國立臺灣大學生物資源暨農學院農藝所生物統計組 碩士論文
Division of Biometry Graduate Institute of Agronomy College of Bioresources and Agriculture
National Taiwan University Master Thesis
應用分數檢定統計量於選擇性基因型試驗 之數量性狀基因座定位研究
Score Test Statistics for QTL Mapping under Selective Genotyping
房佑嬙
Yu-Chuang Fang
指導教授:高振宏 博士、廖振鐸 博士
Advisor: Chen-Hung Kao, Ph.D. & Chen-Tuo Liao, Ph.D.
中華民國 102 年 7 月
July, 2013
口試委員審定書
謝誌
兩年的碩班生涯晃眼即逝,本篇論文如期完成最要感謝一路悉心提點、耐性指導的
高老師,並感謝在生活和學業上均給予諸多鼓勵的廖老師以及專程從花蓮北上指
導口試的靖老師。也謝謝研究室的學長姐們不嫌棄我的愚拙,不論大小事總是熱心 幫忙解惑。最後感謝伴我成長的好夥伴---林昭京,謝謝你。
僅以此獻給育我劬勞的父母
中文摘要
生物上許多重要的經濟、生理、或與生化有關的性狀均為數量性狀。這些控制 數量性狀的基因稱為數量性狀基因座 (Quantitative trait loci),其定位與研究一直是
作物和動物在遺傳育種上的重要課題。利用分子遺傳標誌資料,數量性狀基因座定 位 (QTL mapping)方法可幫助我們了解 QTL 在染色體上的位置及其作用大小。選
擇性基因型鑑定 (Selective genotyping)是一種只針對樣本族群之外表型極大與極
小的部分個體進行基因型鑑定的方法,它除了降低遺傳鑑定的成本外,一般認為也 可以增進定位數量性狀基因座的效率。本篇文章中,利用 Lee et al. (2013) 針對選
擇性基因型鑑定所提出的兩種模式 (事後檢定模式與目前所行的模式),分別推導
其分數檢定統計量(score test statistics)作為另一種定位 QTL 的統計量,並研究其在
選擇性基因型鑑定方法下的顯著性門檻值。結果發現,在單一QTL 存在的假設情
況下,兩種分數檢定統計量表現的一樣好。未來研究中,我們期待將單一QTL 假
設推廣至多個QTL 存在的情況,進行選擇性基因型鑑定之數量性狀基因座定位研
究。
關鍵字: 數量性狀基因座;數量性狀基因座定位;區間定位;分數檢定;選擇性基
因型鑑定
ABSTRACT
The detection of quantitative trait loci (QTL) that govern many biologically and
economically important traits is an important task in plant and animal breeding. Using
genetic marker data, QTL mapping technique has been known to be an efficient tool to
detect QTL location and estimate their effects. In QTL mapping, selective genotyping,
which genotypes only the individuals from high and low phenotypic values, is one of the
most common strategies that can reduce the cost of marker genotyping and at the same
time increase efficiency in QTL detection. In this thesis, with the posterior model of
selective genotyping proposed by Lee et al. (2013), we derived score test statistic for the
model and applied it to QTL detection. Moreover, we compare this score test statistics
with that of the currently used model, and the threshold values of the score test statistics
under selective genotyping are also investigated. As the result, we found out that the two
score test statistics for the posterior model and currently used model perform equally well
under single-QTL model. In the future, we intend to extend the single-QTL posterior
model to multiple-QTL model for QTL detection under selective genotyping.
KEYWORDS: QTL; QTL mapping; interval mapping; score test statistic; selective
genotyping
Contents
口試委員審定書 ... i
謝誌 ... ii
中文摘要 ... iii
ABSTRACT ... iv
1 Introduction ... 1
2 Theory and Methods ... 5
2.1 Population Structures and Selective Genotyping for QTL Detection ... 5
2.2 Statistical Model of QTL Mapping for Complete Data ... 5
2.3 Statistical Model of QTL Mapping for Selective Genotyping ... 6
2.4 Score Test Statistics for Detecting QTL in Selective Genotyping ... 12
3 Simulation and Results ... 14
4 Conclusion and Discussion ... 21
5 References ... 25
6 Abbreviations ... 28
1 Introduction
The traits of peas, such as flower color, seed coat color, observed in Mendel’s
experiment are called qualitative traits, as they can be easily assigned to different
categories. Qualitative traits are usually controlled by one or few genes, and less affected
by environments. Besides the qualitative traits, there is another type of traits such as yield
of crops, body weight of animals, and stress-resistance performance of plants, which can’t
be easily classified into categories. These traits are showing continuous variation and
called “quantitative trait”. Quantitative trait are usually controlled by several genes with
small effects and can be easily modified by environments. The genes control quantitative
traits are named polygenes (MATHER 1941) or called quantitative trait loci (QTL)
(GELDERMANN 1975). In plant and animal breeding, many biologically and economically
important traits are quantitative not qualitative. Therefore, it is essential to study the
inheritance of QTL to modify and improve these traits.
