行政院國家科學委員會專題研究計畫 成果報告
新生鼠給予抗憂鬱劑-百憂解可保護新生兒期窒息腦傷及拮
抗青少年期之古柯鹼成癮(3/3)
計畫類別: 個別型計畫 計畫編號: NSC94-2314-B-006-003- 執行期間: 94 年 08 月 01 日至 95 年 07 月 31 日 執行單位: 國立成功大學醫學系小兒科 計畫主持人: 黃朝慶 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 95 年 10 月 30 日
行政院國家科學委員會補助專題研究計畫
成果報告
新生鼠給予抗憂鬱劑-百憂解可保護新生兒期窒息腦傷及拮抗青少
年期之古柯鹼成癮(3/3)
計畫類別:
□
個別型計畫 □ 整合型計畫
計畫編號:NSC94-2314-B-006 -003
執行期間:
94 年 08 月 01 日至 95 年 07 月 31 日
計畫主持人:黃朝慶
共同主持人:郭余民
計畫參與人員:
成果報告類型(依經費核定清單規定繳交):□精簡報告
□
完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:成功大學醫學院小兒科
中 華 民 國 95 年 10 月 29 日
I 中文摘要 關鍵詞:百憂解、窒息腦傷、水迷宮 外界環境或藥物對發展中的生物可透過其生理狀態之可塑性的改變,進而改變此 生物對環境之適應性。發展中的腦部之可塑性特別敏感,尤其是週產時期更易受到壓 力、內毒素、生長激素或轉錄因子的影響(programming)。cAMP response
element-binding protein (CREB) 為一重要之細胞轉錄因子,在神經系統之發展及其可 塑性扮演一重要角色,且在學習及記憶形成,成癮藥上癮過程及對抗窒息缺氧之細胞
存活機制均極為重要。細胞受到刺激時,很容易活化此因子,在Ser133 之位置受磷
酸化後,進而促進神經信息傳遞,細胞突觸可塑性,細胞內信息傳遞等基因的表現。
抗憂鬱劑之使用可促進成鼠腦部海馬回及大腦皮質CREB 之活化,特別是血清素
(serotonin)回收抑制劑-百憂解,特具此功能,也有研究顯示百憂解透過 CREB 之 活化來促進brain-derived nerve growth factor (BDNF)之作用,而百憂解之抗憂鬱作用 一般認為與此機制有關。 新生鼠使用百憂解是否對其腦部之神經可塑性有長期作用,至今未明。我們在新生兒 期使用低劑量百憂解前處理可在行為上看到其對抗新生鼠缺氧缺血腦傷之神經保護作 用,但在高劑量之下卻有惡化神經功能之表現,我們進一步分析發現低劑量之百憂解 可促進pCREB 之表現以及增加再生神經細胞之存活能力;這些發現均意味著活化 CREB 來促進神經可塑性及再生能力,可為改善新生兒腦傷之重要機制。
II
Abstract
Neuroplasticity after perinatal programming may allow for neuroprotection against hypoxic-ischemia (HI) at birth. The cAMP response element-binding protein (CREB) is a key mediator of stimulus-induced nuclear responses that underlie survival, memory and plasticity of nervous system. Chronic treatment of fluoxetine, a selective serotonin reuptake inhibitor, can up-regulate CREB activation in the hippocampus. We examined whether fluoxetine administration before HI may protect against neonatal HI brain injury through CREB-mediated mechanisms. We found that low-dose fluoxetine pretreatment in a neonatal HI brain injury model significantly reduced functional deficits at adulthood. The
neuroprotective mechanisms were associated with increased CREB phosphorylation, and increased brain-derived neurotrophic factor and synapsin I mRNA expression in the
hippocampus. Neurogenesis also increased because of greater precursor cell survival in the hippocampal dentate gyrus. These findings suggest that functional deficits after HI in the developing brain can be reduced by agents that enhance neural plasticity and neurogenesis through CREB activation.
Keywords: fluoxetine; cAMP response element-binding protein; brain-derived neurotrophic
III
目錄
中文摘要 --- I 英文摘要 --- II 前言 --- 1 研究方法 --- 4 結果 --- 10 討論 --- 16 參考文獻 --- 24 圖 --- 331
Introduction
Hypoxic-ischemia (HI) encephalopathy is a major cause of neonatal mortality and neurological disabilities in childhood (Huang et al, 1999; Volpe, 2001; Ferriero, 2004). Our understanding of the pathogenesis of neonatal HI brain injury has increased considerably over the last decades (Volpe, 2001; Ferriero, 2004); there is, however, no effective clinical
treatment. Perinatal plasticity of physiological systems may allow environmental factors or drugs to alter the “set-point” or to “hard-wire” the developmental plasticity of the
physiological system by which organisms can adapt their physiological responses to better meet environmental demands (Welberg and Seckl, 2001). The brain is a key target for such effects and is sensitive to perinatal programming by agents known to permanently affect brain development, such as stress, enrichment or handling, and steroids (Shanks and Lightman, 2001; Welberg and Seckl, 2001). Neuroplasticity after perinatal programming may allow for neuroprotection against HI at birth.
The cAMP response element-binding protein (CREB), a transcription factor, is a key mediator of stimulus-induced nuclear responses that underlie cell survival, regeneration, learning and memory, and the plasticity of the nervous system (Lonze and Ginty, 2002, Chang and Huang, 2006). CREB is phosphorylated on Ser133 (pCREB) and subsequently binds to the cAMP response element (CRE) of target genes, which controls
CREB-dependent gene expressions, such as brain-derived neurotrophic factor (BDNF) (Walton and Dragunow, 2000; Lonze and Ginty, 2002). Our previous study showed that carotid-artery ligation preconditioning against HI in the neonatal brain is associated with robust and sustained phosphorylation of CREB (Lee et al, 2004). Furthermore,
pharmacological activation of the cAMP-CREB signaling pathway protected the neonatal rat brain through a sustained increase in pCREB expression.
2
action of chronically administered antidepressants against depression (Nibuya et al, 1996; Thome et al, 2000). CREB activation in the hippocampus of both rodents and humans is upregulated by chronic treatment with fluoxetine, a selective serotonin reuptake inhibitor (SSRI) (Nibuya et al, 1996; Dowlatshahi et al, 1998; Thome et al, 2000). Temporal and regional upregulation of BDNF mRNA and its receptor, trkB, parallels CREB activation after chronic fluoxetine administration, and these effects may be important for its
antidepressant effects (Nibuya et al, 1995; Saarelainen et al, 2003). It therefore raises the possibility that chronic fluoxetine treatment may protect against HI injury in the neonatal brain through CREB-mediated mechanisms.
The cellular mechanism underlying the long-term effect of antidepressants in the adult brain include increased neurogenesis, dendritic arborization, and synaptogenesis (Malberg et al, 2000; Nakagawa et al, 2002; Nestler et al, 2002).Furthermore, chronic SSRIs treatment influences the proliferation as well as the maturation, differentiation, and survival of newborn neurons in adult rats (Malberg et al, 2000; Fujioka et al, 2004). Recently, the newborn
neurons in the rodent brain after stroke injury or learning were shown to be actively
integrated into the existing circuitry and to contribute to ameliorating neurological deficits or forming hippocampal-dependent memory (Gould et al, 1999; Nakatomi et al, 2002). Whether a chronic regimen of fluoxetine can protect against HI damage in rat pups by enhancing neurogenesis and synaptogenesis has not been examined.
Despite the strong evidence for the neuroplasticity benefits of the SSRIs in adult rats, the neuroprotective effects of fluoxetine against neonatal HI brain injury have not been investigated. Fluoxetine facilitates motor recovery in stroke patients undergoing
rehabilitation (Dam et al, 1996; Pariente et al, 2000), and improves cognition after traumatic brain injury (Horsfield et al, 2002). Furthermore, it is currently the only SSRI approved for pediatric use in many countries. Therefore, we used an established rat pup model of HI brain
3
injury to determine the effects and underlying mechanisms of early-life exposure to fluoxetine on neural plasticity after HI brain injury in neonatal rats. In this study, we
examined the following hypotheses: 1) fluoxetine pretreatment is neuroprotective against HI damage in the neonatal brain, and 2) the neuroprotective mechanism of fluoxetine
pretreatment involves a) increased phosphorylation of CREB, b) increased CREB-mediated gene expressions, and c) enhanced neurogenesis in the hippocampus.
