行政院國家科學委員會專題研究計畫 成果報告
新發色機團原冰片烯高分子光電材料的合成與特性研究(第 2 年)
研究成果報告(完整版)
計 畫 類 別 : 個別型
計 畫 編 號 : NSC 96-2221-E-011-067-MY2
執 行 期 間 : 97 年 08 月 01 日至 98 年 07 月 31 日 執 行 單 位 : 國立臺灣科技大學化學工程系
計 畫 主 持 人 : 廖德章
計畫參與人員: 碩士班研究生-兼任助理人員:張正宏 碩士班研究生-兼任助理人員:連蔚任 碩士班研究生-兼任助理人員:吳翰宇 博士班研究生-兼任助理人員:陳文祥
報 告 附 件 : 國外研究心得報告
出席國際會議研究心得報告及發表論文
處 理 方 式 : 本計畫涉及專利或其他智慧財產權,2 年後可公開查詢
中 華 民 國 98 年 09 月 14 日
行政院國家科學委員會補助專題研究計畫 □ 成 果 報 告
□期中進度報告
新發色基團原冰片烯高分子光電材料的合成與特性研究
計畫類別:□ 個別型計畫 □ 整合型計畫
計畫編號:NSC 96 - 2221- E - 011 - 067 - MY2 執行期間:96 年 8 月 1 日至 98 年 7 月 31 日
計畫主持人:廖德章 共同主持人:
計畫參與人員:陳文祥、張正宏、吳翰宇、連蔚任
成果報告類型(依經費核定清單規定繳交):□精簡報告 □完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、
列管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□二年後可公開查詢
執行單位:國立台灣科技大學
目錄
摘要………. p.3 計畫緣由與目的………. p.5 儀器與設備………. p.7 實驗………. p.8 結果與討論………. p.11 結論………. p.15 計畫成果自評………. p.16 致謝………. p.16 參考文獻………. p.17 反應式、表格、圖………. p.19
摘要
利用狄耳士-阿德爾(Diels-Alder)及縮合反應成功合成出新型 exo-原冰片烯發 光(芴、芘、咔唑)機能性單體。含芴之新型 exo-原冰片烯發光高活性單體 (exo-1) 可進行活性開環複分解聚合,進一步利用形成雙嵌段共聚合物及數目平均分子量 對 [M]/[I] 比 為 一 線 性 關 係 證 明 之 。 利 用 ”Grubbs catalyst”(I) ({RuCl2(CHPh)[P(C6H11)3]2})聚合新型側鏈含純 exo 形式芴發光機能性基團之新型 原冰片烯聚合物[ Poly(exo-1)],在 90 秒時即可得到高分子量(3.18×104,[M]/[I] = 100)。然而,將含芘(exo-2)、咔唑(exo-3)新型 exo-原冰片烯發光機能性單體進行開 環複分解聚合無法得到活性聚合之現象。這些新型側鏈含發光機能性基團(芴、芘、
咔唑)之新型原冰片烯聚合物具有很好的溶解度在下列的溶劑:氯仿、、1,2-二氯苯。
利用熱示差掃瞄熱卡計(DSC)測量這些新型側鏈含發光機能性基團(芴、芘、咔唑) 之新型原冰片烯聚合物的玻璃轉移溫度,所得到玻璃轉移溫度(Tg = 80 ~109 oC)皆
高於不含側鏈之原冰片烯聚合物(Tg = 35 oC),因為導入懸掛之芳香族基團(芴、芘、
咔唑)於聚合物中會增加其熱穩定性及耐熱性。藉由螢光光譜儀之測量,可得知這 些新型側鏈含發光機能性基團(芴、芘、咔唑)之新型原冰片烯聚合物在可視藍光區 具有很強的激發光。
關鍵詞:發色基團;核磁共振光譜儀;原冰片烯;聚烯烴;開環複分解聚合。
英文摘要
Pure exo-functional norbornene monomers containing various chromophores such as fluorene, pyrene, and carbazole were successfully prepared via the Diels–Alder reaction and condensation reaction. The living ring-opening metathesis polymerization (ROMP) of a fluorene-containing monomer, exo-2-(fluorene-9-ylcarboxymethyl) norborn-5-ene (exo-1), was observed and confirmed by the formation of a diblock copolymer and a linear relationship between the number-average molecular weight and [M]/[I] ratios ([M] = monomer concentration; [I] = initiator concentration). The synthesis and characteristics of novel fluorene-containing polymers based on pure exo-1 are reported with Grubbs catalyst I {RuCl
2(CHPh)[P(C
6H
11)
3]
2} with a high molecular weight of 3.18 × 10
4in 90 s ([M]/[I] = 100). However, the ROMP of pyrene- and
carbazole- containing monomers [exo-5-(pyrene methoxy carbonyl)bicyclo[2.2.1]hept-2-ene and exo-5-(carbazole ethoxy carbonyl)bicyclo[2.2.1]hept-2-ene, respectively] were carried out in a nonliving fashion.
All the chromophore-containing polymers showed excellent solubility in various organic solvents, particularly in chloroform, N-methyl-2-pyrrolidinone, and 1,2-dichlorobenzene. The glass transition temperatures of polynorbornenes containing various chromophores were determined to be 80 – 109
oC (by differential scanning calorimetry) higher than that of ring-opened polynorbornene (glass transition temperature = 35
oC), indicating that the incorporation of the pendant aromatic moieties (e.g., fluorene, pyrene, and carbazole) could enhance the transition temperature for segmental motions of polymer chains. The photoluminescence spectra of all polymer solutions showed a strong emission in the blue region of the visible spectra.