Nowadays, with advanced biotechnology, it is very convenient to gain numerous
molecular genetic markers and construct the genetic maps for various organisms. By
using the genetic marker data, several statistical methods have been applied to the study
of QTL. Lander and Botstein (1989) developed a statistical method called interval
mapping (IM) to systematically detect the genetic locations and estimate the effects of
QTL. In the IM model, because the QTL are unknown and needed to be estimated, a
normal mixture model is used in modeling and a likelihood ratio test (LRT) statistic is
perform to estimate at every position along the genomes. The position with the significant
largest LRT statistics is regarded as the estimated QTL position. Because the likelihood
approach of IM can be computationally slow, Haley and Knott (1992) proposed a
relatively simpler regression version of IM model (REG interval mapping) to
approximate the likelihood approach of IM. However, according to Kao (2000) and
Feenstra et al. (2006) , REG interval mapping can be less powerful and precise as
compared to the likelihood approach of IM. Besides, the IM method considers one
putative QTL at a time in the model, thus the power to detect QTL is lower, and biases
will occur in the estimation of QTL position and effects when there are other QTL exist
on the same chromosome. To conquer this problem, Jansen (1993) and Zeng (1993, 1994)
proposed composite interval mapping (CIM), which combine the IM with multiple
regression analysis in QTL mapping. This approach fits one putative QTL in an interval
and other markers into the model to improve QTL mapping. In the CIM model, the
markers are treated as covariates for reducing the residual variance such that the test for
the putative QTL can be more powerful and the estimation can be improved. Kao et al.
(1999) extend the CIM method to multiple interval mapping (MIM) in a way that QTL
can be directly controlled in the model. The MIM method uses multiple marker intervals
simultaneously to construct multiple putative QTL in the model, it tends to be more
powerful and precise in detecting QTL. In addition to these methods, numerous studies
for the estimation of QTL mapping have been carried out (OOIJEN 1992; DARVASI et al.
1993; HALEY et al. 1994; JIANG and ZENG 1995; KRUGLYAK and LANDER 1995; DOERGE
and CHURCHILL 1996; KNOTT et al. 1996; LYNCH and WALSH 1998; SEN and CHURCHILL
2001).
As the cost of data generation for QTL mapping analysis can be substantial, Lander
and Botstein (1989) claimed that a selective genotyping strategy can reduce the
genotyping cost by only genotyping the extreme progeny in a sample. When analyzing
such selective genotyping data, they also suggested that the other nonextreme progeny
with only phenotypic values still have to be included in the analysis to prevent the bias in
parameter estimation. Later, numerous statistical methods have been proposed to detect
QTL under selective genotyping strategy (DARVASI and SOLLER 1992; MURANTY and
GOFFINET 1997; XU and VOGL 2000). Darvasi and Soller (1992) proposed an ANOVA-
based method to analyze the data by using only the extreme genotyped individuals. They
found that it will almost never be useful to genotype more than the upper and lower 25%
of the population. By including both the genotyped and ungenotyped individuals in the
analysis, Muranty and Goffinet (1997) proposed a mixture normal model to obtain the
estimates of QTL effects. Xu and Vogl (2000) developed a selective genotyping QTL
mapping method based on truncated model when only the extreme genotyped individuals
are included in the analysis. Recently, Lee, Kao and Ho (2013) proposed alternative
likelihood approaches and extended the state statistics model from single-QTL model to
multiple QTL model for selective genotyping. An improvement in QTL detection has
been made by their approaches under selective genotyping.
Score test statistics has been a very popular tool in statistical analysis (COMMENGES
1994; COMMENGES and ANDERSEN 1995; DUDOIT and SPEED 2000; GOLDSTEIN et al.
2001; PUTTER et al. 2002; WANG and HUANG 2002). Compared with likelihood approach,
score test statistic is a simpler and faster statistical method, as the maximum likelihood
approach mapping is relatively difficult in obtaining the estimates and computationally
demanding (CHANG MYRON et al. 2009; GUO 2011; KAO and HO 2012). In this thesis,
with the model proposed by Lee et al. (2013), we derive score test statistic and use this
statistic for QTL mapping in selective genotyping in the F2 population. Moreover, the
threshold values of the score test statistics are also investigated. Simulations were carried
out for illustration.
2 Theory and Methods
2.1 Population Structures and Selective Genotyping for QTL Detection
Various experimental populations have been designed for QTL detection. Among
these populations, backcross and F2 populations are the most widely used designs. In this
thesis, we considered the F2 population as mapping population. Assume that N individuals
are sampled and measured with phenotypic values of y. Among the N individuals, only the upper n2 and the lower n2 extreme individuals are selected for genotyping,
where 𝑛𝑛 ≤ 𝑁𝑁 . The remaining individuals are not genotyping. The n genotyped
individuals and N n ungenotyped individuals are included in the data analysis.
2.2 Statistical Model of QTL Mapping for Complete Data
An interval mapping statistical model for testing a QTL (Q), are assigned to describe
the phenotypic value of the i th individual at any given position, and can be written as a
normal mixture model:
* *
i i i i
y ax dz (1) where i is a random error, we assume i follows N
0 , 2
, a and d are the additive and dominance effects of Q, xi* and z*i defined as*
1 if the genotype of Q is QQ, 0 if the genotype of Q is Qq, 1 if the genotype of Q is qq, xi
and * 12 if the genotype of Q is Qq, 12 otherwise, zi
in section 2.3 mentioned that Q is not observed but can be inferred from interval flanking
markers, the Q can be QQ
x*i 1, zi* 12
, Qq
xi* 1, z*i 12
or qq
xi* 1, zi* 12
for an individual i.At a given position, for a sample of N individuals, the sum of the log likelihood function
of the model in Equation (1) is
2
2
3
21 1 2
, , , log 2 log exp
2 2
N i j
i j ij
N y
l a d p
(2)where pij is the conditional probability of QTL for ith individual, and
2 1
1 1
2 2 2 2
3 3 2 3
with probability , , 2
If Q is with probability , , 2
with probability , , 2
i i i
a d
QQ p N
Qq p N d
qq p N a d
which can be determined by the given position, and need not to be estimated here.
