行政院國家科學委員會補助專題研究計畫
■ 成 果 報 告 □期中進度報告Integrin-associated protein 基因表達之細胞訊息
調控機轉及其蛋白在神經可塑性之角色
計畫類別:■ 個別型計畫 □ 整合型計畫
計畫編號:NSC 92-2320-B -006-035
執行期間: 91 年 08 月 01 日至 93 年 07 月 31 日
計畫主持人:黃阿敏
共同主持人:-
計畫參與人員: 張文騰、邱榮敬、陳貞云
成果報告類型(依經費核定清單規定繳交):□精簡報告 ■完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
■
出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、列管計畫 及下列情形者外,得立即公開查詢 □涉及專利或其他智慧財產權,□一年□二年後可公開查詢執行單位:國立成功大學生理學研究所
中 華 民 國 93 年 10 月 25 日
中英文摘要
中文摘要 IAP 是一個 50 KD 的膜蛋白,在中樞神經系統發現其基因表現與記憶有關。本 研究為兩年期的計劃,第一年計劃探討NGF/MAPK/Sp1 訊息路徑是否參與 IAP 基因 表現的調控。第二年計劃探討IAP 蛋白是否參與神經軸突生長。第一年計畫之研究結 果發現,在人類神經纖維母細胞瘤IMR-32 細胞中,Sp1 並不是調控 IAP 基因的最重 要轉錄因子,但是經由一系列的實驗,我們證實IAP 基因的核心調控子區域位於 ATG前-232 至-12,調控 IAP 基因的重要轉錄因子為 nuclear respiratory factor-1 (NRF-1), 其辨識序列為TGCGCGCTGCGCGC。本結果已發表於 The Journal of Biological Chemistry 279 (15): 14542-14550 (2004)。第二年計畫之研究結果發現,IAP 蛋白及 NRF-1 確實參與神經軸突生長。於 IMR-32 細胞大量表現 NRF-1 時,明顯促進神經軸 突生長。若於此細胞內表現NRF-1 突變蛋白或 IAP 反義 cDNA 序列,則明顯抑制神 經軸突生長,另外發現MAPK 訊息路徑參與 NRF-1/IAP 的活化。 關鍵詞:IAP 基因、啟動子活性、轉錄因子 α-Pal/NRF-1、神經軸突生長、MAPK 訊息路徑 I 英文摘要
Integrin-associated protein (IAP or CD47) is a 50KD membrane glycoprotein expressed in various cell types. Previous studies have shown that IAP is involved in memory formation. This two-year project was planned to delineate signaling pathways that control of the expression of IAP gene and to investigate the role of IAP in neurite outgrowth. In the project of first year, we found that Sp1 plays little role in the regulation of IAP promoter in human neuroblastoma IMR-32 cells. However, we found a nuclear factor, named α-Pal/ nuclear respiratory factor 1 (α-Pal/NRF-1), which locates at 204 to 193 bases upstream of the initiation codon ATG, plays an important in the regulation of IAP gene promoter. These findings were published in the Journal of Biological Chemistry 「279 (15): 14542-14550 (2004)」In the project of second year, we found that IAP and α-Pal/NRF-1 were involved in neurite outgrowth. Stable cell line overexpressed with significant enhance neurite outgrowth in IMR-32 cells. In contrast, expression of IAP antisense cDNA inhibits the enhancement of neurite outgrowth by α-Pal/NRF-1.
Furthermore, we demonstrated that the MAPK signaling is involved in the activation of IAP promoter activity and the enhancement of neurite outgrowth by α-Pal/NRF-1. Key words: Integrin-associated protein, promoter activity, α-Pal/NRF-1, neurite outgrowth, MAPK signaling
目錄
頁數
中英文摘要……… 1
報告內容……… 3
參考文獻……… 16
附圖及說明……….18
計畫成果自評……… 22
附錄……… 23
報告內容
前言及文獻探討
Integrin-associated protein (IAP), also designated as CD47, is a multifunctional membrane protein that is expressed widely in the nervous system, immune system and many other tissues (1, 2). In the adult rat central nervous system, IAP was related to memory formation of an aversive learning task (3). In good-memory rats trained in the inhibitory avoidance learning paradigm, the mRNA level of IAP in the
hippocampus was increased. Injection of antisense oligonucleotides or the monoclonal antibody of IAP into the rat hippocampus impaired memory formation (3, 4).
IAP-deficient mice showed deficits in memory retention in a similar behavioral paradigm (5).
In the peripheral tissues, IAP was first discovered as a cell surface protein associated with integrin αvβ3 and it was involved in the enhancement of neutrophil
adhesion, chemotaxis, and phagocytosis triggered by an extracellular matrix (1, 6, 7). IAP is also a functional component of several processes, including the transepithelial migration of neutrophils (8), chemotaxis of endothelial cells and smooth muscle cells (9, 10), spreading and aggregation of platelets (11), and modulation of T-cell
activation (12, 13). Moreover, IAP functions as a self-check marker on red blood cells to prevent their clearance by macrophages (14).
In cultured cerebellar cells, the P84 neural adhesion molecule, a ligand of IAP, was able to promote neuronal cell adhesion and neurite outgrowth. Monoclonal antibodies to IAP and P84 inhibited these effects (Chuang and Lagenaur, 1990; Jiang et al., 1999). These findings indicate that IAP plays an important role in the process of memory formation and neuronal plasticity. Although the cellular functions of IAP have been intensively studied, the regulation of its expression at the molecular level is still unknown. To answer these questions, we investigated the regulation of IAP gene promoter in human neuroblastoma and hepatoma cell lines by using luciferase reporter and gel electrophoretic mobility shift assays. We found that α-Pal/nuclear respiratory factor 1 (NRF-1) in the core promoter region is a positive regulator of the human IAP gene.
α-Pal/NRF-1 is a transcription factor that was found independently in studying the regulation of different vertebrate genes. α-Pal was first identified on the
eukaryotic initiation factor 2α (eIF2α) gene which product is the rate-limiting
enzyme of protein synthesis (17). Functional α-Pal sites are also found in the E2F1gene which product is essential to cell cycle progression (18). NRF-1 was identified as a nuclear transcription factor that activates the promoter of many genes involved in mitochondrial function and biogenesis (19). These include the cytochrome
c, NADH dehydorgenase, succicinate dehydrogenase, and cytochrome oxidase genes
(20). The chicken homologue of α-Pal/NRF-1 was named as initiation binding receptor (IBF), which bound to the transcription initiation site of the histone h5 gene (21).
GenBank database search reveals that the consensus binding site of
α-Pal/NRF-1/IBF, (T/C)GCGCA(T/C)GCGC(A/G), can be found in the proximal promoter regions of many genes involved in signal transduction, mitochondrial energy transduction, protein metabolism, lipid metabolism, and nucleic acid metabolism (22-24). In addition to housekeeping functions, α-Pal/NRF-1/IBF may also regulate tissue-specific genes, including h5, c-myb, preproenkephalin A, synapsin I, acetylcholine receptor and slow sarcoplasmic reticulum Ca2+-dependent ATPase (24).These lines of evidence suggest that α-Pal/NRF-1 may serve to coordinate a variety of cellular processes by regulation the expression of key proteins in metabolic pathways.
α-Pal/NRF-1 belongs to a novel transcription factor family containing putative basic leucine zipper DNA-binding domain in its N-terminal domain and an activation domain in the C-terminal half (22, 24). The DNA-binding domain of α-Pal/NRF-1 shows strong homology with two invertebrate genes, sea urchin P3A2 and Drosophila erect wing gene (ewg)(22, 23), which have been implicated in embryonic or larval development (25, 26). Furthermore, α-Pal/NRF-1 shows 91% identity to its
homologue in zebrafish, not really finished (nrf) (27). The nrf is primarily located in the brain of zebrafish and is associated with the development of the central nervous system (27).
The functional roles of α-Pal/NRF-1 and IAP in neurons are unknown. Therefore, we hypothesize that α-Pal/NRF-1 may regulate important neuronal process such as neuronal differentiation through the regulation of IAP gene.
研究目的
1. To clarify if the NGF/MAPK/Sp1 signaling cascade involve in the regulation of IAP gene.
2. To clarify if the IAP protein involves in the neurite outgrowth of neuroblastoma IMR-32 cells.
研究方法
Cell Culture—Human neuroblastoma IMR-32 (CCRC 60014) and hepatoma HepG2
(CCRC 60048) cell lines were purchased from Culture Collection and Research Center, Food Industry and Development Institute, Hsinchu, Taiwan. N56R5 and
N56R6 were stable cell lines in which α-Pal/NRF-1 was overexpressed. Cells were grown in minimum essential medium Eagle with Earle's salt base (Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) in a humidified atmosphere containing 5% CO2 at 37 °C.
RNA isolation and Reverse Transcription (RT)-PCR—Total RNA was isolated from
the cultured cells using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA). RT-PCR was performed as described previously (3). Briefly, total RNA (2 µg) was reverse transcribed into cDNA in 20 µl of 1× first strand buffer containing 0.5 µg of oligo(dT) as a primer, 500 µM dNTP, and 200 units of SuperScript II (Invitrogen). PCR was performed in 20 µl of 1× PCR buffer containing 2 µl of RT products, 1 unit of
AmpliTaq DNA polymerase (Roche, Branchburg, NJ, USA), 200 µM dNTP, 1.5 mM
MgCl2, 0.5 µM [35S]dATP (Amersham Pharmacia Biotech, Buckinghamshire, UK),
and 0.4 µM primer pair. We used the primer pair that can distinguish the alternative splicing forms of IAP mRNA, Hiap14: 5'-TAA CCT CCT TCG TCA TTG CC and Hiap15: 5'-CGT AAG GGT CTC ATA GGT G. The PCR parameters were 94 °C for 30 s, 53 °C for 30 s, and 72 °C for 30 s for 30 cycles, followed by a final elongation at 72 °C for 7 min. PCR products were analyzed on a 6% polyacrylamide-urea gel (acrylamide/bisacrylamide 19:1, 8 M urea in 1 × Tris-borate-EDTA buffer). The gel was finally dried and analyzed by autoradiography. The image of cDNA bands was scanned by the ScanJet 4C scanner (Hewlett Packard, Palo Alto, CA, USA). The optical densities of cDNA bands were quantified with the 1-D advanced Universal Software (American Applied Biotechnology, Fullerton, CA, USA).