4
Materials and Methods
This study was approved by the Animal Care Committee at National Cheng Kung University Medical College. Ten to twelve Sprague-Dawley pups per dam were used and housed with a 12/12-hour light/dark schedule in a temperature- and humidity-controlled colony room. The pups were housed with their dams until weaning on postnatal (P) day 21, and then housed in groups of 4-5 per cage.
Experimental Groups
Hypoxic-ischemia. From P1 to P7, rat pups of either sex received daily intraperitoneal injection of fluoxetine (Sigma, St. Louis, MO) or normal saline. Animals received a 100-μl solution containing either saline (Ns-HI group) or fluoxetine: 5 mg/kg (5-Flu-HI group) or 15 mg/kg (15-Flu-HI group) dissolved in saline. On P7, three hours after the last injection, rats pups were subjected to HI (right common carotid artery ligation followed by 8% oxygen hypoxia for 2.5 hours), as described previously (Lee et al, 2004). There were two control groups: sham controls (sham-operated rats without hypoxia) and normal saline controls (Ns group, daily normal saline injection from P1 to P7 without exposure to HI).
On P7, the Ns-HI-, 5-Flu-HI-, and 15-Flu-HI-group animals were anesthetized with 2.5% halothane (balance, room air), and the right common carotid artery was surgically exposed and permanently ligated with 5-0 surgical silk. After surgery, the pups were returned to the dam for a 1-hour recuperation period before HI. The animals were then placed in airtight 500-ml containers partially submerged in a 37°C water bath through which
humidified 8% oxygen (balance, nitrogen) was maintained at a flow rate of 3 L/min for 2.5 hours. After completion of HI, the rat pups were returned to their cages. Behavioral
assessment by Morris water maze (MWM) was performed from P35 to P40, and pathological examination by hemispheric weight reduction determined at P42.
5
long-term behavioral effect of adult rats with early-life fluoxetine exposure, two groups of male animals not subjected to HI but given daily saline (Ns group) or 5-mg/kg fluoxetine (5-Flu group) administrations from P1 to P7 were included. Behavioral assessments included MWM from P35 to P40, open field test on P45, and elevated plus maze test on P50.
Outcome Measure- Behavioral Assessment
Morris water maze (MWM). Rats undertake the Morris water maze test as described previously with some modification (Balduini et al, 2001; Chang et al., 2003; Lee et al., 2004). In brief, rats were trained for 8 sessions in the hidden platform version (4 sessions acquisition phase followed by 4 sessions reversal phase with the platform in a new location). Each session consists of 4 trials with 4 different starting positions. Afterward, rats were tested for 4 trials with the platform marked with a flag (visible test) and move to a new location for every trial. To assess if the animals had remembered the location of platform or whether they used nonspatial strategies to find the platform, each learning phase was followed by a probe trial, in which the platform was removed. Escape distance, escape latency, swimming speed, and swimming patterns of the rats were monitored by a camera mounted above the pool and connected to a computer program (EthoVision, Wageningen, The Netherlands).
Locomotion in open field test. Rat locomotion (including horizontal ambulation and vertical rearing) was assessed in a custom-made transparent Plexiglas box (41 cm × 41 cm × 30 cm) with photocell activity monitors (Opto-Varimex; Columbus Instruments). Two sets of infrared lamps and photocells were mounted on the horizontal and the vertical edges of the
Opto-Varimex. Under illumination from a bright light, rats were individually placed in the center of the box and allowed free navigation for 10 minutes. Each vertical beam break was counted as one incident of vertical rearing, and the horizontal distance traveled was recorded as the ambulation (Ansorge et al, 2004).
6
black Plexiglas runway with two opposing arms (32 cm × 6 cm) closed by 15-cm side walls and the other two arms open. Rats were placed, facing one of the closed arms, at the center of the platform (6 cm × 6 cm) of the maze and allowed to explore the maze for 5 minutes. The latency for the first entry made onto the open arms with two front paws and the time spent in these open arms were individually recorded.
Outcome Measure-Pathological Assessment
Hemispheric weight reduction at P42. Our previous study showed that, in P7-rats with cerebral HI, changes in hemispheric weight reduction were highly correlated not only with changes in the brain areas, and but also with hemispheric volume changes measured on P36 (Lee et al, 2004). Therefore, only the hemispheric weight data are shown in the present study. After they had been anesthetized with pentobarbital (50 mg/kg, i.p.), rats were decapitated and the brain was removed. The brainstem and cerebellum were removed, the forebrain was sectioned at the midline, and left and right hemispheric weights were determined. The percentage of hemispheric weight reduction, measured as [(left hemisphere weight − right hemisphere weight)/left hemisphere weight], was used as the measure of cerebral injury. Immunohistochemical Stain for Neurogenesis
Bromodeoxyuridine (BrdU) labeling. To investigate the effects of fluoxetine treatment specifically on neurogenesis, male rat pups with daily administration of saline or 5-mg/kg fluoxetine from P1 to P7 were given a single intraperitoneal injection of BrdU (100 mg/kg; Sigma, St. Louis, MO) 30 minutes after the last dose of saline or fluoxetine on P7. For the proliferation study, the animals (Ns and 5-Flu groups) were killed 3 hours after BrdU injection to study the number of newly-formed cells in the brain. To determine the survival and cell phenotypes of the newly born cells after HI injury, Ns-HI- and 5-Flu-HI-group rats were not killed until 14 days after the BrdU injection and HI. After perfusion, brains were post-fixed overnight in paraformaldehyde at 4°C and stored at 4°C in 30% sucrose. Serial
7
sections of the brains (30 μm thick) were cut on a freezing microtome.
BrdU immunostaining. BrdU staining was described previously (Tzeng and Wu, 1999). In brief, the sections were heated (37°C for 1 hour), incubated in 50% formaldehyde (65°C for 1 hour), 2 M HCl (37°C for 30 minutes), rinsed in 0.1 M boric acid (pH 8.5) for 10 minutes, incubated in 1% H2O2 in PBS for 30 minutes, and blocked in 3% normal rabbit serum/0.3%
Triton X-100 in PBS (room temperature for 1 hour), followed by incubation with goat anti-BrdU (1:200; Accurate Chemical & Scientific Corporation, Westbury, NY) at 4°C overnight. Subsequently, the sections were incubated with anti-goat secondary antibody (1:200; Santa Cruz Biotechnology, Inc, Santa Cruz, CA) and developed with an ABC kit (Vectastain; Vector Laboratories, Burlingame, CA).
Quantification. BrdU(+) cells in every sixth section in a series of 30-μm coronal sections of dorsal hippocampus was counted by a blinded examiner. Labeled cells were counted in the area encompassing the entire granule cell layer (GCL) and extending approximately one to two cell layers deep into the hilus and the hilus area. For cellular counting, images were magnified 100× and captured digitally using an image analysis system (Image-Pro Plus; Media Cybernetics, Inc., Silver Spring, MD). Area measurements of both the dentate GCL and hilus were made from each slide used for the cell counts. The density of BrdU(+) cells in each section was calculated by dividing the numbers of BrdU(+) nuclei by the area. Density for the five to seven sections was averaged to obtain a mean density value for each animal. The volume of the hippocampal dentate gyrus was determined by summing the traced areas for each section multiplied by the distance between sample sections. We also examined the BrdU(+) cells in the subventricular zone (SVZ) present on every 10th coronal section. Immunofluorescent co-localization analysis by laser-scanning confocal microscopy. To determine phenotype, double immunofluorescence was performed. Each coronal section was first treated with primary BrdU antibody, followed by treatment with cell-specific antibodies:
8
GFAP for astrocytes (1:400; Sigma), Neu-N for neuronal nuclei (1:200; Chemicon International, Inc., CA) staining. The tissue sections were analyzed with a Carl Zeiss LSM510 laser-scanning confocal microscope, and green (FITC) and red (rhodamine)
fluorochromes on the slides were excited by laser beam at 488 and 543 nm, respectively. We counted 100 BrdU(+) cells in the hippocampal GCL per animal to obtain the percentage of BrdU(+) cells co-localized with NeuN.
