中文摘要
視紫質蛋白(rhodopsin)是位於視桿細胞(rod cell)上的感光受器,負責弱光中的視 覺形成。在脊椎動物視紫質蛋白基因的研究中,絕大部分只有一種視紫質蛋白的 cDNA 被報告,然而我們在鯉魚(Cyprinus carpio)卻發現有兩種視紫質蛋白的 cDNA (Tsai et al., 1994; Lim et al., 1997)。在本研究中,藉由聚合 鍊鎖反應(polymerase chain reaction)
以及限制 圖譜分析,證實此兩種視紫質蛋白基因可同時存在於一個鯉魚個體中。在 成功選殖並定序第一型視紫質蛋白的染色體基因(genomic gene)後,與第二型相比較, 獲知此兩型基因在上游調控區(-3434 ~ +94)只有 45.6 %的相似度,但卻含有三段相當 保守的區域 (conserved region):位於 -1262 ~ -614、-539 ~ -419 與 –166 ~ +94,分別 具有 78.0%、79.3 %及 92.3 %的相似度。另一方面,為了研究魚類視紫質基因的調控, 我們在第一型基因的上游調控區域中構築了四種不同長度的 DNA 片段,分別命名為 pDel-6k、pDel-146、pDel-77 與 pDel-53,依次包含–6k~ +66、-146~ +94、-77~ +94 與 –53~ +94 的上游調控區,其後均接上綠色螢光蛋白的 cDNA (green fluorescence protein, GFP)為報導基因。將這些片段分別用顯微注射技術轉殖到稻田魚(Oryzias latipes)單一細胞時期(one-cell stage)的受精卵中。結果顯示:除了 Del-46 片段之外, GFP 在視網膜上都有組織專一性表現。由此,我們提出一個假說:調控第一型視紫 質基因組織專一性表現的片段,可能位於-77~ -53 之間。很有趣的是,以這個片段和
已知調控陸生脊椎動物視網膜組織專一性的片段相比較,彼此之間的核 酸序列相似
Abstr act
Rod opsin, rhodopsin, is one of the photoreceptors but locates at rod cells, which mediates dim-light vision. We have cloned rhodopsin gene from the common carp (Cyprinus carpio). The deduced amino acid sequence of carp rhodopsin showed 95.7, 85.5 and 74.4 % identity with that of goldfish, sand goby and lamprey, respectively. However, few significant homological regions could be found in the 5’ regulatory region compared with corresponding sequences of other terrestrial species. To define the cis-acting DNA elements required for rhodopsin expression, we generated transgenic fish carrying sequences upstream of carp rhodopsin gene fused to the green fluorescence protein cDNA as a reporter gene. Upstream sequences extending from –6000 to +66 bp and from –146 to +94 bp were able to drive retinal-specific expression, whereas from –53 to +94 bp segment was not. This suggests that element which regulates retinal-specific expression may locate within –146~ -53 bp. Interestingly, little homology of polynucleotide sequences was shown between the region (-146~ -53 bp) and any other known retina-specific protein binding sites of terrestrial animals.
Intr oduction
Promoter analysis of retinal genes have recently become possible with the molecular cloning of the visual pigment genes and their flanking sequences from a number of species, such as rhodopsin genes of bovine (Nathans and Hogness, 1983), human (Nathans and Hogness, 1984; Jean et al., 1995), Drosophila (Zuker et al., 1985), mouse (Baehr et al., 1988), chicken (Takao et al., 1988), rat (Morabito et al., 1991), Xenopus (Batni et al., 1996). Characteristically homologous sequences of rhodopsin upstream region can be found among species. A sequence comparison of 2.2-kb upstream of bovine, human and mouse rhodopsin genes shows two regions of homology. The first region encompasses ~250 bp proximal to the transcription start site, containing CCAAT (at –80 bp) and TATA (at –30 bp) sequence elements (Zack et al., 1991). The second region of homology (-1800 in the human, -1500 in the mouse, and –1900 bp in the bovine) is a relatively conserved sequence spanning approximately 100 bp (Nie et al., 1996). In addition, significant homology of rhodopsin upstream sequences of Xenopus and bovine, human, mouse, rat and chicken is found from – 10 to –180 (Batni et al., 1996).