Plasmids— pGL3-Basic and pRL-TK luciferase reporter vectors (Promega, Madison,
WI, USA) were used for IAP promoter reporter assays. A 7.7-Kb human genomic clone (pIAP38) containing exon1 of the IAP gene and 5' upstream region was kindly provided by Dr. F. P. Lindberg. A plasmid, pBSII-445, was generated by inserting the 445-bp SacII fragment from pIAP38 into the pBlueSceipt II (SK-) vector (Stratagene, La Jolla, CA, USA). The SacI/XhoI fragment from pBSII-445 was then inserted into the pCRII vector (Invitrogen) to create pHIAP445 for subsequent constructions.
The reporter construct pGL3-272 was generated by inserting the XmaI/XhoI fragment from pHIAP445 into the pGL3-Basic vector. Different restriction
enzyme-digested fragments from pIAP38 were then ligated into pGL3-272 to create a series of human IAP promoter constructs, including pGL3-1554, pGL3-730, and pGL3-456. Another series of human IAP promoter constructs containing shorter fragments than the insert in pGL3-272—including pGL3-232, pGL3-218, pGL3-209, pGL3-198, pGL3-191, pGL3-159, and pGL3-92—were similarly made by using differential forward primers on IAP promoter with the KpnI site at the 5' end and a common reverse primer (GLprimer2) on pGL3-Basic in the backbone of pGL3-272
for PCR to obtain the shorter fragments, which included differentially truncated IAP promoter regions and a common vector sequence containing the HindIII site. These fragments were then digested by KpnI and HindIII and inserted into pGL3-Basic.
Promoter constructs containing nucleotide substitutions in the sequence motifs of Sp1 and α-Pal/NRF-1 were individually generated by PCR amplification with primer pairs spanning the mutant nucleotides according to the protocol of site-directed mutagenesis by overlap extension (28). The plasmids pGL3-232m1, pGL3-232m2, and pGL3-232m3 were constructed in the backbone of pGL3-232 using primer pairs containing the introduced mutations. The Sp1 site GGGGCGGGGC was mutated into GTTGCTTGGC in the plasmid of pGL3-232m1. The α-Pal/NRF-1 element
TGCGCGTGCGCG was mutated into TTTGCGTGCGCG, and TGCGCGTTTGCG in the plasmid of pGL3-232m2 and pGL3-232m3, respectively.
Transfection and dual-luciferase assay—IMR-32 and HepG2 cells (1.5 × 105) were
plated in each well of six-well plates. Transient transfection was carried out by the calcium phosphate precipitation method (29). The plasmid pRL-TK was cotransfected to normalize the transfection efficiency. After 12 h of transfection, the medium was changed and the cells were incubated at 37 °C for 24 or 48 h. The cells were washed in phosphate-buffered saline (PBS, 137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM dibasic sodium phosphate, and 2 mM monobasic potassium phosphate) and the lysates were prepared by scraping the cells from plates in the presence of 1× passive lysis buffer (Promega). Luciferase assays were performed by using Dual-Luciferase Assay System (Promega) and a Sirius luminometer (Berthold Detection System, Pforzheim, Germany).
Preparation of nuclear extracts—IMR-32 and HepG2 cells were plated onto 6- or
10-cm cultured dishes and incubated for two days. The cells were washed with 2 ml of PBS and collected in 1 ml of PBS. The cells were centrifuged at 2,000 × g for 2 min and the supernatant was discarded. The cell pellet was incubated in 400 µl of buffer A (10 mM HEPES (pH 7.9), 1.5 mM magnesium chloride, 10 mM potassium chloride, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol, 2 µg/ml leupeptin, 10 µg/ml aprotinin, 50 mM sodium fluoride, and 1 mM sodium
orthovanadate) on ice for 10 min and then gently shaken for 10 s. The pellet of the crude nuclei was collected by centrifugation at 12,000 × g for 10 s. The pellet was resuspended in 100 µl of buffer C (20 mM HEPES (pH 7.9), 25% glycerol, 420 mM sodium chloride, 1.5 mM magnesium chloride, 0.2 mM EDTA, 0.5 mM
phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol, 2 µg/ml leupeptin, 10 µg/ml aprotinin, 50 mM sodium fluoride, and 1 mM sodium orthovanadate) by vortex for 15 s, and then incubated on ice for 20 min. After centrifugation at 12,000 × g for 2 min, the supernatant containing the nuclear proteins was collected, quantified with BCA
Protein Assay Reagent (Pierce, Rockford, IL, USA), and stored at −70 °C in aliquots.
Gel electrophoretic mobility shift assays (EMSA)—The EMSA used the following
oligonucleotides: IAP α-Pal/NRF-1 (f), 5'-GAG TGC GCG TGC GCG GCT CT-3'; IAP α-Pal/NRF-1 (r), 3'-TCA CGC GCA CGC GCC GAG AG-5'; IAP α-Pal/NRF-1 mutation (f), 5'-GAG Ttt GCG Ttt GCG GCT CT-3'; IAP α-Pal/NRF-1 mutation (r), 3'-TCA aaC GCA aaC GCC GAG AG-5'; consensus α-Pal/NRF-1 (f), 5'-TTC TTT TGC GCA CGC GCT T-3; consensus α-Pal/NRF-1 (r), 3'-AAG AAA ACG CGT GCG CGA AGA ATC-5'; consensus α-Pal/NRF-1 mutation (f), 5'-TTC TTT TGt aaA CGa atT T-3'; consensus α-Pal/NRF-1 mutation (r), 3'-AAG AAA ACA ttT GCt tAA AGA ATC-5; consensus Sp1 (f), 5'-GTT GCG GGG CGG GGC CGA GTG-3';
consensus Sp1 (r), 3'-AAC GCC CCG CCC CGG CTC ACG-5'; consensus E2F-1 (f), 5'-TGC AAT TTC GCG CCA AAC TTG-3'; and consensus E2F-1 (r), 3'-GTT AAA GCG CGG TTT GAA C-5'. Thirty pmole of each of the forward and reverse
oligonucleotides placed in a volume of 23 µl of 1 × Klenow (DNA polymerase) buffer were heated at 94 °C for 2 min and annealed at room temperature for 30 min. The annealed double-stranded oligonucleotides were end-labeled by a fill-in reaction using DNA polymerase (Klenow) (Promega). One unit of the DNA polymerase (Klenow) and 40 µCi of [α-32P]dCTP (Perkin-Elmer Life Science, Boston, MA, USA) were
added into the annealed oligonucleotides and the mixture was incubated at 30 °C for 15 min. The labeled oligonucleotides were purified by Sephadex G-50 columns (Amersham Pharmacia Biotech). Cold double-stranded oligonucleotides were used as competitors. The DNA-binding reaction was conducted at 4 °C for 30 min in a
mixture containing 3 µg of nuclear extract, 10 mM Tris-Cl (pH 7.5), 50 mM sodium chloride, 0.5 mM dithiothreitol, 0.5 mM EDTA, 1 mM magnesium chloride, 4% glycerol, 0.05 µg poly(dI-dC).poly(dI-dC) (Amersham Pharmacia Biotech), and 2 ×
104 cpm of 32P-labeled double-stranded oligonucleotides. In supershift assays, antibodies were incubated with the reaction mixture at 4 °C for 30 min before the addition of the IAP α-Pal/NRF-1 probes. The anti-NRF-1 goat polyclonal antiserum was kindly provided by Dr. Richard C. Scarpulla. The Sp1 and E2F antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-myc antibody was from Invitrogen. The normal goat serum was from Vector Laboratories (Burlingame, CA, USA). Samples were analyzed on a 4% polyacrylamide gel
(acrylamide/bisacrylamide 29:1 in 0.5 × Tris-borate-EDTA buffer) at 10 V/cm for 2.5 h. The gel was dried and analyzed by autoradiography.
Construction and overexpression of α-Pal/NRF-1— The cDNAs encoding the
full-length and dominant-negative mutant of α-Pal/NRF-1 were constructed. The dominant-negative mutant consisted of the first N-terminal 304 residues of
domains and lacking the activation domain (30, 31). Four α-Pal/NRF-1 cDNA fragments were obtained by RT-PCR. The primer pair ΑΝ-5 (5'-TTAAGCTT GCG CAG CCG CTC TGA GGA A) and ΑΝ-7 (5'-GACTCGAG CAC TGT TCC AAT GTC ACC AC) and primer pair ΑΝ-5 and ΑΝ−11 (5'-GACTCGAG TCA CTG TGA TGG TAC AAG ATG AGC) were used for the untagged full-length and
dominant-negative α-Pal/NRF-1, respectively. Primer pair ΑΝ-5 and ΑΝ-6
(5'-GACTCGAG TCA CTG TTC CAA TGT CAC CA) and primer pair ΑΝ-5 and ΑΝ-10 (5'-GACTCGAG GTC TGT GAT GGT ACA AGA TGA G) were used for the myc-tagged full-length and dominant-negative α-Pal/NRF-1, respectively. The
underlines indicate the restriction enzyme sites. These fragments were digested by HindIII and XhoI and inserted into pcDNA3.1 (B+) vector (Invitrogen). HepG2 cells (8 × 105) were placed onto 6-cm dishes for overexpression of α-Pal/NRF-1 using the
same procedure for transient transfection of the reporter plasmids. After transient transfection, the cells were incubated for 48 h and harvested for nuclear protein extraction.
