M.; Wang, F.; Pan, C.; Wang, P. Efficient Dye-Sensitized Solar Cells with an Organic Photosensitizer Featuring Orderly Conjugated Ethylenediox-ythiophene and Dithienosilole Blocks. Chem. Mater. 2010, 22, 1915− 1925.
(12) Nazeeruddin, M. K.; De Angelis, F.; Fantacci, S.; Seiloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Takeru, B.; Grätzel, M. Combined Experimental and DFT-TDDFT Computational Study of Photo-electrochemical Cell Ruthenium Sensitizers. J. Am. Chem. Soc. 2005, 127, 16835−16847.
(13) Wang, Q.; Ito, S.; Grätzel, M.; Fabregat-Santiago, F.; Mora-Seró, I.; Bisquert, J.; Bessho, T.; Imai, H. Characteristics of High Efficiency Dye-Sensitized Solar Cells. J. Phys. Chem. B 2006, 110, 25210−25221. (14) Huang, W. K.; Wu, H. P.; Lin, P. L.; Lee, Y. P.; Diau, E. W. G. Design and Characterization of Heteroleptic Ruthenium Complexes Containing Benzimidazole Ligands for Dye-Sensitized Solar Cells: The Effect of Fluorine Substituents on Photovoltaic Performance. J. Phys. Chem. Lett. 2012, 3, 1830−1835.
(15) Huang, W. K.; Wu, H. P.; Lin, P. L.; Diau, E. W. G. Design and Characterization of Heteroleptic Ruthenium Complexes Containing Benzimidazole Ligands for Dye-Sensitized Solar Cells: The Effect of Thiophene and Alkyl Substituents on Photovoltaic Performance. J. Phys. Chem. C 2013, 117, 2059−2065.
(16) Banerjee, T.; Kaniyankandy, S.; Das, A.; Ghosh, H. N. Newly Designed Resorcinolate Binding for Ru(II)- and Re(I)-Polypyridyl Complexes on Oleic Acid Capped TiO2 in Nonaqueous Solvent: Prolonged Charge Separation and Substantial Thermalized 3MLCT Injection. J. Phys. Chem. C 2013, 117, 3084−3092.
(17) Huang, J.; Buyukcakir, O.; Mara, M. W.; Coskun, A.; Dimitrijevic, N. M.; Barin, G.; Kokhan, O.; Stickrath, A. B.; Ruppert, R.; Tiede, D. M.; et al. Highly Efficient Ultrafast Electron Injection from the Singlet MLCT Excited State of Copper(I) Diimine Complexes to TiO2 Nanoparticles. Angew. Chem., Int. Ed. 2012, 51, 12711−12715.
(18) De Miguel, G.; Marchena, M.; Ziółek, M.; Pandey, S. S.; Hayase, S.; Douhal, A. Femto- to Millisecond Photophysical Characterization of Indole-Based Squaraines Adsorbed on TiO2 Nanoparticle Thin Films. J. Phys. Chem. C 2012, 116, 12137−12148.
(19) Oum, K.; Lohse, P. W.; Flender, O.; Klein, J. R.; Scholz, M.; Lenzer, T.; Du, J.; Oekermann, T. Ultrafast Dynamics of the Indoline Dye D149 on Electrodeposited ZnO and Sintered ZrO2 and TiO2 Thin Films. Phys. Chem. Chem. Phys. 2012, 14, 15429−15437.
(20) Santra, P. K.; Nair, P. V.; Thomas, K. G.; Kamat, P. V. CuInS2 -Sensitized Quantum Dot Solar Cell. Electrophoretic Deposition, Excited-State Dynamics, and Photovoltaic Performance. J. Phys. Chem. Lett. 2013, 4, 722−729.
(21) Sunahara, K.; Furube, A.; Katoh, R.; Mori, S.; Griffith, M. J.; Wallace, G. G.; Wagner, P.; Officer, D. L.; Mozer, A. J. Coexistence of Femtosecond- and Nonelectron-Injecting Dyes in Dye-Sensitized Solar Cells: Inhomogeniety Limits the Efficiency. J. Phys. Chem. C 2011, 115, 22084−22088.