Due to the lack of suitable retinal cell lines, transgenic approaches are usually applied in defining the cis-acting DNA elements required for rhodopsin expression. Upstream sequences of the bovine rhodopsin gene extending from –2174 to +70 bp, from –734 to +70 bp and from –222 to +70 bp are able to direct photoreceptor-specific expression in the transgenic mice (Zack et al., 1991, Gouras et al., 1994). A similar result is shown on transgenic mice carrying 4.4 kb or 500 bp of mouse rhodopsin 5’ flanking sequence fused to the E. coli lacZ gene (Lem et al., 1991). Likewise, upstream sequences of Xenopus rhodopsin gene from –5500 to +41 bp and from –508 to +41 are both able to direct rod-specific expression (Knox et al., 1998). The results show that the proximal upstream portion (~300 bp) is both necessary and sufficient for the correct tissue and developmental expression of the reporter gene, however, sequences in more upstream regions appear to be needed for higher and more uniform levels of expression, and more precise temporal expression.
Furthermore, other approaches such as electrophoretic mobility shift assays, DNase I footprinting analysis, in vitro transcription analysis are used to more fully define the DNA regulatory elements and protein factors involved in regulating rhodopsin transcription. The
Ret 1 element, for example, binds developmentally retinal nuclear proteins and is suggested to be able and necessary to drive gene expression in rod photoreceptors in rat (Morabito et al., 1991; Yu et al., 1996). The Ret 2 and Ret 3 sites, which are defined by footprinting analysis, can interact with the same retinal nuclear proteins (Yu et al., 1993). The Ret 4 element, which is identified with the bovine retina in vitro transcription system, is bound by both retina-specific and ubiquitously expressed protein factors (Chen and Zack, 1996). There is also a putative binding site, Eopsin-1, binding with the basic helix-loop-helix
transcription factor Mash-1, which plays a significant role in mammalian neurogenesis (Ahmad, 1995). Transient expression studies with CV-1 cells and primary chick retinal cultures have implicated the bZIP family member neural retinal leucine zipper protein (Nrl) as a trans acting regulator of rhodopsin expression and have identified its response cis element, NRE, which is relevant to stimulating rhodopsin promoter activity (Rehentulla et al., 1996; Kumar et al., 1996). The nucleotide sequences and position of these cis-acting DNA elements are more or less conserved among species.
There are three conserved segments between the upstream regions of the carp type I and type II rhodopsin gene. Besides, some putative retina-specific protein binding sites are also found. Among those sequences, the third conserved segments (–166~ +94) exhibited the highest similarity (92.3%) between the two types and may be crucial to tissue specific regulation. To identify the roles of this segment in regulating rhodopsin gene expressing in fish, chimeric gene constructs with nested deletion fragments of rhodopsin upstream region fused to the reporter gene, green fluorescence protein (GFP), were generated and microinjected into the one-cell stage eggs of medaka (Oryzias latipes). This is the first study of visual promoter of fish and thus can be very informative to understand the mechanism of visual gene regulation of aquatic animals.
Microinjection foreign DNA into fertilized medaka eggs is a relative simple technique and can yield transformants in high frequency (Kinoshita and Ozato, 1995). GFP fluorescence is stable, species-independent and can be monitored non-invasively in living cells or transparent animals. Besides, it is more sensitive than standard reporter proteins, such as β-galactosidase, which utilize enzymatic amplification (Zhang et al., 1996). These methods are therefore very suitable for dynamic promoter analysis of fish rhodopsin gene.
Mater ials and methods
Prepar ation of the upstream fr agment from type I r hodopsin
Generation of unidirectional deletion fragments Plasmid DNA, pRHO1-8kb,containing type I rhodopsin gene 5’ regulatory region and partial cDNA (+659) was purified via Plasmid Miniprep Kit (Viogene). Because there were no restriction enzyme cutting sites suitable for excising upstream fragment from the vector and the cDNA, unidirectional deletion fragments were generated to remove the cDNA portion of the plasmid. To generate linear DNA with one 5’ protruding cohesive end and 3’ protruding cohesive end, the plasmid DNA, pRHO1-8kb was digested with Xho I and Apa I. Before exonuclease III (Exo III) digestion, the restriction enzyme activities were heat inactivated at 75℃ for 10 minutes without purification by phenol/ chloroform/ isoamyl alcohol. Eleven eppendorfs with 3 μl of S1 solution (S1 nuclease 400~ 600 units/ ml, 50 mM potassium acetate, pH 4.6, 316 mM NaCl, 1.6 mM ZnSO4, 8% glycerol) were prepared. The linearized plasmid DNA was
digested with 3 units of Exo III in total 25μl Exo III solution (66 mM Tris-HCl, pH 8.0, 0.6 mM MgCl2) at 30℃. Every 2 minutes, 2μl of the reaction solution was transferred into the
S1 solution incubating on ice. Afterward, all 11 eppendorfs were transferred from ice and incubated at room temperature to generate blunt-end termini. The S1 nuclease reactions were terminated by adding 1 μl of S1 stop solution (303 mM Tris base, 50 mM EDTA) and heat-inactivated at 65℃ for 10 minutes. 3μl of each sample was analyzed with 0.5 % agarose gel (SeaKem Gold Agrose, FMC). Samples with desired sizes were circularized with T4 ligase and trasnformed into competent cells, DH5α(described below). The above procedures were performed according to the instruction of Double-Stranded Nested Deletion Kit (Pharmacia Biotech). Colonies selected with ampicillin (50μg/ ml) were cultured in 3 ml LBA at 37℃ overnight to perform minipreparation. Sequence analysis was performed as described previously using specific primer 5’-GGCTGCGGTTGGATGTTGT-3’ (EPA) to select desired clones.
