BioMed Central
Page 1 of 6 (page number not for citation purposes)
BMC Research Notes
Open Access
Technical Note
Prediction of protein secondary structures with a novel kernel
density estimation based classifier
Darby Tien-Hao Chang
1, Yu-Yen Ou
2,3, Hao-Geng Hung
4, Meng-Han Yang
4,
Chien-Yu Chen
5and Yen-Jen Oyang*
4,6,7,8Address: 1Department of Electrical Engineering, National Cheng Kung University, Tainan, 70101, Taiwan, ROC, 2Graduate School of
Biotechnology and Bioinformatics, Yuan Ze University, Chung-Li, 320, Taiwan, ROC, 3Department of Computer Science and Engineering, Yuan Ze University, Chung-Li, 320, Taiwan, ROC, 4Department of Computer Science and Information Engineering, National Taiwan University, Taipei, 106, Taiwan, ROC, 5Department of Bio-Industrial Mechatronics Engineering, National Taiwan University, Taipei, 106, Taiwan, ROC, 6Graduate Institute of Biomedical Electronics and Bioinformatics, National Taiwan University, Taipei, 106, Taiwan, ROC, 7Graduate Institute of Networking and Multimedia, National Taiwan University, Taipei, 106, Taiwan, ROC and 8Center for Systems Biology and Bioinformatics, National Taiwan University, Taipei, 106, Taiwan, ROC
Email: Darby Tien-Hao Chang - darby@ee.ncku.edu.tw; Yu-Yen Ou - yien@saturn.yzu.edu.tw; Hao-Geng Hung - hghung@mars.csie.ntu.edu.tw; Meng-Han Yang - d95001@csie.ntu.edu.tw; Chien-Yu Chen - cychen@mars.csie.ntu.edu.tw; Yen-Jen Oyang* - yjoyang@csie.ntu.edu.tw * Corresponding author
Abstract
Background: Though prediction of protein secondary structures has been an active research
issue in bioinformatics for quite a few years and many approaches have been proposed, a new challenge emerges as the sizes of contemporary protein structure databases continue to grow rapidly. The new challenge concerns how we can effectively exploit all the information implicitly deposited in the protein structure databases and deliver ever-improving prediction accuracy as the databases expand rapidly.
Findings: The new challenge is addressed in this article by proposing a predictor designed with a
novel kernel density estimation algorithm. One main distinctive feature of the kernel density estimation based approach is that the average execution time taken by the training process is in the order of O(nlogn), where n is the number of instances in the training dataset. In the experiments reported in this article, the proposed predictor delivered an average Q3 (three-state prediction accuracy) score of 80.3% and an average SOV (segment overlap) score of 76.9% for a set of 27 benchmark protein chains extracted from the EVA server that are longer than 100 residues.
Conclusion: The experimental results reported in this article reveal that we can continue to
achieve higher prediction accuracy of protein secondary structures by effectively exploiting the structural information deposited in fast-growing protein structure databases. In this respect, the kernel density estimation based approach enjoys a distinctive advantage with its low time complexity for carrying out the training process.
Published: 23 July 2008
BMC Research Notes 2008, 1:51 doi:10.1186/1756-0500-1-51
Received: 14 June 2008 Accepted: 23 July 2008 This article is available from: http://www.biomedcentral.com/1756-0500/1/51
© 2008 Chang et al; licensee BioMed Central Ltd.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
BMC Research Notes 2008, 1:51 http://www.biomedcentral.com/1756-0500/1/51
Page 5 of 6 (page number not for citation purposes)
2) R(si) is the maximum distance between si and its k near-est training instances;
3) Γ (·) is the Gamma function [20];
4) β and k are parameters to be set either through cross
validation or by the user.