Primary cortical culture—Primary cortical cells were prepared according to the
protocol from Dichter (32). In brief, pregnant ICR mice, 15 days post conception, were anesthetized with pentobarbital and embryos were removed. The cortices were removed and collected in MEM. The tissue was triturated three times with a
fire-polished Pasteur pipette. Dissociated cells were plated onto 6-well plates coated with 0.09 mg/ml poly-L-lysine and grown in MEM supplemented with 10% fetal bovine serum (MEM 10), 50 U/ml penicillin and streptomycin. After 24 h, plating media were replaced with MEM 10 and cells were treated with 3 µM of cytosine arabinoside. Twenty-four hours later, the medium was replaced with MEM 10 again and cells were cultured for another 48 h for the transfection experiments.
Isolation of stable cell lines and analysis of α-Pal/NRF-1 expression
The α-Pal/NRF-1 cDNA was inserted in the sense orientation under the control of the CMV promoter of the mammalian expression vector pcDNA3.1 containing the neomycin resistance gene (Chang and Huang, 2004). Following transfection into IMR-32 cells, stable transfectants were selected by supplementation of the culture medium with G418 to a final concentration of 1 mg/ml, and individual clones were isolated after 20 to 35 days of selection. RNA was isolated from cells by the TRIzol reagent (Invitrogen) and used as template for cDNA synthesis. Resultant cDNA was amplified by PCR using different pairs of primers. To confirm the expression of exogenous construct, PCR was performed using a forward primer located in the coding sequence of α-Pal/NRF-1 (AN-1, 5'-GAG TCC AAG ATG CTA ATG) and a reverse primer internal to the vector sequence (BGH, 5'-TAG AAG GCA CAG TCG AGG) so as not to amplify endogenous α-Pal/NRF-1 mRNA. The endogenous
α-Pal/NRF-1 mRNA was amplified by the primer pairs of AN-1 and AN-2 (5'-GAA AAT CAA GAG TGG TGC). Total α-Pal/NRF-1 mRNA was amplified by the primer pairs of AN-1 and ΑΝ-7 (5'-GACTCGAG TCA CTG TTC CAA TGT CAC CA).
Plasmid constructs
The pCMS-EGFP (Clontech) was used as the vector. The full-length and truncated α-Pal/NRF-1 cDNA fragments were amplified by primer pair ΑΝ-12 (5'-CTAGCTAGC GCG CAG CCG CTC TGA GGA A) and ΑΝ-7 and primer pair ΑΝ-12 and ΑΝ−11 (5'-GACTCGAG TCA CTG TGA TGG TAC AAG ATG AGC), respectively, by using the pcDNA3.1-NRF-1 (Chang and Huang, 2004) as the template. cDNA fragments were cut with NheI and XhoI and ligated into the
linearized pCMS-EGFP. The correct constructs were confirmed by DNA sequencing and named as pCMS-α-Pal/NRF-1-FL and pCMS-α-Pal/NRF-1-DN. The IAP/CD47 cDNA was amplified by primer pair hIAPF2 (5’-TTTGCGGCCGC AGA CAC CTG CGG CGG CGG CGG) and hIAPR1 (5’-GCTCTAGA TAC TTT TCT TGT TTC TTC TCC CCA). The cDNA fragments were digested by NotI and XbaI and inserted in a reverse orientation into the pCMS-EGFP vector. These constructs contains the α-Pal/NRF-1 or IAP/CD47 cDNAs under the control of the CMV promoter and the GFP cDNA under the control of the SV40 promoter.
Transient transfection
The plasmids pCMS-α-Pal/NRF-1-FL and pCMS-α-Pal/NRF-1-DN were transfected into various cell lines by the calcium phosphate precipitation method (Jordan et al., 1996). Cells (1.5 × 105) were cultured on 6-well plates for 24 h. The
medium was exchanged with 2 ml of fresh medium and the cells were incubated for additional 1 h for the subsequent transfection. Plasmid DNA (1 µg) was mixed with 5 µl of 2.5 M calcium chloride in a volume of 50 µl containing 0.1× TE buffer. The mixture was then mixed with one volume of 2 × HEPES buffer and incubated for 1 min at room temperature. The mixture was added to the cells for transfection. After 12-h transfection, the medium was exchanged with fresh medium and the cells were grown for 24, 48 or 72-h for neurite measurement.
Measurement of neurite outgrowth
To determine the neurite process length, GFP-positive cells were observed under the fluorescent microscope (Leica). The length of the longest process for each of >100 cells was determined for each sample by the software of Metamorph. The process longer than 20 µm in PC12 and 25 µm in IMR-32, and Neuro-2A cells, respectively, is defined as a neurite.
Promoter assay
The IAP promoter activity was assayed as described previously (Chang and Huang, 2004). IMR-32 cells were sub-cultured for 24 h and co-transfected with 50 ng
of the IAP promoter construct pGL3-232, 10 ng of the pRL-TK, and 2 µg of the pNRF-FL construct or the control vector pcDNA3.1. After 12 h of transfection, the medium was changed with fresh medium containing U0126 or vehicle. The cells were incubated for 48 h and the promoter activity was determined by the Dual-Luciferase Assay System (Promega).
Statistics—Statistical analysis was performed by unpaired t test for pairwise
comparisons. A P value <0.05 was regarded as significant. 結果與討論
Expression of integrin-associated protein gene in human IMR-32 and HepG2 cells
To use IMR-32 and HepG2 cells to study the IAP gene promoter, we examined firstly the expression of IAP transcripts in these cells by RT-PCR. We used primers that can detect alternative splicing forms of IAP mRNAs. As shown in Fig. 1A, both form 1 and form 2 IAP mRNAs were expressed in these two cell lines at a similar level. The major form of IAP mRNA in IMR-32 was form 4. But form 4 is not expressed in HepG2 cells at all. Total IAP transcripts were quantified. The level of total IAP transcripts expressed in IMR-32 cells was approximately three-fold of that expressed in HepG2 cells (Fig. 1B). These results confirmed the expression of the human IAP gene in these two cell lines.
Determination of IAP promoter activity in IMR-32 and HepG2 cells
To define the boundaries of a minimal IAP promoter region and identify cis elements that regulate the expression of IAP, we generated a series of 5'- IAP
promoter deletion constructs and transfected them into IMR-32 and HepG2 cells. All plasmid constructs were defined relative to the translation initiation codon (Fig. 2A). The reporter constructs were cotransfected into IMR-32 and HepG2 cells with an internal control Renilla luciferase vector. The firefly luciferase activity of each reporter was normalized with the internal control to correct transfection efficiency. Results were represented as a fold-increase in activity with respect to that of the pGL3-Basic vector (Fig. 2B and 2C) or the relative activity compared with that of the -232 construct (Fig. 4).
In IMR-32 cells, the shortest reporter that still retained the basal promoter activity was the -232 construct, whereas deletion for another 41 bp (the -191 construct), 73 bp (the -159 construct) or 140 bp (the -92 construct) resulted in markedly loss of reporter activity. Addition of 40 bp to the -232 construct generated the -272 construct and stimulated the reporter activity by approximately 25%. In the construct containing additional 184 bp (the -456 construct), the promoter activity was not increased. Interestingly, the reporter activity decreased to about the basal proximal
promoter activity in the construct containing additional 274 bp (the -730 construct) and in the longest construct (the -1554 construct) (Fig. 2B). A similar pattern of promoter activity was observed when the reporter constructs were transfected into HepG2 cells, but with three exceptions (Fig. 2C). Firstly, overall promoter strength relative to pGL3-Basic was much lower in HepG2 cells, approximately 1/6 to 1/4 compared with IMR-32 cells. Secondly, the constructs of -272 and -456 produced maximal activity in IMR-32 cells; in HepG2 cells, however, the construct of –730 produced maximal activity. Thirdly, there were negative regulators located between −457 and −730 in IMR-32 cells; in HepG2 cells, however, there were negative
regulators located between −730 and −1554. Results suggested a core promoter of the human IAP gene located between −232 and −12 upstream of the translation initiation codon in both IMR-32 and HepG2 cells.
The sequence from –272 to ATG was searched for homology to previously described regulatory elements in several databases. There are many putative binding sites for transcription factor in this region (Fig. 3), including activating enhancer binding protein 2 (AP-2), myc-associated zinc finger protein (Maz), cyclic-AMP responsive element binding protein (CREB), transcription factor Sp1, E2 promoter binding factor (E2F), and α-Pal/nuclear respiratory factor 1 (α-Pal/NRF-1).
Identification of cis-elements in the core promoter of the IAP gene
To determine more precisely the core promoter of the IAP gene, we generated several shorter or point-mutation constructs. When 14 and 23 bp were deleted from the 5' end of the -232 construct to generate the constructs of -218 and -209
respectively, the promoter activity of these two constructs was identical to that of the -232 construct in IMR-32 cells. This indicated that the sequence between −232 and −209 played no significant role in IAP gene expression under the then current experimental conditions. When 34 bp were deleted from the 5' end of the -232
construct to generate the -198 construct, however, the promoter activity was markedly reduced by 90% in IMR-32 cells, indicating that the sequence between −209 and −198 might be required for the IAP promoter activity (Fig. 4). As shown in Fig. 3, the region from −232 to −198 consists of a GC-rich sequence that includes the putative Sp1 and α-Pal/NRF-1 sites. Point mutations were introduced into these sites in IMR-32 cells to determine whether these sites were necessary for IAP promoter activity. When four bases of the Sp1 site were substituted with four T residues to generate the -232m1 construct, no significant effect on the IAP promoter activity was observed (Fig. 4), indicating that Sp1 site in this region was not required for the IAP promoter activity under this condition. The putative α-Pal/NRF-1 site in the IAP promoter is a 12-base tandem-repeat sequence, TGCGCGTGCGCG. When two bases in each of the repeat sequence were replaced by two T residues to generate the
-232m2 and -232m3 constructs respectively, an 80% and a 90% drop in promoter activity was observed (Fig. 4). These results suggested that the consensus
α-Pal/NRF-1 sequence, but not the consensus Sp1 sequence, is a functional regulatory element in the IAP promoter in IMR-32 cells.