Competent cell preparation & transformation (Inoue et al., 1990) Frozen stock DH5α cells were thawed, streaked on an LB plate, and cultured overnight at 37℃. About 3 colonies were inoculated to 50 ml of LB medium in a 250-ml side arm flask, and grown to
an A600 of 0.6 at 18℃ (about two to three days) with vigorous shaking (200- 250 rpm). The
flask was removed from the incubator and placed on ice for 10 minutes. The culture was transferred to two 50-ml centrifuge bottles and spun at 2500×g for 10 minutes at 4℃. The pellet was resuspended in 16 ml TB (10 mM Hepes, 55 mM MnCl2, 15 mM CaCl2, 250 mM
KCl) and incubated on ice for 10 minutes. After centrifugation, the pellet was resuspended in 4 ml TB/ 7% DMSO and dispensed by 100 μl into 1.5 ml eppendorfs. The competent cells were stored at –70℃. As to transformation, 1-5 μl of the plasmid solution (less than 10 ng) was added to the 100 μl competent cells and then incubated on ice for 30 minutes. The cells were heat-shocked without agitation at 42℃ for 30 seconds and put on ice. After 0.9 ml of LB was added, the tubes were incubated at 37℃ for 1 hour. A desired portion of the mixture was transferred onto proper selective medium plates.
Isolating insert fragments Plasmid DNA of desired unidirectional deletion clone was digested with restriction enzyme BssH II, which is on the multiple cloning site of the vector, pBluescript II SK (+/-), to excise the insert fragment. The digested DNA was subject to agarose gel electrophoresis and the insert fragment was gel-recovered by Jetsorb Gel Extraction kit (Genomed).
Prepar ation of type I r hodopsin upstream deletion fr agments
Phosphorylation of deletion primers Because deletion primers were synthesized with hydroxyl group on both 5’ and 3’ termini, it is necessary to add phosphate group on the 5’ end before ligation. 1 nmole of each deletion primer (Del-100, Del-50, Del-25, EPA) was phosphorylated with 10 units of T4 polynucleotide kinase (Gibo BRL) in 2 nmole of r-ATP and 1X forward reaction buffer ( 70 mM Tris-HCl, pH 7.6, 10 mM MgCl2, 100 mM KCl, 1
mM 2-mercaptoethanol) at 37℃ for 2.5 hours. Afterward, T4 polynucleotide kinase was heat inactivated at 70℃ for 10 minutes. The primers were purified via phenol/ chloroform/ isoamyl alcohol (25: 24: 1) extraction and ethanol precipitation. The primers were resuspended in 100μl ddH2O and the concentration was about but less than 10μM.
PCR generation of deletion fragments Three nested deletion fragments, del-146 (from – 146 to +94), del-77 (from –77 to +94) and del-53 (from –53 to +94), of type I rhodopsin gene upstream region were generated via PCR. For every 50μl PCR reaction, it contained 50 ng template, 200μM of dNTPs (dATP, dCTP, dGTP and dTTP), 5μM of deletion primer set, 1X ThermolPol buffer (10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris-HCl, pH
8.8, 2 mM MgSO4, 0.1% Triton X-100) and 2 units of high fidelity thermophilic DNA
polymerase (VentR
DNA Polymerase, NEB). The PCR conditions were 94℃ 1 minute, 58
℃ 1 minute and 72℃ 1 minute for 25 cycles (DNA Thermal Cycler, PERKIN ELMER CENTUS). 10μl of PCR products were subject to 0.8% agarose gel electrophoresis.
