國 立 交 通 大 學
生 物 資 訊 研 究 所
碩士論文
建置人類核糖核酸編輯點資料庫
EdRNA: a RNA editing database comprising experimentally validated
and putative RNA editing sites in human genome
研 究 生:王威霽
指導教授:黃憲達 博士
建置人類核糖核酸編輯點資料庫
EdRNA: a RNA editing database comprising experimentally
validated and putative RNA editing sites in human genome
研 究 生:王威霽 Student:Wei-Chi Wang 指導教授:黃憲達 博士 Advisor:Dr. Hsien-Da Huang
國立交通大學 生物資訊研究所
碩士論文
A Thesis
Submitted to Institute of Bioinformatics College of Biological Science and Technology
National Chiao Tung University in partial Fulfillment of the Requirements
for the Degree of Master
in Bioinformatics
July 2006
建置人類核糖核酸編輯點資料庫
學生: 王威霽 指導教授 : 黃憲達 博士國立交通大學生物資訊研究所碩士班
中文摘要
核糖核酸編輯 (RNA Editing) 是核糖核酸序列的修改,其中主要有核甘酸的 刪除、插入、以及取代三種。核糖核酸編輯機制導致訊息核糖核酸前驅物剪接、 架構位移、核醣核酸結構改變和訊息核醣核酸轉譯。A-to-I 和 C-to-U 核糖核酸編 輯機制是目前已經被了解和研究的,但是,目前為止沒有任何的資料庫是關於哺 乳類核糖核酸編輯。因此,本研究建立ㄧ個儲存核糖核酸編輯相關資訊的資料庫 系統。這個資料庫系統所儲存的資料包含不同的核糖核酸編輯類型,以及其詳細 地註解,亦即未轉譯區域、重複區域、單一核酸多樣性和經由實驗驗證的核糖核 酸編輯點。此外,本研究也進行跨物種的比較分析,用以來探討演化上的意義, 並且針對每一種核糖核酸編輯類型,找尋潛在相關的核糖核酸結構。 主要的研究方法是利用序列分析 (Sequence analysis) 來預測核糖核酸編輯 點。本研究的方法是藉由對準基因、訊息核醣核酸和表現序列片段的序列的結果 來找到核糖核酸編輯點。本研究也收集實驗驗證過的核糖核酸編輯點來確認我們 預 測 的 核 糖 核 酸 編 輯 點 。 本 研 究 所 建 置 的 EdRNA 資 料 庫 目 前 提 供 於 http://EdRNA.mbc.nctu.edu.tw/。EdRNA: a RNA editing database comprising
experimentally validated and putative RNA editing
sites in human genome
Student: Wei-Chi Wang Advisor : Dr. Hsien-Da Huang
Institute of Bioinformatics, National Chiao Tung University
Abstract
RNA editing is the modification of RNA sequence through nucleotide deletion, insertion, or base modification mechanisms. RNA editing mechanisms affect the pre-mRNA splicing, frameshfits, RNA structure changes and mRNA translation. Recently, two types of RNA editing such as A-to-I and C-to-U were investigated. A database comprising RNA editing sites in mammalian genomes is crucial for deciphering the RNA editing mechanism. In this thesis, we established a database which comprises both the experimentally validated RNA editing sites and computationally predicted RNA editing sites. Experimentally validated sites ware obtained by literature survey. A computational method based on sequence comparing analysis is implemented for identifying putative RNA editing sites. Additionally, cross-species comparison is performed to confer the evolution meanings of the RNA editing site. Motif discovery method is applied to discover RNA structural motifs, which are potentially related to each type of editing site. The database is now available in http://EdRNA.mbc.nctu.edu.tw/.
誌 謝
首先,我要感謝指導教授黃憲達博士在這兩年的日子裡
對於我的細心指導,使我得以在生物資訊這個領域裡慢慢地
從無到有,一點一滴地累積許多生物資訊相關的知識。並且
也在學術研究上有顯著的進步和成長;另外,我也要感謝中
國醫藥大學的張建國醫師在研究過程中的指導,讓我在生物
的領域內的知識能夠快速的成長。
實驗室的學長姊們,謝謝你們對學弟的細心指導,實驗
室的同學們,謝謝大家在這兩年的互相幫忙及鼓勵,和大家
一起討論的日子,是我成長的動力,實驗室內的點點滴滴更
是美好的回憶。
最後,我要特別感謝我的家人給予我的支持,讓我能在
研究上全力以赴、完成學業。能夠完成碩士論文,是大家的
指導、支持、與鼓勵,誠心的感謝大家,將這份成果與關心
我的所有一同分享。
王威霽 于交通大學 2006Table of Contents
中文摘要...i
Abstract ...ii
誌 謝... iii
Table of Contents ...iv
List of Figures ...vi
List of Tables... viii
List of Tables... viii
Chapter 1 Introduction ...1
1.1 Background ...1
1.1.1 The Central Dogma...1
1.1.2 RNA editing ...2
1.1.2.1 Substitution RNA editing...4
1.1.2.2 Insertion/deletion RNA editing ...6
1.1.3 Functions of RNA editing ...7
1.1.4 RNA editing affects diseases ...9
1.2 Motivation...10
1.3 Goal... 11
Chapter 2 Related Works ...12
2.1 Identification of RNA editing sites ...12
2.2 Related Biological Databases ...13
2.2.1 GenBank ...13
2.2.2 Ensembl...13
2.2.3 dbEST ...14
2.2.4 UniGene ...14
2.2.5 UCSC Genome Browser Database ...15
2.2.6 dbSNP ...16 2.3 Related Software...17 2.3.1 Blast ...17 2.3.2 ClustalW ...18 2.3.3 SIM4 ...18 2.3.4 MEME...19 2.3.5 Mfold...20
Chapter 3 Materials and Methods ...21
3.2 Materials ...22
3.3 Methods...24
3.3.1 RNA editing site prediction ...24
3.3.2 Cross-Reference...27
3.3.3 Cross-species comparison...30
3.3.4 Motifs discovery ...31
3.3.5 Web interface ...33
Chapter 4 Results ...37
4.1 The statistics of EdRNA database...37
4.2 The analysis of EdRNA database...43
4.2.1 Different genes have the same motifs ...44
4.2.2 Gene family have the same motifs...46
4.2.3 The summary of analysis result ...49
Chapter 5 Discussion ...50 5.1 Comparison ...50 5.2 Drawbacks...50 5.3 Future Works...51 5.4 Experimentally Confirmation ...52 Chapter 6 Conclusions ...53 References...54
List of Figures
Figure 1.1 Central dogma of molecular biology. ...2
Figure 1.2 ADARs recognize duplex RNA that is formed between the editing site and the ECS that is often located in a downstream intron...5
Figure 1.3 In this figure, pre-mRNA containing Alu repeat form dsRNA structure. And ADAR binding on dsRNA region edits some of the adenosines into inosines. ...5
Figure 1.4 APOBE protein complex binds on mRNA and edits some the cytidines into uridines. ...6
Figure 1.5 The instance of insertion/deletion editing. ...7
Figure 1.6 The influence of RNA editing...8
Figure 1.7 A single C-to-U RNA editing produces different protein. ...9
Figure 1.8 Insertion/deletion editing causes ORFs changing. Then, ORFs variation leads to translate different protein...10
Figure 2.1 The UCSC Genome Browser...16
Figure 3.1 The system flow of EdRNA. ...22
Figure 3.2 Clustering gene, mRNA and EST by UniGene information...24
Figure 3.3 The flow of finding correct RNA editing site position in mRNA and EST. ...25
Figure 3.4 The flow of find editing site position in gene...26
Figure 3.5 The flow of computing editing site position in chromosome. ...27
Figure 3.6 The flow of annotation of RNA editing site. ...29