2.3 Statistical Model of QTL Mapping for Selective Genotyping
2.3.1 Statistical Model
Applying the statistical model to selective genotyped data analysis, we have to
separate the individuals into two parts: one of them is genotyped individuals
n , the other is ungenotyped individuals
N n
. Then the sum of the log likelihood functionbecomes
3 2
2 2
1 1 2
3 2
1 1 2
, , , log 2 log exp
2 2
log exp
2
n i j
i j ij
N i j
i n j j
N y
l a d p
q y
(3)
where pij is the conditional probability of QTL for ith individual in selective genotyped
individual, which can be determined by the given position and need not to be estimated.
qj is the conditional probability of QTL for ungenotyped individuals, which have to be inferred from the posterior probability of ungenotyped flanking markers. According to
the model proposed by Lee et al. (2013), we derived posterior model to estimate qj in
comparison with the prior model by using the score test statistic. Note that the log
likelihood for ungenotyped individuals have the same mixing proportions, qj.
2.3.2 The Genotypic Structure of QTL in Ungenotyped Individuals
In the data analysis of selective genotyping, however, we only have the genotyped
data of extreme individuals. For those ungenotyped individuals, we have to speculate the
genotypic distribution of QTL according some rules.
Consider a QTL (Q), in the F2 population in which the frequency of genotypes QQ, Qq and qq are 14, 12and 14, respectively. In general, Q is not observed but can
be inferred from the interval flanking markers according to the principle of conditional
probability as
| , P MQN P Q M N
P MN . (4)
In F2 population, the two flanking markers have nine different genotypes, and for each
one of them, the genotype of the flanked Q can be QQ, Qq or qq. Thus, when considering
the flanking markers and the QTL (M, N and Q) together, there are 27 different
conditional probabilities. For example, the conditional probability for QTL given the
marker genotype MNMN is P QQ| MN MN
, P Qq| MN MN
and P qq| MN MN
. For the genotyped individuals, we used conditional probability which is proposed by Kao and
Zeng (1997) in Table 1 as the mixing proportion (pij) for the frequency of putative QTL
genotypes in the genotyped individuals. For the ungenotyped individuals, Xu and Vogl
(2000) applied currently used method ( (P QQ P Qq P qq ) : ( ) : ( ) 1: 2 :1) to represent the
mixing proportion (qj ) for genotypic distribution of QTL based on ungenotyped
individuals. However, the frequencies of QTL genotype for ungenotyped individuals do
not follow the prior model under selective genotyping, for the reason that the extreme
individuals includes the same genotype practically which lead to the movement of the
frequencies of QTL for ungenotyped individuals and break the assumption of prior model
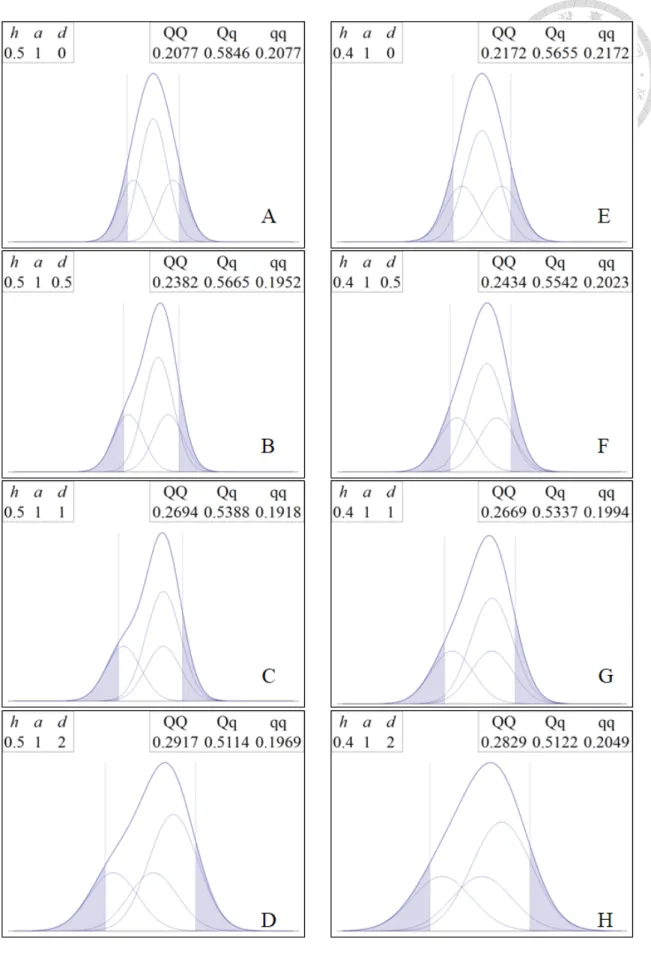
(Figure 1).
Figure 1: Normal mixture of phenotypic value. Based on different ℎ2, 𝑎𝑎 𝑎𝑎𝑛𝑛𝑎𝑎 𝑎𝑎.