Western blot analysis
Tissue was homogenized in cold lysis buffers as described previously (Lee et al, 2004; Chang et al, 2005). Fifty-microgram samples were resolved in 10% SDS-PAGE gels and blotted electrophoretically to nitrocellulose membranes. Membranes were incubated with primary antibodies, and immunoreactivity was detected using horseradish-conjugated secondary antibody or mouse immunoglobulin G antibody (1:5000; Chemicon) and
visualized with enhanced chemiluminescence. The optical density of pCREB was normalized to that of the total CREB in each sample. The following primary antibodies were used: anti-CREB (1:1000; Upstate Biotechnology, Charlottesville, VA), anti-pCREB (1:1000; Upstate Biotechnology), and β-actin (1:1000; Chemicon).
Reverse transcription (RT)-PCR
Total RNA was extracted according to the TRIzol protocol (Invitrogen, San Diego, CA), as described previously (Lee et al, 2004). The purity and integrity of the RNA samples were assessed by OD260/OD280 spectrophotometric measurements and by 1% agarose gel electrophoresis. A 5-μg portion of total RNA and 1.5 μg oligo-dT primers were incubated at 70°C for 10 minutes and gradually cooled to room temperature. Each RT mixture containing 25 units of M-MLV reverse transcriptase (Promega, Madison, WI), 10 μl 5× reaction buffer, 0.5 mM dNTP, and nuclease-free distilled water was added to a final volume of 50 μl. The samples were incubated at 37°C for 90 minutes, followed by enzyme denaturation at 95°C for
9
10 minutes. Each PCR (20 μl) contained 2 μl of RT product, 1 unit of Taq DNA polymerase (Viogene, Taipei, Taiwan), 2 μl of 10× PCR buffer plus MgCl2, 0.2 mM dNTP, and 0.5 μM of
gene-specific primers (BDNF exon V: forward 5’-GACAAGGCAACTTGGCCTAC-3’, reverse 5’-CTGTCACACACGCTCAGCTC-3’, product size: 356 bp; BDNF exon III: forward 5’-ATGAAATCTCCCAGTCTC-3’, reverse 5’-AGTAAACATCAAGGCAGC-3’, product size:264 bp; Synapsin I: forward 5’-CTCAGAAACCCAGCCAGG-3’, reverse 5’-AGAAATGGACTCAGGACCC-3’, product size 250 bp; GAPDH: forward
5’-ACATTGTTGCCATCAACGAC-3’, reverse 5’-ACGCCAGTAGACTCCACGAC-3’, product size: 216 bp). The amplified reaction was performed using a thermocycler for a single 3-minute initial denaturation at 94°C followed by 32 cycles (BDNF exon III), 30 cycles (BDNF exon V), and 31 cycles (Synapsin I) or 20 cycles (GAPDH) under the
conditions: 94°C (20 seconds), 53°C (20 seconds), 72°C (20 seconds); and a final extension at 72°C for 4 minutes. The PCR products were separated on 1.5% agarose gels containing ethidium bromide, and quantified using scanning densitometry. The PCR products of BDNF and synapsin I were normalized to that of the PCR product of GAPDH in each sample. Statistics
Repeated measures ANOVA was employed for MWM learning where days or quadrants were treated as a within-subject factor and groups as a between-subject factor. The effects of sex on MWM learning were examined as covariant in repeated measures ANOVA analysis. Statistical significance (p < 0.05) was determined using one-way ANOVA. When data were not normally distributed, analyses were performed using a non-parametric ANOVA by ranks (Kruskal-Wallis one way test) followed by Dunn's multiple comparison tests. Continuous data are expressed as mean ± standard error of the mean (SEM) unless indicated otherwise.
10
Results
High-dose fluoxetine pretreatment exaggerated, but low-dose fluoxetine protects against, hypoxic brain damage at the behavioral level
Abnormal behaviors were seen immediately after fluoxetine injection in 70% of the 15-mg/kg-fluoxetine-treated-group rats. The rat pups became agitated and showed
hyperactivity, head shaking, clonic twitching of hind-limbs, wide-running, myoclonic jerk, ataxic walking, circling, or falling, which gradually subsided within 30 minutes after
injection. In contrast, none of these adverse behaviors were seen in the 5-mg/Kg-fluoxetine- and Ns-treated group rats. There were no significant differences in the degree of rectal temperature measured before and after HI among the three HI groups. There were also no significant differences in body weight from P1 to P7 or from P14 to P28 between the Ns-HI, 5-Flu-HI, 15-Flu-HI, sham control, and Ns groups.
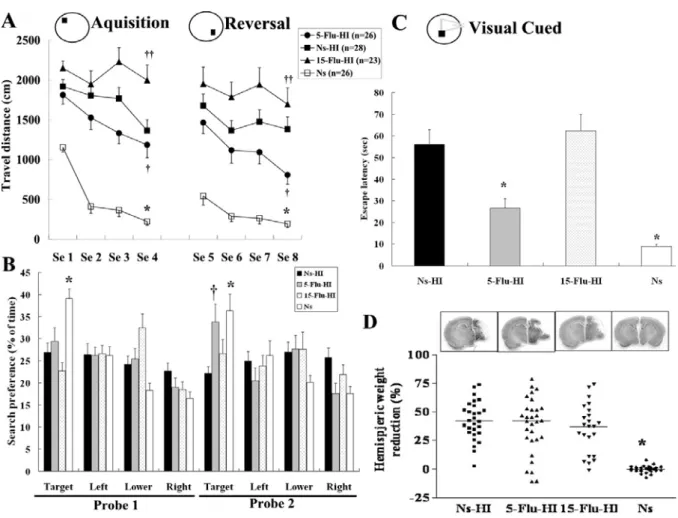
During the acquisition phase of the MWM, all groups except the 15-Flu-HI group learned to find the submerged platform (p < 0.001, F=55.8). The 15-Flu-HI-group rats had to be categorized as complete non-learners because their travel distance did not decrease over time. The total distance covered to reach the submerged platform was significantly different between the Ns-HI, 5-Flu-HI, 15-Flu-HI, sham control, and Ns groups (p < 0.001, F=74.2), and there was no interaction between the training sessions and treatments. Post hoc multiple comparisons revealed that the Ns-HI-group rats traveled significantly longer distances when finding the platform than did the sham control and Ns- (both p < 0.001) or the
5-Flu-HI-group (p < 0.05) rats. In contrast, the 15-Flu-HI group traveled a significantly longer distance searching for the submerged platform than did the Ns-HI group (p < 0.01). There was also a significant difference between the 5-Flu-HI and the Ns groups (p < 0.001). During the reversal learning phase, all the three HI groups traveled significantly longer distances than did the sham control and Ns-group (p < 0.001, F=54.8). The 5-Flu-HI-group
11
traveled a significantly shorter distance finding the platform than the Ns-HI group did (p < 0.05). The 15-Flu-HI group, however, travel a significantly longer distance than the Ns-HI group (p < 0.01) (Fig. 1A).
During probe test 1, Ns-group and sham control rats spent more time in the target quadrant than did the three HI-group rats (p < 0.001, F=12.9). There was no significant difference between the 5-Flu-HI, 15-Flu-HI and Ns-HI groups. However, in probe test 2, the 5-Flu-HI group showed much improvement and spent significantly more time searching in the new target quadrant than did the Ns-HI group (p < 0.05). There was no significant difference between the 5-Flu-HI and the Ns groups. There was no significant difference between the 15-Flu-HI and Ns-HI groups in either probe test (Fig. 1B). In the cued version, there was a significant difference (p < 0.001, F=19.8) between the five groups in time spent reaching the visible platform. The 5-Flu-HI group spent significantly (p < 0.001) less time than the Ns-HI group finding the visible platform, but they spent significantly (both p < 0.01) more time than the Ns and sham control groups. There was no significant difference between the 15-Flu-HI and Ns-HI groups in time spent reaching the visible platform (Fig. 1C).