Keyword:chromophores; NMR; norbornene; polyolefins; ROMP
計畫緣由與目的
烯類複分解反應(Olefin metathesis)在近年來成為頗受重視的碳-碳鍵合成方 法,隨著各種新型觸媒開發,環烯類精密複分解聚合與非環二烯類及炔類的複分 解聚合日趨重要。1 Robert H. Grubbs (Caltech) 和 Richard R. Schrock (MIT) 更在 2005年因對與有機化合物複分解合成方面有卓越的成果而得到諾貝爾化學獎。其 中 , 以 原 冰 片 烯(Norbornene) 衍 生 物 進 行 開 環 複 分 解 聚 合 反 應 (Ring-opening metathesis polymerization,ROMP)成為合成聚合物的一種新方法2,對於帶有各式 官能基的高分子合成材枓也越來越多,成為重要的功能性高分子材枓合成方法之 一。
有機金屬觸媒用於複分解聚合已有一段時日,但對於有官能基之單體在聚合 時會有所限制,同時水份或氧氣的存在對聚合也相當敏感。例如鎢(W)、鈦(Ti)、
鉬(Mo)及釕(Ru)等金屬化合物為較常用的環烯類精密複分解聚合觸媒,其中釕金屬 觸媒 (Ru catalyst) 對水份或氧氣的安定性 (Stability) 或相容度 (Tolerance) 較
佳,甚至可於水溶液中進行聚合反應。3 例如1996年 Grubbs 等人新開發的釕碳烯
配位觸媒 {Cl2Ru(CHPh)[P(C6H11)3]2},4 對環烯類精密複分解聚合非常有效,尤其 在空氣中還算安定,可進行具有官能基單體的聚合。另外亦具有高聚合速率及高 分子量之特性,一般而言均具有活性聚合 (Living polymerization) 的現象。
利用環烯類衍生物為單體所進行的精密複分解聚合之相關研究越來越多,主 要有側鏈型液晶,5-7兩步法 (Two step) 合成三嵌段 (Triblock) 共聚合物,8各種官
能基導入之聚合物。9, 10其中官能基的導入,使聚合物具有光電性質11及生化活性。
12
聚原冰片烯 (Polynorbornene) 及其衍生物是第一個藉精密複分解聚合之商業
化產品,為重要的工程塑膠之一。13因其透明性良好、耐衝擊性佳 (橡膠添加劑) 、
廣泛之溫度使用範圍、良好之機械物性和加工性。廣用於照明器具、機械、電子 零件、管件和食品包裝等。又其衍生物如酸及酯類的聚合物更被當作電子產業的
光阻劑。14-16國內有關精密複分解聚合的研究,據筆者所知還很少,尤其在大學之
學術機構更少,對於此一極具潛力的高分子材料之合成方法更顯示出其重要性。
由於近年來光電產業蓬勃發展,(Organic Light-Emitting Display,OLED)或 (Polymer Light-Emitting Display,PLED)在未來的發展將佔有舉足輕重的地位,相 關領域皆投入大量物力與人力積極開發新型發光材料,而精密複分解聚合技術有
利於發展均相或嵌段聚合物,若單體具有活性聚合的特性,聚合物的分子量將可 被控制,將是非常具有工業化潛力的光電材料。
本計畫主要是藉由狄耳士-阿德爾反應 (Diels-Alder Reaction)17-22 之分子設計 合成出多種新的具不同功能性基團的高活性原冰片烯衍生物,進一步合成出新型 機能性之原冰片烯聚合物及原冰片烯共聚合物,並研究其聚合行為與物性,對原 冰片烯衍生物的精密複分解聚合相關領域有更深入的了解與貢獻。本計劃第一年 度預計合成出末端為純 exo 形式的高活性原冰片烯化合物,本計劃藉由狄耳士-阿 德爾反應 (Diels-Alder Reaction)得到 endo/exo 混合形式的原冰片烯甲酸酯單體 (endo/exo-NBMAM),甲酸酯基團在金屬納的的作用下形成甲酸基,由於 endo 形 式的原冰片甲酸單體其甲酸基與原冰片烯的雙鍵距離接近,藉由雙鍵和碘的自由 基反應形成原冰片烷環內酯碘化合物,進一步將疏水性的原冰片烷環內酯碘化合 物分離後得到純 exo 形式的原冰片烯甲酸。原冰片烯甲酸經過還原反應後得到高活 性的原冰片烯甲醇,再利用脫水反應得到所設計的新型功能性原冰片烯單體,利 用開環複分解聚合反應得到新型的具有純 exo 形式側鏈的原冰片烯機能性高分子 材料。第二年度將原冰片烯甲酸經過醯氯化反應後得到高活性的原冰片烯甲醯 氯,再利用脫氯化氫反應得到本計劃的所設計的新型功能性原冰片烯單體,利用 開環複分解聚合反應得到新型的具有純 exo 形式側鏈的原冰片烯機能性高分子材 料。
儀器與設備
(1) 紅外線光譜儀(IR),型號為 JASCO IR-9700 測試範圍為 4000-400cm-1,測試
法為利用KBr 打片測試而得。
(2) 13C 及1H NMR 核磁共振光譜儀,型號為 Bruker DRX-500,測試範圍為 125 MHz 到 500 MHz 之間。所使用的溶劑為重氫氯仿(CDCl3)。
(3) 示差熱分析儀(DSC),型號為 Du pont 9000 DSC system,設定升溫速度為每 分鐘10℃。
(4) 毛細管熔點測定儀,型號為 Model BüCHI 535。
(5) 元素分析儀(Elemental analzer),型號為 Perkin-Elmer 2400。
(6) 熱重分析儀(TGA),型號為 ULVAC (Sinku-Riko) model 7000,升溫速率為每
分鐘10℃,氮氣流率為每分鐘 60 立方公分。
(7) 凝膠滲透層析儀(GPC),由五根 Waters (Ultrastyragel) 所構成 [300 × 7.7 mm (guard, 500, 103, 104, 105 Å in a series)],四氫呋喃為移動相,使用 RI 偵測器測 定樣品之分子量,聚苯乙烯(Polystyrene)作為標準品。
(8) 螢光光譜儀,廠牌型號為 Shimadzu RF-5031。
Monomer Synthesis
Synthesis of exo-1A mixture of 9H-fluorene-9-carboxylic acid (7.74 g, 0.0368 mol) and
exo-5-(hydroxyl methyl)bicyclo[2.2.1]hept-2-ene (5.48 g, 0.044 mol)
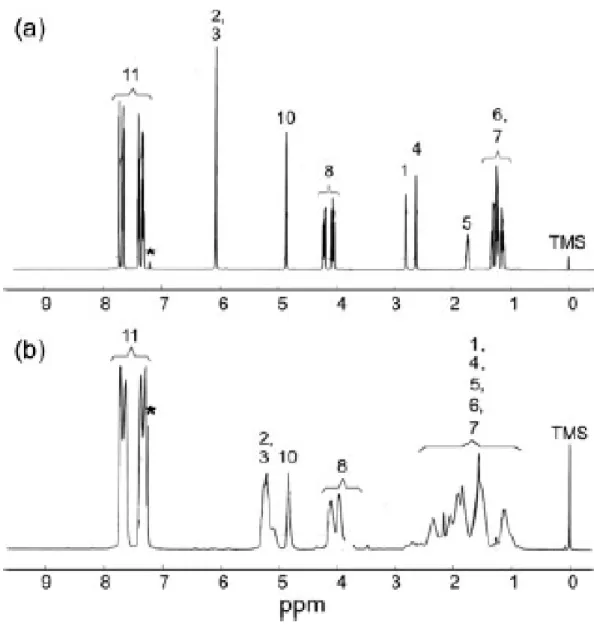
23 in toluene was charged into a flask [250 mL; Scheme 1(a)]. The p-toluenesulfonic acid was added as a catalyst. The reaction mixture was refluxed at 140 oC for 5 h, and water was removed as an azeotropic mixture via refluxing with toluene. After the completion of the reaction, the solvent was removed in a rotary evaporator. The reaction mixture was purified by column chromatography (ethyl acetate/n-hexane = 1:3) and then recrystallized from cold n-hexane, giving a white solid (mp = 44–45 oC). IR (KBr pellet, cm-1): 1587 (νc=c, vinylic), 1647, 1478 (νc=c, aromatic stretching). The 1H NMR (CDCl3) spectrum ofexo-1 exhibited signals at δ = 1.18–1.21 (H
6, 2H), 1.27–1.37 (H7, 2H), 1.77–1.81 (H5, 1H), 2.68 (H4, 1H), 2.86 (H1, 1H), 4.09–4.28 (H8, 2H), 4.92 (H10, 1H), 6.11 (H2 and H3, 2H), and 7.37–7.80 ppm (H11, 8H) [Fig. 1(a)]. The 13C NMR (CDCl3) spectrum of exo-1 exhibited signals at δ = 29.49 (C6), 37.95 (C5), 41.52 (C1), 43.58 (C4), 44.92 (C7), 53.47 (C10), 69.34 (C8), 136.21 (C3), 137.99 (C2), 119.94, 125.53, 127.21, 127.99, 140.67, 141.36 (C11), and 170.75 ppm (C9).Synthesis of exo-2
A mixture of 1-pyrene methanol (3 g, 0.013 mol) in dry tetrahydrofuran (THF; 50 mL) and triethylamine (1.58 g, 0.015 mol) was added to a flask (250 mL). The
exo-5-(carbonyl chloride)bicyclo[2.2.1]hept-2-ene (2.03 g, 0.013 mol) was dissolved in
dry THF (10 mL) and then added dropwise at the ambient temperature to the above reaction mixture, which was stirred continuously for 4 h. After the completion of the reaction, the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:3). Then, the product was recrystallized from n-hexane, and a white solid was obtained (mp = 75 – 77 oC). The 1H NMR (CDCl3) spectrum of exo-2 exhibited signals at δ = 1.41 and 2.08 (H6, 2H), 1.44 and 1.67 (H7, 2H), 2.38 (H5, 1H), 2.97 (H1, 1H), 3.17 (H4, 1H), 5.86 (H9, 2H), 6.11 (H2, 1H), 6.16 (H3, 1H), and 7.37–7.80 ppm (H10, 9H). The 13C NMR (CDCl3) spectrum ofexo-2 exhibited signals at δ = 30.33 (C
6), 41.57 (C1), 43.19 (C5), 46.33 122.82, 124.51, 124.56, 124.78, 125.31, 125.38, 125.95, 127.26, 127.56, 127.65, 128.01, 129.08, 130.61,Synthesis of exo-3
A mixture of 9H-carbazole-9-ethanol (3 g, 0.014 mol) in dry THF (50 mL) and triethylamine (1.7 g, 0.017 mol) was added to a flask (250 mL). The exo-5-(carbonyl chloride)bicyclo[2.2.1]hept-2-ene (2.19 g, 0.014 mol) was dissolved in dry THF (10 mL) and then added dropwise at the ambient temperature to the above solution, which was stirred continuously for 4 h. After the completion of the reaction, the solvent was removed in a rotary evaporator. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:3). Then, the product was recrystallized from n-hexane, and a white solid was obtained (mp 72–73 oC). The 1HNMR (CDCl3) spectrum of exo-3 exhibited signals at δ = 1.16 and 1.68 (H6, 2H), 1.18 and 1.32 (H7), 1.71 (H5, 1H), 2.76 (H1 and H4, 2H), 4.51 (H9, 2H), 5.98 and 6.05 (H2 and H3, 2H), 7.25, 7.42, and 8.09 ppm (H11, 8H). The 13C NMR (CDCl3) spectrum of exo-3 exhibited signals at δ = 30.11 (C6), 41.44 (C1), 41.56 (C10), 42.87 (C5), 46.19 (C7), 46.26 (C4), 61.98 (C9), 108.55, 119.14, 120.25, 122.92, 125.62, 140.30 (C11), 135.50 (C2), 137.85 (C3), and 175.89 ppm (C8) [Fig. 2(c,d)]