In this thesis we used posterior probability, which is proposed by Lee et al. (2013),
as the mixing proportion (qj) trying to estimate the approximate probability of the QTL
genotypes from ungenotyped individuals. First, we have to estimate the flanking marker
genotypes for ungenotyped individuals, the rule of posterior probability:
1 1 2
1 1 2
1 1 2
flanking marker genotypes
, , , ,
| , , ,
, , , ,
n n
n n
n n
P G MN s s s
MN MN
P G s s s
MN P G s s s
9 | |
1 1
1 2 9
9 9
| | 1, ,9 \ 1
1
1 1 2
1 ( ) ( )
| | 1,| |, ,|
| , |
1 ( ) ( )
|
, ,
| 1,(| | )
k
k
s k u
k
s k u i
i n
k i
n
j j i
P G MN s
n p
N n
M p p
s
p
s
g
g
g g
g g g
g g
g g
9 1
1, flanking makrer genotypes
1 | 1 2
1
| | 1
| 1
, 1
|
, ,
i i
n MN n
P
M
G s s s
MN
P N MN
P G
gg
(5)
where the nine different flanking marker genotypes (λ) are the same as these listed in
Table 1. And in section 1 ,
n
is the total size of selective genotyped individual.9 1
i|
i
n
| g ,| g
i|
represent the number of each flanking marker genotypes in selectivegenotyped individuals respectively. Second, the QTL conditional probabilities of
ungenotyped individuals ( 𝑞𝑞𝑗𝑗) can be inferred from those posterior probabilities of
flanking markers by using Table 1.
Table 1: Conditional probabilities of a putative QTL given the flanking marker genotypes for an F2
population (KAO and ZENG 1997)
Marker genotype
Expected frequency
QTL genotype
QQ Qq qq
MNMN
1 2 4
r
1 0 0
MNMn 12
r r 1 p p 0
MnMn 42
r
1 p
2 2 1p
p
p2MNmN 12
r r p 1 p 0
MN or Mn
mn mN 12 22
r r
cp
1p
1 2 1 cp
p
cp
1p
Mnmn 12
r r 0 1 p p
mNmN 42
r p2 2 1p
p
1 p
2mNmn 12
r r 0 p 1 p
mnmn
1 2 4
r
0 0 1
MQ MN
pr r , where rrMN is the recombination fraction between the two flanking markers M and N, rMQ is the recombination fraction between the left marker M and the putative QTL.
2
2 1 2
MN
MN MN
c r
r r
. The possibility of a double recombination event in the interval is ignored.
2.4 Score Test Statistics for Detecting QTL in Selective Genotyping
Under our proposed model (Equation (3)), score test statistic can be constructed to
test for the hypothesis of H a0: 0 and d0 for the model at any given putative position along the whole genome. The score functions of a and d are the first derivatives
of the log likelihood (Equation (5)) with respect to a and d, and using ˆ yi
N and
22 ˆ
ˆ yi
N
(the MLEs of and 2) evaluated at a given position x underH0:a0 and d0.
Let u x1
and u x2
represent the score functions of a and d. The two score functions are1 2 1 3
1 3
1 1
1 ˆ
n N
i i i i
i i n
u x p p y y q q y y
, (6) and
2 2 1 2 3 1 2 3
1 1
1 2ˆ
n N
i i i i i
i i n
u x p p p y y q q q y y
, (7) respectively, and under the null hypothesis, the variances of u x1
and u x2
are
1
12 2 1 1var ˆ
N N
i i j
i i j
u x k N k k
N N
, (8) and
2
12 2 1 1var 4ˆ
N N
i i j
i i j
u x c N c c
N N
, (9) respectively, where1 3
1 3
i i
i
p p
k q q
and 1 2 3
1 2 3
i i i
i
p p p
c q q q
, where 1,..., 1,...,
i n
i n N
and the covariance between u x1
and u x2
is
1 2
21
1 1 1
cov ,
2ˆ
N N
i i i i
i i j
u x u x k c N k c
N N
(10) If take only additive or dominance effect into consideration, then under the nullhypothesis, the score test statistic is
1 1
var 1
U x u x
u x or
2 2
var 2
U x u x
u x
If both additive and dominance effects are taken into consideration simultaneously,
the score test statistic become
1 2 1
1 2
2
U x u x u x V u x
u x
(11)
where V is the variance-covariance matrix of u x
1 1 2
1 2 2
var cov ,
cov , var
u x u x u x
V u x u x u x
Here we can also use the maximum of U x2
under the null hypothesis to assess the threshold value for QTL detection, which is simpler and faster to obtain the thresholdvalue, as it avoids the iterative procedures in retraining the estimations of the parameter
in the normal mixture likelihood. (COX and HINKLEY 1979; CHANG MYRON et al. 2009;
GUO 2011; KAO and HO 2012)
3 Simulation and Results
We performed computer simulation to evaluate score test statistics for the two
selective genotyping models. The QTL mapping results of complete data are also
presented for comparisons. The issue of determining threshold values for both statistical
models was also investigated by simulations. All simulations were done by using
Mathematica program (WOLFRAM RESEARCH 2012).