We then tested whether there was gender-dependent effect on how fluoxetine pretreatment affects functional recovery after perinatal HI brain injury. The male/female distribution was similar in each group. In both acquisition and reversal learning, the treatment effects across time existed in both male and female but the effects across time were similar in both sex. There were also no significant effects of sex on the probe tests and visual-cued tests by two-way ANOVA analysis.
The degree of brain injury, as measured by hemispheric brain weight reduction, showed no significant difference between the 5-Flu-HI, 15-Flu-HI, and Ns-HI groups (Fig. 1D). The degree of hemispheric weight reduction was unaffected by sex, in concordance with the recent results showing that no sex differences were found in the extent of brain lesion after HI
12
injury in neonatal mice (Zhu et al, 2006) and rats (Yager et al, 2005). Therefore, we found that a low dose of fluoxetine protected rat pups from HI brain damage at functional levels, but that a high dose exaggerated the damage.
Low-dose fluoxetine pretreatment increased posthypoxia CREB phosphorylation in the hippocampus and cortex
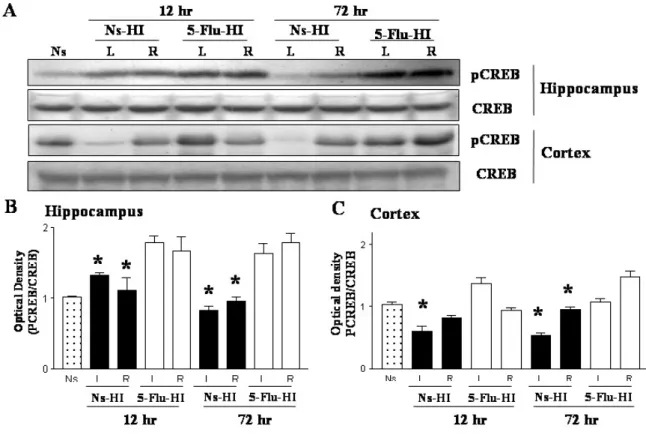
Chronic fluoxetine treatment in adult rats increases CREB phosphorylation, which induces cell survival signals and promotes synaptic plasticity (Thome et al, 2000). We therefore investigated, using Western blotting, the levels of CREB phosphorylation after HI in rat pups with or without 5-mg/kg fluoxetine pretreatment. The 5-Flu-HI group had
significantly higher levels of pCREB expression in the ipsilateral hippocampus 12 hours and 72 hours after HI than did the Ns-HI group at the respective time points (both p < 0.05). The 5-Flu-HI group also had significantly higher pCREB levels in the ipsilateral cortex 72 hours after HI than did the Ns-HI group (p < 0.05) (Figs. 2A-C). There were no significant gender differences in the pCREB expression levels after fluoxetine pretreatment in the ipsilateral cortex and hippocampus 72 hours after HI injury. Compared with the Ns-HI group, the 5-Flu-HI group also had significantly higher pCREB levels in the contralateral hippocampus and cortex 12 hours and 72 hours after HI (both p < 0.05). Total CREB was unchanged in the hippocampus and cortex between the two HI groups.
Low-dose fluoxetine pretreatment increased posthypoxia BDNF transcription in the hippocampus
We next analyzed whether BDNF gene expression, the specific target gene of CREB, after HI was affected by fluoxetine pretreatment. The transcriptional levels of both BDNF exon III and V in the cortex were comparable between the Ns-HI and 5-Flu-HI groups (Figs. 3A-C). HI caused a significant reduction of BDNF exon III and exon V transcription in the
13
ipsilateral hippocampus 72 hours after HI. In contrast, compared with the Ns-HI group, the 5-Flu-HI group expressed significantly more BDNF exon III mRNA and exon V mRNA in the ipsilateral hippocampus 72 hours after HI (both p < 0.05) (Figs. 3D-F).
Low-dose fluoxetine pretreatment increased posthypoxia synapsin I transcription in the hippocampus
CREB also regulates expression of synapsin I, a vesicle-associated protein that plays a crucial role in regulating neurotransmission and synaptogenesis (Lonze and Ginty, 2002). We examined whether the transcriptional level of synapsin I after HI was affected by fluoxetine pretreatment. There were 2 cDNA fragments in our PCR products. The fragments were recovered from the gels, reamplified with the same set of primer pairs, and cloned into the pGEM-T vector using the TA cloning system (Promega). Plasmid DNA containing the correct inserts was sequenced using the automated DNA sequencing technology. Sequence comparison revealed that these two cDNA bands were identical to different alternative splicing forms of rat synapsin I, i.e., Ia and Ib. The levels of synapsin I mRNA in the cortex were comparable between the two HI groups (Figs. 4A, B). Again, we observed that 72 hours after HI, the 5-Flu-HI group expressed significantly more synapsin Ia and Ib mRNA in the ipsilateral hippocampus than the Ns-HI group did (Figs. 4C, D).
Low-dose fluoxetine pretreatment increased posthypoxia neurogenesis in the hippocampal dentate gyrus
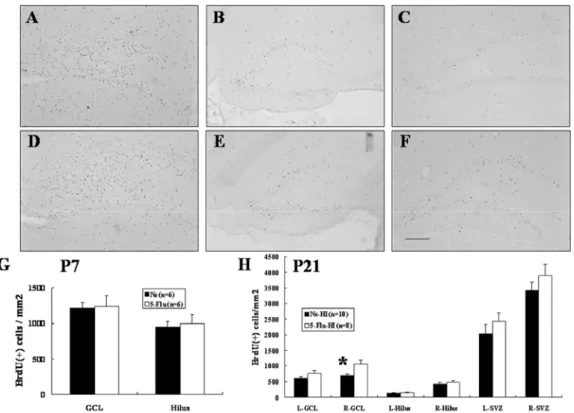
Increased precursor cell proliferation, survival, or differentiation can up-regulate
neurogenesis. We first assessed progenitor cell proliferation by investigating BrdU labeling 3 hours after the last fluoxetine or saline administration on P7. Unbiased cell counts revealed no significant differences in the numbers of BrdU(+) cells in the SVZ between the 5-Flu and Ns groups (data not shown). BrdU(+) cell density in the hippocampal GCL and hilus of
14
dentate gyrus in the 5-Flu group also did not differ from that of the Ns group (Figs. 5A, D, G). These data indicated that the 7 days of low-dose fluoxetine treatment did not alter progenitor cell proliferation in the immature rat brain.
To address the survival of newly born cells after HI, a second group of rats was killed 14 days after BrdU injection and HI.The density of BrdU(+) cells in the ipsilateral
hippocampal GCL of the 5-Flu-HI group was significantly higher (~50%) than in the Ns-HI group (p < 0.01) (Figs. 5B, E, H). The 5-Flu-HI group also had a non-significant trend of increased BrdU(+) cell density in the contralateral hippocampal GCL than the Ns-HI group (Figs. 5C, F, H). There were no differences in the newly born cell density in the hippocampal dentate hilus or the SVZ between the two groups in either cerebral hemisphere (Fig. 5H). There was also no significant difference in volume of the hippocampal GCL or dentate hilus in either cerebral hemisphere between the two groups (data not shown). These results suggested that fluoxetine pretreatment increased neurogenesis by increasing precursor cell survival in the ipsilateral hippocampal GCL after HI.
To determine the neuronal identity of BrdU(+) cells in the hippocampus, brain sections were co-labeled with antibodies to BrdU and NeuN (neuronal marker) or BrdU and GFAP (astroglial marker). The BrdU(+) cells that co-expressed NeuN in the hippocampal GCL had the morphological characteristics of granular cells (a round or oval cell body ~20 μm in diameter with long dendrites extending toward the molecular layer) (Figs. 6D-F). The morphology of these newly born cells was not different between the 5-Flu-HI and Ns-HI groups. The percentage of newly born cells that differentiated into neurons in the
hippocampal GCL was similar in the 5-Flu-HI and Ns-HI groups (5-Flu-HI group left side: 78.9 ± 4 %, right side: 75.3 ± 6 %; Ns-HI group left side: 78.5 ± 4 %, right side: 73.5 ± 4 %), indicating that the differentiation was not influenced by fluoxetine treatment.