α-Pal/NRF-1 is a transcription factor regulating the IAP promoter activity To demonstrate that the consensus α-Pal/NRF-1 sequence was functional, i.e., that there were endogenous nuclear proteins binding to this region, we performed the EMSA experiment. Nuclear extracts from IMR-32 cells were combined with
32P-fill-in-labeled double-stranded oligonucleotides in vitro. A major band of
DNA-protein complex was found in all lanes when the nuclear extracts were incubated with the wild type IAP α-Pal/NRF-1 probes (Fig. 5A, lane 3 and lanes 5-12), but not with the mutant IAP α-Pal/NRF-1 probes (Fig. 5A, lane 4). No band was found when nuclear extracts were not added into the probes (Fig. 5A, lanes 1 and 2). Competition analysis using a 10- or 60-fold molar excess of unlabeled probes was used to characterize the factor which specifically binds to this sequence. As expected, the addition of a 10- or 60-fold molar excess of published wild type consensus
α-Pal/NRF-1 element reduced the intensity of these complexes (Fig. 5A, lanes 5 and 6), whereas the addition of the mutant consensus α-Pal/NRF-1 sequence did not (Fig. 5A, lanes 7 and 8). Results suggested that α-Pal/NRF-1 proteins might bind to the IAP α-Pal/NRF-1 element. However, the α-Pal/NRF-1 site is GC-rich and might therefore interact with factors other than α-Pal/NRF-1, such as Sp1 and E2F. We therefore used the unlabeled consensus Sp1 and E2F sequence for the competition experiment. The intensity of the migrating bands was not significantly reduced (Fig. 5A, lanes 9-12). Supershift assays using the anti-α-Pal/NRF-1 antiserum were used to further confirm the binding of the α-Pal/NRF-1 on its DNA element. The migrating bands were weakened when increasing amounts of α-Pal/NRF-1 antiserum were added and supershifted bands appeared (Fig. 5B, lanes 3-5). However, the Sp1 or E2F antibody did not generate any supershifted band (Fig. 5B, lanes 6 & 7), neither did the normal goat serum (Fig. 5B, lane 8). The EMSA experiments also revealed that the DNA-binding activity of α-Pal/NRF-1 in IMR-32 cells (Fig. 5C, lanes 1-3) was much higher than that in HepG2 cells (Fig. 5C, lanes 4-6). The oligonucleotide probes and competitors used in the EMSA experiments were shown in Fig. 5D. These results strongly suggested that α-Pal/NRF-1 but not Sp1 or E2F binds to the IAP
α-Pal/NRF-1 site.
To further confirm that α-Pal/NRF-1 is the major transcription factor that binds to the IAP α-Pal/NRF-1 element, plasmids encoding the full-length or
dominant-negative mutant of α-Pal/NRF-1 with or without a myc tag were transiently transfected into HepG2 cells. The addition of a myc tag in the C-terminal of
α-Pal/NRF-1 is useful for the supershift assay in the EMSA experiments. In HepG2 cells, overexpression of the full-length α-Pal/NRF-1 (Fig. 6A, lanes 4 to 6) and myc-tagged α-Pal/NRF-1 (Fig. 6B. lanes 3 to 5) enhanced the binding of DNA-protein complex in a dose-dependent manner as compared with the mock controls (Fig. 6A, lane 3 and Fig. 6B, lane 2). Overexpression of the
dominant-negative mutant of α-Pal/NRF-1, which contains only the N-terminal DNA binding domain, did not affect endogenous DNA-protein binding in HepG2 cells but generated an additional band of DNA-protein complex with a smaller molecular weight. The DNA binding activity was strongly enhanced by the overexpression of the dominant-negative mutant (Figs. 6A and 6B, lanes 7 to 9). This discrepancy may be related to the higher transfection efficiency of the plasmid that contained the dominant-negative mutant. In supershift assays, the band of DNA-protein complex containing the α-Pal/NRF-1-myc fusion protein was supershifted when monoclonal anti-myc antibody was used (Fig. 6B, lane 6). The band of DNA-protein complex containing the dominant-negative mutant of α-Pal/NRF-1 fused with the myc protein fragment was also completely supershifted by the monoclonal anti-myc antibody (Fig. 6B, lane 10). These data strongly suggested that α-Pal/NRF-1 binds to this region (–204 to –193) of the human IAP gene promoter in vitro.
If the α-Pal/NRF-1 protein binds to the α-Pal/NRF-1 element in the IAP
promoter in vitro, it should functionally regulate the promoter activity of the IAP gene
in vivo. To test this possibility, the full-length or dominant-negative α-Pal/NRF-1
constructs were cotransfected with the reporter construct -232 into IMR-32 and HepG2 cells, and the luciferase activity was measured. As shown in Fig. 7A, overexpression of dominant-negative α-Pal/NRF-1 significantly reduced the IAP promoter activity in a dose-dependent manner in both cell lines, as compared with the mock controls. At the highest dose (50 ng) the promoter activity of the reporter construct was reduced markedly up to 50% and 92% in IMR-32 and HepG2 cells respectively. In contrast, overexpression of full-length α-Pal/NRF-1 significantly increased the IAP promoter activity in a dose-dependent manner (Fig. 7B). At highest dose (2 µg) the promoter activity of the reporter construct increased up to 3.9- and 5.3-fold at the highest dose (2 µg) in IMR-32 and HepG2 cells respectively. In Fig. 7B, the relative activity was represented as the percentage of firefly luciferase activity of mock control but not normalized with the Renilla luciferase activity, because the full-length α-Pal/NRF-1 could enhance the activity of the Renilla luciferase by
unknown effects. These results confirm that α-Pal/NRF-1 interacts with IAP promoter and regulates its downstream gene expression in vivo.
Regulation of the IAP promoter activity by α-Pal/NRF-1 in primary cells
conditions, we transfected the truncated or mutant human IAP promoter constructs into the mouse primary cortical cells. Results are shown in Fig. 8. Similar to the results observed in IMR-32 and HepG2 cells (Fig. 2), the -232 construct retained the basal promoter activity, but the activity of shorter constructs, -198 and -195 constructs, was reduced 76 and 62%, respectively. The promoter activity of point-mutation
constructs, -232m2 and m3, was significantly decreased about 40%, but not the -232m1 construct. Overexpression of dominant-negative α-Pal/NRF-1 in the primary cortical cells decreased the IAP gene promoter activity in a dose-dependent manner (Fig. 8B). Overexpression the wild type α-Pal/NRF-1 in the primary cortical cells significantly enhanced the IAP gene promoter activity (Fig. 8C). These results indicate that the function of α-Pal/NRF-1 in primary cells is similar to that in cell lines.
Stable transfection of α-Pal/NRF-1 enhances neurite outgrowth
To investigate the function of α-Pal/NRF-1 in neurons, we overexpressed full-length α-Pal/NRF-1 in neuroblastoma IMR-32 cells. Stable cell lines expressed the exogenous α-Pal/NRF-1 were screened by resistance to the antibiotics neomycin. When cultured in the medium containing 10% serum for three days, extended neurites were found in cell lines N56R5 and N56R6. The parental IMR-32 cells showed no or short neurites in such culture condition. The IMR-32 cells grew long neurites when they were cultured in medium containing 0% serum. In such serum-free medium, the growth of neurites in N56R5 and N56R6 cells was potentiated (Fig. 9A). Exogenous expression of α-Pal/NRF-1 was characterized by RT-PCR analyses. The exogenous α-Pal/NRF-1 mRNA was significantly expressed in four selected stable cell lines. The levels of endogenous of α-Pal/NRF-1 mRNA in all cell lines and wild type cells were similar. However, the total mRNA levels in the four cell lines were about two to three folds of that in the parental IMR-32 cells (Fig. 9B). These results suggested that α-Pal/NRF-1 functions to enhance neurite outgrowth in neuroblastoma cells.
Dominant negative α-Pal/NRF-1 inhibits neurite outgrowth
To investigate the importance of the endogenous α-Pal/NRF-1 on neurite outgrowth, the dominant-negative mutant of α-Pal/NRF-1 was transfected into IMR-32 and N56R5 cells. The transfectants were cultured in the serum-free medium for two days and observed under fluorescence microscope. Ectopic expression of dominant-negative α-Pal/NRF-1 inhibited the neuronal-like morphology in both IMR-32 and N56R5 cells (Fig. 10A). The curve of cumulative percentage of cells bearing variance length of neurites in α-Pal/NRF-1 tranfectants shifted to left, suggesting that dominant negative α-Pal/NRF-1 decreased the average length of neurites (Fig. 10B). Ectopic expression of dominant negative α-Pal/NRF-1 decreased the number of neurite-bearing cells from 40% to 28% of total transfected IMR-32
cells. Dominant negative α-Pal/NRF-1 decreased the number of neurite-bearing cells from 60% to 30% of total transfected N56R5 cells (Fig. 10C). These results revealed that endogenous α-Pal/NRF-1 is required for the induction of neurites in IMR-32 cells.