Constr uction of type I r hodopsin upstream-GFP chimer ic genes
Vector preparation pEGFP-1 (Clontech) is an expression vector that encodes the EGFP (enhanced green fluorescence protein) variant for monitoring the activity of promoters cloned into the MCS (multiple cloning site). The map of the plasmid was shown in Appendix I. Ten micrograms of pEGFP-1 was linearized by EcoR I or Age I and then filled-in by Klenow fragment, and dephosphrylated by CIAP (calf filled-intestfilled-ine alkalfilled-ine phosphotase, Promega) to generate blunt end termini with hydroxyl group on both end. Enzyme treatment of each step was heat inactivated and the DNA was purified via phenol/ chloroform/ isoamyl alcohol (25: 24: 1) extraction and ethanol precipitation.
Ligation and transformation Type I rhodopsin upstream fragment and del-146, del-77
and del-53 were ligated into ~25 ng plasmid vector, pEGFP-1/ EcoRI/ fill-in/ CIAP, by T4 DNA ligase with 1.2μM Co(NH3)6Cl3 and 5% PEG8000 at 4℃ for 16 hours. 2μl of the
ligation mixture (10μl) was used to transform 100μl of competent cells, DH5α as described previously. Colonies with plasmid were selected via selection medium, LBK plates (LB plates with 30μg/ ml kanamycin), to inoculate 3 ml LBK broth. After overnight culture at 37℃, plasmid DNA of putative clones was purified via minipreparation using alkaline-lysis method and subject to PCR reaction as templates.
Examination of insert existence and orientation Insert existence and orientation were
determined via PCR using primers CAGCTCG TCCATGCCATGTG-3’ (GFP-R) and 5’-GGTAATCCCAAGCGAGCCTC-3’ (Del-25), which were on the vector and insert,
respectively. Only plasmid containing insert fragment with desired orientation could generate PCR products. For every 50μl PCR reaction, it contained 100 ng template, 200μ M of dNTPs (dATP, dCTP, dGTP and dTTP), 0.5μM of GFP-R and Del-25, 1X buffer (10 mM Tris-HCl, pH 8.8 at 25℃, 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100) and 2.5
units of thermostable DNA polymerase (Viotaq, Viogene). The PCR conditions were 94℃ 1 minute, 58℃ 1 minute and 72℃ 1 minute for 25 cycles (DNA Thermal Cycler, PERKIN ELMER CENTUS). 10 μ l of PCR products were subject to 0.8% agarose gel electrophoresis.
Gener ation of tr ansgenic fish
Preparation DNA for microinjection pGREEN LANTERN-1 (Gibco BRL) contains the cytomegalovirus (CMV) immediate early promoter for high expression of GFP and is suitable as a reporter plasmid for transgenic animals. The map of the plasmid was shown in Appendix II. pGREEN LANTERN-1 and pEGFP-1 were linearized withSca I and Age I, respectively, as positive and negative controls. Plasmid DNA of type I upstream-GFP chimeric gene constructs were linearized outside the promoter-GFP region with Xho I. Restriction enzyme activities were heat inactivated at 65℃ for 15 minutes. The linearized plasmid DNA was purified via phenol/ chloroform/ isoamyl alcohol (25: 24: 1) extraction and ethanol precipitation. The purified DNA was resuspended in sterile ddH2O and
quantified via absorbance measurements at 260 nm with GeneQuant II, RNA/ DNA Calculator (Pharmacia Biotech) and agarose gel electrophoresis.
Cytoplasmic microinjection (Kinoshita and Ozato, 1995) Linearized plasmid DNA was diluted into 10 ng/μl in 1X PBS (1.47 mM KH2PO4, 8.06 mM Na2HPO4, 137 mM
NaCl, 2.68 mM KCl)/ 0.1% phenol red before microinjection. The manipulator, injector and microscope were set up and the loading pipettes and injection needles were prepared according to the reference. Fertilized eggs before the first cleavage of medaka were collected. After removing of the attaching filaments, the eggs were soaked in ddH2O at 6℃.
To perform cytoplasmic microinjection, the egg was oriented on the egg holder to present the animal pole of the cytoplasm under a stereoscopic microscope. After orienting, the egg was brought under the field of a microscope and injected with 10~ 100 pl of DNA solution
by increasing pressure level of the injector. After injection, the egg was transferred from the egg holder to a glass dish containing ddH2O and incubated at 26℃.
F luorescence microscopy Transgenic embryos were selected using a stereo dissecting microscope (MZ12, Leica, Germany) equipped with a fluorescence module having an enhanced GFP filter cube (Kramer Scientific). Embryos were observed everyday until hatching out (about 10 to 14 days post-fertilization). Photographs were taken using MPS 60 camera (Leica, Germany) and FUJI 400 ASA film.