15
Low-dose fluoxetine treatment per se in rat pups did not cause long-term adverse effects in adulthood
Transient inhibition of serotonin transporter with fluoxetine during early development may produce abnormal emotional behaviors in adult mice (Ansorge et al, 2004). Therefore, we examined the long-term cognitive and behavioral effect of early-life repetitive
administration of 5-mg/kg fluoxetine. Since anxiety related tests may give different results in male and female animals (Renard et al, 2005), we test male rats only in these behavioral tests. There were no significant differences in learning, memory, or visual motor
performance in the MWM between the 5-Flu and Ns groups (Figs. 7A-C). The anxiety state was assessed in the open field test and elevated plus maze. The 5-Flu group did not differ from the Ns group in exploratory behavior, as demonstrated by rearing, ambulation and locomotion in the open field (Fig. 7D). There was also no significant difference in time spent in the open arms between the two groups in the elevated plus maze, although the
5-Flu-group rats showed a non-significant trend of shorter latency for the first entry onto the open arms (Fig. 7E). These findings suggest that early-life short-term exposure of low-dose fluoxetine in rat pups did not have adverse impacts on cognition or emotional functioning at adulthood.
16
Discussion
Our study suggests that low-dose fluoxetine, a widely used SSRI antidepressant, reduces functional deficits in adult rats with neonatal HI brain injury. The neuroprotective
mechanisms of fluoxetine treatment before HI are associated with 1) sustained increased CREB phosphorylation in both the ipsilateral and contralateral hippocampus after HI, 2) increased expression of BDNF and synapsin I mRNA in the ipsilateral hippocampus after HI, and 3) increased neurogenesis by raising newly-born cell survival in the ipsilateral
hippocampal GCL after HI. Furthermore, exposure to a low dose of fluoxetine in early life does not cause long-term adverse effects in cognition or behavior. These findings provide evidence that functional recovery after HI injury in the developing brain can be induced by agents that increase neurogenesis and neural plasticity through the CREB activation
mechanism, which may suggest a potential strategy for influencing the cognitive outcome of HI injury in neonates.
Sparing neurons from death may not necessarily translate into sparing them from dysfunction. Furthermore, differences in cognitive function deficits may occur despite no difference in the degree of brain injury at morphological levels. Therefore, in addition to pathological evaluation, sophisticated behavioral outcome is important for evaluating the efficacy of potential treatment that may improve the consequences of experimental brain insult (Corbett and Nurse, 1998). In this study, animals were subjected to a sequence of MWM tasks that measured different aspects of learning, such as spatial orientation, long-term retention, reversal learning, and visual-oriented motor learning. Although fluoxetine pretreatment, at a low or high dose, did not provide neuroprotection at
pathological levels, we found that low-dose fluoxetine reduced and, in contrast, high-dose fluoxetine exacerbated functional deficits in adult rats with neonatal HI brain injury.
17
pretreatment in neonatal HI brain injury may be related to increased neurogenesis in the hippocampal GCL. Fluoxetine has been shown, by modulating the serotonergic systems, to decrease the degree of inflammation (Abdel-salam et al. 2004), which plays an important role in neonatal HI brain injury (Ferriero, 2004).But recent data suggests that the behavioral and neuroprotective effects of chronic SSRIs in adult rats are mediated by the stimulation of neurogenesis in the hippocampus (Malberg et al, 2000; Santarelli et al, 2003). There is also evidence that neurogenesis plays a contributory role in functional recovery after cerebral ischemia in adult rats (Raber et al, 2004). Most experimental studies of perinatal HI brain injury are concerned with how cell death can be prevented or at least minimized. However, many investigators found strong increases in neurogenesis in adult rats following a variety of brain injury paradigms, including ischemia and seizures (Liu et al, 1998; Madsen et al, 2000). Furthermore, regenerated neurons are actively integrated into the existing brain circuitry and contribute to forming hippocampal-dependent memory or ameliorating neurological deficits (Nakatomi et al, 2002). These findings suggest that instead of
minimizing cell death, functional outcome after injury can be improved by the stimulation of endogenous neurogenesis. Our results suggest that strategies aimed at stimulating
neurogenesis can provide new avenues for the treatment of neonatal HI brain injury. We found that fluoxetine pretreatment was specifically beneficial for the survival, but not proliferation, of precursor cells that differentiated into new neurons in the hippocampal GCL after HI in rat pups. It may be related to significantly sustained increases in CREB phosphorylation, especially in the hippocampus, after HI in fluoxetine-pretreated rats. Several signals control the proliferation, differentiation, and survival of endogenous
progenitors (Barnabe-Heider and Miller, 2003). Of these, CREB activation seems to be both necessary and sufficient for the survival of precursor cells in the hippocampal GCL of adult rats (Zhu et al, 2004). Our results were consistent with other studies in which chronic
18
treatment of adult rats with fluoxetine, which increases the levels of pCREB in the dentate gyrus, increased the survival of immature neurons (Malberg et al, 2000; Nakagawa et al, 2002). CREB activation and its facilitation of the survival of neuronal precursor cells may also be responsible for the hippocampus-associated learning-dependent changes of dentate gyrus neurogenesis (Gould et al, 1999; Tully et al, 2003). All our data taken together suggested that fluoxetine pretreatment increased CREB activation and neurogenesis by facilitating the survival of neuronal precursor cells after HI injury in rat pups.
The neuronal-precursor-cell survival regulated by CREB activation may involve expression of BDNF, which provides a growth environment for precursor cells resident in the hippocampal dentate gyrus (Zhu et al, 2004). This hypothesis is supported by our results showing significantly increased BDNF transcription after HI in the ipsilateral hippocampus of the fluoxetine-pretreated rats compared with that in the saline-pretreated rats. In vitro and in vivo data provide direct evidence that CREB and BDNF are key mediators of the
therapeutic response to SSRIs (D’Sa and Duman, 2002). Thus, BDNF expression induced by fluoxetine-mediated CREB activation may facilitate functional recovery by increasing the survival of newly born neurons in the hippocampus after HI injury.
In addition to its role in cell survival, BDNF is a critical modulator of
synaptic-plasticity, especially in the hippocampus (Poo, 2001). Studies have shown that BDNF regulates the phosphorylation and synthesis of synapsin I (Jovanovic et al, 2000), a neuron-specific phosphoprotein with two isoforms: Ia and Ib. The rat synapsin I gene also carries CRE consensus sequences in its promoter regions (Lonze and Ginty, 2002),
suggesting that CREB might also regulate synapsin I activity at the level of gene expression and thus contribute to synaptic plasticity. In the present study, fluoxetine-pretreated rats, with their significantly higher CREB activation and BDNF transcription levels, also had increased synapsin I mRNA expression in the ipsilateral hippocampus than that of the
19
saline-pretreated rats after HI. Synapsins regulate neurotransmitter release and synaptic plasticity, and are essential for the proper assembly of vesicle clusters in presynaptic
terminals (Greengard et al, 1993). Locally high levels of synapsin I are considered a marker of synaptic plasticity and neurogenesis and are thought to maintain high rates of synaptic activity, such as during learning (Gomez-Pinilla et al, 2001). Thus, elevated synapsin I appears to have contributed to facilitated functional recovery after HI injury in
fluoxetine-treated rats.
The present study also demonstrated that fluoxetine regulation of neurogenesis and synaptogenesis in immature brains may be region-specific. Long-term SSRIs treatment specifically increases neurogenesis in the hippocampus but not in the SVZ (Malberg et al, 2000), which suggests that SSRIs do not have a general effect on neurogenesis. CREB regulation of neurogenesis seems to be specific to the hippocampal dentate gyrus because overexpression of CREB did not alter the survival of BrdU-labeled cells in the SVZ (Zhu et al, 2004). We also found a significant increase in CREB phosphorylation, especially in the hippocampus, and an increase in the transcription of BDNF and synapsins only in the hippocampus after HI injury. Taken together, these findings suggest that BDNF, the downstream target of CREB activation, plays a contributory role in region-specific neurogenesis and synaptogenesis.