Polymerization
Polymerization of exo-1
A solution of the catalyst was prepared by the dissolution of RuCl2(CHPh)[P(C6H11)3]2 (0.0075 g, 9.11 × 10-3 mmol) in 0.2 mL of anhydrous THF under an argon-filled drybox. exo-1 (0.29 g, 0.911 mmol) was added to 3 mL of THF and then degassed via a freeze–pump–thaw cycle. After complete degassing, the catalyst solution was injected into the monomer solution by a syringe. The pink solution was vigorously stirred at 35 oC for 90 s. The reaction was terminated by the addition of 1 mL of ethyl vinyl ether. After the termination, the polymer was precipitated in an excess of methanol and dried in a vacuum system at the ambient temperature. The 1H NMR (CDCl3) spectrum of poly(exo-1) exhibited signals at δ = 1.14– 2.71 (H1, H4, H5, H6, H7), 3.75–4.13 (H8), 4.85 (H10), 5.10–5.27 (H2, H3), and 7.26–7.73 ppm (H11) [Fig.
1(b)]. The 13C NMR (CDCl3) spectrum of poly(exo-1) exhibited signals at δ =
ٛ
29.58–46.27 (C1, C4, C5, C6, C7), 53.56 (C10), 69.90 (C8), 131.37–132.79 (C3, C2), 120.02, 125.63, 127.29, 128.10, 140.72, 141.43 (C11), and 170.95 ppm (C9; trans/cis = 85/15).Polymerization of exo-2
Poly(exo-2) was prepared by the adoption of a procedure similar to the synthesis of poly(exo-1), with [M]/[I] = [exo-2]/[RuCl2(CHPh)[P(C6H11)3]2] = 1000 ([M] = monomer concentration; [I] = initiator concentration), via polymerization in chloroform at 35 oC for 1 h with a yield of 73%. The polymer was insoluble in THF. The 1H NMR (CDCl3) spectrum of poly(exo-2) exhibited signals at δ = 0.40–2.91 (H1, H4, H5, H6, H7), 4.50–5.18 (H9), 5.64–5.66 (H2, H3), and 7.84–8.29 ppm (H10). The 13C NMR (CDCl3) spectrum of poly (exo-2) exhibited signals at δ = 41.82–50.34 (C1, C4, C5, C6, C7), 64.53 (C9), 131.37–132.79 (C3, C2), 122.83–138.05 (C10), and 176.20 ppm (C8).
Polymerization of exo-3
Poly(exo-3) was prepared by the adoption of a procedure similar to the synthesis of poly(exo-1), with [M]/[I] = [exo-3]/[RuCl2(CHPh)[P(C6H11)3]2] = 1000, via polymerization at 35 oC for 1 h. A number-average molecular weight of 6.15 × 104 (the theoretical value was 3.31 × 105) and polydispersity index (PDI) values close to 1.60 were observed, with a yield of 14%. The 1H NMR (CDCl3) spectrum of poly(exo-3) exhibited signals at δ = 0.50–2.70 (H1, H4, H5, H6, H7), 4.20–4.80 (H9, H10), 4.90–5.30 (H2, H3), and 7.84–8.29 ppm (H11). The 13C NMR (CDCl3) spectrum of poly (exo-3) exhibited signals at δ = 35.01–51.48 (C1, C4, C5, C6, C7, C10), 63.53 (C9), 131.37–132.79 (C3, C2), 122.83–138.05 (C11), and 176.20 ppm (C8).
結果與討論:
A series of functional monomers, exo-1, exo-2, and exo-3, were synthesized and isolated (Scheme 1). The structures of these monomers (exo-1, exo-2, and exo-3) were confirmed by 1H and 13C NMR spectroscopy. 13C NMR was used to differentiate varieties of aliphatic carbons by use of the distortionless enhancement by polarization transfer (DEPT) technique because of some overlapped peaks. Figure 2 shows the 13C NMR/DEPT spectra of exo-2 and exo-3. For instance, the DEPT spectrum of exo-2 [Fig.
2(a,b)] shows three upward resonances at 41.57, 43.19, and 46.58 ppm in the region of 30–70 ppm, which can be assigned to the methine carbons (CH; C1, C5, and C4, respectively), and three downward signals at 30.33, 46.33, and 64.63 ppm can be attributed to the three methylene (CH2) carbons (C6, C7, and C9, respectively) of the aliphatic part of exo-2 in the region of 30–70 ppm. The DEPT spectrum of exo-2 [Fig.
2(a,b)] shows no peaks at 124.45, 124.65, 128.98, 129.24, 130.50, 131.01, and 131.45 ppm, which can be assigned to the aromatic carbons without attached protons (CH0) and observed in the 13C NMR spectrum of exo-2 [Fig. 2(b)]. There are nine upward resonances at 122.69, 124.41, 125.21, 125.27, 125.84, 127.15, 127.41, 127.53, and 127.89 ppm, which can be assigned to the methine carbons (CH; C10) of the pyrene group. Furthermore, the 13C NMR/DEPT spectrum of exo-3 [Fig. 2(c,d)] shows three upward resonances at 41.44, 42.86, and 46.26 ppm in the region of 30–70 ppm, which can be assigned to the methine carbons (CH; C1, C5, and C4, respectively), and four downward signals at 30.11, 41.50, 46.19, and 61.98 ppm can be attributed to the four methylene (CH2) carbons (C6, C10, C7, and C9, respectively) of the aliphatic part of exo-3 in the region of 30–70 ppm. The DEPT spectrum of exo-3 [Fig. 2(c,d)] shows no peaks at 122.92 and 140.30 ppm, which can be assigned to the aromatic carbon without attached protons [CH0; C and Cβ; Scheme 1(c) and Fig. 2(d)]. There are four upward resonance at 108.55, 119.14, 120.25, and 125.62 ppm [Fig. 2(c,d)], which can be assigned to the methine carbons (CH; C11) of the carbazole group. This experiment clearly indicates the assignments of carbons and even overlapped carbon peaks. ROMP of exo-1 was carried out with the RuCl2(CHPh)[P(C6H11)3]2 catalyst. Poly(exo-1) with a high molecular weight of 3.18 × 104 ([M]/[I] = 100) was obtained in a high yield above 99%, with a narrow PDI ( ~ 1.10) for a short polymerization time, which was as short as 90 s in THF at 35 oC [Table 1 and Fig. 3(a)]. THF was employed as a reaction solvent to determine the molecular weight of resulting poly(exo-1) easily because the
polymerization could be carried out in a short time. A linear relationship of the number-average molecular weight of poly(exo-1) increasing with the [M]/[I] ratio, a narrow polydispersity between 1.05 and 1.10, and a yield above 99% for all ratios ([M]/[I] < 250) were observed [Figs. 3(b,c) and 4(a)], and this was an indication of a living polymerization. Moreover, the linear relationship of the number-average molecular weight of poly (exo-1) and the polymerization time is shown in Figure 4(b).