Results and Discussion
Chimeric gene constructs Plasmid, pRHO1-66, has been generated from 3’
unidirectional deletion of pRHO1-8kb (Fig. 1, lane 5). It contained a insert fragment of type I rhodopsin gene ranging from –7.5 kb to +66 bp. A 6 kb segment containing the proximal portion of type I rhodopsin upstream region was isolated after BssHII digestion and gel-recovery (Fig. 2). Moreover, three nested deletion fragments were generated via PCR, which were from –146 to +94, from –77 to +94 and from –53 to +94, respectively (Fig. 3). These DNA fragments were blunt-end ligated into pEGFP-1 to generate four chimeric gene constructs, pDel-6k, pDel-146, pDel-77 and pDel-53, which were named according to the position of the 5’-most base pair. The maps of the constructs relative to the type I rhodopsin gene upstream region were shown. Note that putative Ret 1, which is crucial to gene expression in rod photoreceptors in rat, was absent in pDel-77 and pDel-53 (Fig. 4).
Generation of transgenic fish Transgenic fish was generated via cytoplasmic
microinjection of foreign DNA into medaka fertilized eggs. For each construct (pDel-6k, pDel-146, pDel-77 and pDel-53), about 100 eggs were microinjected; as to positive control (pGREEN LANTERN) and negative control (pEGFP-1), about 30 eggs were microinjected.
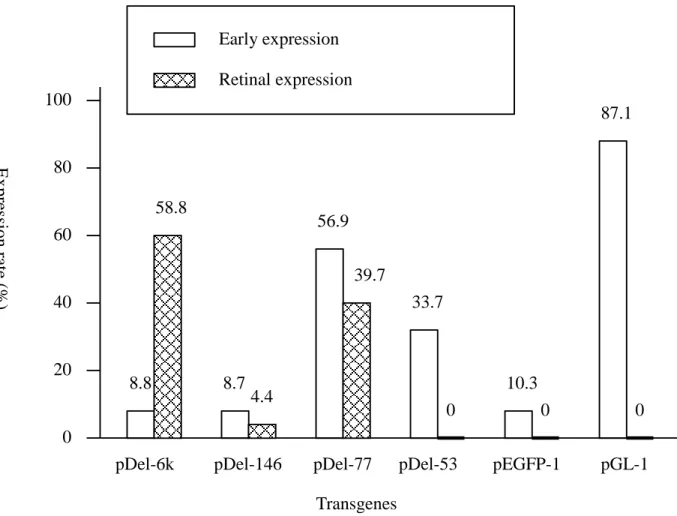
Early expression The expression of GFP could be easily observed on the second day
after microinjection on the yolk sac or other portions of the embryos, i.e. early expression, of transgenic fish carrying pGREEN LANTERN-1, pDel-53 and pDel-77 (Fig. 5). As to the other three constructs, pDel-6k, pDel-143 and pEGFP-1, only very weak GFP expression could be observed on some embryos and could merely last 3 to 4 days (stage 29 to stage 33) (Iwamatsu, 1994). pGREEN LANTERN-1 (Gibco BRL), which contains the cytomegalovirus (CMV) immediate early promoter for high expression of GFP, had an expression rate (number of expressing eggs/ number of injected eggs) of 87.1%. The GFP was expressed randomly on the embryos and could last even after the fish hatched out. Nonetheless, no transgenic fish carrying pGREEN LANTERN-1 expressed GFP on retina. The early expression levels of pDel-77 and pDel-53, on the other hand, decreased as embryos developed. GFP expression on the yolk sac or other portion (not on retina) of the embryos could hardly be seen after the fish hatched out.
Tissue specific expression Among the four constructs containing type I rhodopsin
upstream region, pDel-6k, pDel-146 and pDel-77 could direct GFP expression specific on retina (Fig. 6), while pDel-53 could not (Fig. 7). The results suggested that the element, which regulates tissue specific expression of rhodopsin gene, located within nucleotide position from –77 to –53. As to the tissue specific expression rate, pDel-6k was the highest (58.8%), while pDel-146 was the lowest (4.4%) and pDel-77 was in the middle (39.7%). Since pDel-146 contained a larger upstream fragment than pDel-77, the lower retinal expression rate may be attributed to the inhibiting activity within nucleotide position from – 146 to –77. Furthermore, since high level of early expression could be observed on pDel-77 (56.9%) but not on pDel-146 (8.7%), another inhibitor may exist within the same region (-146 to –77) to constrain rhodopsin gene early expression. However, the polynucleotide sequences of neither –77~ -53 nor –146~ -77 showed significant homology with any reported retinal protein binding sites of other terrestrial species. In addition, pDel-77, which lacked putative Ret 1 core region, could still drive tissue specific expression in transgenic fish. The above results implied that the regulatory mechanism of visual gene expression of fish may be different from that of the terrestrial animals.