In the present study, it seems unlikely that the neuroprotective mechanisms of
fluoxetine pretreatment in rat pups were sex-dependent. Firstly, the degree of HI brain injury in rat pups was similar in both sexes. Secondly, the functional recovery by fluoxetine
pretreatment after HI injury, as measured by MWM test, was not affected by sex using two-way ANOVA analysis. Thirdly, there was no sex-related difference in post-hypoxia pCREB expression in the hippocampus and cortex after fluoxetine pretreatment. It has been well established that adult brain injury is sexually dimorphic. Many studies clearly
20
demonstrate less tissue damage after cerebral ischemia in females versus male adults (Hall et al, 1991; Alkayed et al, 1998; Carswell et al, 1999). However, in immature brains, the
severity of damage was linearly linked to the duration of HI, but not affected by sex (Hurn et al, 2005; Yager et al, 2005; Zhu et al, 2006), in agreement with our findings. In experimental adult animal models, reduced sensitivity to ischemic injury in females has been attributed to circulating estrogen, a factor not relevant to prepubertal animals. The sexual dimorphism for HI brain injury emerges between P21-P60, a timeframe coincident with the onset of sexual maturation and sex steroid production (Zhu et al, 2006). Estrogen has been shown to have a host of beneficial mechanisms, including enhanced cerebral reperfusion, antioxidant activity, and excitatory amino acid blockade (Hurn and Brass, 2003; Hurn et al., 2005). One potential pathway by which estrogen can protect against cell death is via the phosphorylation of CREB (Murphy and Segal., 1997; Auger, 2003). Estrogen has been shown to increase the pCREB and BDNF expression in hippocampus and amygdala of adult rats (Zhou et al, 2005). Studies employed to stratify the sex to determine the underlying neuroprotective
mechanisms for different gender will potentially allow for the development of sex-specific therapeutic interventions for ischemic brain injury in the mature brain. In rat pups, however, there was sex-related difference in pCREB expression on P0 and P1 only in the sexually dimorphic areas, such as ventromedial hypothalamus, but not in other areas (Auger et al, 2001). The transient sex difference in pCREB expression is most probably the results of testosterone surge, which is soon metabolized into estradiol, in male rat pups that occurs immediately following birth (Auger, 2003). Therefore, the functional recovery and sustained pCREB expression in rat pups in the current study is most likely treatment-related instead of gender-induced difference. Although emerging findings suggests that some ischemic cell death pathways might be sex-specific in the immature brain (Hagberg et al, 2004; Zhu et al, 2006), more studies are needed to be performed to demonstrate that the effectiveness of
21
therapeutic intervention in the neonatal hypoxic-ischemic brain injury is gender-related. Human fetal exposure to SSRIs may cause transient behavioral disturbances in the
newborn period (Zeskind and Stephens, 2004). Our data in rat pups shows that the manifestations of serotonergic symptoms are related with the treatment of high-dose fluoxetine: 70% of the 15-mg/kg-fluoxetine-group rats presented with serotonergic
symptoms, while none of the 5-mg/kg-fluoxetine- or saline-group rats did. The occurrence of the seizure-like serotonergic symptoms during high-dose fluoxetine injection may be related with worsened neurologic deficits after HI in the 15-Flu-HI group. Although clinical studies suggest that children with prenatal or perinatal exposure to fluoxetine did not have an adverse long-term outcome in their neurocognition and language development (Nulman et al, 1997), fluoxetine treatment during early brain development altered emotional behavior in adult mice (Ansorge et al, 2004). Our data show no long-term adverse cognitive or emotional effects of early-life low-dose fluoxetine treatment, as revealed by the MWM, open field, and elevated plus maze tests in adult rats. This discrepancy in animal studies may be because we used a lower dose (5 mg/kg vs. 10 mg/kg) and a shorter period of treatment (7 days vs. 18 days).
In adult rats, only long-term (> 14 days) treatment with fluoxetine can increase neurogenesis, and CREB and BDNF expression in the dentate gyrus. Fourteen days is consistent with the time required for the initiation of therapeutic action of fluoxetine in human adults. However, our results indicated that short-term (7 days) fluoxetine pretreatment reduced functional deficits after HI injury in rat pups. Furthermore, there were corresponding increases in neurogenesis, synaptogenesis, CREB phosphorylation, and BDNF transcription after HI in the hippocampus. The delayed effects of fluoxetine in adult rats suggest the slow structural and long-term gene changes taking place within the limbic target areas. It appears that the upregulated cAMP-CREB signaling cascade can accelerate the time course of gene
22
alterations (Thome et al, 2000). The shortened interval for the molecular and cellular
alterations after fluoxetine treatment in immature brains may be related to the development of cAMP signaling, including the developmental changes of receptor adenylate cyclase and phosphodiesterase activity (Lannutti and Schneider, 2001; Slotkin et al, 2001). Therefore, we hypothesize that it will be more advantageous to induce neurogenesis and neuroplasticity for cell survival using low-dose fluoxetine in immature brains than in mature brains. To our knowledge, these are the first data to demonstrate that fluoxetine contributes to protecting against HI brain injury in rat pups through activating CREB-mediated cascades. Our findings raise the possibility that short-term perinatal low-dose fluoxetine treatment may have
23
Acknowledgments
This study was supported by grants from the Taiwan National Health Research Institute (NHRI-EX94-9131NN), National Science Counsel (NSC: 92-2314-B-006-068,
92-2314-B-006-069), andChang Gung Memorial Hospital Research Program
(CGMHG-83020). The authors thank Dr. Hung Li and Ching-Yuan Su in the Academia Sinica for technical support in the laser-scanning confocal microscopy.
24 References
Abdel-Salam, O.M., Baiuomy, A.R., Arbid, M.S., 2004. Studies on the anti-inflammatory effects of fluoxetine in the rat. Pharmacol. Res. 49, 119-131.
Alkayed, N.J., Harukuni, I., Kimes, A.S., London, E.D., Traystman, R.J., Hurn, P.D., 1998. Gender-linked brain injury in experimental stroke. Stroke 29, 159-166.
Ansorge, M.S., Zhou, M., Lira, A., Hen, R., Gingrich, J.A., 2004. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science 306, 879-881.
Auger, A.P., 2003. Sex differences in the developing brain: crossroads in the
phosphorylation of cAMP response element binding protein. J. Neuroendocrinology 15, 622-627.
Auger, A.P., Hexter, D.P., Mccarthy, M.M., 2001. Sex difference in the phosphorylation of cAMP response element binding protein (CREB) in neonatal rat brain. Brain Res. 890, 110-117.
Balduini, W., De Angelis, V., Mazzoni, E., Cimino, M., 2001. Simvastatin protects against long-lasting behavioral and morphological consequences of neonatal hypoxic/ischemic brain injury. Stroke 32, 2185-2191.
Barnabe-Heider, F., Miller, F.D., 2003. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J.
25 Neurosci. 23, 5149-5160.
Carswell, H.V., Anderson, N.H., Clark, J.S., Graham, D., Jeffs, B., Dominiczak, A.F., Macrae, I.M., 1999. Genetic and gender influences on sensitivity to focal cerebral ischemia in the stroke-prone spontaneously hypertensive rat. Hypertension 33, 681-685.
Chang, Y.C., Huang, A.M., Kuo, Y.M., Wang, S.T., Chang, Y.Y., Huang, C.C., 2003. Febrile seizures impair memory and cAMP response-element binding protein activation. Ann. Neurol. 54, 706-718.
Chang, Y.C., Kuo, Y.M., Haung, A.M., Huang, C.C., 2005. Repetitive febrile seizures in rat pups cause long-lasting deficits in synaptic plasticity and NR2A tyrosine phosphorylation. Neurobiol. Dis. 18, 466-475.
Chang, Y.C., Huang, C.C., 2006. Perinatal brain injury and regulation of transcription. Curr. Opin. Neurol. 19, 141-147.
Corbett, D., Nurse, S., 1998. The problem of assessing effective neuroprotection in experimental cerebral ischemia. Prog. Neurobiol. 54, 531-548.
D’Hooge, R., De Deyn, P.P., 2001. Applications of the Morris water maze in the study of learning and memory. Brain Res. Rev. 36, 60-90.