The conversion was over 95% after 90 s [Fig. 5(a)]. The semilogarithmic plot of the monomer conversion versus the time was linear for the ROMP of exo-1 [Fig. 5(b)]. One equivalent of the Ru catalyst {Cl2Ru(CHPh)[P(C6H11)3]2} and 35 equiv of exo-1 were polymerized for 90 s at 35 oC in THF. One portion of the polymerization solution exhibited a number-average molecular weight of 1.14 × 104, which was close to the calculated molecular weight of 1.10 × 104 {[M]/[I] = 35; Fig. 3(d)}. Another portion of 35 equiv of exo-1 was added and stirred for an additional 90 s before termination. The number-average molecular weight of 2.55 × 104 (the theoretical value was 2.21 × 104) and PDI values around 1.10 were observed [Fig. 3(e)]. A clear increase in the molecular weight was accomplished, indicating that no chain termination processes had occurred, and the living ROMP of exo-1 was confirmed [Fig. 3(b,c)]. In the NMR measurement of the solution of exo-1 and the Ru catalyst, the signals of olefinic protons at δ = 5.5–6.0 ppm (norbornene group of monomer exo-1) were shifted to 5.1–5.2 ppm (polynorbornene main chain, trans/cis ratio = 85/15), completely indicating that exo-1 was consumed within a short time of 90 s, which is another piece of evidence supporting living polymerization. The living ROMP of exo-1 was confirmed. In addition, the GPC trace of poly(exo-1) after a long polymerization time (e.g., >2 h) was observed, with a small shoulder appearing on the high-molecular weight side of the GPC curves, which might be due to traces of oxygen in the system. ROMP of several
exo-norbornene derivatives has recently been carried out using Grubbs catalyst I in a
living fashion.24,25 Although such exo-norbornene derivatives react more quickly to give a quantitative yield of the polymer than endo-rich or endoisomers, they need to spend more than 25 min and even 5 h for living ROMP using Grubbs catalyst I. 25-28 As the ROMP of exo-1 with [M]/[I] = 125 was carried out at 35 oC in THF with the RuCl2(CHPh)[P(C6H11)3]2 catalyst, the number average molecular weight (3.90×104; PDI = 1.06) of poly(exo-1) was close to the calculated molecular weight (3.96 × 104)after the addition of another monomer, NBCbz ([M]/[I] = 50), for another 2 h was observed, which is close to the calculated molecular weight [5.23 × 104; Fig. 3(g)].26 The NMR spectra of poly(exo-1) [Fig. 1(b)] showed the aromatic proton at δ = 7.26 – 7.73 ppm (Ha ~ Hd; 1H NMR) and the aromatic carbon at δ = 118.63, 125.60, 127.28, 128.31 (Ca ~ Cd), 140.38, and 140.37 ppm (Ce, Cf, 13C NMR), which are due to aromatic fluorene groups within poly(exo-1) chains. After copolymerization, the NMR spectra of diblock copolymer poly(exo-1)-b-(NBCbz) showed a new signal at δ = 8.11 ppm and signals at δ = 7.26–7.73 ppm (1H NMR) and the aromatic carbon at δ = 109.79, 118.54, 120.28, 125.43 (Ca’ ~ Cd’), 122.81, and 138.49 ppm (Ce’, Cf’), which are due to the aromatic carbazole group. However, the ROMP of pyrene- and carbazole-containing monomers (exo-2 and exo-3, respectively) was carried out in a nonliving fashion, as shown in Table 1. Similar behaviors for other monomers with ester linkage groups have been observed by Weck et al.,29 Endo et al.,30 Abdelaziz et al.,31 and Slugovc and coworkers.32,33 The observed effects are explained by different tendencies of the anchor groups to coordinate to the ruthenium center during the resting state of the propagation.
32,33 Slugovc and coworkers32,33 reported that neither eight-membered chelate systems with oxo oxygen nor six-membered rings with the oxy oxygen of the ester group are formed. Consequently, the ROMP of exo-1 is the fastest of the three monomers (exo-1,
exo-2, and exo-3). According to the model study of Slugovc and coworkers,
32,33 the anchor groups (C=O) of exo-2 and exo-3 are easy to coordinate to the ruthenium center during the resting state of the propagation. This is the reason that the ROMP behaviors of exo-2 and exo-3 are different from that of exo-1, and poor ROMP behaviors of exo-2 and exo-3 are shown. Similar behavior has been observed by Slugovc and coworkers.32,33
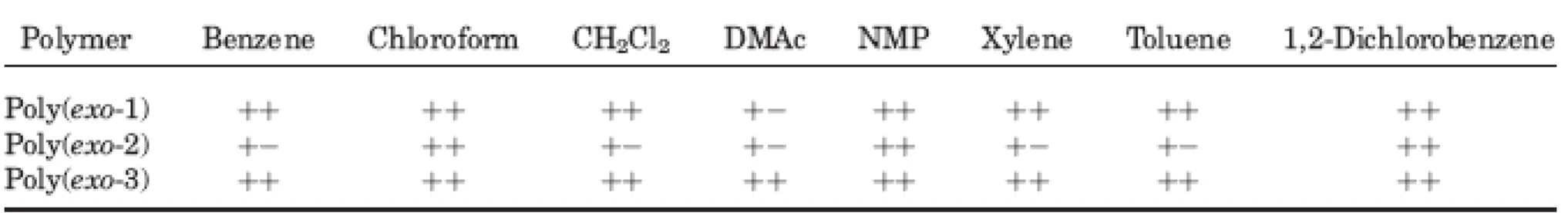
Solubility of Polynorbornenes Containing Various Chromophores
The excellent solubility of these polymers was observed in various organic solvents such as benzene, chloroform, THF, CH2Cl2, N-methyl-2-pyrrolidinone (NMP), xylene, toluene, 1,2-dichlorobenzene, and N,N-dimethylacetamide (DMAc; Table 2).
Poly(exo-1) and poly(exo-3) exhibited better solubility than poly(exo-2).
Thermal Properties of Polynorbornenes Containing Various Chromophores
The glass transition temperatures of polynorbornenes containing various chromophores were determined to be 80–109 oC (by differential scanning calorimetry),
which is higher than that of the commercially available ring-opened polynorbornene (35
oC), indicating that the incorporation of the pendant aromatic moieties (e.g., fluorene, pyrene, and carbazole) could enhance the transition temperature for segmental motions of polymer chains. The polymers exhibited glass transitions at 80, 109, and 91 oC for poly(exo-1), poly(exo-2), and poly(exo-3), respectively.