Figure 3.7 The example conserved region format of UCSC Genome Browser Database...30
Figure 3.8 The flow of cross-species conserved region comparison. ...31
Figure 3.9 The instance of motif discovery flow chart. ...33
Figure 3.10 EdRNA web interface. ...34
Figure 3.11 The example of filtering results which contain gene and mRNA information...35
Figure 3.12 The example of filtering results which contain gene, mRNA and ESTs alignment surrounding the editing site...36
Figure 4.1 The distribution of the number of editing site which has more than how many EST sequences support. ...37
Figure 4.2 The distribution of chromosome length and the distribution of the number of editing site in each chromosome per mbps...39
Figure 4.4 The motifs are located in the two mRNA. Editing site locates in front of the motifs. ...46 Figure 4.5 The motifs are in the three mRNA. Editing site locates in motif 2. ...47 Figure 4.6 The motifs are in the three mRNA. Editing site locates in motif 2. ...48
List of Tables
Table 1.1 Editing distribution: substitution editing. ...3
Table 1.2 Editing distribution: insertion/deletion editing...4
Table 3.1 Data source of EdRNA. ...23
Table 3.2 Tool list of EdRNA...23
Table 3.3 The list of filtering term...32
Table 3.4 The filtering option of EdRNA interactive browser interface ...34
Table 4.1 The number of editing site and gene of each editing type...38
Table 4.2 The number of editing site in each repeat type...40
Table 4.3 The number of editing site in UTR, CDS...40
Table 4.4 The number of editing site is SNP site or not...41
Table 4.5 The number of editing site in cross-species conserved region (human and mouse)...41
Table 4.6 The number of editing site of each RNA editing type in coding region, 5’UTR and 3’UTR. ...42
Table 4.7 The number of editing site of each RNA editing type in cross-species conserved region (human and mouse). ...42
Table 4.8 The number of editing site in each editing type through some filtering conditions...44
Table 4.9 Gene information in A-to-T editing type (two different genes). ...44
Table 4.10 Gene information in C-to-G editing type (two different genes)...45
Table 4.11 Gene information in A-to-C editing type (three genes). ...46
Chapter 1 Introduction
1.1
Background
1.1.1
The Central Dogma
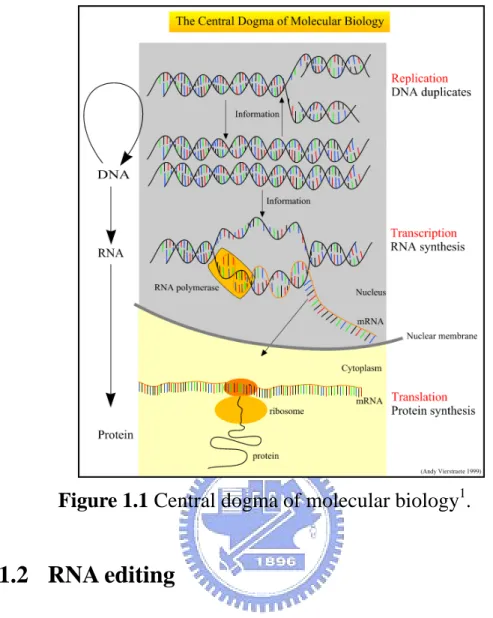
The central dogma is the biological process of protein synthesis. As shown in Fig. 1.1, DNA is first transcribed to RNA through DNA duplicates and RNA synthesis in nucleus. This particular kind of RNA is called messenger RNA (mRNA), and other kinds of RNA also exist such as transfer RNA (tRNA) and ribosomal RNA (rRNA). After DNA is transcribed to RNA, RNA is conveyed from nucleus to cytoplasm. Then, RNA is translated to protein by ribosome.
Gene expression is the process that a gene is transcribed from DNA to mRNA which then is translated to protein. The amount of gene expression is controlled by some of bridle mechanisms such as alternative splicing, frameshifting, RNA editing, and so on. The process of transcription is more complicated in eukaryotes, especially in higher organisms. In many eukaryotic organisms, there are three different types of RNA polymerases, which catalyze the production of three different classes of RNA molecules.
Figure 1.1 Central dogma of molecular biology1.
1.1.2
RNA editing
RNA editing is the co- or post- transcriptional modification of RNA primary sequence from that encoded in the genome through nucleotide deletion, insertion, or base modification mechanisms [1]. RNA editing is mainly classified into two classes. One is substitution editing (chemical alteration of individual nucleotides), and the other is insertion/deletion editing (insertion or deletion of nucleotides in the RNA). The distribution of the forms of RNA editing is listed in Table 1.1 (substitution editing) and Table 1.2 (insertion/deletion editing) [2]. RNA editing is quite
widespread occurring in mammals, viruses, marsupials, plants, flies, frogs, worms, squid, fungi, slime molds, dinoflagellates, kinetoplastid protozoa, and other unicellular eukaryotes. It should be noted that the list most likely represents only the tip of the iceberg [2].
Table 1.2 Editing distribution: insertion/deletion editing.
1.1.2.1
Substitution RNA editing
In mammalian, two the substitution editing sites have been found such as A-to-I RNA editing and C-to-U RNA editing.
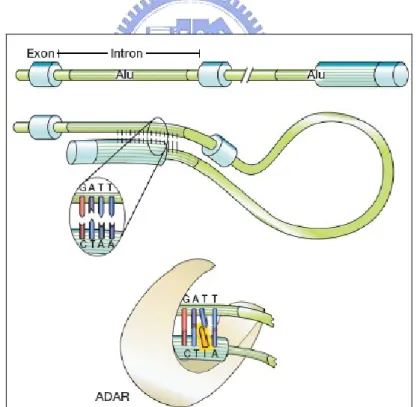
About A-to-I RNA editing, the pre-mRNA has double-stranded RNA (dsRNA) structure and the enzyme ADAR (adenosine deaminases acting on RNA) recognizes this double-stranded region. The ADAR binds on the region that is called editing site complementary sequence (ECS) as shown in Fig. 1.2 [3]. And this ADAR binding region usually contains Alu repeat patterns. When ADAR binds on ECS region, ADAR edits some of the adenosines into inosines Fig. 1.3 [4].
Figure 1.2 ADARs recognize duplex RNA that is formed between the
editing site and the ECS that is often located in a downstream intron.
Figure 1.3 In this figure, pre-mRNA containing Alu repeat form dsRNA
structure. And ADAR binding on dsRNA region edits some of the adenosines into inosines.
Figure 1.4 APOBE protein complex binds on mRNA and edits some the
cytidines into uridines.
As to C-to-U RNA editing, an eleven nts length sequence (the ‘mooring sequence’) and the flanking nucleotides within the apolipoprotein B (apoB) mRNA are recognized by Apobec-1 and ACF1 (Apobec-1 complementing factor) [3]. When APOB protein complex binds on APOB mRNA, it edits some the cytidines into uridines Fig. 1.4 [3] .