26 183-194.
Dam, M., Tonin, P., De Boni, A., Pizzolato, G., Casson, S., Ermani, M., Freo, U., Piron, L., Battistin, L., 1996. Effects of fluoxetine and maprotiline on functional recovery in poststroke hemiplegic patients undergoing rehabilitation therapy. Stroke 27, 1211-1214.
Dowlatshahi, D., MacQueen, G.M., Wang, J.F., Young, L.T., 1998. Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet 352,
1754-1755.
Ferriero, D.M., 2004. Neonatal brain injury. N. Engl. J. Med. 351, 1985-1995.
Fujioka, T., Fujioka, A., Duman, R.S., 2004. Activation of cAMP signaling facilitates the morphological maturation of newborn neurons in adult hippocampus. J. Neurosci. 24, 319-328.
Gomez-Pinilla, F., So, V., Kesslak, J.P., 2001. Spatial learning induces neurotrophin receptor and synapsin I in the hippocampus. Brain Res. 904, 13-19.
Gould, E., Beylin, A., Tanapat, P., Reeves, A., Shors, T.J., 1999. Learning enhances adult neurogenesis in the hippocampal formation. Nature Neurosci. 2, 260-265.
Greengard, P., Valtorta, F., Czernik, A.J., Benfenati, F. 1993. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259, 780-785.
27
Hagberg, H., Wilson, M.A., Matsushita, H., Zhu, C., Lange, M., Gustavsson, M., Poitras, M.F., Dawson, T.M., Dawson, V.L., Northington, F., Johnston, M.V., 2004. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J. Neurochem. 90, 1068-1075.
Hall, E.D., Pazara, K.E., Linseman, K.L., 1991. Sex differences in postischemic neuronal necrosis in gerbils. J. Cereb. Blood Flow Metab. 11, 292-298.
Horsfield, S.A., Rosse, R.B., Tomasino, V., Schwartz, B.L., Mastropaolo, J., Deutsch, S.I., 2002. Fluoxetine's effects on cognitive performance in patients with traumatic brain injury. Int. J. Psychiatry Med. 32, 337-344.
Huang, C.C., Wang, S.T., Chang, Y.C., Lin, K.P., Wu, P.L., 1999. Measurement of the urinary lactate:creatinine ratio for the early identification of newborn infants at risk for hypoxic-ischemic encephalopathy. N. Engl. J. Med. 34, 328-335.
Hurn, P.D., Brass, L.M., 2003. Estrogen and stroke: a balance analysis. Stroke 34, 338-341.
Hurn, P.D., Vannucci, S.J., Hagberg, H., 2005. Adult or perinatal brain injury. Does sex matter? Stroke 36, 193-195.
Jovanovic, J.N., Czernik, A.J., Fienberg, A.A., Greengard, P., Sihra, T.S., 2000. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nature Neurosci. 3, 323-329.
28
Lannutti, B.J., Schneider, L.E., 2001. Gprk2 controls cAMP levels in Drosophila development. Dev. Biol. 233, 174-185.
Lee, H.T., Chang, Y.C., Wang, L.Y., Wang, S.T., Huang, C.C., Ho, C.J., 2004. cAMP response element-binding protein activation in ligation preconditioning in neonatal brain. Ann. Neurol. 56, 611-623.
Liu, J., Solway, K., Messing, R.O., Sharp, F.R., 1998. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J. Neurosci. 18, 7768-7778.
Lonze, B.E., Ginty, D.D., 2002. Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605-623.
Madsen, T.M., Treschow, A., Bengzon, J., Bolwig, T.G., Lindvall, O., Tingstrom, A., 2000. Increased neurogenesis in a model of electroconvulsive therapy. Biol. Psychiatry. 47, 1043-1049.
Malberg, J., Eisch, A., Nestler, E., Duman, R.S., 2000. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104-9110.
Murphy, D.D., Segal, M., 1997. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc. Natl. Acad. Sci. U.S.A. 94, 1482-1487.
29
Nakagawa, S., Kim, J.E., Lee, R., Malberg, J.E., Chen, J., Steffen, C., Zhang, Y.J., Nestler, E.J., Duman, R.S., 2002. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J. Neurosci. 22, 3673-3682.
Nakatomi, H., Kuriu, T., Okabe, S., Yamamoto, S., Hatano, O., Kawahara, N., Tamura, A., Kirino, T., Nakafuku, M.,2002. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110, 429-441.
Nestler, E.J., Barrot, M., Dileone, R.J., Eisch, A.J., Gold, S.J., Monteggia, L.M., 2002. Neurobiology of depression. Neuron 34, 13-25.
Nibuya, M., Morinobu, S., Duman, R.S., 1995. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539-7547.
Nibuya, M., Nestler, E.J., Duman, R.S., 1996. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in the rat hippocampus. J. Neurosci. 16, 2365-2372.
Nulman, I., Rovet, J., Stewart, D.E., Wolpin, J., Gardner, H.A., Theis, J.G., Kulin, N., Koren, G., 1997. Neurodevelopment of children exposed in utero to antidepressant drugs. N. Engl. J. Med. 336, 258-262.
30
Chollet, F., 2001. Fluoxetine modulates motor performance and cerebral activation of patients recovering from stroke. Ann. Neurol. 50, 718-729.
Poo, M.M., 2001. Neurotrophins as synaptic modulators. Nature Rev. Neurosci. 2, 24-32.
Raber, J., Fan, Y., Matsumori, Y., Liu, Z., Weinstein, P.R., Fike, J.R., Liu, J., 2004.
Irradiation attenuates neurogenesis and exacerbates ischemia-induced deficits. Ann. Neurol. 55, 381-389.
Renard, G. M., Suarez, M. M., Levin, G. M., Rivarola, M. A., 2005. Sex differences in rats: effects of chronic stress on sympathetic system and anxiety. Physiology Behavior. 85, 363-369.
Saarelainen, T., Hendolin, P., Lucas, G.., Koponen, E., Sairanen, M., MacDonald, E.,
Agerman, K., Haapasalo, A., Nawa, H., Aloyz, R., Ernfors, P., Castren, E., 2003. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J. Neurosci. 23, 349-357.
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., Belzung, C., Hen, R., 2003. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805-809.
Shanks, N., Lightman, S.L., 2001. The maternal-neonatal neuro-immune interface: are there long-term implications for inflammatory or stress-related disease? J. Clin. Invest. 108,
31 1567-1573.
Slotkin, T.A., Tate, C.A., Cousins, M.M., Seidler, F.J., 2001. Beta-adrenoceptor signaling in the developing brain: sensitization or desensitization in response to terbutaline. Dev. Brain. Res. 131, 113-125.
Thome, J., Sakai, N., Shin, K., Steffen, C., Zhang, Y.J., Impey, S., Storm, D., Duman, R.S., 2000. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J. Neurosci. 20, 4030-4036.
Tully, T., Bourtchouladze, R., Scott, R., Tallman, J., 2003. Targeting the CREB pathway for memory enhancers. Nature Rev. Drug. Discovery 2, 267-277.
Tzeng, S.F., Wu, J.P., 1999. Responses of microglial and neural progenitors to mechanical brain injury. Neuroreport 10, 2287-2292.
Volpe, J.J., 2001. Neurology of the newborn. 4th ed. Saunders, Philadelphia.
Walton, M.R., Dragunow, I., 2000. Is CREB a key to neuronal survival? Trends Neurosci. 23, 48-53.
Welberg, L.A., Seckl, J.R., 2001. Prenatal stress, glucocorticoids and the programming of the brain. J. Neuroendocrinol. 13, 113-128.
32
for determining the influence of age and sex on functional recovery following hypoxic-ischemic brain damage. Dev. Neurosci. 27, 112-120.
Zeskind, P.S., Stephens, L.E., 2004. Maternal selective serotonin reuptake inhibitor use during pregnancy and newborn neurobehavior. Pediatrics 113, 368-375.
Zhou, J., Zhang, H., Cohen, R.S., Pandey, S.C., 2005. Effects of estrogen treatment on expression of brain-derived neurotrophic factor and cAMP response element-binding protein expression and phosphorylation in rat amygdala and hippocampal structures.