MAPK involves in the function of α-Pal/NRF-1
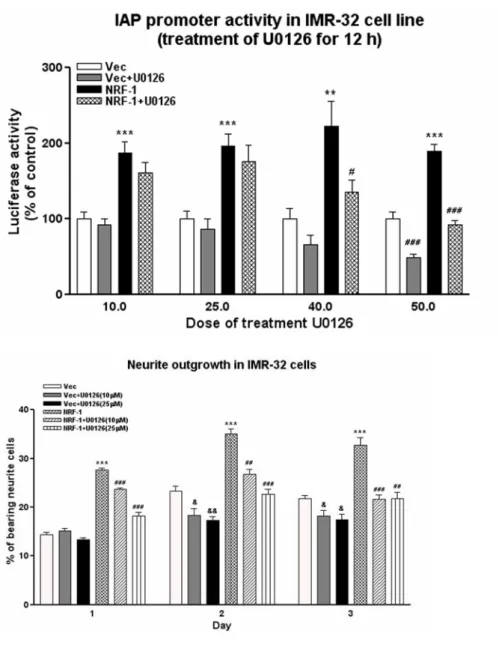
MAPK is known to be involved in the outgrowth of neurites. We tested the possibility that MAPK is involved in the function of α-Pal/NRF-1, the positive regulator of the promoter activity of human integrin-associated protein/CD47
(IAP/CD47) gene (Chang and Huang, 2004). We found that U0126, inhibitor of MEK, dose-dependently inhibited the IAP promoter activity in IMR-32 cells cotransfected with the empty vector or the vector containing the α-Pal/NRF-1 (Fig. 11A). U0126 inhibited neurite outgrowth in IMR-32 cells both in the transfection of the empty vector and the α-Pal/NRF-1 construct (Fig. 11B).
Anti-sense IAP cDNAs reduces neurite outgrowth
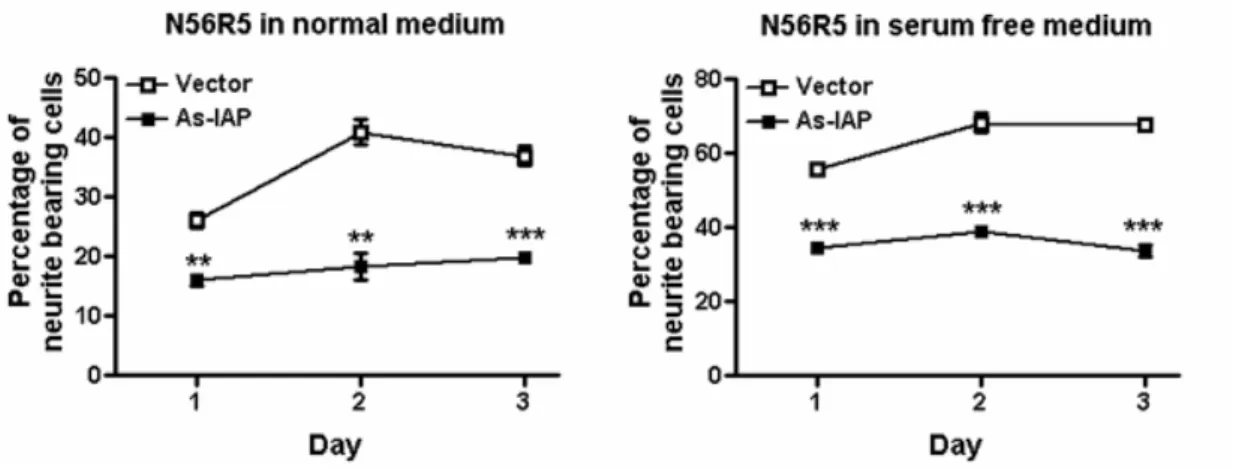
Since α-Pal/NRF-1 is a critical regulator of IAP/CD47 gene (Chang and Huang, 2004), we asked if IAP/CD47 is a downstream gene of α-Pal/NRF-1 in the regulation of neurite outgrowth. We used the strategy of antisense cDNA to knockdown the IAP level in N56R5 cells. Antisense IAP cDNA significantly reduced the percentage of cells bearing neurites in N56R5 cells when cultured in 10% or 0% serum for 1, 2 and 3 days (Fig. 12A). Similar results were obtained when cells were cultured in medium containing without serum (Fig. 12B)
參考文獻
1. Brown, E., Hopper, L., Ho, T., and Gresham, H. (1990) J. Cell Biol. 111, 2785-2794
2. Lindberg, F. P., Gresham, H. D., Schwarz, E., and Brown, E. J. (1993) J. Cell Biol.
123, 485-496
3. Huang, A. M., Wang, H. L., Tang, Y. P., and Lee, E. H. Y. (1998) J. Neurosci. 18, 4305-4313
4. Chang, H. P., Ma, Y. L., Wan, F. J., Tsai, L. Y., Lindberg, F. P., and Lee, E. H. Y. (2001) Neuroscience 102, 289-296
5. Chang, H. P., Lindberg, F. P., Wang, H. L., Huang, A. M., and Lee, E. H. Y. (1999)
Learn. Mem. 6, 448-457
6. Gresham, H. D., Goodwin, J. L., Allen, P. M., Anderson, D. C., and Brown, E. J. (1989) J. Cell Biol. 108, 1935-1943
7. Senior, R. M., Gresham, H. D., Griffin, G. L., Brown, E. J., and Chung, A. E. (1992) J. Clin. Invest. 90, 2251-2257
8. Parkos, C. A., Colgan, S. P., Liang, T. W., Nusrat, A., Bacarra, A. E., Carnes, D. K., and Madara, J. L. (1996) J. Cell Biol. 132, 437-450
9. Gao, A.-G., Lindberg, F. P., Finn, M. B., Blystone, S. D., Brown, E. J., and Frazier, W. A. (1996) J. Biol. Chem. 271, 21-24
10. Wang, X.-Q., and Frazier, W. A. (1998) Mol. Biol. Cell 9, 865-874
11. Chung, J., Gao, A.-G., and Frazier, W. A. (1997) J. Biol. Chem. 272, 14740-14746 12. Ticchioni, M., Deckert, M., Mary, F., Bernard, G., Brown, E. J., and Bernard, A.
(1997) J. Immunol. 158, 677-684
13. Waclavicek, M., Majdic, O., Stulnig, T., Berger, M., Baumruker, T., Knapp, W., and Pickl, W. F. (1997) J. Immunol. 159, 5345-5354
14. Oldenborg, P.-A., Zheleznyak, A., Fang, Y.-F., Lagenaur, C. F., Gresham, H. D., and Lindberg, F. P. (2000) Science 288, 2051-2054
15. Chuang, W., and Lagenaur, C. F. (1990) Dev. Biol. 137, 219-232
16. Jiang, P., and Lagenaur, C. F., and Narayanan, V. (1999) J. Biol. Chem. 274, 559-562
17. Jacob, W. F., Silverman, T. A., Cohen, R. B., and Safer, B. (1989) J. Biol. Chem.
264, 20372-20384
18. Efiok, B. J. S., and Safer, B. (2000) Biochim. Biophy. Acta 1495, 51-68 19. Evans, M. J., and Scarpulla, R. C. (1989) J. Biol. Chem. 264, 14361-14348 20. Kelly, D. P., and Scarpulla, R. C. (2004) Genes Dev. 18, 357-368
21. Gómez-Cuadrado, A., Rousseau, S., Renaud, J. and Ruiz-Carrillo, A. (1992)
22. Virbasius, C. A. Virbasius, J. V., and Scarpulla, R. C. (1993) Genes Dev. 7, 2431-2445
23. Efiok, B. J. S., Chiorini, J. A., and Safer, B. (1994) J. Biol. Chem. 269, 18921-18930
24. Gómez-Cuadrado, A., Martín, M., Noël, M., and Ruiz-Carrillo, A. (1995) Mol.
Cell. Biol. 15, 6670-6685
25. Calzone, F. J., Höög, C., Teplow, D. B., Cutting, A. E., Zeller, R. W., Britten, R. J., and Davidson, E. H. (1991) Development 112, 335-350
26. DeSimone, S. M., and White, K. (1993) Mol. Cell. Biol. 13, 3641-3649
27. Becker, T. S., Burgess, S. M., Amsterdam, A. H., Allende, M. L., and Hopkins, N. (1998) Development 125, 4369-4378
28. Aiyar, A., Xiang, Y., and Leis, J. (1996) Methods Mol. Biol. 57, 177-191 29. Jordan, M., Schallhorn, A., and Wurm, F. M. (1996) Nucleic Acids Res. 24,
596-601
30. Dichter, M. A. (1978) Brain Research 149, 279-293
附圖與說明
Fig.1 to Fig. 8 請見附錄
B
A
Figure 9. Enhancement of neurite outgrowth in stable cell lines overexpressed
α-Pal/NRF-1. A, morphology of stable cell lines overexpressed α-Pal/NRF-1. Cells were cultured in medium containing 10% or 0% serum for two to three days. B, detection of exogenous α-Pal/NRF-1 transcripts in stable cell lines by RT-PCR. The cDNAs that
represents the exogenous, endogenous and total α-Pal/NRF-1 transcripts were amplified by different pairs of primer. The cDNA fragments were analyzed by 1.5% agarose gel and stained with ethedium bromide.
Figure 10. The dominant-negative α-Pal/NRF-1 inhibits neurite outgrowth in
IMR-32 and N56R5 cells. A, morphology of truncated α-Pal/NRF-1 cDNA
transfected IMR-32 and N56R-5 cells. IMR-32 and N56R5 cells were transfected for 12 hr with GFP-empty vector (1 µg) or vector containing truncated
α-Pal/NRF-1 cDNA (1 µg). Cells were cultured for 48 hr in the medium
containing 0% serum and observed under microscope. B, cumulative percentage of cells bearing various length of neurite. The curve of dominant-negative
α-Pal/NRF-1-overexpressed IMR-32 cells from one experiment. C, quantification of cells carrying extended neurites. Cells carrying extended neurites (> 25 µm) among > 100 GFP-positive cells were counted. Each value represents the mean ± S.E. (n = 4). *, p<0.05; **, p<0.01; unpaired t test.