(22) Tiwana, P.; Docampo, P.; Johnston, M. B.; Snaith, H. J.; Herz, L. M. Electron Mobility and Injection Dynamics in Mesoporous ZnO, SnO2, and TiO2Films Used in Dye-Sensitized Solar Cells. ACS Nano 2011, 5, 5158−5166.
(23) Imahori, H.; Kang, S.; Hayashi, H.; Haruta, M.; Kurata, H.; Isoda, S.; Canton, S. E.; Infahsaeng, Y.; Kathiravan, A.; Pascher, T.; et al. Photoinduced Charge Carrier Dynamics of Zn-Porphyrin-TiO2 Electrodes: The Key Role of Charge Recombination for Solar Cell Performance. J. Phys. Chem. A 2011, 115, 3679−3690.
(24) Tachibana, Y.; Moser, J. E.; Grätzel, M.; Klug, D. R.; Durrant, J. R. Subpicosecond Interfacial Charge Separation in Dye-Sensitized Nanocrystalline Titanium Dioxide Films. J. Phys. Chem. 1996, 100, 20056−20062.
(25) Watson, D. F.; Meyer, G. J. Electron Injection at Dye-Sensitized Semiconductor Electrodes. Annu. Rev. Phys. Chem. 2005, 56, 119−156. (26) Anderson, N. A.; Lian, T. Ultrafast Electron Transfer at the Molecule-Semiconductor Nanoparticle Interface. Annu. Rev. Phys. Chem. 2005, 56, 491−519.
(27) Listorti, A.; O’Regan, B.; Durrant, J. R. Electron Transfer Dynamics in Dye-Sensitized Solar Cells. Chem. Mater. 2011, 23, 3381−3399.
(28) Pijpers, J. J. H.; Ulbricht, R.; Derossi, S.; Reek, J. N. H.; Bonn, M. Picosecond Electron Injection Dynamics in Dye-Sensitized Oxides in the Presence of Electrolyte. J. Phys. Chem. C 2011, 115, 2578−2584. (29) Bräm, O.; Cannizzo, A.; Chergui, M. Ultrafast Fluorescence Studies of Dye Sensitized Solar Cells. Phys. Chem. Chem. Phys. 2012, 14, 7934−7937.
(30) Pellnor, M.; Myllyperkiö, P.; Korppi-Tommola, J.; Yartsev, A.; Sundström, V. Photoinduced Interfacial Electron Injection in RuN3 -TiO2Thin Films: Resolving Picosecond Timescale Injection from the Triplet State of the Protonated and Deprotonated Dyes. Chem. Phys. Lett. 2008, 462, 205−208.
(31) Ito, S.; Chen, P.; Comte, P.; Nazeeruddin, M. K.; Liska, P.; Péchy, P.; Grätzel, M. Fabrication of Screen-Printing Pastes from TiO2 Powders for Dye-Sensitized Solar Cells. Prog. Photovoltaics 2007, 15, 603−612.
(32) Wu, H. P.; Lan, C. M.; Hu, J. Y.; Huang, W. K.; Shiu, J. W.; Lan, Z. J.; Tsai, C. M.; Su, C. H.; Diau, E. W. G. Hybrid Titania Photoanodes with a Nanostructured Multi-Layer Configuration for Highly Efficient Dye-Sensitized Solar Cells. J. Phys. Chem. Lett. 2013, 4, 1570−1577.
(33) Huang, G. J.; Cheng, C. W.; Hsu, H. Y.; Prabhakar, C.; Lee, Y. P.; Diau, E. W. G.; Yang, J. S. Effects of Hydrogen Bonding on Internal Conversion of GFP-like Chromophores. I. The para-Amino Systems. J. Phys. Chem. B 2013, 117, 2695−2704.
(34) Ellingson, R. J.; Asbury, J. B.; Ferrere, S.; Ghosh, H. N.; Sprague, J. R.; Lian, T.; Nozik, A. J. Dynamics of Electron Injection in Nanocrystalline Titanium Dioxide Films Sensitized with [Ru(4,4′-dicarboxy-2,2′-bipyridine)2(NCS)2] by Infrared Transient Absorption. J. Phys. Chem. B 1998, 102, 6455−6458.