Neuroendocrinology 81, 284-310.
Zhu, C., Xu, F., Wang, X., Shibata, M., Uchiyama, Y., Blomgren, K., Hagberg, H., 2006. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxic-ischemic. J. Neurochem. 96, 1016-1027.
Zhu, D.Y., Lau, L., Liu, S.H., Wei, J.S., Lu, Y.M., 2004. Activation of
cAMP-response-element-binding protein (CREB) after focal cerebral ischemia stimulates neurogenesis in the adult dentate gyrus. Proc. Natl. Acad.Sci.U.S.A.101, 9453-9457.
33
Figure legends
Fig. 1 High-dose fluoxetine pretreatment exaggerated, but low-dose fluoxetine reduced,
functional deficits after hypoxic-ischemia (HI) as measured by the Morris water maze. (A) The total distance covered to reach the submerged platform in both the acquisition and reversal training was significantly different between the normal-saline-pretreated (Ns-HI), 5-mg/kg fluoxetine-pretreated (5-Flu-HI), 15-mg/kg fluoxetine-pretreated (15-Flu-HI), normal-saline only (Ns) and the sham-operated (sham) groups (both p < 0.001, F=74.2 and 54.8, respectively). Ns-HI-group rats traveled significantly longer distances when finding the platform than did the Ns- and sham- (*: p < 0.001) or the 5-Flu-HI-group (†: p < 0.05) rats in both the acquisition and reversal training. In contrast, the 15-Flu-HI group traveled a
significantly longer distance searching for the submerged platform than did the Ns-HI group (††: p < 0.01). The travel distance searching for the submerged platform in the 15-Flu-HI group rats did not decrease over time. (B) In probe test 1, Ns- and sham control rats spent more time in the target quadrant than did the three HI-group rats (*: p < 0.001, F=12.9). There was no significant difference between the 5-Flu-HI, 15-Flu-HI and Ns-HI groups. However, in probe test 2, the 5-Flu-HI group showed much improvement and spent
significantly more time searching in the new target quadrant than did the Ns-HI group (: p < 0.05). There was no significant difference between the 5-Flu-HI, Ns and sham control groups. There was no significant difference between the 15-Flu-HI and Ns-HI groups in either probe test. (C) In the cued version, there was a significant difference (p < 0.001, F=19.8) between the five groups in time spent reaching the visible platform. The 5-Flu-HI group spent
significantly (*: p < 0.001) less time than the Ns-HI group finding the visible platform. There was no significant difference between the 15-Flu-HI and Ns-HI groups in time spent reaching the visible platform. (D) The degree of brain injury, as measured by hemispheric weight reduction, showed no significant difference between the Ns-HI, 5-Flu-HI, and 15-Flu-HI
34
groups. Se: session. ( * : p <0.001; †: p < 0.05, ††: p < 0.01 vs. the Ns-HI group).
Fig. 2 Fluoxetine pretreatment increased CREB phosphorylation (pCREB) in the
hippocampus and cortex after hypoxic-ischemia (HI). (A-C) Representative western blots and the corresponding densitometry of pCREB/CREB ratio showed that the 5-mg/kg
fluoxetine-pretreated (5-Flu-HI) group had significantly higher pCREB levels in the
ipsilateral hippocampus 12 hours and 72 hours after HI than did the normal-saline-pretreated (Ns-HI) group at the respective time point (* both p < 0.05). The 5-Flu-HI group also had significantly higher pCREB levels in the ipsilateral cortex 72 hours after HI than did the Ns-HI group (*p < 0.05). Compared with the Ns-HI group, the 5-Flu-HI group had
significantly higher pCREB levels in the contralateral hippocampus and cortex 12 hours and 72 hours after HI (*all p < 0.05). Total CREB was unchanged between the two HI groups. Data are from n= 5 experiments in each group.L: left hemisphere, R: right hemisphere, Ns:
35 normal saline controls.
Fig. 3 Fluoxetine pretreatment increased brain-derived neurotrophic factor (BDNF)
transcription in the hippocampus after hypoxic-ischemia (HI). The levels of both BDNF exon III and exon V mRNA in the cortex were comparable between the normal-saline-pretreated (Ns-HI) and 5-mg/kg fluoxetine-pretreated (5-Flu-HI) groups (A, B, C). In contrast,
compared with the Ns-HI group, the 5-Flu-HI group had significantly higher expressions of BDNF exon III and exon V mRNA in the ipsilateral hippocampus 72 hours after HI (both *p < 0.05) (D, E, F). Data are from n= 4-6 experiments in each group. The PCR products of BDNF were normalized to corresponding GAPDH products in each sample. L: left hemisphere, R: right hemisphere,Ns: normal saline controls.
36
Fig. 4 Fluoxetine pretreatment increased synapsin I transcription in the hippocampus after
hypoxic-ischemia (HI). The levels of synapsin I mRNA in the cortex were comparable between the normal-saline-pretreated (Ns-HI) and 5-mg/kg fluoxetine-pretreated (5-Flu-HI) groups (A, B). In contrast, the 5-Flu-HI group had significantly higher expressions of synapsin Ia and Ib mRNA in the ipsilateral hippocampus than did the Ns-HI group 72 hours after HI (both *p < 0.05) (C, D). Data are from n= 4-6 experiments in each group. The PCR products of synapsin I were normalized to corresponding GAPDH products in each sample. L: left hemisphere, R: right hemisphere, Ns: normal saline controls.
37
Fig. 5 Fluoxetine pretreatment enhanced precursor cell survival in the ipsilateral
hippocampal granule cell layer (GCL) after hypoxic-ischemia (HI). Unbiased cell counts revealed no significant differences in the density of bromodeoxyuridine (BrdU)(+) cells in the hippocampal GCL and hilus of the dentate gyrus between the normal-saline (Ns) (A) and 5-mg/kg fluoxetine (5-Flu) groups before HI (D, G). However, 14 days after
hypoxic-ischemia (HI), the 5-mg/kg fluoxetine-pretreated (5-Flu-HI) group had an ~50% higher density of BrdU(+) cells in the ipsilateral hippocampal GCL (E) than did the
normal-saline-pretreated (Ns-HI) group (B) (H) (*p < 0.01). The 5-Flu-HI group also had a non-significant trend of increased BrdU(+)-cell density in the contralateral hippocampal GCL (F) than the Ns-HI group did (C) (H). There were no differences in the density of the newly born cells in the hippocampal dentate hilus or the subventricular zone (SVZ) (H) between the two groups in either cerebral hemisphere. Scale bar = 100 μm. L: left hemisphere, R: right hemisphere.
38
Fig. 6 The neuronal identity of bromodeoxyuridine (BrdU)(+) cells in the hippocampal
dentate gyrus after hypoxic-ischemia (HI). Double immunofluorescent staining showed that the BrdU(+) cells (B, E) were neuron-specific nuclear-protein (NeuN) immunoreactive (D), not glial-fibrillary acidic-protein (GFAP) immunoreactive (A). Merged image F (from D and
E) shows the co-localization of BrdU(+) (red) with NeuN(+) (green) in cells indicated as
yellow nuclei (inset). Merged image C (from A and B) shows that most of the BrdU(+) (red) cells in the granular cell layer were not co-localized with GFAP(+) cell (green).
Co-localization of BrdU with GFAP is shown in cells in the hilus with green cytoplasm surrounding red nuclei (inset). Scale bar = 50 μm in A-F, 20 μm in insets.
39
Fig. 7 Low-dose fluoxetine treatment per se in rat pups did not cause long-term adverse
effects on cognition or behavior in adulthood. (A-C) There were no significant differences in the learning, memory, or visual motor performance in the Morris water maze between the 5-mg/kg fluoxetine (5-Flu) and normal saline (Ns) groups. The 5-Flu group did not differ from the Ns group in exploratory behavior, as demonstrated by rearing, ambulation, and locomotion in the open field test (D). These two groups also showed no significant difference in time spent in the open arms in the elevated plus maze (E), despite a
non-significant trend of shorter latency for the first entry onto the open arms found in the 5-Flu group. Se: session.