1.1.2.2
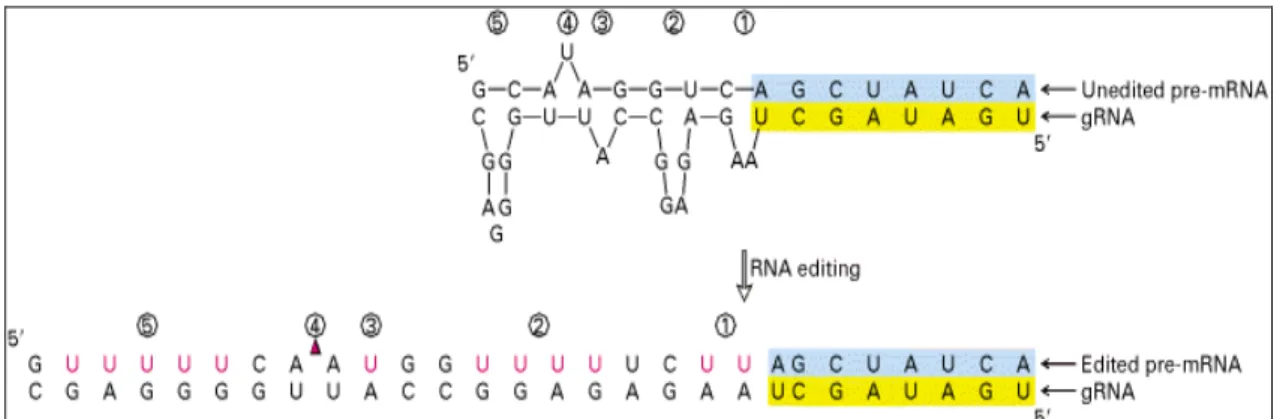
Insertion/deletion RNA editing
The insertion/deletion editing mechanism occurs when a guide RNA (gRNA) hybridizes with an unedited pre-mRNA. As shown in Fig. 1.5, an anchor sequence (blue) in the unedited pre-mRNA hybridizes to the 5’
anchor sequence (yellow) of a guide RNA. In order to perfect hybridization, some area is inserted nucleotides in and some area is deleted nucleotides from unedited pre-mRNA.
Figure 1.5 The instance of insertion/deletion editing.
1.1.3
Functions of RNA editing
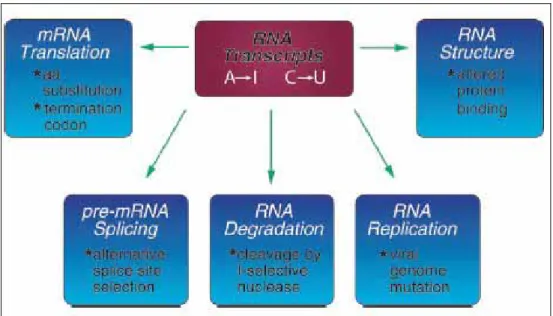
The RNA editing mechanism causes functional activities different from the unedited transcripts. As shown in Fig. 1.6 [5], RNA editing may alter processes including mRNA translation by changing codons; RNA editing may alter pre-mRNA splicing patterns by changing splice site recognition sequences; RNA editing affect RNA degradation by modifying RNA sequences involved in nuclease recognition; RNA editing may effect viral RNA genome stability by changing template and hence product sequences during RNA replication; and RNA editing potentially may affect RNA structure dependent activities that entail binding of RNA by proteins[5].
Figure 1.6 The influence of RNA editing.
Substitution RNA editing often leads to amino acids change when mRNA is translated to protein. For instance, a single C-to-U change within the apolipoprotein B mRNA changes a glutamine codon (CAA) to a stop codon (UAA), leading to the production of two proteins from a single gene Fig. 1.7 [2].
Figure 1.7 A single C-to-U RNA editing produces different protein.
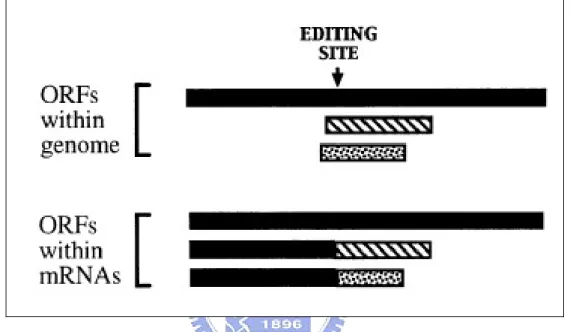
Insertion/deletion RNA editing usually results in frameshifting in mRNA. When frameshifting occurs, open reading frames (ORFs) would change and produce different protein Fig. 1.8 [2].
1.1.4 RNA editing affects diseases
RNA editing is essential for normal life and developmental stages in both invertebrates and vertebrates [4, 6-9]. Hyper editing caused by over expression of Apobec-1 leads to carcinomas in model systems [2, 10], while hyper editing of meals transcripts has been observed in patients with subacute sclerosing panencephalitis and measles inclusion body encephalitis [2, 11, 12]. In mouse, ADAR1 knockout mice die embryonically and ADAR2 null mice are born at full term but die
prematurely [4, 6, 9]. In human, altered editing levels have also been observed in malignant gliomas [2, 13], schizophrenic patients [2, 14] and suicide victims [2, 15], and may be affected in patients with Alzheimer’s and Huntington’s disease [2, 16].
Figure 1.8 Insertion/deletion editing causes ORFs changing. Then, ORFs
variation leads to translate different protein.
1.2
Motivation
RNA editing plays an important role during the post-transcriptional regulation of gene expression. This mechanism affects a variety of biological processes such as mRNA translation, pre-mRNA splicing, RNA degradation, and so on. RNA editing site are usually discovered by accident, through the comparison of genomic and cDNA sequences. Therefore, there are only a few experimentally validated RNA editing
sites. Computational method is necessary for systematically identifying putative RNA editing sites.
Besides, a biological database comprising RNA editing site information is curial for the investigators who interested in the regulatory mechanism of RNA editing.
1.3 Goal
In this thesis, we designed a systematic method to identify RNA editing sites based on sequence comparing analysis between genomics sequence, mRNA sequence, EST sequences, and proteins. A database, namely EdRNA, is established to deposit both the experimentally validated RNA editing sites collected from literatures and the putative RNA editing sites identified in this thesis. Furthermore, the cross-species comparison is performed to confer the evolution meanings of the RNA editing sites. A user-friendly web interface is also designed for the access to the data of RNA editing sites in EdRNA database.
Chapter 2 Related Works
2.1 Identification of RNA editing sites
In mammalian genome, A-to-I and C-to-U RNA editing are understood and researched. A-to-I RNA editing sites are identified and experimentally validated in human [4]. They collect gene, mRNA, EST sequence from genbank and perform alignment. Then, they obtain putative A-to-I editing sites by some filtering condition according to A-to-I RNA editing characteristics. They offer 12,723 putative A-to-I editing sites in 1,637genes. And they experimentally validated 26 editing sites from 30 genes. Another A-to-I RNA editing sites identification works [17] unlike the front, their data resource is dbSNP (SNP database). This is because they think that in SNP database, some SNP maybe RNA editing site. They provide 102 RNA editing previously annotated in dbSNP as SNPs and experimentally validate seven of these. Unlike these two works, we focus on each editing type such as A-to-I, C-to-U, and other possible editing type. Moreover, each RNA editing site would be annotated detailed information such as UTR, repeat and so on. We perform analysis in each editing type to discovery RNA structure motifs. And these two works only focus on human genome, we perform
cross-species comparison to find evolution meanings and discovery potentially related RNA structural motifs for each editing type.
2.2
Related Biological Databases
2.2.1
GenBank
GenBank [18] is a database of nucleotid sequences from > 130,000 organisms. Records that are annotated with coding region (CDS) features also include amino acid translations. GenBank belongs to an international collaboration of sequence databases, which also includes EMBL [19] and DDBJ [20].