For prediction of protein secondary structures, one kernel density estimator is created to approximate the distribu-tion of each class of training instances. As mendistribu-tioned ear-lier, in our experiment, each residue is associated with a PSSM computed with the PSI-BLAST software package, and is labeled as one of the three types of secondary struc-ture elements: alpha-helix, beta-strand, or coil, as deter-mined by DSSP. Then, a query instance located at v is predicted to belong to the class that gives the maximum value with the likelihood function defined as follows:
where |Sj| is the number of class-j training instances, and is the kernel density estimator corresponding to class-j training instances. In our current implementation, in order to improve the efficiency of the predictor, we include only a limited number, denoted by k', of the near-est class-j training instances of v while computing . With the predictions made by the RVKDE based algorithm for the query protein chain, Prote2S carries out a smooth-ing process as the last step before outputtsmooth-ing the results. The smoothing process includes two phases. In the first phase, each single-residue segment of secondary struc-tures with its two neighboring residues belonging to the same secondary structure is examined to determine whether switching the prediction of the single-residue seg-ment to the same secondary structure as its neighbors can form a new segment containing 4 or more residues. If yes, then the switching is carried out. Otherwise, nothing will
happen. In the second phase, all the remaining single-res-idue segments of secondary structures except those pre-dicted to be a coil are located and the prediction of each segment is switched to the secondary structure of its longer neighboring segment.
Parameter tuning
In the experiments reported in this article, the 4 parame-ters in the RVKDE algorithm were set as m = 1, β = 3, k =
38, and k' = 60, through a validation process. The valida-tion dataset was derived from the 1903 protein chains deposited into the PDB between June 1 and August 31 in 2007. In order to remove redundancy, BLAST was invoked to guarantee that the BLAST-computed e-value similarity score between any two protein chains in the validation dataset is larger than 0.1. Furthermore, we removed those protein chains that are homologous to one or more of the protein chains used to generate the training dataset with a BLAST-computed sequence identity larger than 25%. As a result, a total of 302 protein chains remained. Among these 302 protein chains, we then included those 45 chains that are longer than 100 residues to generate the validation dataset.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
YJO proposed the RVKDE algorithm and conceived the study. DTHC and YYO implemented the Prote2S predic-tor. HGH, MHY, and CYC designed the experiments reported in this article. All authors read and approved the final manuscript.
Acknowledgements
This research has been supported by the National Science Council of R.O.C. under the contracts NSC 95-3114-P-002-005-Y and NSC 96-2627-B-002-003 to Y.J. Oyang, 96-2320-B-006-027-MY2 and 96-2221-E-006-232-MY2 to D.T.H. Chang.
References
1. Eidhammer I, Jonassen I, Taylor WR: Protein Bioinformatics: An
Algorithmic Approach to Sequence and Structure Analysis.
Chichester: John Wiley & Sons Ltd; 2004.
L S j f j Sh fh h j( ) | | ( ) | | ( ), v v v = ⋅ ⋅ ∑ ˆ ( ) fj v ˆ ( ) fj v
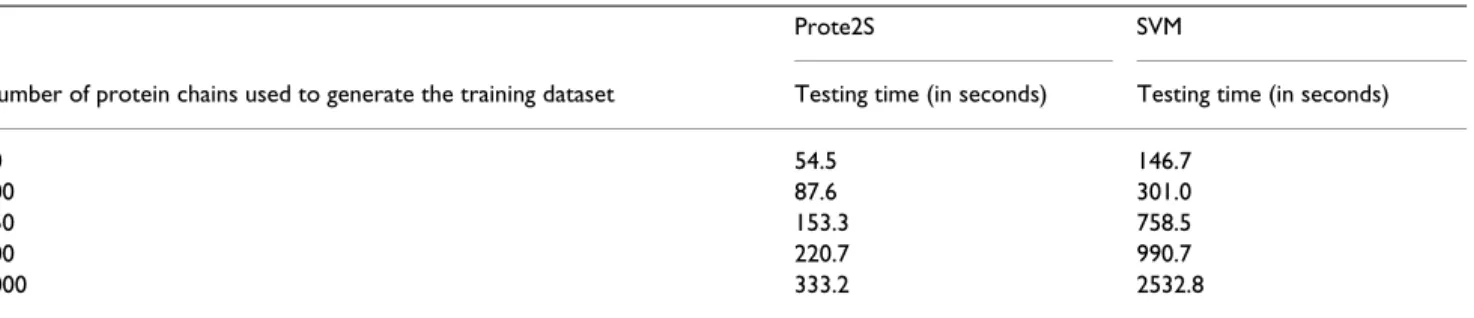
Table 5: Size of the training dataset vs. execution times taken by Prote2S and the SVM for making predictions.