(35) Benkö, G.; Kallioinen, J.; Korppi-Tommola, J. E. I.; Yartsev, A. P.; Sundström, V. Photoinduced Ultrafast Dye-to-Semiconductor Electron Injection from Nonthermalized and Thermalized Donor States. J. Am. Chem. Soc. 2002, 124, 489−493.
(36) Asbury, J. B.; Anderson, N. A.; Hao, E.; Ai, X.; Lian, T. Parameters Affecting Electron Injection Dynamics from Ruthenium Dyes to Titanium Dioxide Nanocrystalline Thin Film. J. Phys. Chem. B 2003, 107, 7376−7386.
(37) Kallioinen, J.; Benkö, G.; Myllyperkiö, P.; Khriachtchev, L.; Skårman, B.; Wallenberg, R.; Tuomikoski, M.; Korppi-Tommola, J.; Sundström, V.; Yartsev, A. P. Photoinduced Ultrafast Dynamics of Ru(dcbpy)2(NCS)2-Sensitized Nanocrystalline TiO2 Films: The Influence of Sample Preparation and Experimental Conditions. J. Phys. Chem. B 2004, 108, 6365−6373.
(38) Verma, S.; Kar, P.; Das, A.; Palit, D. K.; Ghosh, H. N. The Effect of Heavy Atoms on Photoinduced Electron Injection from Non-thermalized and Thermalized Donor States of MII-Polypyridyl (M=Ru/Os) Complexes to Nanoparticulate TiO2 Surfaces: An Ultrafast Time-Resolved Absorption Study. Chem.Eur. J. 2010, 16, 611−619.
(39) Katoh, R.; Furube, A.; Fuke, N.; Fukui, A.; Koide, N. Ultrafast Relaxation as a Possible Limiting Factor of Electron Injection
Efficiency in Black Dye Sensitized Nanocrystalline TiO2Films. J. Phys. Chem. C 2012, 116, 22301−22306.
(40) Ambre, R.; Chen, K. B.; Yao, C. F.; Luo, L.; Diau, E. W. G.; Hung, C. H. Effects of Porphyrinic meso-Substituents on the Photovoltaic Performance of Dye-Sensitized Solar Cells: Number and Position of p-Carboxyphenyl and Thienyl Groups on Zinc Porphyrins. J. Phys. Chem. C 2012, 116, 11907−11916.
(41) Asbury, J. B.; Hao, E.; Wang, Y.; Ghosh, H. N.; Lian, T. Ultrafast Electron Transfer Dynamics from Molecular Adsorbates to Semi-conductor Nanocrystalline Thin Films. J. Phys. Chem. B 2001, 105, 4545−4557.
(42) Gao, Y. Q.; Georgievskii, Y.; Marcus, R. A. On the Theory of Electron Transfer Reactions at Semiconductor Electrode/Liquid Interfaces. J. Chem. Phys. 2000, 112, 3358−3369.
(43) Kittel, C. Introduction to Solid State Physics, 8thed.; Wiley: New York,U.S., 2004.
(44) Huang, J.; Stockwell, D.; Boulesbaa, A.; Guo, J.; Lian, T. Comparison of Electron Injection Dynamics from Rhodamine B to In2O3, SnO2, and ZnO Nanocrystalline Thin Films. J. Phys. Chem. C 2008, 112, 5203−5212.
(45) Ai, X.; Anderson, N. A.; Guo, J.; Lian, T. Electron Injection Dynamics of Ru Polypyridyl Complexes on SnO2 Nanocrystalline Thin Films. J. Phys. Chem. B 2005, 109, 7088−7094.
(46) Anderson, N. A.; Lian, T. Ultrafast Electron Injection from Metal Polypyridyl Complexes to Metal-Oxide Nanocrystalline Thin Films. Coord. Chem. Rev. 2004, 248, 1231−1246.
(47) Katoh, R.; Furube, A.; Barzykin, A. V.; Arakawa, H.; Tachiya, M. Kinetics and Mechanism of Electron Injection and Charge Recombination in Dye-Sensitized Nanocrystalline Semiconductors. Coord. Chem. Rev. 2004, 248, 1195−1213.