2.2.2
Ensembl
Ensembl [21] is a joint project between EMBL-EBI and Sanger Institute to develop a software system which produces and maintains automatic annotation on eukaryotic genomes. Ensembl is primarily funded by the Welcome Trust. Access to all the data produced by the project, and to the software used to analysis and present it, it provided free and without constraints. Ensembl presents up-to-date sequence data and the best possible automatic annotation for eukaryotic genomes.
2.2.3
dbEST
dbEST [22] is a database of expressed sequence tags; short, single pass read cDNA (mRNA) sequences. Also includes cDNA sequences from differential display experiments and RACE experiments. EST sequences are available from two sources: dbEST and the EST division of GenBank. In another word, dbEST is a division of GenBank that contains sequence data and other information on “single-pass” cDNA sequences, or Expressed Sequence Tags, from a number of organisms.
Before the 1990s, the human gene sequence data that were available were largely those derived from academic studies of individual genes and just a few extend regions. The advent of ESTs was a milestone that promised that nearly the entire expressed genome would be expressed within a few years, which prompted a flurry of bioinformatics activity in both the public and private sectors.
2.2.4
UniGene
Unigene [23] is an experimental system for automatically partitioning GenBank sequences into a non-redundant set of gene-oriented clusters. ESTs and full-length mRNA sequences organized into clusters that each represents a unique known or putative gene within the organism from
which the sequences were obtained. Each UniGene cluster contains sequences that represent a unique gene, as well as related information such as the tissue types in which the gene has been expressed and map location. UniGene clusters are annotated with mapping and expression information when possible, and include cross-references to other resources.
Currently in UniGene, sequences from the animals human, rat, mouse, cow, zebrafish, clawed frog, fruit fly and mosquito have been processed. Plant organisms are wheat, rice, barley, maize and cress. These species were chosen because they have the greatest amounts of EST data available and represent a variety of species. Additional organisms may be added in the future.
2.2.5
UCSC Genome Browser Database
UCSC Genome Browser Database [24] is an up to date source for genome sequence data integrated with a large collection of related anntotations. UCSC Genome Browser Database also offers the genome browser which zooms and scrolls over chromosomes, showing the work of annotators worldwide Fig. 2.1.
Figure 2.1 The UCSC Genome Browser.
UCSC Genome Browser Database offers cross-species conserved region information which can let biologists easily confer evolution meanings in different species.
2.2.6
dbSNP
dbSNP [25] is a database which contains SNP (single nucleotide polymorphism) information.
In collaboration with the National Human Genome Research Institute, The National Center for Biotechnology Information has established the dbSNP database to serve as a central repository for both single base
nucleotide subsitutions and short deletion and insertion polymorphisms. Once discovered, these polymorphisms could be used by additional laboratories, using the sequence information around the polymorphism and the specific experimental conditions. (Note that dbSNP takes the looser 'variation' definition for SNPs, so there is no requirement or assumption about minimum allele frequency.) The data in dbSNP will be integrated with other NCBI genomic data. As with all NCBI projects, the data in dbSNP will be freely available to the scientific community and made available in a variety of forms.
dbSNP distinguishes a report of how to assay a SNP from the use of that SNP with individuals and populations. This separation simplifies some issues of data representation. However, these initial reports describing how to assay a SNP will often be accompanied by SNP experiments measuring allele occurrence in individuals and populations.
2.3
Related Software
2.3.1
Blast
The Basic Local Alignment Search Tool (BLAST) [26] finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the
statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.
2.3.2
ClustalW
ClustalW [27] is a general purpose multiple sequence alignment program for DNA or proteins. It produces biologically meaningful multiple sequence alignments of divergent sequences. It calculates the best match for the selected sequences, and lines them up so that the identities, similarities and differences can be seen. Evolutionary relationships can be seen via viewing Cladograms or Phylograms.
2.3.3
SIM4
SIM4 [28] is a similarity-based tool for aligning an expressed DNA sequence (EST, cDNA, mRNA) with a genomic sequence for the gene. It also detects end matches when the two input sequences overlap at one end (i.e., the start of one sequence overlaps the end of the other).
SIM4 employs a blast-based technique to first determine the basic matching blocks representing the "exon cores". In this first stage, it detects all possible exact matches of W-mers (i.e., DNA words of size W) between the two sequences and extends them to maximal scoring
gap-free segments. In the second stage, the exon cores are extended into the adjacent as-yet-unmatched fragments using greedy alignment algorithms, and heuristics are used to favor configurations that conform to the splice-site recognition signals (GT-AG, CT-AC).
2.3.4
MEME
MEME [29] is a software which uses an algorithm to discovery several different motifs with differing numbers of occurrences in a single dataset.
The algorithm discovers one or more motifs in a collection of DNA or protein sequences by using the technique of expectation maximization to fit a two-component finite mixture model to the set of sequences. Multiple motifs are found by fitting a mixture model to the data, probabilistically erasing the occurrences of the motif thus found, and repeating the process to find successive motifs. The algorithm requires only a set of unaligned sequences and a number specifying the width of the motifs as input. It returns a model of each motif and a threshold which together can be used as a Bayes-optimal classifier for searching for occurrences of the motif in other databases. The algorithm estimates how many times each motif occurs in each sequence in the dataset and outputs an alignment of the occurrences of the motif.
2.3.5
Mfold
Mfold [30] is a tool for predicting the secondary structure of RNA and DNA, mainly by using thermodynamic methods. The core algorithm predicts a minimum free energy as well as minimum free energies for folding that must contain any particular base pair. Base pair within this free energy increment are chosen either automatically or else by the user.
They also provided a web site [31] for the prediction of secondary structure of single stranded nucleic acids. The objective of this web server is to provide easy access to RNA and DNA folding and hybridization software to the scientific community at large.
Secondary structure annotation has been described by Zuker and Jacobson [32]. Bases in plotted structures may be annotated by ‘p-num’, which represent the number of ways that a base pair in folding from the minimum energy. Low values indicate ‘well-defined’ base. Values of 0 or 1 indicate that a base is always single stranded or always paired to a unique partner. The number of times that a base is single stranded in the computed folding is called its ‘ss-count’ number, and structure plots may also be annotated using these numbers.
Chapter 3
Materials and Methods
3.1
Overview
The system flow of the proposed method is shown in Fig. 3.1. EdRNA has five components which are external databases, editing site prediction, cross-reference, motif discovery and web interface. The first step, EdRNA collects gene, mRNA and EST information and sequences from Ensembl, GenBank and EMBL. The second step, we use the information of UniGene which offers gene clusters that contains mRNA and EST sequences to group gene, mRNA and EST. In each group, we use blast and the program which is developed by us to perform alignment with gene, mRNA, EST sequence to find correct editing site position. The third step, each editing site which we predicted is annotated by some cross-references such as UTR, repeat, SNP, and so on. And we add experimentally validated RNA editing site to improve our editing site believable level. Moreover, we perform cross-species comparison to detect the evolution meanings. The fourth step, we filter the editing site according to some standards like UTR, repeat, SNP, EST sequence number, each editing type to different group. And then, each group data is used motifs discovery tool MEME to find motifs and use Mfold to see the
structure of these motifs. The final step, we develop an interactive web interface to let users access the whole information of EdRNA based on their interest.
Figure 3.1 The system flow of EdRNA.