Figure 11. MAPK involves in the function of α-Pal/NRF-1. A, The MEK
inhibitor U0126 inhibits the enhancement of the IAP promoter activity of α-Pal/NRF-1 in IMR-32 cells. IMR-32 cells were cotransfected with the IAP promoter construct pGL3-232 (50 ng), pRL-TK (10 ng) and pNRF-FL (0.5 µg) or pcDNA3.1 vector (0.5 µg). After 48 hr, the cells were treated with 10 to 50 µM of U0126 for 12 hr. The cell lysates were harvest and the luciferase and renilla activity were determined. B, U0126 abolishes the enhancement of neurite
outgrowth of α-Pal/NRF-1. IMR-32 cells were transfected with pCMS-GFP vector (1µg) or pNRF-GFP (1 µg) and grown in 10% serum containing vehicle or U0126 (10 or 25 µM) for 1 to 3 days. Quantification of neurite outgrowth was determined as described above.
Figure 12. IAP mediates the function of α-Pal/NRF-1. Antisense IAP cDNA
inhibits neurite outgrowth in N56R-5 cells. N56R5 cells were transfected with µg IAP antisense cDNA or µg empty vector and grown in medium containing 10% serum (A) or without serum (B) for 1 to 3 days. Cells carrying extended neurites (> 25 µm) among > 100 GFP-positive cells were counted. Each value represents the mean ± S.E. **, p<0.01; ***, p<0.001; unpaired t test.
計劃成果自評
1. 本研究為兩年期的計劃,第一年計劃探討 NGF/MAPK/Sp1 訊息路徑是否參與 IAP 基因表現的調控。第二年計劃探討 IAP 基因是否參與神經軸突生長。第
一年研究結果證實Sp1 並不是調控 IAP 基因的最重要轉錄因子。雖然 Sp1 並
不參與IAP 基因的調控,但是透過一系列實驗我們證實在 IAP 基因的 core
promoter 中 NRF-1 是一個重要的轉錄因子。這部分的結果已發表於 The Journal of Biological Chemistry 279 (15): 14542-14550 (2004),成果極佳。 2. 第二年計畫之研究結果發現,IAP 蛋白及 NRF-1 確實參與神經軸突生長。於
IMR-32 細胞大量表現 NRF-1 時,明顯促進神經軸突生長。若於此細胞內表
現NRF-1 突變蛋白或 IAP 反義 cDNA 序列,則明顯抑制神經軸突生長,另
外發現MAPK 訊息路徑參與 NRF-1/IAP 的活化。此成果吻合計畫預期成果,
附錄
第一年計畫研究成果已發表於The Journal of Biological Chemistry 279 (15): 14542-14550 (2004)。PDF 如附。
␣-Pal/NRF-1 Regulates the Promoter of the Human
Integrin-associated Protein/CD47 Gene*
Received for publication, September 4, 2003, and in revised form, January 20, 2004 Published, JBC Papers in Press, January 27, 2004, DOI 10.1074/jbc.M309825200
Wen-Teng Chang‡ and A-Min Huang‡§¶
From the ‡Institute of Basic Medical Sciences and the §Department of Physiology, National Cheng Kung University Medical College, Tainan 701, Taiwan
Integrin-associated protein (IAP or CD47) is ex-pressed in a variety of tissues, including the nervous system and immune system. To understand how cells control the expression of the IAP gene, we cloned the 5ⴕ-proximal region of the human IAP gene and investi-gated IAP promoter activity by transient transfection. RT-PCR confirmed the expression of IAP transcripts in human neuroblastoma IMR-32 and hepatoma HepG2 cells. Deletion analysis identified a core promoter of the human IAP gene located between nucleotide positions
ⴚ232 and ⴚ12 relative to the translation initiation codon
in these two cell lines. Site-directed mutagenesis and gel electrophoretic mobility shift assay identified a ␣-Pal/ NRF-1 binding element within the IAP core promoter. Supershift assays using the␣-Pal/NRF-1 antiserum con-firmed the binding of this transcription factor on the
␣-Pal/NRF-1 site. Overexpression of the DNA binding
domain of ␣-Pal/NRF-1 in cells enhanced DNA-␣-Pal/ NRF-1 binding in vitro. Furthermore, overexpression of full-length␣-Pal/NRF-1 significantly enhanced IAP pro-moter activity while overexpression of dominant-nega-tive mutant reduced promoter activity both in the cul-tured human cell lines and primary mouse cortical cells. These results revealed that␣-Pal/NRF-1 is an essential transcription factor in the regulation of human IAP gene expression.
Integrin-associated protein (IAP),1also designated as CD47,
is a multifunctional membrane protein that is expressed widely in the nervous system, immune system and many other tissues (1, 2). In the adult rat central nervous system, IAP was related to memory formation of an aversive learning task (3). In good memory rats trained in the inhibitory avoidance learning par-adigm, the mRNA level of IAP in the hippocampus was in-creased. Injection of antisense oligonucleotides or the mono-clonal antibody of IAP into the rat hippocampus impaired memory formation (3, 4). IAP-deficient mice showed deficits in memory retention in a similar behavioral paradigm (5).
In the peripheral tissues, IAP was first discovered as a cell
surface protein associated with integrin ␣v3 and it was
in-volved in the enhancement of neutrophil adhesion, chemotaxis, and phagocytosis triggered by an extracellular matrix (1, 6, 7). IAP is also a functional component of several processes, in-cluding the transepithelial migration of neutrophils (8), che-motaxis of endothelial cells and smooth muscle cells (9, 10), spreading and aggregation of platelets (11), and modulation of T-cell activation (12, 13). Moreover, IAP functions as a self-check marker on red blood cells to prevent their clearance by macrophages (14).
IAP mRNA and protein sequences are conserved among hu-mans, mice, and rats (3, 15), suggesting that it is evolutionally important to biological functions. IAP mRNA has five alterna-tive splicing forms. These forms are also conserved in evolution (3, 15–17). In humans and mice, different forms of IAP mRNA were expressed at varying levels in different tissues; macro-phage and endothelial cells expressed predominantly form 2 mRNA and brain tissues expressed predominantly form 4. Why different tissues express different levels and forms of IAP mRNA and how the IAP gene is regulated are, however, un-known. To answer these questions, we investigated the regu-lation of IAP gene promoter in human neuroblastoma and hepatoma cell lines by using luciferase reporter and gel elec-trophoretic mobility shift assays. We found that␣-Pal/NRF-1 in the core promoter region is a positive regulator of the human IAP gene.
EXPERIMENTAL PROCEDURES
Cell Culture—Human neuroblastoma IMR-32 (CCRC 60014) and
hepatoma HepG2 (CCRC 60048) cell lines were purchased from Culture Collection and Research Center, Food Industry and Development Insti-tute, Hsinchu, Taiwan. Cells were grown in minimum essential me-dium Eagle with Earle’s salt base (Sigma) supplemented with 10% fetal bovine serum (HyClone, Logan, UT) in a humidified atmosphere con-taining 5% CO2at 37 °C.
RNA Isolation and Reverse Transcription (RT)-PCR—Total RNA was
isolated from the cultured cells using TRIzol reagent (Invitrogen). RT-PCR was performed as described previously (3). Briefly, total RNA (2 g) was reverse-transcribed into cDNA in 20 l of 1⫻ first strand buffer containing 0.5g of oligo(dT) as a primer, 500 MdNTP, and 200 units of SuperScript II (Invitrogen). PCR was performed in 20l of 1⫻ PCR buffer containing 2l of RT products, 1 unit of AmpliTaq DNA polym-erase (Roche Applied Science), 200MdNTP, 1.5 mMMgCl2,0.5M
[35S]dATP (Amersham Biosciences), and 0.4
Mprimer pair. We used the primer pair that can distinguish the alternative splicing forms of IAP mRNA, Hiap14: 5⬘-TAA CCT CCT TCG TCA TTG CC and Hiap15: 5⬘-CGT AAG GGT CTC ATA GGT G. The PCR parameters were 94 °C for 30 s, 53 °C for 30 s, and 72 °C for 30 s for 30 cycles, followed by a final elongation at 72 °C for 7 min. PCR products were analyzed on a 6% polyacrylamide-urea gel (acrylamide/bisacrylamide 19:1, 8Murea in 1⫻ Tris borate-EDTA buffer). The gel was finally dried and analyzed by autoradiography. The image of cDNA bands was scanned by the Scan-Jet 4C scanner (Hewlett Packard). The optical densities of cDNA bands were quantified with the one-dimensional advanced Universal Software (American Applied Biotechnology, Fullerton, CA).
* This work was supported by Research Grants NSC90-2320-B-006-029 from the National Science Council and 89-B-FA08-1-4 from the Academic Excellence Program of the Ministry of Education, Executive Yuan, Taiwan. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
¶To whom correspondence should be addressed: Dept. of Physiology, National Cheng Kung University Medical College, Tainan 701, Taiwan. Tel.: 886-6-235-3535 (ext. 5457); Fax: 886-6-236-2780; E-mail: [email protected].
1The abbreviations used are: IAP, integrin-associated protein;
THEJOURNAL OFBIOLOGICALCHEMISTRY Vol. 279, No. 15, Issue of April 9, pp. 14542–14550, 2004 © 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
mega, Madison, WI) were used for IAP promoter reporter assays. A 7.7-kb human genomic clone (pIAP38) containing exon 1 of the IAP gene and 5⬘-upstream region was kindly provided by Dr. F. P. Lindberg. A plasmid, pBSII-445, was generated by inserting the 445-bp SacII frag-ment from pIAP38 into the pBlueScript II (SK⫺) vector (Stratagene, La Jolla, CA). The SacI/XhoI fragment from pBSII-445 was then inserted into the pCRII vector (Invitrogen) to create pHIAP445 for subsequent constructions.