Refer ences
Ahmad, I. (1995). Mash-1 is expressed during ROD photoreceptor differentiation and binds an E-box, Eopsin-1 in the rat opsin gene. Developmental Brain Research 90, 184-189.
Batni, S., Scalzetti, L., Moody, S. A. and Knox, B. E. (1996). Characterization of the Xenopus rhodopsin gene. The Journal of Biological Chemistry 271, 3179-3186.
Baehr, W., Falk, J. D., Bugra, K., Triantaafyllos, J. T. and McGinnis, J. F. (1988). Isolation and analysis of the mouse opsin gene. FEBS Letters 238, 253-256.
Chen, N., Ma, J. X., Corson, D. W., Hazard, E. S. and Grouch, R. K. (1996). Molecular cloning of a rhodopsin gene from salamander rods. Investigative Ophthalmology & Visual Science 37, 1907-1913.
Gouras, P., Kjeldbye, H. and Zack, D. J. (1994). Reporter gene expression in cones in transgenic mice carrying bovine rhodopsin promoter/ lacZ transgenes. Visual Neuroscience
11, 1227-1231.
Iwamatsu, T. (1994). Stages of normal development in the medaka Oryzias latipes. Zoological Science 11, 828-839.
Knox, B. E., Schlueter, C., Sanger, B. M., Green, C. B. and Besharse, J. C. (1998). Transgene expression in Xenopus rods. FEBS Letters 423, 117-121.
Kumar, R., Chen, S., Scheurer, D., Wang, Q. L., Duh, E., Sung, C. H., Rehemtulla, A., Swaroop, A., Adler, R. and Zack, D. J. (1996). The bZIP transcription factor Nrl stimulates rhodopsin promoter activity in primary retinal cell cultures. The Journal of Biological Chemistry 271, 29612-29618.
Lem, J., Applebury, M. L., Falk, J. D., Flannery, J. G. and Simon M. I. (1991). Tissue-specific and developmental regulation of rod opsin chimeric genes in transgenic mice. Neuron 6, 201-210.
Morabito, M. A., Yu, X. and Barnstable, C. J. (1991). Characterization of developmentally regulated and retina-specific nuclear protein binding to a site in the upstream region of the rat opsin gene. The Journal of Biological Chemistry 266, 9667-9672.
Nathans, J. and Hogness, D. S. (1983). Isolation, sequence analysis, and intron-exon arrangement of the gene encoding bovine rhodopsin. Cell 34, 807-814.
Nathans, J. and Hogness, D. S. (1984). Isolation and nucleotide sequence of the gene encoding human rhodopsin. Proceedings of the National Academy of Science of the United States of American 81,4851-4855.
Nie, Z., Chen, S., Kumar, R. and Zack, D. J. (1996). RER, an evolutionary conserved sequence upstream of the rhodopsin gene, has enhancer activity. The Journal of Biological Chemistry 271, 2667-2675.
Rehemtulla, A., Warwar, R., Kumar, R., Ji, X., Zack, D. J. and Swaroop, A. (1996). The basic motif-leucine zipper transcription factor Nrl can positively regulate rhodopsin gene expression. Proceedings of the National Academy of Science of the United States of American 93, 191-195.
Takao, M., Yasui, A. and Tokunaga, F. (1988). Isolation and sequence determination of the chicken rhodopsin gene. Vision Research 28, 471-480.
Yu, X., Chung, M., Morabito, M. A and Barnstable, C. J. (1993). Shared nuclear protein binding sites in the upstream region of the rat rod opsin gene. Biochemical and Biophysical Research Communications 191, 76-82.
Yu, X., Leconte, L., Martinez, J. A. and Barnstable C. J. (1996). Ret 1, a cis-acting element of the rat opsin promoter, can direct gene expression in rod photoreceptors. Journal of Neurochemistry 67, 2494-2504.
Zack, D. J,. Bennett, J., Wang. Y., Davenport, C,. Klaunberg, B., Gearhart, J. and Nathans, J. (1991). Unusual topology of bovine rhodopsin promoter-lacZ fusion gene expression in transgenic mouse retinas. Neuron 6, 187-199.