3.2
Materials
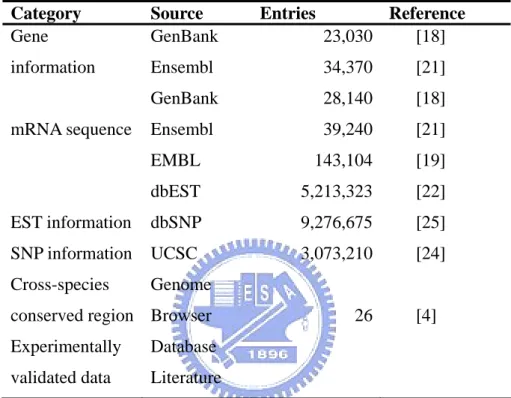
As given in Table 3.1, EdRNA collects the gene, mRNA, EST sequences and information from GenBank, Ensembl and EMBL. Moreover, we obtain the SNP information from dbSNP, repeat information from Ensembl, UTR information from UCSC Genome Browser Database, cross-species conserved region information from
UCSC Genome Browser Database and experimental evidence data from literature [4].
The analyzing tools used in this work are given in Table 3.2.
Table 3.1 Data source of EdRNA.
Category Source Entries Reference Gene information mRNA sequence EST information SNP information Cross-species conserved region Experimentally validated data GenBank Ensembl GenBank Ensembl EMBL dbEST dbSNP UCSC Genome Browser Database Literature 23,030 34,370 28,140 39,240 143,104 5,213,323 9,276,675 3,073,210 26 [18] [21] [18] [21] [19] [22] [25] [24] [4]
Table 3.2 Tool list of EdRNA.
Software URL Reference
Blast ClustalW SIM4 MEME Mfold http://www.ncbi.nlm.nih.gov/BLAST/ http://www.ebi.ac.uk/clustalw/ http://globin.cse.psu.edu/html/docs/sim4.html http://meme.sdsc.edu/meme/meme.html http://bioweb.pasteur.fr/seqanal/interfaces/mfold-simple. html [26] [27] [28] [29] [30]
3.3
Methods
3.3.1
RNA editing site prediction
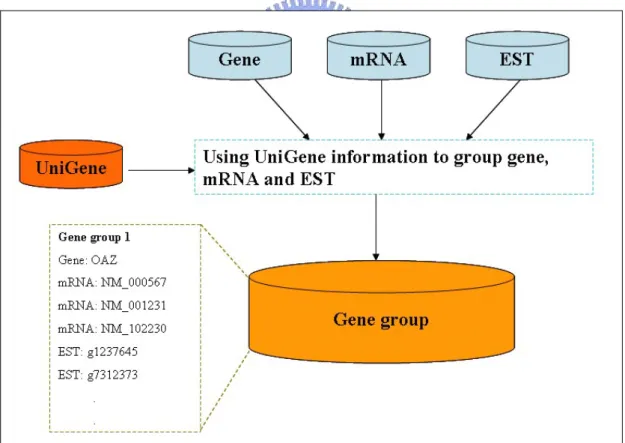
In this step, we first cluster gene, mRNA, and EST sequences to different clusters by the gene group information which comes from UniGene. As shown in Fig. 3.2, after using UniGene information to cluster gene, mRNA and EST, we obtain different gene groups which contain gene, mRNAs and ESTs.
Figure 3.2 Clustering gene, mRNA and EST by UniGene information.
After getting each gene group, we perform the alignment of each gene group to predict RNA editing site. Briefly, it can divide into three
steps.
The first step is the alignment of mRNA and EST sequence. In this step, we first perform mRNA and EST alignment by Blast to find mRNA and EST matching blocks. Blast parameters are identity >90% and <100%. Blast is a tool that does alignment using heuristic methods to find matching blocks [26]. Its execution time is short but the correctness may be not good. Therefore, we develop a program which is based on semi-global alignment to find correct editing site position in mRNA and EST through matching blocks, as given in Fig. 3.3.
Figure 3.3 The flow of finding correct RNA editing site position in
mRNA and EST.
The second step is the alignment of gene and mRNA sequence. In this step, we obtain gene and mRNA mapping by using a tool SIM4 [28]. SIM4 is a tool which aligns a cDNA sequence with genomic DNA
sequence. Then, the editing site position in mRNA and EST can be mapped to gene by the result generated by SIM4. After mapping to the gene, the position of editing site in gene can be determined, as shown in Fig. 3.4.
Figure 3.4 The flow of find editing site position in gene.
The third step is to find the chromosomal position of editing site. Editing site position in chromosome must be computed. This is because some information like as UTR, SNP, repeat and cross-species conserved region is recorded position in chromosome. Gene position in chromosome
information comes from Ensembl and GenBank. This information contains that gene start position and end position in chromosome and in straight strand or opposite strand. The editing site position in chromosome is computed by the gene information which comes from Ensembl and GenBank and the editing site position in gene which comes from step two, as given in Fig. 3.5.
Figure 3.5 The flow of computing editing site position in chromosome.
3.3.2
Cross-Reference
In this step, each putative editing site is annotated by cross-referencing to other biological signals. There are four kinds of
cross-reference.
The first one is the reference to UTR (untranslated region). UTR information comes from UCSC Genome Browser Database. UTR information contains that 5’UTR end position and 3’UTR start position in chromosome. And then, we using this information and editing site position in chromosome which comes from our computation to determine that the editing site is in 5’UTR or 3’UTR or coding region. RNA editing site occurs in different region leading to distinct influence in mRNA or protein level.
The second is the reference to SNP (single nucleotide polymorphism). SNP information comes from dbSNP. SNP information contains that SNP position in chromosome. And then, we use this information and editing site position in chromosome which comes from our computation to annotate that the editing site may be SNP or not. Some literature catalogs that the SNP information in dbSNP is not always correct. Some SNP site in dbSNP is RNA editing site [17].
The third is the reference to the repeat region. Repeat region information comes from Ensembl. It contains that repeat positions on chromosome and the type of repeat. And then, we using this information and editing site position on chromosome which comes from our computation to annotate that the editing site is in which kind of repeat region or not in repeat region. Some literature catalogs that the editing
site of some RNA editing type such as A-to-I is in Alu repeat region [4, 17, 33].
The fourth is the reference to prediction editing site from literature. Prediction editing site information comes from literature [4]. Prediction editing site information contains that editing type and editing site position in chromosome. Then, we using this information and editing site position in chromosome which comes from our computation to determine that the editing site could perfectly map to prediction data. By this method, we can confer the effect of our method that predicts RNA editing site.
Figure 3.6 The flow of annotation of RNA editing site.
Figure 3.6 shows the flow that how we annotate information by editing site position in chromosome.
3.3.3
Cross-species comparison
In this step, we perform cross-species comparison by editing site position which comes from our computation. Cross-species conserved region information comes from UCSC Genome Browser Database. The information format is shown in Fig. 3.7.
Figure 3.7 The example conserved region format of UCSC Genome
Browser Database.
There are two steps for doing cross-species comparison. The first step is computing that the position in other species which is match to the editing site in human. In this step, we first find the editing site in human in which conserved region. After obtaining the conserved region, we compute the position in other species according to the position in human by the result of conserved region alignment. The second step is determining that the position in other species is RNA editing site or not. In this step, we first collect the gene, mRNA and EST sequences which contain this position. Then, gene, mRNA and EST perform alignment to determine that this position is RNA editing site or not.
Figure 3.8 The flow of cross-species conserved region comparison.
3.3.4
Motifs discovery
In this step, we discover the RNA motifs which occur in the flanking regions of the editing site. There are third parts in finding motifs.
The first part is filtering the editing site in EdRNA by some constraints to obtain the data sets which are used to motif finding tool as input. The terms of filtering constraints are listed in Table 3.3.
Table 3.3 The list of filtering term.