Optical Properties of Polynorbornenes Containing Various Chromophores
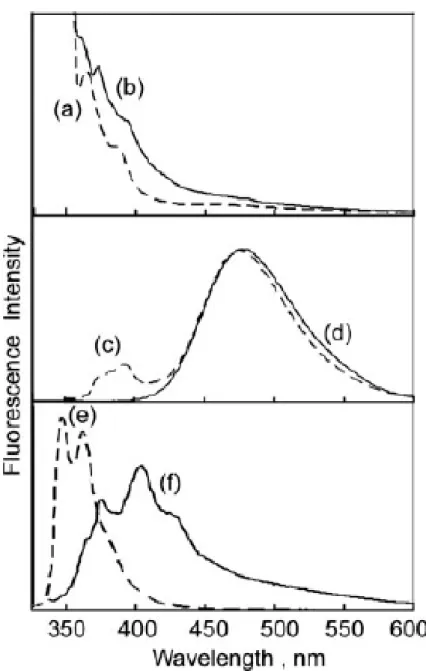
The photoluminescence spectra of poly(exo-1), poly(exo-2), and poly(exo-3) are shown in Figure 6. The photoluminescence emission of poly(exo-1) with two peaks at 360 and 375 nm and a significant shoulder extending from 391 to 500 nm were observed in a CHCl3 solution [Fig. 6(a)]. In the photoluminescence spectrum of poly(exo-1) film, the maxima and shoulder shifted to 375 and 395 nm, respectively, implying some aggregations of the poly(exo-1) film [Fig. 6(b)]. The photoluminescence spectra of the poly(exo-1) solution and poly (exo-1) film showed strong emissions in the blue region of the visible spectra. From photoluminescence spectra of poly(exo-2), poly(exo-2) in a CHCl3 solution exhibited a monomer emission at 378 and 394 nm and a strong excimer emission near 476 nm [Fig. 6(c)]. The poly(exo-2) film exhibited only an excimer emission near 480 nm, implying aggregations of the poly(exo-2) film [Fig.
6(d)].34,35 From photoluminescence spectra of poly(exo-3), poly(exo-3) in a CHCl3
solution exhibited an emission at kmax values of 347 and 363 nm [Fig. 6(e)]. The poly(exo-3) film exhibited a strong fluorescence monomer emission of the carbazole group occurring in near-UV at 377 nm and a significant excimer emission extending from 390 to 500 nm, which exhibited a blue-green color [Fig. 6(f)]. The photoluminescence spectra of all the polymer solutions showed a strong emission in the blue region of the visible spectra.
結論:
The synthesis and characteristics of novel polymers based on pure exo-isomers with ROMP have been reported. The monomer of exo-5-(fluorine carboxyl methyl)bicyclo[2.2.1]hept-2-ene under ROMP conditions, with the concentration ratio of the monomer to the initiator being 100, yielded polymers with a high molecular weight of 3.18 × 104 in a reaction time of 90 s. The living ROMP of
exo-2-(fluorene-9-ylcarboxymethyl)norborn-5-ene was confirmed by the formation of a
diblock copolymer and the linear relationship of the numberaverage molecular weight increasing with the [M]/[I] ratio. However, the ROMP of pyrene- and carbazole-containing monomers was carried out in a nonliving fashion. The polymers showed excellent solubility in most of the organic solvents.計畫成果自評:
本計畫完全依照計畫內容執行,執行之相關內容已發表或投稿在下列著名期 刊及專利中:
(1) J. Polym. Sci., Part A: Polym. Chem., Vol. 45, pp. 3022-3031 (2007) (2) 美國專利申請中(Application NO.: 11/905,939)
本計畫之新型含新發色基團原冰片烯單體、聚合物及共聚合物皆使用儀器鑑 定其結構,鑑定結果均符合結構,另外其聚合物皆具有優良物性(如溶解性、熱
性質及機械性質),可供學術界及工業界參考,為一具有發展潛力之高分子材料。
致謝:
感謝行政院國家科學委員會補助專題計劃NSC 96-2221-E-011-067-MY2 之經 費補助,使本兩年期計劃之期末報告得以順利執行。
參考文獻:
1. Demonceau, A., A. W. Stumpf, E. Savive and A. F. Noels, Macromolecules, 30, 3127 (1997)
2. Buchmeiser, M. R. Chem. Rev. 2000, 100, 1565
3. Schwab, P., R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 118, 100 (1996) 4. Lynn, D. M., S. Kanaka and R. H. Grubbs, J. Am. Chem. Soc., 118, 784 (1996) 5. Pugh, C., J. Dharoa and S. V. Arehart, Macromolecules, 30, 4520 (1997)
6. Maughon, B. R., M. Weck, B. Mohrand and R. H. Grubbs, Macromolecules, 30, 257 (1997)
7. Arehart, S. V. and C. Pugh, J. Am. Chem. Soc., 119, 3027 (1996)
8. Weck, M., P. Schwab and R. H. Grubbs, Macromolecules, 29, 1789 (1996) 9. Maughon, B. R. and R. H. Grubbs, Macromolecules, 30, 3459 (1997) 10. Deffieux, A. and M. Schappacher, Polym. Adv. Tec., 7, 122, (1996) 11. Gratt, J. and R. E. Cohen, Macromolecules, 30, 3137 (1997)
12. Gibson, V. C., E. L. Marshall, M. North, D. A. Robson and P. J. William, Chem.
Commun., 1095 (1997)
13. Japan Plastic, 8, 11 (1974)14. Liaw, D. J., A. Soum, M. Fontanille, A. Parlier and H. Rudler, Makromol. Chem.
Rapid Comm., 6, 309 (1985)
15. 木尾田徹 日本合成橡膠 第七回光反應電子用材料研究會講座(東京)1998 年 1 月21 日
16. Okoroanyanwu, U., T. Shimokawa, J. Byers, D. Medeiros, C. G. Willson, Q. J. Niu and J. M. J. Frechet, SPIE, 3049, 92
17. Bachmann, W. E., L. F. Fieser, A. H. Blatt and J. R. Johnson, “Organic Reaction”
Vol. 4, Roger Adams, New York, (1948), pp.60-173.
18. Ivin, K. J., L. M. Lam and J. J. Rooney, Macromol. Chem. 195, 3245 (1994)
19. Heroguez, V., Y. Gnanou and M. Fontanille, Macromol. Rapid Commun, 17, 137 (1996)
20. Zhang, X., J. Xia and K. Matyjaszewski, Macromolecules, 31, 5167 (1998)
21. Ivin, K. J. and J. C. Mol, Olefin Metathesis and Metathesis Polymerization, Academic Press Limited, London, UK, (1997)
22. Liaw, D. J., C. Lucas, A. Soum, M. Fontanille, A. Parlier and H. Rudler, "Transition
Metal Catalyzed Polymerization", R.P. Qurik (Ed.), Cambridge University Press,
U.S.A. pp. 671 (1985)23. Berson, J. A., J. S. Walia, A. Remanick, S. Suzuki, P. Reynolds-Warnhoff and D.
Willnerv, J. Am. Chem. Soc., 83, 3986 (1961).
24. Pollino, J. M., L. P. Stubbs and M. Weck, Macromolecules, 36, 2230 (2003).
25. Maynard, H. D., S. Y. Okada and R. H. Grubbs, Macromolecules, 33, 6239 (2000).
26. Carlise, J. R. and M. Weck, J. Polym. Sci. Part A: Polym. Chem., 42, 2973 (2004).
27. Liaw, D. J. and C. H. Tsai, Polymer, 41, 2773 (2000).
28. Demel, S., W. Schoefberger, C. Slugovc and F. Stelzer, J. Mol. Catal. A, 200, 11 (2003).
29. Kriegel, R. M., W. S. Rees and M. Weck, Macromolecules, 37, 6644 (2004).
30. Hino, T., N. Inoue and T. Endo, J. Polym. Sci. Part A: Polym. Chem., 44, 395 (2006).
31. Abdelaziz, A. S., R. M. Okasha, L. I. May and J. Hurd, J. Polym. Sci. Part A:
Polym. Chem., 44, 3053 (2006).
32. Slugovc, C., S. Demel, S. Riegler, J. Hobisch and F. Stelzer, Macromol. Rapid
Commun., 25, 475 (2004).