A single QTL is assumed to be located at 25cM of a 100-cM chromosome covered
by different marker densities in the 𝐹𝐹2 population. The marker densities, represented by
the gap between two adjacent markers, are assigned to 5, 10 and 20cM. The genetic effects
of the QTL are set at (𝑎𝑎 = 1, 𝑎𝑎 = 0.5), (𝑎𝑎 = 1, 𝑎𝑎 = 1) and (𝑎𝑎 = 1, 𝑎𝑎 = 2), representing
different levels of dominance effect. The heritability of all these cases is assumed to be
0.05 (ℎ2 = 0.05). The number of selectively genotyped individuals was fixed at 100,
from 200 and 1000 individuals, which lead to 50% and 10% selective genotyping
proportions, respectively. The QTL location was estimated at the chromosomal position
with the largest value of the test statistic computed every 1cM. For each case, 100
replicates are simulated. Meanwhile, the score test statistics based on 10,000 simulated
replicates under null (nonexistence of QTL) are also computed for investigating the
behavior of the statistics under selective genotyping. These 10,000 maxima of the score
test statistics along the chromosome are ordered to have us obtain the approximated distribution of
2
sup0, ( )
x DU x
. In the meantime, the threshold values at significant level α = 0.05 can be determined. Results are shown in Figure 2 and Tables 2 to 5.
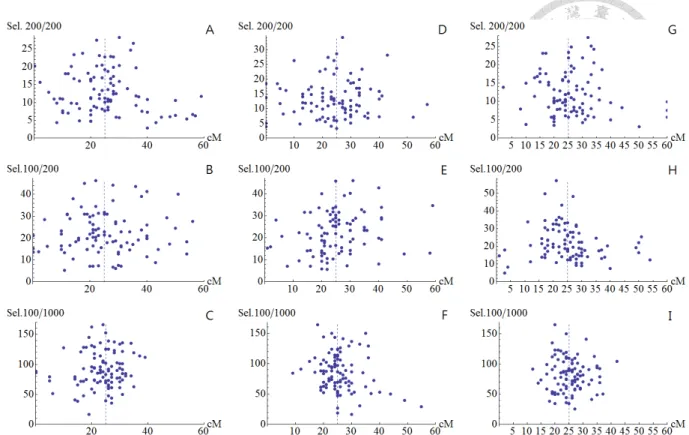
Figure 2 presents the scatter plots of the maxima of score test statistics of the posterior model under different selective proportions and marker densities. We can see
that the scatter points are more concentrated around the true position as the marker
becomes denser and the selective proportion gets more intense. Table 2 shows the score
test statistics and their thresholds of QTL mapping for full genotyping data, we found that
both of them become greater with denser markers. However, the powers to detect QTL
are slightly higher as marker becomes denser. For example, when the genetic effects are
set at 𝑎𝑎 = 1 , 𝑎𝑎 = 1, the score test statistics would be 11.34, 12.43 and 13.54, while their
thresholds would be 9.68, 10.35 and 10.99 respectively with marker densities being 20cM,
10cM and 5cM. There are increasing trends in both statistics and thresholds with the
denser markers. But the power to detect the QTL are stable at 58%, 61% and 63%,
respectively.
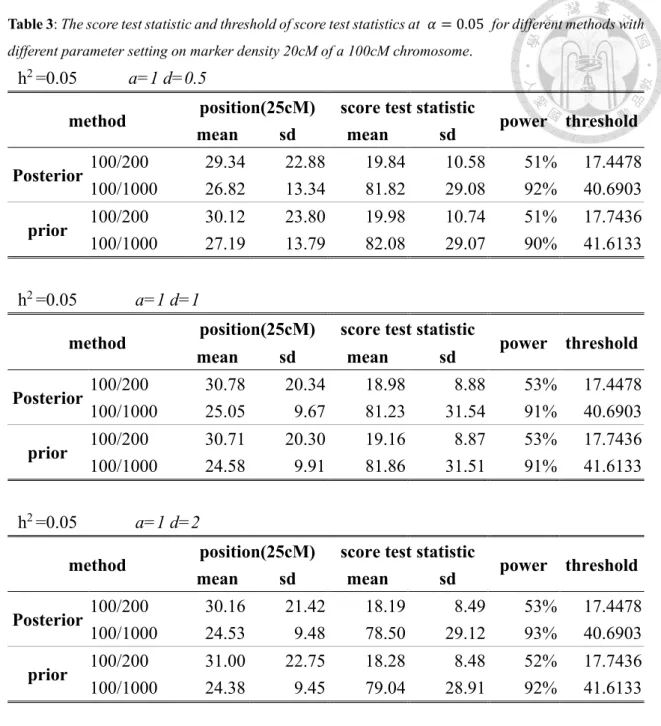
Tables 3 to 5 shows the score test statistics for both posterior and prior models as
well as their thresholds under marker densities 20cM, 10cM and 5cM. We can find that
the two score test statistics both increased when marker becomes denser and selective
proportion becomes more intense. For example, with posterior model, when selective
proportion is 50% (10%) and the marker densities are 20cM, 10cM and 5cM, the score
test statistics are 19.84 (81.82), 20.97 (85.92) and 22.54 (92.51), while the thresholds are
17.4478 (40.6906), 18.769 (43.782) and 19.8247 (45.7761). On the other hand, the score
test statistics for prior model are 19.98 (82.08), 20.87 (85.78) and 22.29 (93.32), while
the thresholds are 17.7436 (41.6133), 18.831 (44.315), 19.8896 (46.9538). Both score test
statistics for each model have the similar sizes and the similar thresholds.