The reporter construct pGL3–272 was generated by inserting the XmaI/XhoI fragment from pHIAP445 into the pGL3-Basic vector. Dif-ferent restriction enzyme-digested fragments from pIAP38 were then ligated into pGL3–272 to create a series of human IAP promoter con-structs, including pGL3–1554, pGL3–730, and pGL3– 456. Another se-ries of human IAP promoter constructs containing shorter fragments than the insert in pGL3–272, including pGL3–232, pGL3–218, pGL3– 209, pGL3–198, pGL3–191, pGL3–159, and pGL3–92, were similarly made by using differential forward primers on the IAP promoter with the KpnI site at the 5⬘-end and a common reverse primer (GLprimer2) on pGL3-Basic in the backbone of pGL3–272 for PCR to obtain the shorter fragments, which included differentially truncated IAP pro-moter regions and a common vector sequence containing the HindIII site. These fragments were then digested by KpnI and HindIII and inserted into pGL3-Basic.
Promoter constructs containing nucleotide substitutions in the se-quence motifs of Sp1 and␣-Pal/NRF-1 were individually generated by PCR amplification with primer pairs spanning the mutant nucleotides according to the protocol of site-directed mutagenesis by overlap exten-sion (18). The plasmids pGL3–232m1, pGL3–232m2, and pGL3–232m3 were constructed in the backbone of pGL3–232 using primer pairs containing the introduced mutations. The Sp1 site GGGGCGGGGC was mutated into GTTGCTTGGC in the plasmid of pGL3–232m1. The ␣-Pal/NRF-1 element TGCGCGTGCGCG was mutated into TTTGCGT-GCGCG, and TGCGCGTTTGCG in the plasmid of pGL3–232m2 and pGL3–232m3, respectively.
Transfection and Dual-luciferase Assay—IMR-32 and HepG2 cells
(1.5⫻ 105) were plated in each well of six-well plates. Transient
trans-fection was carried out by the calcium phosphate precipitation method (19). The plasmid pRL-TK was cotransfected to normalize the transfec-tion efficiency. After 12 h of transfectransfec-tion, the medium was changed, and the cells were incubated at 37 °C for 24 or 48 h. The cells were washed in phosphate-buffered saline (137 mMsodium chloride, 2.7 mM potas-sium chloride, 10 mMdibasic sodium phosphate, and 2 mMmonobasic potassium phosphate) and the lysates were prepared by scraping the cells from plates in the presence of 1⫻ passive lysis buffer (Promega). Luciferase assays were performed by using Dual-Luciferase Assay Sys-tem (Promega) and a Sirius luminometer (Berthold Detection SysSys-tem, Pforzheim, Germany).
Preparation of Nuclear Extracts—IMR-32 and HepG2 cells were
plated onto 6- or 10-cm cultured dishes and incubated for 2 days. The cells were washed with 2 ml of phosphate-buffered saline and collected in 1 ml of phosphate-buffered saline. The cells were centrifuged at 2,000⫻ g for 2 min, and the supernatant was discarded. The cell pellet was incubated in 400l of buffer A (10 mMHEPES (pH 7.9), 1.5 mM
magnesium chloride, 10 mMpotassium chloride, 0.5 mM phenylmethyl-sulfonyl fluoride, 0.5 mMdithiothreitol, 2g/ml leupeptin, 10 g/ml aprotinin, 50 mMsodium fluoride, and 1 mMsodium orthovanadate) on ice for 10 min and then gently shaken for 10 s. The pellet of the crude nuclei was collected by centrifugation at 12,000⫻ g for 10 s. The pellet was resuspended in 100l of buffer C (20 mMHEPES (pH 7.9), 25% glycerol, 420 mMsodium chloride, 1.5 mMmagnesium chloride, 0.2 mM
EDTA, 0.5 mMphenylmethylsulfonyl fluoride, 0.5 mMdithiothreitol, 2 g/ml leupeptin, 10 g/ml aprotinin, 50 mMsodium fluoride, and 1 mM
sodium orthovanadate) by vortex for 15 s, and then incubated on ice for 20 min. After centrifugation at 12,000⫻ g for 2 min, the supernatant containing the nuclear proteins was collected, quantified with BCA Protein Assay Reagent (Pierce), and stored at⫺70 °C in aliquots.
Gel Electrophoretic Mobility Shift Assays (EMSA)—The EMSA used
the following oligonucleotides: IAP␣-Pal/NRF-1 (f), 5⬘-GAG TGC GCG TGC GCG GCT CT-3⬘; IAP␣-Pal/NRF-1 (r), 3⬘-TCA CGC GCA CGC GCC GAG AG-5⬘; IAP␣-Pal/NRF-1 mutation (f), 5⬘-GAG Ttt GCG Ttt GCG GCT CT-3⬘; IAP␣-Pal/NRF-1 mutation (r), 3⬘-TCA aaC GCA aaC GCC GAG AG-5⬘; consensus␣-Pal/NRF-1 (f), 5⬘-TTC TTT TGC GCA CGC GCT T-3; consensus␣-Pal/NRF-1 (r), 3⬘-AAG AAA ACG CGT GCG CGA AGA ATC-5⬘; consensus␣-Pal/NRF-1 mutation (f), 5⬘-TTC TTT TGt aaA CGa atT T-3⬘; consensus␣-Pal/NRF-1 mutation (r), 3⬘-AAG
CCA AAC TTG-3⬘; and consensus E2F-1 (r), 3⬘-GTT AAA GCG CGG TTT GAA C-5⬘. 30 pmol of each of the forward and reverse oligonucleo-tides placed in a volume of 23l of 1⫻ Klenow (DNA polymerase) buffer were heated at 94 °C for 2 min and annealed at room temperature for 30 min. The annealed double-stranded oligonucleotides were end-labeled by a fill-in reaction using DNA polymerase (Klenow) (Promega). One unit of the DNA polymerase (Klenow) and 40 Ci of [␣-32P]dCTP
(PerkinElmer Life Sciences) were added into the annealed oligonucleo-tides and the mixture was incubated at 30 °C for 15 min. The labeled oligonucleotides were purified by Sephadex G-50 columns (Amersham Biosciences). Cold double-stranded oligonucleotides were used as com-petitors. The DNA binding reaction was conducted at 4 °C for 30 min in a mixture containing 3g of nuclear extract, 10 mMTris-Cl (pH 7.5), 50 mMsodium chloride, 0.5 mMdithiothreitol, 0.5 mMEDTA, 1 mM
mag-nesium chloride, 4% glycerol, 0.05g poly(dI-dC)䡠poly(dI-dC) (Amer-sham Biosciences) and 2⫻ 104cpm of 32P-labeled double-stranded
oligonucleotides. In supershift assays, antibodies were incubated with the reaction mixture at 4 °C for 30 min before the addition of the IAP ␣-Pal/NRF-1 probes. The anti-NRF-1 goat polyclonal antiserum was kindly provided by Dr. Richard C. Scarpulla. The Sp1 and E2F anti-bodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-Myc antibody was from Invitrogen. The normal goat serum was from Vector Laboratories (Burlingame, CA). Samples were analyzed on a 4% polyacrylamide gel (acrylamide/bisacrylamide 29:1 in 0.5⫻ Tris borate-EDTA buffer) at 10 V/cm for 2.5 h. The gel was dried and analyzed by autoradiography.
Construction and Overexpression of␣-Pal/NRF-1—The cDNAs
en-coding the full-length and dominant-negative mutant of␣-Pal/NRF-1 were constructed. The dominant-negative mutant consisted of the first N-terminal 304 residues of␣-Pal/NRF-1, which contained the proposed DNA binding and nuclear localization domains and lacking the activa-tion domain (20, 21). Four␣-Pal/NRF-1 cDNA fragments were obtained by RT-PCR. The primer pair〈N-5 (5⬘-TTAAGCTT GCG CAG CCG CTC TGA GGA A) and〈N-7 (5⬘-GACTCGAG CAC TGT TCC AAT GTC ACC AC) and primer pair〈N-5 and 〈N-11 (5⬘-GACTCGAG TCA CTG TGA TGG TAC AAG ATG AGC) were used for the untagged full-length and dominant-negative␣-Pal/NRF-1, respectively. Primer pair 〈N-5 and 〈N-6 (5⬘-GACTCGAG TCA CTG TTC CAA TGT CAC CA) and primer pair AN-5 and AN-10 (5⬘-GACTCGAG GTC TGT GAT GGT ACA AGA TGA G) were used for the Myc-tagged full-length and dominant-nega-tive␣-Pal/NRF-1, respectively. The underlined region indicates the restriction enzyme sites. These fragments were digested by HindIII and XhoI and inserted into pcDNA3.1 (B⫹) vector (Invitrogen). HepG2 cells (8⫻ 105) were placed onto 6-cm dishes for overexpression of␣-Pal/
NRF-1 using the same procedure for transient transfection of the re-porter plasmids. After transient transfection, the cells were incubated for 48 h and harvested for nuclear protein extraction.
Primary Cortical Culture—Primary cortical cells were prepared
ac-cording to the protocol from Dichter (22). In brief, pregnant ICR mice, 15-days postconception, were anesthetized with pentobarbital, and em-bryos were removed. The cortices were removed and collected in MEM. The tissue was triturated three times with a fire-polished Pasteur pipette. Dissociated cells were plated onto 6-well plates coated with 0.09 mg/ml poly-L-lysine and grown in MEM supplemented with 10% fetal
bovine serum (MEM 10), 50 units/ml penicillin, and streptomycin. After 24 h, plating media were replaced with MEM 10 and cells were treated with 3Mof cytosine arabinoside. Twenty-four hours later, the medium was replaced with MEM 10 again, and cells were cultured for another 48 h for the transfection experiments.