33. Slugovc, C. Macromol. Rapid Commun., 25, 1283 (2004).
34. Liaw, D. J., C. C. Huang, H. C. Sang and E. T. Kang, Langmuir, 14, 3195 (1998).
35. Liaw, D. J., C. C. Huang, H. C. Sang and E. T. Kang, Langmuir, 15, 5204 (1999).
CH2 O C O C
HO O CH2 OH
1 8 2 9
7
3
6
4 5 10
11
10 11 4
5 6
7 3 2
9
8 1
CH2 O C O CH2 O C
O
C O
O CH2 C
O
Cl HO H2C
4 10 5
6 3
7 2
9 8 1
C O CH2 O
C O
O CH2
1
8
2 7 3
6 5
4 9
10
C O
O CH2 CH2 N N
H2C H2C C HO
O
Cl 11
10
4 5
6 3
7 2
9 8 1
C O
O CH2 CH2 N C O CH2 CH2 N
O
9 10 4
6 5 7 3 2
8
1 11
Scheme 1. Synthesis and polymerization of a series of exo-norbornene monomers containing
various chromophores: (a) exo-1, (b) exo-2, (c) exo-3exo-1
對甲苯磺酸exo-原冰片烯甲醇
甲苯、迴流 芴酸
釕金屬觸媒 四氫呋喃,35 ℃ 精密複分解聚合
Poly(exo-1)
exo-1
(a)
exo-2
三乙胺exo-原冰片
烯甲醯氯四氫呋喃
1-甲基醇芘 (b)
三乙胺 四氫呋喃
9-乙基醇咔唑
exo-3
(c)
exo-原冰片
烯甲醯氯四氫呋喃,35℃
精密複分解聚合
Poly(exo-2)
exo-2
四氫呋喃,35℃
精密複分解聚合
Poly(exo-3)
exo-3
CH
2N CH
2O C O
CH2 N
Ru
CH
2O C O
H Ph Cl
Cl PCy
3PCy
3CH
2O C O
1
8
2 7 3
6 5
4 4'
6' 5' 7' 3' 2'
8' 1'
a
c b
f
e d
d' e'
f'
b' c' a'
Scheme 2. Synthesis of a fluorene-containing diblock copolymer [poly(exo-1)-b-(NBCbz)]
釕金屬觸媒 四氫呋喃,35 ℃
精密複分解聚合
側鏈含咔唑(Carbazole)及含芴發 光基團之新型原冰片烯共聚合物 [Poly(exo-1-b-NBMCbz)]
endo/exo-NBMCbz
Table 1. Results for the Polymerization Times, Molecular Weights, Polydispersities, and Yields for the Homopolymerization of Norbornene
Monomers Using Grubbs Catalyst I
Table 2. Solubility of New Polynorbornenes Containing Various Chromophores in Various Solvents
aFigure 1. The
1H-NMR spectra of (a) exo-1 and (b) poly(exo-1) (solvent = CDCl3)Figure 2. (a) DEPT-135 and (b)
13C-NMR spectra of exo-2 and (c) DEPT-135 and (d)13C-NMR spectra of exo-3 (solvent = CDCl ). The structures of the exo-
Figure 3. GPC traces of the polymerization of exo-1 with (a) [M]/[I] = 100, (b) [M]/[I]
= 180, and (c) [M]/[I] = 250; the polymerization of (d) exo-1 ([M]/[I] = 35) and (e) additional exo-1 ([M]/[I] = 35); (f) prepoly(exo-1) ([M]/[I] = 125) of the diblock copolymer; and (g) the diblock copolymer of exo-1 and poly(exo-1)-b-(NBCbz).
Figure 4. (a) Molecular weight dependence of poly (exo-1) on the [M]/[I] ratio ([M] =
concentration of exo-1; [I] = concentration of the initiator) and (b)
molecular weight dependence of poly(exo-1) on the polymerization time.
Figure 5. Homopolymerization of exo-1 via ROMP: (a) the conversion versus the
polymerization time and (b) the semilogarithmic plot of the monomer
concentration versus the polymerization time ([I] = 0.000867 mol L
-1).
Figure 6. Fluorescence spectra of (a) fluorene-containing poly(exo-1) in a CHCl
3solution (dashed line), (b) a fluorene-containing poly(exo-1) film (solid
line), (c) pyrene-containing poly(exo-2) in a CHCl
3solution (dashed line),
(d) a pyrene-containing poly(exo-2) film (solid line), (e)
carbazole-containing poly(exo-3) in a CHCl
3solution (dashed line), and (f)
a carbazole-containing poly(exo-3) film (solid line).
出國參訪及參加國際會議心得報告
計畫執行期間: 2008 年 8 月 1 日至 2009 年 7 月 31 日 報告人:國立台灣科技大學化工系 廖德章
本人於計畫執行期間,為提升個人研究能力及增進國際交流。故 至國外多所大學參訪,同時也多次參加國際高分子會議,並同時受邀 於會議中演講個人研究領域。
其中本人前往交流的學校有中國科學技術大學及廈門大學,而參 加 的 國 際 會 議 有 : 在 土 耳 其 舉 辦 的 “NATO ASI on New Smart Materials via Metal Mediated Macromolecular Engineering: From Complex to Nano Structures”,在日本東京舉辦的”Polycondensation 2008” 和 在 印 尼 舉 辦 的 ”International Conference on Advanced and Sustainable Polymer “。
本人於 2008 年 8 月 4 日及 8 月 5 日期間參加於印尼舉辦之國際 會議,本人同時受邀擔任國際會議 Plenary lecture。演講的題目內容 為具吡啶及三苯胺功能基團之有機可溶聚醯亞胺。在演講後有教授提 問到“聚醯亞胺一般常用於半導體及電子封裝材料上,而此類聚醯亞 胺是否有其他應用?”,筆者回答,“此類聚醯亞胺左側苯(Phenyl)基團 視為電子施體(Electron-Donor); 右側酞醯亞胺(Phthalimide)基團視為 電子受體(Electron-Acceptor),而導入不對稱蒎(Pyrene)等光電基團,
增加電子施體的電子施與能力,所以此類聚醯亞胺含記憶行為,也可 應用於記憶材料”。而相關之研究筆者也曾與新加坡大學之康燕堂教 授合作發表於 J. Am. Chem. Soc. 2006, 128, 8732 及 Polymer 2007, 48, 5182 及 J. Appl. Phys. 2007, 102, 024502 等國際著名期刊中。
n
Ar
CN
C C
N
CO O
O O
N
A
A: Pyrene, naphthalene, etc
Ar: Pendent, bulky, flexible isopropylidene, unsymmetric, noncoplanar, polar sulfonyl, trifluoromethyl groups, etc.