In comparison with full genotyping data (Table 2), the score test statistics and their
thresholds under selective genotyping data (Tables 3-5) inflate. In addition, the results for
the two score test statistics (posterior and prior model) were similar. From Tables 2 to 5,
it is clear that the two models of score test statistics have similar performances. For
example, when genetic effects are (𝑎𝑎 = 1, 𝑎𝑎 = 0.5) and marker density is 10 cM, under
50% selective proportion, the score test statistics of posterior model (prior model) has a
mean of 20.97 (20.87) and the power to detect QTL is 50% (49%). When the selective
proportion is 10%, the score test statistics of the posterior model (prior model) and the
power for the posterior model (prior model) under the same settings become 85.92 (85.78)
and 92% (92%), respectively. It showed clearly that the more intense the selective
genotyping is, the greater the statistics inflate.
Figure 2: The scatter plots of the maximum score test statistics with a QTL at position 25cM on a 100-cM chromosome and genetic effect 𝑎𝑎 = 1, 𝑎𝑎 = 0.5 and ℎ2= 0.05. X-axis presents the marker positions (in cM). The marker density was 20cM (A-C), 10cM (D-F), 5cM (G-I) with the total population size 200, 200, 1000, and selective proportion 100%, 50%, 10% respectively. Each plots used 100 simulation replicates.
The dashed vertical lines indicate the QTL position (25 cM).
Table 2: the score test statistics and threshold of full genotyping data for different genetic effect and marker density 20cM, 10cM and 5cM on a 100cM chromosome.
h2 =0.05 𝒂𝒂 = 𝟏𝟏 Sample size: 200/200
marker
distance d position(25cM) score test statistic
power threshold
mean sd mean sd
20cM
0.5 30.66 21.03 11.73 6.15 60%
9.6771
1 32.59 22.01 11.34 5.54 58%
2 31.68 23.04 11.04 5.46 57%
10cM
0.5 27.92 17.52 12.35 5.85 56%
10.351
1 30.60 16.94 12.43 5.84 61%
2 29.64 18.28 12.14 5.45 56%
5cM
0.5 30.98 19.09 13.23 6.08 57%
10.9863
1 30.76 17.85 13.54 6.27 63%
2 29.30 17.20 13.08 5.60 61%
Table 3: The score test statistic and threshold of score test statistics at 𝛼𝛼 = 0.05 for different methods with different parameter setting on marker density 20cM of a 100cM chromosome.
h2 =0.05 a=1 d=0.5
method position(25cM) score test statistic
power threshold
mean sd mean sd
Posterior 100/200 29.34 22.88 19.84 10.58 51% 17.4478 100/1000 26.82 13.34 81.82 29.08 92% 40.6903 prior 100/200 30.12 23.80 19.98 10.74 51% 17.7436 100/1000 27.19 13.79 82.08 29.07 90% 41.6133
h2 =0.05 a=1 d=1
method position(25cM) score test statistic
power threshold
mean sd mean sd
Posterior 100/200 30.78 20.34 18.98 8.88 53% 17.4478 100/1000 25.05 9.67 81.23 31.54 91% 40.6903 prior 100/200 30.71 20.30 19.16 8.87 53% 17.7436 100/1000 24.58 9.91 81.86 31.51 91% 41.6133
h2 =0.05 a=1 d=2
method position(25cM) score test statistic
power threshold
mean sd mean sd
Posterior 100/200 30.16 21.42 18.19 8.49 53% 17.4478 100/1000 24.53 9.48 78.50 29.12 93% 40.6903 prior 100/200 31.00 22.75 18.28 8.48 52% 17.7436 100/1000 24.38 9.45 79.04 28.91 92% 41.6133
Table 4: The score test statistic and threshold of score test statistics at 𝛼𝛼 = 0.05 for different methods with different parameter setting on marker density 10cM of a 100cM chromosome.
h2 =0.05 a=1 d=0.5
method position(25cM) score test statistic
power threshold
mean sd mean sd
posterior 100/200 29.10 20.52 20.97 10.65 50% 18.769 100/1000 27.54 12.71 85.92 29.99 92% 43.782 prior 100/200 28.26 19.26 20.87 10.68 49% 18.831 100/1000 27.61 12.65 85.78 30.01 92% 44.315
h2 =0.05 a=1 d=1
method position(25cM) score test statistic
power threshold
mean sd mean sd
posterior 100/200 30.45 17.38 20.87 9.96 54% 18.769 100/1000 25.90 10.29 86.78 31.70 93% 43.782 prior 100/200 30.99 18.77 20.73 9.89 53% 18.831 100/1000 25.87 10.23 86.62 31.68 93% 44.315
h2 =0.05 a=1 d=2
method position(25cM) score test statistic
power threshold
mean sd mean sd
posterior 100/200 30.32 20.30 19.96 8.55 60% 18.769 100/1000 24.52 5.95 83.77 31.17 91% 43.782 prior 100/200 30.32 20.27 19.89 8.57 56% 18.831 100/1000 24.51 5.94 83.62 31.03 91% 44.315
Table 5: The score test statistic and threshold of score test statistics at α=0.05 for different methods with different parameter setting on marker density 5cM of a 100cM chromosome.