Filtering term Condition SNP UTR EST number EST quality Repeat Editing type
Editing site is SNP site or not
Editing site is in 3’UTR, or 5’UTR or CDS Editing site contain which number of EST The identity of EST aligning with mRNA, the identity must be not low.
Editing site is in repeat region or not
Editing site is include in which editing type such as A-to-I, C-to-U…etc
For instance about filtering like this, we choose A-to-I RNA editing type, editing site in repeat region, the site in 3’UTR region, editing site is not SNP site, and each editing site must have ten or more EST sequences support (It means that there are ten or more EST sequences matching mRNA block which contains editing site) and EST quality must be >99% (Quality means that the identity of EST aligns with mRNA) as the constraints, the result data set must conform these after filtering.
The second part is finding motifs through the data sets which come from part one. In each data set, we choose editing sites surrounding two hundred nts to take as input for MEME (MEME is a statistical base tool which can find motif from many sequence). Then, the results of MEME would give us the possible motif sets.
The third part is predicting the secondary structure of the motifs which obtain from the second part. We use the secondary structure
prediction tool Mfold to see the structure of the motifs.
The motifs discovery flow is shown in Fig. 3.9. According to the result of motifs discovery, we could find possible new characteristics in different editing type. And we may suggest biologists the new mechanism of RNA editing through the characteristics surrounding the editing site.
Figure 3.9 The instance of motif discovery flow chart.
3.3.5
Web interface
In order to let users to access EdRNA, we build an interactive web site. In this web site, it offers the interactive browser interface, some statistics charts, tutorials and key words search function. Table 3.4 gives that the filtering options of interactive browser interface and Figure 3.10 shows the interactive browser interface.
Table 3.4 The filtering option of EdRNA interactive browser interface
Option Condition Description
Chromosome 1 to 22, X and Y Which chromosome the editing sites locates in Editing type A to I, C to U, A to C...C to G The type of substitution RNA editing
EST quality >90%, >91%, >92%..., >99% How well that all of the EST aligns to mRNA1 EST support >10, >5, >1 The EST amount that cover each editing site UTR covering 3’ UTR, 5’ UTR, both, none Indicate if all the editing site locates in 3’, 5’ UTR
or both/none
SNP evidence Yes, No All the editing sites should/shouldn’t be a SNP2 Repeat covering Yes, No All the editing sites should/shouldn’t locate in
repeat region Cross-species
conservation
Mouse All the editing sites should be conserved in selected species
1
If its quality is respectively low, than it might implies that the editing site on this EST is an sequencing error
2
SNP is defined in dbSNP, the editing site which is annotated as a SNP is not 100% a SNP, see [17].
Figure 3.10 EdRNA web interface.
By selecting the desired types of RNA editing, EST quality, EST support, UTR covering, SNP evidence, repeat covering and cross-species conservation, the program will filter out the unsuitable editing sites.
After the system retrieves the data required, the multiple sequence alignment between ESTs, mRNA and DNA can be shown by click on the editing region. System would run ClustalW to demonstrate the editing site and its neighboring sequences. Fig. 3.11 and Fig. 3.12 show the graph view of the result through users’ option. Figure 3.11 shows that the editing site in which gene and mRNA.
Figure 3.11 The example of filtering results which contain gene and
mRNA information.
Figure 3.12 shows the alignment result of gene and mRNA and ESTs which contains RNA editing site.
Figure 3.12 The example of filtering results which contain gene, mRNA
and ESTs alignment surrounding the editing site.
Statistics charts are generated dynamically by graph view, which mean that users can set up the specific filtering conditions and system will render it online.
Chapter 4
Results
4.1
The statistics of EdRNA database
First of all, the number of editing site which has more than 1 EST sequence support is 1,008,736 and has more than 100 EST sequences support is 8,889. Each editing site has 14.22 EST sequences support in average. The distribution of the number of editing site which has more than how many EST sequences supports is shown in Figure 4.1.
Figure 4.1 The distribution of the number of editing site which has more
than how many EST sequences support.
support belonging 6,064 genes in EdRNA database. This means that about 17% genes occur RNA editing (There are 34,270 genes now) and each gene has 11.58 editing site in average. Table 4.1 gives the number of editing site of each editing type and the number of gene of each editing site.
Table 4.1 The number of editing site and gene of each editing type.
RNA editing type The number of editing site
The number of gene A/G C/T C/G A/T G/T A/C 22,738 20,406 10,108 5,198 9,051 7,977 3,890 3,598 2,091 1,448 2,218 1,947 As shown in Fig. 4.2, we can find that the distribution of chromosome and the distribution of the number of editing site in each chromosome per mbps are different. For instance, the length of chromosome one is 245,522,847 nts and the length of chromosome nineteen is 63,811,651 nts. The distribution of the number of editing site in one and nineteen per mbps are 1,535 and 3,982. This suggests that RNA editing site occurrence is not random and is meaningful.
Figure 4.2 The distribution of chromosome length and the distribution of
the number of editing site in each chromosome per mbps.
Table 4.2 gives the number of editing site in each repeat type. According to the table result, we can see that there are 67,911 editing site not in repeat regions (about 96.4% of total editing site). This means that the editing site in repeat region is more significant. This is because only 3.6% editing sites are in repeat region.
Table 4.2 The number of editing site in each repeat type.
Repeat type The number of editing site in repeat region SINE/Alu SINE/MIR Simple_repeat LINE/L2 LINE/L1 LTR/ERV1 LTR/ERVL LTR/MaLR trf dust none(not in repeat region) 122 53 284 53 23 16 4 13 321 1,177 67,911
Table 4.3 gives the number of editing site in 3’UTR, 5’UTR and coding region. According to the table result, there are 49,026 editing sites in coding region (about 69.8% of total editing site), 17,475 editing sites in 3’UTR (about 24.8% of total editing site) and 3,745 editing sites in
5’UTR (about 5.3% of total editing site). This suggests that the greater part of RNA editing site occurs in coding region and maybe cause the influence in mRNA translation. In UTR region, the number of editing site in 3’UTR is greater than in 5’UTR.
Table 4.3 The number of editing site in UTR, CDS.
Region(UTR,CDS) The number of editing site in region Coding region 3’UTR 5’UTR 49,026 17,475 3,745
table result, there are 44,034 editing sites which are not SNP site (means the editing site is not SNP). And there are 26,212 editing sites that are SNP site.
Table 4.4 The number of editing site is SNP site or not.
Editing site is SNP or not The number of editing site SNP site
Not SNP site
26,212 44,034
Table 4.5 gives the number of editing in cross-species conserved region (human and mouse). According to the table result, there are 38,401 editing sites in cross-species conserved region (identity>=70%) (about 54.5% of total editing sites). This suggests that over half of total editing sites have evolution meanings. Another word, RNA editing is a very important mechanism in organisms. .
Table 4.5 The number of editing site in cross-species conserved region
(human and mouse). Editing site in cross-species conserved
region or not
The number of editing site
In conserved region Not in conserved region
38,401 32,025
Table 4.6 gives the number of editing site of each RNA editing type in coding region, 5’UTR and 3’UTR. In A/G editing type, the number of editing site in coding region is 16,641(about 73.2% of total editing site)
which is little greater than the percentage of total number of editing site of each editing type in coding region (69.8%). In A/T editing type, the number of editing site in coding region is 3,168 (about 60.9% of total editing site) which is smaller than the percentage of total number of editing site of each editing type in coding region (69.8%). Approximately, the percentage of editing site of each editing type in coding region, 5’UTR and 3’UTR is similar to the percentage of editing site of total editing type in coding region, 5’UTR and 3’UTR.