而其他多位來自各地的傑出學者均有很多很好的研究成果。如來 自日本的 Hiroyuki Nishide 教授利用自由基高分子成功合成組裝成電 池。其利用的自由基基團為 tetramethylpiperidinoxyl, galvinoxyl 及
nitronylnitroxide.此種新型之電池材料具有高安全性、低能量損耗之濕 式製程、可拋棄式等優點,具有很好之發展潛力。
另外,於 2008 年 9 月 1 日至 7 日,本人前往土耳其參加了國際
Macromolecular Engineering: From Complex to Nano Structures”。本次 會議的主題是利用各種技術,合成新型智慧型材料。會議中多為利用”
陰離子轉移自由基聚合”(ATRP)”、開環複分解聚合”(ROMP) 或是
Click 反應等技術,以合成高智慧型之材料。而本人同時也有發表個 人之演說,內容即是利用 ROMP 來合成新型高分子材料,並同時搭 配 ATRP 之技術,其分子之設計可有更多應用變化。如先合成含 ATRP 起始物側鏈之原冰片烯單體,先利用 Grubbs 釕金屬觸媒經由開環複 分解反應聚合,最後在進行 ATRP。反應式如下所示:
此外,來自英國 Durham University 的 Ezat Khosravi 教授也是 ROMP 領域的國際著名學者,並發表了很多關於 ROMP 之新技術及其研究
成果。如 Ezat Khosravi 教授首先將具生物可吸收/降解之 PLA 巨分子 導入原冰片烯側鏈,之後再利用開環複分解聚合(ROMP)合成出生物 可吸收/分解之高分子材料,相關內容發表於 Macromolecules 2007, 40, 1444-1452 期刊中,反應式如下所示:
辦一次的高分子會議 ”POLYCONDENSATION 2008”。本人不僅學習 到來自世界各地之優秀學者之作品,同時也有發表自己之成果演說,
可謂收穫豐富。而台大的劉貴生教授,其研究領域和本人十分接近,
其所發表的成果也相當具有參考交流之價值。其合成一系列含三苯胺 之聚醯胺及聚醯亞胺,且由於三苯胺具有很好之電洞傳遞之能力及優 良之電致變色性質。故其相關衍生物於 PLED、電致變色材料上均有 很大潛能,相關內容發表於 Macromolecules 2008, 41, 1667-1674 期刊 中,結構及反應式如下所示:
另外,來自南韓的學者 Chang-Sik Ha,利用全脂環族之二胺單 體與二酸酐進行聚縮合反應,得到一系列無色具高穿透度之聚醯亞 胺,相關內容發表於 J. Appl. Polym. Sci. 2006, 102, 3316–3326 期刊 中,結構及反應式如下所示:。
而除了參加多次國際會議之外,本人於 2008 年 10 月 13 日至 18 日前往廈門大學,拜訪了多位教授。如黃本立院士,黃院士於 2001 年曾任中國化學會會長。本人在那受到他熱情款待,與他相處交流學 術多日,收穫豐富,並獲贈兩本”院士春秋”。而本人也同時拜訪同為 院士的趙玉芬院士,與她討論了很多有關含磷之高分子材料。
此外,由於本人之研究領域也涵蓋了複分解反應(metathesis)之聚 合。因此本人也與夏海平教授及他的博士生討論了利用複分解反應 (metathesis)合成含鋨(Os)之金屬觸媒。
情款待,並也互相交流學術心得及知識,可謂不虛此行。
本人也於 2008 年 10 月 22 日至 11 月 4 日前往大陸合肥的中國科 大作交流訪問,並與中國科大劉士勇、潘才元及張廣照教授(均為國 際知名期刊 Macromolecules 之編輯委員)進行學術之相互交流與探 討。同時進行相關合作研究,以我方擅長之合成製出新的高分子材 料,而對方進一步的作物理上之研究與探討。其中我方提供了新型高 分子材料包括:含雙蒎螢光基團之原冰片烯聚合物及具有感溫和 pH 感測性質的 POLY(NBDMAEMA)、POLY(NBNIPAM)等聚合物。
CH3
C O O CH2
CH2
N CH3
H3C CH2 C
CH3
C O O CH2
CH2
N CH3
H3C
CH3
C O O CH2
CH2 N CH3
H3C
n CH3
C O O CH2
CH2
N H3C CH3
POLY(NBDMAEMA)
n
C O NH CH H3C CH3
C O NH CH H3C CH3
C C O NH CH H3C CH3
C
+
C O NH CH H3C CH3
POLY(NBNIPAM)
本人於計畫期間,前往世界各地參訪之經歷與多位教授交流之心
得,不僅有助於本人日後之研究工作,更可間接提升台灣國際化程度 及知名度,最後,感謝國科會在經費上之支持,使筆者得以順利進往。
Novel Organosoluble Poly(pyridine-imide) with Pendent Pyrene Group: Synthesis, Thermal, Optical, Electrochemical, Electrochromic, and Protonation Characterization
Der-Jang Liaw,*
,†Kun-Li Wang,
‡and Feng-Chyuan Chang
†Department of Chemical Engineering, National Taiwan UniVersity of Science and Technology, Taipei, 106 Taiwan, and Department of Chemical Engineering and Biotechnology, National Taipei UniVersity of Technology, Taipei 106, Taiwan
ReceiVed NoVember 4, 2006; ReVised Manuscript ReceiVed February 16, 2007
ABSTRACT: A new diamine containing a pyridine heterocyclic group and a pyrene substituent, 4-(1-pyrene)- 2,6-bis(4-aminophenyl)pyridine (PBAPP), was synthesized and used in a preparation of poly(pyridine-imide) by direct polycondensation with 4,4′-hexafluoroisopropylidenediphathalic anhydride (6FDA) in N-methyl-2- pyrrolidinone (NMP). The poly(pyridine-imide) derived from diamine (PBAPP) was highly soluble in several solvents such as THF, NMP, DMAc, DMF, pyridine, DMSO, and cyclohexanone at room temperature or upon heating at 70°C and exhibited good thermal stability both in nitrogen and air (Td10>520°C) and a high dielectric constant of 4.32 at 1 kHz. The poly(pyridine-imide) could be cast into a flexible and tough film from DMAc solution. The poly(pyridine-imide) film had a tensile strength of 118 MPa and a tensile modulus of 2.2 GPa.
The optical properties exhibited the UV-vis absorption bands at the region of 200-400 nm and possessed strong orange fluorescent (560 nm) after protonated with protic acid.
Introduction
Polyimides constantly attract wider interest because of their unique mechanical properties, thermal stability, and morphologi- cal properties.
1-7Conventional polyimides like Kapton produced by DuPont have been applied to microelectronic devices and aerospace fields. However, these polyimides are generally insoluble in organic solvent, exhibit low optical transparency, and have an intense yellow color. Soluble polyimides are needed as coating materials on specific space components. In order to overcome this drawback, either bulky lateral substituents, flexible alkyl side chains, unsymmetric, alicyclic or kinked structure have been attached along the backbone.
8-14The rigid- rod polyimides with high organic solubility have attracted some research efforts in recent years. These efforts have been focused on designing and synthesizing new rigid diamines that resulted in soluble and processable polyimides without deterioration of their positive properties.
In particular, polymers have attracted considerable attention because of their good scalability, mechanical strength, flexibility, and most important of all, ease of processing. In the recent years, there has been a considerable interest in the photoluminescent and electroluminescent properties of conjugated polymers.
15-17These polymer materials are used for several electronic ap- plications, including light-emitting devices (LEDs), transistors, photovoltaic cells, polymer memory, and switches.
16,18-24However, certain non-conjugated polymers, e.g., polyimides, in combination with electron transporting layers, also rank among efficient electroluminophores.
22-24Recently, some re- ports have concerned the incorporation of pyridine and its derivatives into polymeric frameworks.
25Compared to a benzene ring, pyridine is an electron-deficient aromatic heterocycle, with
a localized lone pair of electrons in sp
2orbital on the nitrogen atom; consequently, the derived polymers have increased electron affinity,
26improved electron-transporting properties,
27and offer the possibility of protonation or alkylation of the lone pair electrons as a way of modifying their properties.
28,29On the other hand, the pyrene unit is an efficient fluorescent probe because it has a long singlet lifetime and readily forms excimer.
30A literature survey revealed that there are a limited number of investigations concerning the attachment of pyrene to polyimides.
31,32Pyrene containing polymers have been used as acceptors for energy transfer from various donors.
33,34Additionally, the bulky condensed aromatic ring of pyrene is expected to enhance the solubility and thermal stability of polyimides.
The present investigation deals with the synthesis and characterization of a new poly(pyridine-imide) derived from a new monomer, 4-(1-pyrene)-2,6-bis(4-aminophenyl)pyridine (PBAPP), containing heterocyclic pyridine and pyrene substit- uents. The solubility, electrochemical stability, mechanical, thermal and optical properties of the obtained poly(pyridine- imide) were investigated.
Experimental
Materials. The materials, 1-pyrenecarboxaldehyde, 4′-nitroac- etophenone, ammonium acetate, hydrazine monohydrate, and 10%
palladium on activated carbon were purchased from Merck and used as received. Glacial acetic acid was purchased from Aldrich Chemical Co. and used as received. 4,4′-Hexafluoroisopropylidene- diphathalic anhydride (6FDA, TCI) was recrystallized twice from acetic anhydride, and then sublimated before use. The solvents, N-methyl-2-pyrrolidinone (NMP), tetrahydrofuran (THF), N,N′- dimethylacetamide (DMAc), N,N-dimethylformamide (DMF) were purchased from Merck and purified by distillation under reduced pressure over calcium hydride and store over 4 Å molecular sieves.