h=0.05 a=1, d=0.5
method position(25cM) score test statistic
power threshold
mean sd mean sd
posterior 100/200 29.67 18.06 22.54 11.02 54% 19.8247 100/1000 28.22 12.15 92.51 29.90 91% 45.7731 prior 100/200 29.57 18.02 22.29 11.04 54% 19.8896 100/1000 28.23 12.31 93.32 29.96 91% 46.9538
h=0.05 a=1, d=1
method position(25cM) score test statistic
power threshold
mean sd mean sd
posterior 100/200 30.88 19.59 22.67 10.66 53% 19.8247 100/1000 24.80 5.75 95.39 32.62 96% 45.7731 prior 100/200 31.42 20.14 22.40 10.62 53% 19.8896 100/1000 25.12 6.03 96.01 32.68 96% 46.9538
h=0.05 a=1, d=2
method position(25cM) score test statistic
power threshold
mean sd mean sd
posterior 100/200 31.10 20.77 21.34 8.90 53% 19.8247 100/1000 24.96 2.94 95.66 31.43 97% 45.7731 prior 100/200 31.19 20.66 21.14 8.85 52% 19.8896 100/1000 24.99 3.25 96.63 31.13 96% 46.9538
4 Conclusion and Discussion
In selective genotyping, only the individuals with upper and lower extreme trait
values are genotyped, while the remaining individuals are not. The score test statistic is
simple in derivation and computation in comparison to the likelihood approach. Based on
the posterior model proposed by Lee et al. (2013), which takes both genotyped and
ungenotyped individual into account in the analysis, we derived the score test statistic for
this posterior model for QTL mapping under selective genotyping. We also derived the
score test statistics for the model proposed by Xu and Vogl (2000) and Muranty and
Goffinet (1997) for comparisons. Moreover, we studied the threshold values of QTL
mapping in score test statistics of both models. The results show that the score test
statistics for posterior model and currently used model perform equally well under single-
QTL model. Given a significance level and a genome size, the threshold values are higher
in denser marker maps and extremer selective proportions when score test statistics are
used for QTL detection.
The results for full genotyping and for selective genotyping were compared. The test
statistics and thresholds from the maximum likelihood approach are similar, but these
results from their score test statistics have significant differences. The test statistics and
thresholds of score test statistics under selective genotyping are significantly inflated as
compared to those under full genotyping. However, the statistics (LRT) based on
maximum likelihood approach obtained by Lee et al. (2013) do not possess the trend of
enlargement as in the score test statistics under extremely selective genotyping. Reasons
for the inflation of statistics and thresholds for the score test statistics might be due to the
decrement of the variance when selection is more intense. Table 7 showed the mean
values of the score and their variances and covariance (u x1
、u x2
and the variance- covariance matrix), we found that the score test statistic would raise under extremelyselective proportion and the variance of 𝜇𝜇1(𝑥𝑥) and 𝜇𝜇2(𝑥𝑥) would decrease with the
increasing number of total individuals. Moreover, the determinant of variance-covariance
matrix also becomes decreasing to enlarge the statistics gravely. However, the exact
reason for the inflations of the score test statistics and threshold values under selective
genotyping has not been well studied and deserves to be further investigated.
Table 6:Comparison of the factor values of score test statistic in different selective proportion with the total phenotyping individuals number
ℎ2 = 0.05, (𝑎𝑎 = 1, 𝑎𝑎 = 0.5) Method: Posterior
Selective size
Position (25cM)
Score test
statistics 𝑢𝑢1(𝑥𝑥) var1 𝑢𝑢2(𝑥𝑥) var2 cov V-Cov mean sd mean Sd Det
200/200 27.92 17.52 12.35 5.5 8.68 8.51 1.89 3.99 -0.01 34.18 100/200 29.10 20.52 20.97 10.65 8.05 4.43 1.86 2.08 0.00 9.28
1000/1000 24.61 4.30 47.89 13.41 40.82 41.18 9.85 18.81 -0.13 775.79 100/1000 25.90 10.29 86.78 31.70 13.48 3.35 6.08 1.50 0.08 5.01 Each value based on 100 replicates with marker distance 10-cM along a 100-cM chromosome. (𝑢𝑢1(𝑥𝑥) and 𝑢𝑢2(𝑥𝑥) represent the score functions of a and d, var1 and var2 are the variance of 𝑢𝑢1(𝑥𝑥) and 𝑢𝑢2(𝑥𝑥).)
In this thesis, we only considered the cases of a single QTL with small effects. As
shown in Tables 2 to 4, the results from the score test statistics of the currently used and
posterior models is similar because the frequencies of QTL genotypes in ungenotyped
individuals are close to the frequencies in the whole population ( 1/4、1/2 and 1/4 ) for
small QTL effect. Their differences will become more significant when the QTL has large
effects (not shown). Most quantitative traits are believed to be influenced by multiple
QTL and their interaction. In the cases of multiple QTL, the frequencies of multiple QTLs
genotypes among the ungenotyping individuals may deviate from the population
frequencies, and the differences between posterior model and currently used model might
become remarkable and is worth pursuing. In the future works, we intend to extend the
one-QTL posterior model to multiple-QTL posterior model for QTL detection when
selective genotyping is implemented in QTL experiments.
5 References
CHANG MYRON,N., R.WU, S.WU SAMUEL and G.CASELLA, 2009 Score Statistics for Mapping Quantitative Trait Loci, pp. 1 in Statistical Applications in Genetics and Molecular Biology.
COMMENGES, D., 1994 Robust genetic linkage analysis based on a score test of homogeneity: The weighted pairwise correlation statistic. Genetic Epidemiology 11: 189-200.
COMMENGES,D., and P.K.ANDERSEN, 1995 Score test of homogeneity for survival data.