Zhang, G., Gurtu, V. and Kain, S. R. (1996). An enhanced green fluorescent protein allows sensitive detection of gene transfer in mammalian cells. Biochemical and Biophysical Research Communications 227, 707-711.
Zuker, C. S., Cowman, A. F. and Rubin, G. M. (1985). Isolation and structure of a rhodopsin gene from D. melanogaster. Cell 40, 851-858.
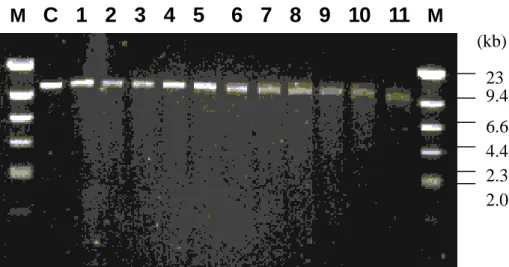
Figur e 1. Unidirectional deletion fr agments of pRHO1-8kb. Lane M: molecular weight
marker, λDNA/Hind III; lane C: plasmid, pRHO1-8kb, linearized with Apa I & Xho I; lanes 1~11: successive deletion fragments from the beginning (time = 0) with a 2-minute time interval. M
C 1 2 3 4 5 6 7 8 9 10 11
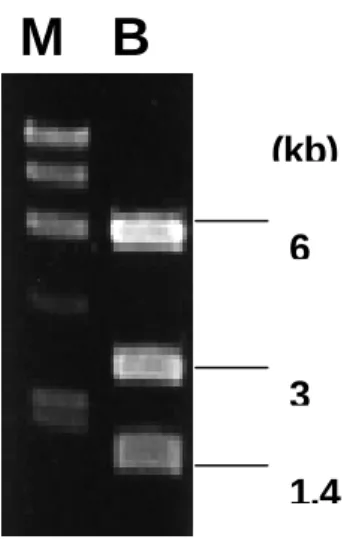
M 23 (kb) 9.4 6.6 4.4 2.3 2.0Figur e 2. Agar ose gel electrophoresis of pRHO1-66 digested by BssHII. Lane M:
molecular weight marker, λDNA/Hind III; lane B: plasmid, pRHO1-66, linearized with BssHII. The 6 kb and 1.4 kb fragments contained the proximal and distal portions of the insert, respectively. The 3 kb fragment was the vector, pBluescriptII SK (+/-).
M B
6
3 1.4 (kb)
Figur e 3. PCR gener ated deletion fr agments. Lanes 1~3: three nested deletion fragments,
from –146 to +94, from -77 to +94 and from –53 to +94, respectively; lane M: molecular weight marker, φX174/Hae III.
1 2 3 M
240 169 146 (bp)
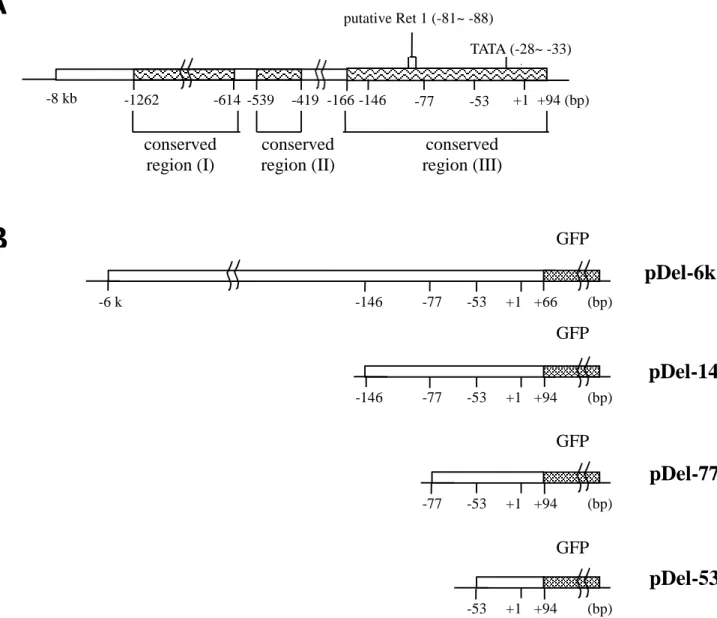
Figur e 4. Maps of the type I r hodopsin gene upstream-GFP fusion constr uct. (A):
Schematic diagram of type I rhodopsin gene upstream region. Numbers indicate nucleotide position relative to transcription start site (+1). “ “: conserved region between type I and type II upstream polynucleotide sequences; putative Ret-1: sequences shared 87.5% identity with Ret-1 core region (CTAATTAG). (B): Schematic diagram of the deletion constructs. “ “ : green fluorescence protein (GFP) cDNA, The constructs were named according to the position of the 5’-most base pair, which is on the left of each construct.