Table 4.6 The number of editing site of each RNA editing type in coding
region, 5’UTR and 3’UTR. RNA editing type The number of
editing site in coding region The number of editing site in 5’UTR The number of editing site in 3’UTR A/G C/T C/G A/T G/T A/C 16,641 14,773 7,034 3,168 5,666 5,365 1,063 1,171 681 175 558 289 5,034 4,462 2,393 1,855 2,827 2,325
Table 4.7 The number of editing site of each RNA editing type in
cross-species conserved region (human and mouse). RNA editing type The number of editing
site in cross-species conserved region
Total number of editing site A/G C/T C/G A/T G/T A/C 12,791 11,564 5,175 2,611 4,703 4,338 22,738 20,406 10,108 5,198 9,051 7,977
Table 4.7 gives the number of editing site of each RNA editing type in cross-species conserved region (identity>=70%). In A/G editing type, the number of editing site in cross-species conserved region is 12,791 (about 56.2% of total editing site) which is little greater than the percentage of total number of editing site of each editing type in cross-species conserved region (54.5%). In C/G editing type, the number of editing site in cross-species conserved region is 5,175 (about 51.2% of total editing site) which is little smaller than the percentage of total number of editing site of each editing type in cross-species conserved region (54.5%). Approximately, the percentage of editing site of each editing type in cross-species conserved region is similar to the percentage of editing site of total editing type in cross-species conserved region.
4.2 The analysis of EdRNA database
In this section, we perform some analysis from EdRNA database. We filter each type editing site from EdRNA database by some conditions. The conditions are that the identity which the alignment of mRNA and EST must be > 95%, the editing site must have more than 30 EST sequence support, the editing site must be in the coding region and not the SNP site. Table 4.6 gives the editing site number of each editing type through filtering condition.
In each editing type set, we adopt two hundred nts around editing site sequence to the MEME (Motif discovery tool) inputs and obtain some analysis result.
Table 4.8 The number of editing site in each editing type through some
filtering conditions.
RNA editing type The number of editing site A/G A/C A/T C/T C/G G/T 447 97 87 356 134 113
4.2.1 Different genes have the same motifs
According to the result, we find some different genes have the same motifs.
In A-to-T editing type, we find two genes have the same motifs. The two genes information is listed in Table 4.9. And the motifs are shown in Fig. 4.3. In this case, the editing site is contained in the motifs. The length of motifs is 80 nts. One gene is NDUFV2 and the other is UBE1C.
Table 4.9 Gene information in A-to-T editing type (two different genes).
Gene Symbol mRNA NDUFV2
UBE1C
CR456928 CR533537
Figure 4.3 The motifs are in the two mRNA. Editing site locates in
motif1.
In C-to-G editing type, we find two genes have the same motifs. The two genes information is listed in Table 4.10. And the motifs are shown in Fig. 4.4. In this case, the motifs are behind the editing site. The length of motifs is 50 nts. One gene is GTPBP4 and the other gene is ATP5J.
Table 4.10 Gene information in C-to-G editing type (two different genes).
Gene Symbol mRNA GTPBP4
ATP5J
AF325353 M73031
Figure 4.4 The motifs are located in the two mRNA. Editing site locates
in front of the motifs.
4.2.2 Gene family have the same motifs
In addition, we find some gene family have the same motifs surrounding the RNA editing site.
In A-to-C editing type, we find three genes have the same motifs. The three genes information is listed in Table 4.11. And the motifs are shown in Fig. 4.5.
Table 4.11 Gene information in A-to-C editing type (three genes).
Gene symbol mRNA HLA-B HLA-C HLA-E D49820 U29083 BC004297
Figure 4.5 The motifs are in the three mRNA. Editing site locates in
motif 2.
The three genes are HLA (human leukocyte antigen) genes. The HLA system is genetically encoded in humans by the major histocompatibility complex (MHC), which is found on chromosome 6, and plays a determining role in immunity and in self-recognition in virtually all cells and tissues, with the exception of erythrocytes.2
For instance, HLA-B7 antigen is associated with the diseases of narcolepsy and idiopathic hermochromatosis. If the RNA editing occurs, the influence of RNA editing may cause impact in HLA-B7 expression.
In G-to-T editing type, we find three genes which have the same motifs. The three genes information is listed in Table 4.12. And the
2
motifs are shown in Fig. 4.6.
Table 4.12 Gene information in G/T editing type(three genes).
Gene symbol mRNA KRT14 KRT 16 KRT 19 BC002690 S72493 Y00503
Figure 4.6 The motifs are in the three mRNA. Editing site locates in
motif 2.
The three genes are KRT (keratin) genes. The keratins are intermediate filament proteins responsible for the structural integrity of epithelial cells and are subdivided into cytokeratins and hair keratins. Most of the type I cytokeratins consist of acidic proteins which are arranged in pairs of heterotypic keratin chains and are clustered in a region of chromosome 17q12-q21. This keratin has been co-expressed with keratin 14 in a number of epithelial tissues, including esophagus, tongue, and hair follicles3.
4.2.3 The summary of analysis result
According to the analysis result, we could find the same motifs in different genes or in gene family. And some motifs are behind the editing site or some motifs contain editing site. The same motifs occurring in different genes are more meaningful than the same motifs occurring in gene family. This is because different genes occurs RNA editing by confirming the same motifs. This suggests that if we find the same motifs in some other genes, these genes maybe also occur RNA editing. Oppositely, the same motifs occur in gene family is as a matter of course. But these maybe imply that gene family is regulated by the same RNA editing mechanism.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene&cmd=Retrieve&dopt=full_report&list_uids= 3868
Chapter 5 Discussion
5.1 Comparison
In A-to-I RNA editing, EdRNA database compares with the prediction site from the web site4 [4]. EdRNA can map 828 editing site from 1104 editing site. The reason of other editing site which can not be mapped is the mRNA and EST sequence version. In these editing site that can not be mapped, many old mRNA sequence are different with the mRNA sequence which store in database and many EST sequence can not be found in database now. The literature [4] describes that their prediction false positive rate is very low (near 1.9%). This means that our prediction may have many verily possible real editing sites. And EdRNA have more editing sites that this literature not offers.
5.2 Drawbacks
EdRNA exploited lots of available experiment verified sequence data from renowned databases (dbEST, NCBI, EMBL, ensembl) to reduce the time in doing redundant sequencing. However, the quality of sequencing is not perfect, so spurious RNA editing sites are unavoidable.
However, this work is not complete enough to cover all possible
substitution RNA editing sites among human, since there are still lots of genomic region lacked of the coverage of EST sequences. Nevertheless, some of the RNA editing events might occur in very low level, which implies that the materials are still not ample enough to provide the sufficient sensitivity (i.e., certain site is edited, but no data shown this happened).
5.3 Future Works
In many other species, like some literature cataloging ([34],[35]), are easily to use machine learning approaches to predict novel RNA editing sites since there are abundant known RNA editing sites for training the classifiers. These approaches provide more possibility in finding RNA editing sites. Once the verified RNA editing sites is getting increasing because of the promising experiment materials providing from EdRNA, the plenty data will be adequate for performing knowledge discovery and data mining approaches. This can complete the missing part of the methodology that used by EdRNA, since EdRNA cannot detect the RNA editing without any evidence, i.e. no any existing sequences show the variance.
By knowledge discovery, data mining and machine learning, a much more omnipotent classifier will show up and predict all possible putative
RNA editing sites and contribute more in understanding the mechanism. So the perspective goal is focus on verifying the data stored in our database to provide further material doing data mining.