Lifetime Data Analysis 1: 145-156.
COX,D.R., and D.V.HINKLEY, 1979 Theoretical statistics. Chapman & Hall/CRC.
DARVASI,A., and M.SOLLER, 1992 Selective genotyping for determination of linkage between a marker locus and a quantitative trait locus. Theoretical and Applied Genetics 85: 353-359.
DARVASI,A., A.WEINREB, V.MINKE, J.WELLER and M.SOLLER, 1993 Detecting marker- QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics 134: 943-951.
DOERGE,R.W., and G.A.CHURCHILL, 1996 Permutation tests for multiple loci affecting a quantitative character. Genetics 142: 285-294.
DUDOIT,S., and T.P.SPEED, 2000 A score test for the linkage analysis of qualitative and quantitative traits based on identity by descent data from sib-pairs. Biostatistics 1:
1-26.
FEENSTRA,B., I.M.SKOVGAARD and K.W.BROMAN, 2006 Mapping Quantitative Trait Loci by an Extension of the Haley–Knott Regression Method Using Estimating Equations. Genetics 173: 2269-2282.
GELDERMANN, H., 1975 Investigations on inheritance of quantitative characters in animals by gene markers I. Methods. Theoretical and Applied Genetics 46: 319- 330.
GOLDSTEIN,D.R., S.DUDOIT and T.P.SPEED, 2001 Power and robustness of a score test for linkage analysis of quantitative traits using identity by descent data on sib pairs.
Genetic Epidemiology 20: 415-431.
GUO,Y.-T., 2011 進階回交族群之數量性狀基因座定位門檻值研究, pp. 1-55 in 臺灣 大學農藝學研究所學位論文. 臺灣大學.
HALEY, C. S., and S. A. KNOTT, 1992 A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315-324.
HALEY,C. S., S.A. KNOTT and J. M. ELSEN, 1994 Mapping quantitative trait loci in
crosses between outbred lines using least squares. Genetics 136: 1195-1207.
JANSEN,R. C., 1993 Interval mapping of multiple quantitative trait loci. Genetics 135:
205-211.
JIANG,C., and Z.B.ZENG, 1995 Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140: 1111-1127.
KAO,C.-H., 2000 On the Differences Between Maximum Likelihood and Regression Interval Mapping in the Analysis of Quantitative Trait Loci. Genetics 156: 855- 865.
KAO,C.-H., and Z.-B.ZENG, 1997 General Formulas for Obtaining the MLEs and the Asymptotic Variance- Covariance Matrix in Mapping Quantitative Trait Loci When Using the EM Algorithm. Biometrics 53: 653-665.
KAO, C.-H., Z.-B. ZENG and R. D. TEASDALE, 1999 Multiple Interval Mapping for Quantitative Trait Loci. Genetics 152: 1203-1216.
KAO,C. H., and H. A. HO, 2012 A score-statistic approach for determining threshold values in QTL mapping. Front Biosci (Elite Ed) 4: 2770-2782.
KNOTT,S.A., J.M.ELSEN and C.S.HALEY, 1996 Methods for multiple-marker mapping of quantitative trait loci in half-sib populations. Theoretical and Applied Genetics 93: 71-80.
KRUGLYAK, L., and E. S. LANDER, 1995 A nonparametric approach for mapping quantitative trait loci. Genetics 139: 1421-1428.
LANDER, E. S., and D. BOTSTEIN, 1989 Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185-199.
LEE,H.I., C.H.KAO and H.A.HO, 2013 A Novel Statistical Approach to QTL Mapping under Selective Genotyping: QTL Detection and Threshold Determination.
Unpublished.
LYNCH,M., and B.WALSH, 1998 Genetics and analysis of quantitative traits.
MATHER,K., 1941 Variation and selection of polygenic characters. Journal of Genetics 41: 159-193.
MURANTY,H., and B.GOFFINET, 1997 Selective Genotyping for Location and Estimation of the Effect of a Quantitative Trait Locus. Biometrics 53: 629-643.
OOIJEN, J., 1992 Accuracy of mapping quantitative trait loci in autogamous species.
Theoretical and Applied Genetics 84: 803-811.
PUTTER,H., L.A.SANDKUIJL and J.C. VAN HOUWELINGEN, 2002 Score test for detecting linkage to quantitative traits. Genetic Epidemiology 22: 345-355.
SEN, Ś., and G. A. CHURCHILL, 2001 A Statistical Framework for Quantitative Trait Mapping. Genetics 159: 371-387.
WANG, K., and J. HUANG, 2002 A Score-Statistic Approach for the Mapping of Quantitative-Trait Loci with Sibships of Arbitrary Size. The American Journal of
Human Genetics 70: 412-424.
WOLFRAM RESEARCH,I., 2012 Mathematica pp. Wolfram Research, Inc., Champaign, Illinois.
XU,S., and C.VOGL, 2000 Maximum likelihood analysis of quantitative trait loci under selective genotyping. Heredity 84: 525-537.
ZENG,Z. B., 1993 Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci. Proceedings of the National Academy of Sciences 90: 10972-10976.
ZENG,Z.B., 1994 Precision mapping of quantitative trait loci. Genetics 136: 1457-1468.
6 Abbreviations
Abbreviations Term
QTL Quantitative trait loci
IM Interval mapping
LRT Likelihood ratio test
REG interval mapping Regression interval mapping
CIM Composite interval mapping
MIM Multiple interval mapping