pDel-6k
pDel-146
pDel-77
pDel-53
conserved region (I) conserved region (II) conserved region (III) +94 (bp) -53 -77 -146 -419 -1262 -8 kb -614 -539 TATA (-28~ -33) putative Ret 1 (-81~ -88) GFP +66 (bp) -6 k GFP +94 (bp) -146 GFPB
+1 +1 +1 +94 (bp) -77 -53 +1 -53 -53 -146 -77 -77A
GFP +94 (bp) -53 +1 -166Figur e 5. Compar ison of expression patter ns in the tr ansgenic fish embr yos microinjected with type I r hodopsin upstream-GFP chimer ic genes . pDel-6k, pDel-146,
pDel-77 and pDel-53: type I rhodopsin upstream-GFP chimeric gene constructs; pEGFP-1: negative control; pGL-1 (pGREEN LANTERN-1): positive control. Early expression: GFP expressed on the yolk sac or other portions of the embryo before expressing on retina. Retinal expression: GFP expressed specifically on retina.
Transgenes 100 80 60 40 20 0 33.7 0 56.9 8.7 58.8 87.1 0 0 39.7 4.4 8.8 10.3
pDel-6k pDel-146 pDel-77 pDel-53 pEGFP-1 pGL-1
Early expression Retinal expression Ex pre ssion rate (% )
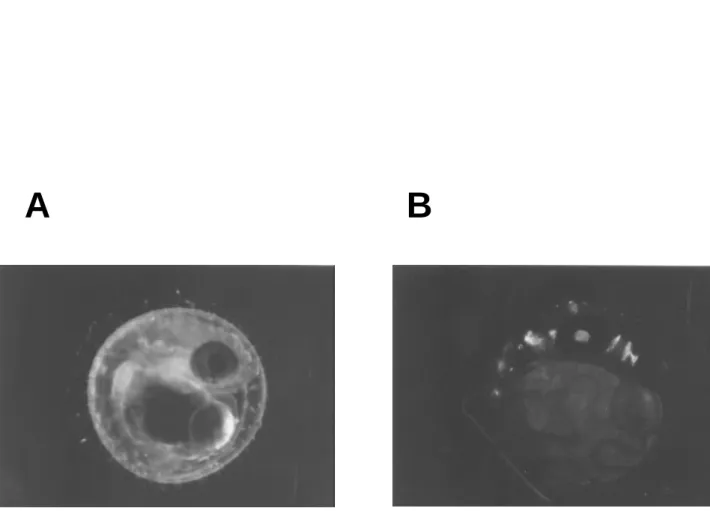
Figur e 6. GFP expr ession on the retina of the tr ansgenic fish embr yo micr oinjected with pDel-6k. A: bright field illumination. B: blue light. The bright green fluorescence was
seen exclusively in the eye of the medaka embryo (stage 36).
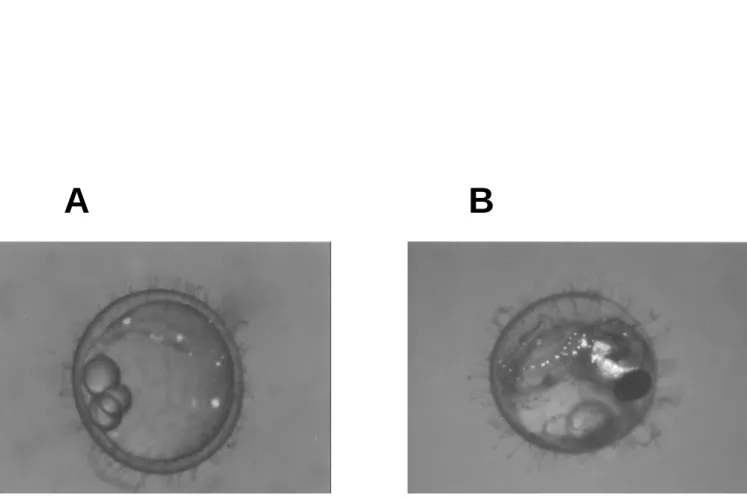
Figur e 7. GFP expr ession on yolk sac and embr yos of tr ansgenic fish microinjected with pDel-53. A: stage 17; B: stage 34. The bright green fluorescence from the transgene
was seen under blue light and dim bright field illumination. The yellow color came from endogenous fluorescence of medaka.