Based on the ample RNA editing sites from EdRNA, advanced investigation about relative biological process is consequently proposed. The relationship between editing and alternative splicing or other gene regulation mechanisms now acquired much more quantity and quality of information for doing assay in depth.
5.4 Experimentally Confirmation
EdRNA plans to use some filter conditions such as EST number, EST quality (the identity of EST aligning with mRNA) and so on to establish most possible believable RNA editing site sets. Furthermore, we plan to perform experiment to confirm the EdRNA database through these editing site sets. We think that if the editing site can be proved by experiment, we can not only convince others the correctness of EdRNA but also maybe discovery RNA editing mechanism such as A-to-C, C-to-G, and so on.
Chapter 6 Conclusions
EdRNA is the first and most comprehensive database in the world contained abundant possible substitution RNA editing sites in all types by examining all available human nucleotide sequences and provides wide annotations. The interactive web site interface is convenient and efficient to retrieve the annotated RNA editing sites and needn’t bother to perform the additional filtering process by users. EdRNA provides biologists the reliable materials to discover novel RNA editing sites by experimental methodology. Also, the motif discovery of different type of RNA editing proposes a promising sequence pattern, which suggest biologist the possible RNA editing mechanism. There are now no other existing and well-known databases contributing equally to this work.
Furthermore, human RNA editing types which are proven in lately research are relatively fewer than other species, only A-to-I and C-to-U editing. Albeit there is no existing evidence supports the occurrence of other types of RNA editing, this work still found large amount of editing sites with this kind of types, and most of them are supported with many EST sequences. Therefore, it is reasonable to assume that other types of RNA editing (e.g. C-to-A, T-to-C) perform take place in human. Further, sequence motif and structure motif discovery are both perform to suggest promising patterns in different types of RNA editing.
References
1. Smith, H.C., J.M. Gott, and M.R. Hanson, A guide to RNA editing. Rna, 1997. 3(10): p. 1105-23.
2. Gott, J.M., Expanding genome capacity via RNA editing. C R Biol, 2003.
326(10-11): p. 901-8.
3. Keegan, L.P., A. Gallo, and M.A. O'Connell, The many roles of an RNA editor. Nat Rev Genet, 2001. 2(11): p. 869-78.
4. Levanon, E.Y., et al., Systematic identification of abundant A-to-I editing sites
in the human transcriptome. Nat Biotechnol, 2004. 22(8): p. 1001-5.
5. Samuel, C.E., RNA editing minireview series. J Biol Chem, 2003. 278(3): p.
1389-90.
6. Higuchi, M., et al., Point mutation in an AMPA receptor gene rescues lethality
in mice deficient in the RNA-editing enzyme ADAR2. Nature, 2000. 406(6791):
p. 78-81.
7. Palladino, M.J., et al., A-to-I pre-mRNA editing in Drosophila is primarily
involved in adult nervous system function and integrity. Cell, 2000. 102(4): p.
437-49.
8. Tonkin, L.A., et al., RNA editing by ADARs is important for normal behavior
in Caenorhabditis elegans. Embo J, 2002. 21(22): p. 6025-35.
9. Wang, Q., et al., Requirement of the RNA editing deaminase ADAR1 gene for
embryonic erythropoiesis. Science, 2000. 290(5497): p. 1765-8.
10. Yamanaka, S., et al., Apolipoprotein B mRNA-editing protein induces
hepatocellular carcinoma and dysplasia in transgenic animals. Proc Natl
Acad Sci U S A, 1995. 92(18): p. 8483-7.
11. Cattaneo, R., et al., Measles virus editing provides an additional cysteine-rich
protein. Cell, 1989. 56(5): p. 759-64.
12. Cattaneo, R., et al., Biased hypermutation and other genetic changes in
defective measles viruses in human brain infections. Cell, 1988. 55(2): p.
255-65.
13. Maas, S., et al., Underediting of glutamate receptor GluR-B mRNA in
malignant gliomas. Proc Natl Acad Sci U S A, 2001. 98(25): p. 14687-92.
14. Sodhi, M.S., et al., RNA editing of the 5-HT(2C) receptor is reduced in
schizophrenia. Mol Psychiatry, 2001. 6(4): p. 373-9.
15. Niswender, C.M., et al., RNA editing of the human serotonin 5-HT2C receptor.
alterations in suicide and implications for serotonergic pharmacotherapy.
Neuropsychopharmacology, 2001. 24(5): p. 478-91.
16. Akbarian, S., M.A. Smith, and E.G. Jones, Editing for an AMPA receptor
subunit RNA in prefrontal cortex and striatum in Alzheimer's disease, Huntington's disease and schizophrenia. Brain Res, 1995. 699(2): p. 297-304.
17. Eisenberg, E., et al., Identification of RNA editing sites in the SNP database. Nucleic Acids Res, 2005. 33(14): p. 4612-7.
18. Benson, D.A., et al., GenBank. Nucleic Acids Res, 1998. 26(1): p. 1-7.
19. Kulikova, T., et al., The EMBL Nucleotide Sequence Database. Nucleic Acids Res, 2004. 32(Database issue): p. D27-30.
20. Shin'i, T. and T. Gojobori, [DDBJ database and genetic information analysis
softwares]. Tanpakushitsu Kakusan Koso, 1994. 39(11): p. 1927-43.
2002. 30(1): p. 38-41.
22. Boguski, M.S., T.M. Lowe, and C.M. Tolstoshev, dbEST--database for
"expressed sequence tags". Nat Genet, 1993. 4(4): p. 332-3.
23. Wheeler, D.L., et al., Database resources of the National Center for
Biotechnology Information: update. Nucleic Acids Res, 2004. 32(Database
issue): p. D35-40.
24. Karolchik, D., et al., The UCSC Genome Browser Database. Nucleic Acids Res, 2003. 31(1): p. 51-4.
25. Sherry, S.T., et al., dbSNP: the NCBI database of genetic variation. Nucleic Acids Res, 2001. 29(1): p. 308-11.
26. Altschul, S.F., et al., Basic local alignment search tool. J Mol Biol, 1990. 215(3): p. 403-10.
27. Thompson, J.D., D.G. Higgins, and T.J. Gibson, CLUSTAL W: improving the
sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic
Acids Res, 1994. 22(22): p. 4673-80.
28. Florea, L., et al., A computer program for aligning a cDNA sequence with a
genomic DNA sequence. Genome Res, 1998. 8(9): p. 967-74.
29. Bailey, T.L. and C. Elkan, Fitting a mixture model by expectation
maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol
Biol, 1994. 2: p. 28-36.
30. Mathews, D.H., et al., Expanded sequence dependence of thermodynamic
parameters improves prediction of RNA secondary structure. J Mol Biol, 1999.
288(5): p. 911-40.
31. Zuker, M., Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res, 2003. 31(13): p. 3406-15.
32. Zuker, M. and A.B. Jacobson, Using reliability information to annotate RNA
secondary structures. Rna, 1998. 4(6): p. 669-79.
33. Blow, M., et al., A survey of RNA editing in human brain. Genome Res, 2004. 14(12): p. 2379-87.
34. Gott, J.M., N. Parimi, and R. Bundschuh, Discovery of new genes and deletion
editing in Physarum mitochondria enabled by a novel algorithm for finding edited mRNAs. Nucleic Acids Res, 2005. 33(16): p. 5063-72.
35. Thompson, J. and S. Gopal, Genetic algorithm learning as a robust approach