Prote2S SVM
Number of protein chains used to generate the training dataset Testing time (in seconds) Testing time (in seconds)
50 54.5 146.7
100 87.6 301.0
250 153.3 758.5
500 220.7 990.7
Publish with BioMed Central and every scientist can read your work free of charge "BioMed Central will be the most significant development for disseminating the results of biomedical researc h in our lifetime."
Sir Paul Nurse, Cancer Research UK Your research papers will be:
available free of charge to the entire biomedical community peer reviewed and published immediately upon acceptance cited in PubMed and archived on PubMed Central yours — you keep the copyright
Submit your manuscript here:
http://www.biomedcentral.com/info/publishing_adv.asp
BioMedcentral BMC Research Notes 2008, 1:51 http://www.biomedcentral.com/1756-0500/1/51
Page 6 of 6 (page number not for citation purposes)
2. Lesk A: Introduction to Bioinformatics. Oxford: Oxford Univer-sity Press; 2005.
3. Baldi P, Brunak S: Bioinformatics: the Machine Learning
Approach. Cambridge: MIT Press; 2001.
4. Cuff JA, Barton GJ: Application of multiple sequence alignment
profiles to improve protein secondary structure prediction.
Proteins-Structure Function and Genetics 2000, 40(3):502-511.
5. McGuffin LJ, Bryson K, Jones DT: The PSIPRED protein
struc-ture prediction server. Bioinformatics 2000, 16(4):404-405.
6. Ward JJ, McGuffin LJ, Buxton BF, Jones DT: Secondary structure
prediction with support vector machines. Bioinformatics 2003, 19(13):1650-1655.
7. Wu KP, Lin HN, Chang JM, Sung TY, Hsu WL: HYPROSP: a hybrid
protein secondary structure prediction algorithm – a knowl-edge-based approach. Nucleic Acids Research 2004,
32(17):5059-5065.
8. Lin HN, Chang JM, Wu KP, Sung TY, Hsu WL: HYPROSP II – A
knowledge-based hybrid method for protein secondary structure prediction based on local prediction confidence.
Bioinformatics 2005, 21(15):3227-3233.
9. Montgomerie S, Sundararaj S, Gallin WJ, Wishart DS: Improving
the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinformatics 2006, 7:301.
10. Dor O, Zhou Y: Achieving 80% Ten-fold Cross-validated
Accu-racy for Secondary Structure Prediction by Large-scale Training. Proteins: Structure, Function, and Bioinformatics 2007, 66:.
11. Przybylski D, Rost B: Alignments grow, secondary structure
prediction improves. Proteins-Structure Function and Genetics 2002, 46(2):197-205.
12. Oyang YJ, Hwang SC, Ou YY, Chen CY, Chen ZW: Data
classifica-tion with radial basis funcclassifica-tion networks based on a novel ker-nel density estimation algorithm. IEEE Transactions on Neural
Networks 2005, 16(1):225-236.
13. EVA [http://cubic.bioc.columbia.edu/eva/]
14. Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation
of protein database search programs. Nucleic Acids Research
1997, 25(17):3389-3402.
15. Li WZ, Godzik A: CD-HIT: a fast program for clustering and
comparing large sets of protein or nucleotide sequences.
Bio-informatics 2006, 22(13):1658-1659.
16. Kabsch W, Sander C: Dictionary of Protein Secondary
Struc-ture – Pattern-Recognition of Hydrogen-Bonded and Geo-metrical Features. Biopolymers 1983, 22(12):2577-2637.
17. Chang CC, Lin CJ: LIBSVM: a library for support vector
machines. 2001 [http://www.csie.ntu.edu.tw/~cjlin/libsvm].
18. Hsu CW, Lin CJ: A comparison of methods for multiclass
sup-port vector machines. IEEE Transactions on Neural Networks 2002, 13(2):415-425.
19. Silverman BW: Density Estimation for Statistics and Data
Analysis. Boca Raton: Chapman & Hall/CRC; 1986.
20. Artin E: The Gamma Function. New York: Holt, Rinehart and Winston; 1964.