Statistics—The relative activity of different reporter constructs was
compared. Statistical analysis was performed by unpaired Student’s t test for pairwise comparisons. A p value ⬍0.05 was regarded as significant.
RESULTS
Expression of Integrin-associated Protein Gene in Human IMR-32 and HepG2 Cells—To use IMR-32 and HepG2 cells to study the IAP gene promoter, we examined firstly the expres-sion of IAP transcripts in these cells by RT-PCR. We used primers that can detect alternative splicing forms of IAP mRNAs. As shown in Fig. 1A, both form 1 and form 2 IAP mRNAs were expressed in these two cell lines at a similar level. The major form of IAP mRNA in IMR-32 was form 4. But form
1B). These results confirmed the expression of the human IAP gene in these two cell lines.
Determination of IAP Promoter Activity in IMR-32 and HepG2 Cells—To define the boundaries of a minimal IAP pro-moter region and identify cis elements that regulate the ex-pression of IAP, we generated a series of 5⬘-IAP promoter deletion constructs and transfected them into IMR-32 and HepG2 cells. All plasmid constructs were defined relative to the translation initiation codon (Fig. 2A). The reporter constructs were cotransfected into IMR-32 and HepG2 cells with an inter-nal control Renilla luciferase vector. The firefly luciferase ac-tivity of each reporter was normalized with the internal control to correct transfection efficiency. Results were represented as a fold-increase in activity with respect to that of the pGL3-Basic vector (Fig. 2, B and C) or the relative activity compared with that of the⫺232 construct (Fig. 4).
In IMR-32 cells, the shortest reporter that still retained the basal promoter activity was the⫺232 construct, whereas dele-tion for another 41 bp (the⫺191 construct), 73 bp (the ⫺159 construct), or 140 bp (the⫺92 construct) resulted in markedly loss of reporter activity. Addition of 40 bp to the⫺232 construct generated the⫺272 construct and stimulated the reporter ac-tivity by⬃25%. In the construct containing additional 184 bp (the⫺456 construct), the promoter activity was not increased. Interestingly, the reporter activity decreased to about the basal proximal promoter activity in the construct containing addi-tional 274 bp (the⫺730 construct) and in the longest construct (the⫺1554 construct) (Fig. 2B). A similar pattern of promoter activity was observed when the reporter constructs were trans-fected into HepG2 cells, but with three exceptions (Fig. 2C). First, overall promoter strength relative to pGL3-Basic was
cells, however, the construct of⫺730 produced maximal activ-ity. Third, there were negative regulators located between ⫺457 and ⫺730 in IMR-32 cells; in HepG2 cells, however, there were negative regulators located between ⫺730 and ⫺1554. Results suggested a core promoter of the human IAP gene located between⫺232 and ⫺12 upstream of the translation initiation codon in both IMR-32 and HepG2 cells.
The sequence from⫺272 to ATG was searched for homology to previously described regulatory elements in several data-bases. There are many putative binding sites for transcription factor in this region (Fig. 3), including activating enhancer binding protein 2 (AP-2), Myc-associated zinc finger protein (Maz), cyclic-AMP responsive element-binding protein (CREB), transcription factor Sp1, E2 promoter binding factor (E2F), and ␣-Pal/nuclear respiratory factor 1 (␣-Pal/NRF-1).
Identification of cis-Elements in the Core Promoter of the IAP Gene—To determine more precisely the core promoter of the IAP gene, we generated several shorter or point mutation con-structs. When 14 and 23 bp were deleted from the 5⬘-end of the ⫺232 construct to generate the constructs of ⫺218 and ⫺209, respectively, the promoter activity of these two constructs was identical to that of the⫺232 construct in IMR-32 cells. This indicated that the sequence between⫺232 and ⫺209 played no significant role in IAP gene expression under the current ex-perimental conditions. When 34 bp were deleted from the 5 ⬘-end of the ⫺232 construct to generate the ⫺198 construct; however, the promoter activity was markedly reduced by 90% in IMR-32 cells, indicating that the sequence between⫺209 and⫺198 might be required for the IAP promoter activity (Fig. 4). As shown in Fig. 3, the region from⫺232 to ⫺198 consists of a GC-rich sequence that includes the putative Sp1 and
FIG. 1. Expression of the human integrin-associated protein gene in IMR-32 and HepG2 cells. A, RT-PCR analysis of the IAP transcripts. Total RNA was isolated from cells grown 3 days in MEM medium and primers were used to distinguish the alternative splicing forms of the IAP gene. B, comparison of the level of total IAP transcripts. The optical densities of cDNA bands shown in A were quantified, and the total IAP transcript in HepG2 cells was shown as percentage of that in IMR-32 cells.
␣-Pal/NRF-1 Regulates the Human IAP Promoter 14544
Sp1 site were substituted with four T residues to generate the ⫺232m1 construct, no significant effect on the IAP promoter
moter is a 12-base tandem-repeat sequence, TGCGCGT-GCGCG. When two bases in each of the repeat sequence were replaced by two T residues to generate the ⫺232m2 and ⫺232m3 constructs respectively, an 80% and a 90% drop in promoter activity was observed (Fig. 4). These results sug-gested that the consensus␣-Pal/NRF-1 sequence, but not the consensus Sp1 sequence, is a functional regulatory element in the IAP promoter in IMR-32 cells.
␣-Pal/NRF-1 Is a Transcription Factor Regulating the IAP Promoter Activity—To demonstrate that the consensus␣-Pal/ NRF-1 sequence was functional, i.e. that there were endoge-nous nuclear proteins binding to this region, we performed the EMSA experiment. Nuclear extracts from IMR-32 cells were combined with32P-fill-in-labeled double-stranded
oligonucleo-tides in vitro. A major band of DNA-protein complex was found in all lanes when the nuclear extracts were incubated with the wild-type IAP␣-Pal/NRF-1 probes (Fig. 5A, lane 3 and lanes 5–12), but not with the mutant IAP␣-Pal/NRF-1 probes (Fig. 5A, lane 4). No band was found when nuclear extracts were not added into the probes (Fig. 5A, lanes 1 and 2). Competition analysis using a 10- or 60-fold molar excess of unlabeled probes was used to characterize the factor, which specifically binds to this sequence. As expected, the addition of a 10- or 60-fold molar excess of published wild-type consensus ␣-Pal/NRF-1 element reduced the intensity of these complexes (Fig. 5A, lanes 5 and 6), whereas the addition of the mutant consensus ␣-Pal/NRF-1 sequence did not (Fig. 5A, lanes 7 and 8). Results suggested that ␣-Pal/NRF-1 proteins might bind to the IAP ␣-Pal/NRF-1 element. However, the ␣-Pal/NRF-1 site is GC-rich and might therefore interact with factors other than␣-Pal/ NRF-1, such as Sp1 and E2F. We therefore used the unlabeled consensus Sp1 and E2F sequence for the competition experi-ment. The intensity of the migrating bands was not signifi-cantly reduced (Fig. 5A, lanes 9 –12). Supershift assays using the anti-␣-Pal/NRF-1 antiserum were used to further confirm the binding of the␣-Pal/NRF-1 on its DNA element. The mi-grating bands were weakened when increasing amounts of ␣-Pal/NRF-1 antiserum were added and supershifted bands appeared (Fig. 5B, lanes 3–5). However, the Sp1 or E2F anti-body did not generate any supershifted band (Fig. 5B, lanes 6 and 7), neither did the normal goat serum (Fig. 5B, lane 8). The EMSA experiments also revealed that the DNA binding
activ-FIG. 2. Identification of the core promoter region of the human IAP gene in IMR-32 and HepG2 cells. A, promoter constructs. The restriction map of the 5⬘-flanking region of the human IAP gene is shown at the top. Positions are indicated relative to the translation initiation codon. The promoter constructs are named by the left boundary and have a common 3⬘-end at ⫺12. B and C, luciferase activity of the promoter constructs in IMR-32 and HepG2 cells. 250 ng of independent constructs and pRL-TK plasmids were cotransfected into cells by the calcium phosphate precipitation method. The firefly luciferase activity after 48-h incubation was normalized to Renilla luciferase activity as a transfection control. These values were then normalized to those of pGL3-Basic vector without any insert. Note the different scales of the x-axes in B and C. Values are presented as the mean⫾ S.E. obtained from three independent experiments, each conducted with triplicate cultures. *, p ⬍ 0.05; **,
p⬍ 0.01; ***, p ⬍ 0.001; unpaired Student’s t test. n.s., not significant.
FIG. 3. The sequence of 5ⴕ-flanking region of the human IAP
gene. The sequence spanning from⫺272 to the translation initiation
codon ATG is shown. This region contains putative binding sites for transcription factors such as AP-2, Maz, CREB, Sp1, E2F, and␣-Pal/ NRF-1. However, two Sp1 (TGCGC) and one E2F sites share partial sequences with the␣-Pal/NRF-1 site (23). The positions of these sites are indicated by underlines. The plasmids for the luciferase assay were constructed by inserting fragments spanning from nucleotides (indi-cated by arrows and numbers) to nt⫺12, relative to ATG, into the firefly luciferase expression vector (pGL3-Basic).
FIG. 4. Identification of functional cis elements in the IAP core
promoter. Shorter promoter fragments were made by PCR (the⫺218,
⫺209, and ⫺198 constructs). Mutant constructs were made by site-directed mutagenesis (the⫺232m1, m2, and m3 constructs). The puta-tive Sp1 and␣-Pal/NRF-1 sites are indicated. Independent constructs and pRL-TK plasmids were cotransfected into IMR-32 cells using the same protocol as shown in Fig. 2. The promoter activity was expressed with respect to the⫺232 construct. *, p ⬍ 0.05; ***, p ⬍ 0.001; unpaired Student’s t test.