Acetic anhydride and pyridine were also purchased from Merck but used as received.
* Corresponding author. Fax: 886-2-23781441 or 886-2-27376644.
E-mail: [email protected] [email protected].
†Department of Chemical Engineering, National Taiwan University of Science and Technology.
3568 Macromolecules 2007, 40, 3568-3574
flask, a mixture of 15 g (65 mmol) of 1-pyrenecarboxaldehyde, 21.5 g (130 mmol) of 4′-nitroacetophenone, 100.4 g (1.3 mol) of ammonium acetate, and 300 mL of glacial acetic acid was refluxed for 16 h. Upon cooling, the precipitated light yellow solid was collected by filtration and washed with cold N,N′-dimethylacetamide (DMAc). The crude product was recrystallized from DMAc five times to afford 8.1 g (57%) of light yellow needles; mp 333°C (by DSC). FTIR (KBr): 1523 and 1345 cm-1(NO2).1H NMR (500 MHz, DMSO-d6): δ8.67-8.65 (d, 4H), 8.50 (s, 2H),8.48 (s, 1H), 8.42-8.40 (d, 4H), 8.39 (s, 1H), 8.37-8.35 (d, 1H), 8.31 (s, 2H), 8.30-8.29 (d, 1H), 8.26-8.24 (d, 2H), 8.16-8.13 (t, 1H). Anal.
Calcd for C33H19N3O4: C, 76.00; H, 3.67; N,8.06. Found: C, 76.00;
H, 3.86; N, 7.98.
4-(1-Pyrene)-2,6-bis(4-aminophenyl)pyridine (PBAPP, 2). A mixture of 2.7 g (5.1 mmol) of PBNPP, 0.13 g of 10% Pd/C, 2.5 mL of hydrazine monohydrate, and 200 mL of ethanol was placed in a flask. The reaction was heating at 90°C for 24 h and then removed the ethanol under reduce pressure. THF (50 mL) used as a solvent was added to the mixture, filtered to remove Pd/C and removed the THF using rotation evaporator. The light yellow solid was recrystallized from THF/ethanol (10/1, v/v) twice and dried under vacuum. The yield was 67%; mp 266°C (by DSC). FTIR (KBr): 3472, 3379, 1247 cm-1.1H NMR (500 MHz, DMSO-d6):
δ 8.40-8.38 (d, 1H), 8.33-8.31 (d, 1H), 8.28-8.26 (d, 1H), 8.23 (s, 2H), 8.17-8.14 (m, 3H), 8.10-8.06 (t, 1H), 8.03-8.02 (d, 4H), 7.75 (s, 2H), 6.71-6.70 (d, 4H), 5.45 (s, 4H).13C NMR (125 MHz, DMSO-d6): δ 156.1, 150.0, 149.4, 135.6, 130.9, 130.7, 130.4, 128.1, 127.8, 127.7, 127.6, 127.3, 127.2, 126.5, 126.4, 125.5, 125.2, 125.0, 124.3, 124.1, 124.0, 116.5, 113.7. Anal. Calcd for C33H23N3: C, 85.87; H, 5.03; N, 9.10. Found: C, 85.94; H, 5.01;
N, 9.05.
Synthesis of Poly(pyridine-imide). To a stirred solution of 0.6183 g (1.3 mmol) of (PBAPP) in 5 mL of N-methyl-2- pyrrolidinone (NMP), 0.5775 g (1.3 mmol) of 4,4′-hexafluoroiso- propylidenediphathalic anhydride were gradually added. The mix- ture was stirred at ambient temperature overnight (ca. 12 h) to form the poly(amic acid). Chemical cyclodehydration was carried out by addition of 1 mL of acetic anhydride and 0.5 mL of pyridine into the above-mentioned poly(amic acid) solution with stirring at room temperature for 1 h, and then heating at 110 °C for 4 h (Scheme 2). The polymer solution was poured into methanol. The precipitate was filtered, washed with methanol, and dried at 100°C under vacuum. The inherent viscosity of the polymer in N,N-dimethylacetamide was 0.58 dL‚g-1, measured at a concentra- tion 0.5 g dL-1at 30°C. FTIR (KBr): 1784, 1724, 1372 cm-1.1H NMR (500 MHz, DMSO-d6): δ 8.46 (s, 4H), 8,27-8.08 (m, 15H), 7.91 (s, 2H), 7.65-7.64 (d, 4H).13C NMR (125 MHz, DMSO-d6):
δ 166.6, 157.0, 152.1, 139.6, 139.4, 136.8, 136.7, 136.1, 134.3, 134.0, 133.9, 132.5, 132.0, 129.3, 129.2, 128.9, 128.4, 128.2, 128.1, 127.4, 127.2, 126.5, 126.2, 125.9, 125.8, 125.7, 125.4, 124.7, 123.7, 121.7, 121.4, 66.3. Anal. Calcd for C52H25N3O4F6: C, 71.81; H,
chromatography (GPC). Calibration was made by using polystyrene as standard with molecular weight in the range of 1680-402100 g/mol. Four Waters (UltraStyragel) columns (300× 7.7 mm, guard, 105, 104, 103, and 500 Å in a series) were used for GPC analysis with tetrahydrofuran (THF; 1 mL min-1) as the eluent. The eluents were monitored with a UV detector (JMST Systems, VUV-24, USA) at 254 nm. Thermogravimetric data were obtained on a TA Instruments Dynamic TGA 2950 under nitrogen and air flowing condition at a rate of 50 cm3min-1 and a heating rate of 10°C min-1. Melting points of monomers and glass transition temperature of polymer were performed on a differential scanning calorimeter (TA Instruments TA 910) under nitrogen flowing conditions at a rate of 50 cm3min-1and a heating rate of 10°C min-1. Dielectric constants of polyimide thin film were measured by the parallel- plate capacitor method using a dielectric analyzer (TA Instrumenents DEA 2970) at the range of frequency 1-10 kHz. Gold electrodes were vacuum-deposited on both surfaces of dried film, and measurement were made at 25°Cunder N2atmosphere. Average tensile properties were determined at room temperature with five Scheme 1. Synthesis of New Monomer
4-(1-Pyrene)-2,6-bis(4-aminophenyl)pyridine (2)
Scheme 2. Preparation of Poly(pyridine-imide)
Scheme 3. Ref-1
Macromolecules, Vol. 40, No. 10, 2007 Novel Organosoluble Poly(pyridine-imide) 3569

![Figure 3. GPC traces of the polymerization of exo-1 with (a) [M]/[I] = 100, (b) [M]/[I]](https://thumb-ap.123doks.com/thumbv2/9libinfo/9126116.410455/26.892.224.680.109.933/figure-gpc-traces-polymerization-exo-m-i-m.webp)
![Figure 4. (a) Molecular weight dependence of poly (exo-1) on the [M]/[I] ratio ([M] = concentration of exo-1; [I] = concentration of the initiator) and (b) molecular weight dependence of poly(exo-1) on the polymerization time](https://thumb-ap.123doks.com/thumbv2/9libinfo/9126116.410455/27.892.220.650.100.880/molecular-dependence-concentration-concentration-initiator-molecular-dependence-polymerization.webp)
![Figure 5. Homopolymerization of exo-1 via ROMP: (a) the conversion versus the polymerization time and (b) the semilogarithmic plot of the monomer concentration versus the polymerization time ([I] = 0.000867 mol L -1 )](https://thumb-ap.123doks.com/thumbv2/9libinfo/9126116.410455/28.892.184.706.111.477/figure-homopolymerization-conversion-polymerization-semilogarithmic-monomer-concentration-polymerization.webp)