科技部補助專題研究計畫成果報告
期末報告
腸病毒A71型病毒準種與毒力因子之研究(第3年)
計 畫 類 別 : 個別型計畫 計 畫 編 號 : MOST 105-2320-B-006-032-MY3 執 行 期 間 : 107年08月01日至108年07月31日 執 行 單 位 : 國立成功大學醫學檢驗生物技術學系 計 畫 主 持 人 : 王貞仁 共 同 主 持 人 : 王世敏 計畫參與人員: 碩士級-專任助理:黃乙惠 博士後研究-博士後研究:黃聖文中 華 民 國 108 年 10 月 30 日
中 文 摘 要 : 腸病毒A71型感染在近十幾年內持續地在亞洲環太平洋地區增加,腸 病毒A71型除了引發輕微的疾病,如:手足口症與疱疹性咽峽炎外 ,也會伺機造成嚴重的神經性併發症,如:無菌性腦膜炎、腦炎、急 性無力肢體麻痺甚至死亡,目前並沒有可使用的藥物或疫苗可以使 用。腸病毒A71型病毒是一個高度突變率的病毒,因此,在病毒族群 的組成中,並非為單一種病毒序列,而為一群帶有不同突變的病毒 所組成,此種現象稱為病毒準種(Quasispecies)。當天擇壓力出現 在病毒族群中,例如:細胞趨性、或需克服免疫反應與抗病毒藥物時 ,經過篩選後,可能只有某些病毒能夠存活,並成為主要的群別。 近來的研究指出病毒準種的多樣性,決定了病毒在宿主體內的毒力 以及致病機轉,因此,進一步了解在不同宿主中腸病毒A71型病毒準 種將可以提供我們了解病毒毒力以及致病機轉一個嶄新的見解。 我們試圖透過對一位解剖病人各組織的病毒進行次世代定序和病毒 突變型別分析,來定義腸病毒A71感染人類後進入中樞神經系統的適 應性的進化途徑。我們發現在呼吸和消化系統等初始感染部位觀察 到具有廣泛的突變型別組成,在病毒進一步入侵後,會發生突變型 別的轉換並有一顯性突變型─在VP1第31個鹼基處帶有甘氨酸的病毒 顆粒為傳播到皮膚和中樞神經系統主要突變型。此外,在死亡病例 中VP1-31G比例也比手足口症輕症患者高。我們進一步利用基因重組 病毒探討VP1-31突變位點對於病毒的影響,發現在神經細胞中VP1-31G的病毒生長速率較快及有較好的適應性,然而VP1-31D的病毒則 是在腸道細胞中生長較好,進一步以老鼠實驗探討VP1-31突變點在 宿主體內的毒力與致病機轉,結果顯示VP1-31突變位點會影響腸病 毒A71在宿主體內的毒力。這些研究將提供關於腸病毒A71致病機轉 寶貴的資訊。總結,腸病毒A71型經常造成全球大眾健康問題的潛在 威脅,了解此病毒的致病機轉將有助於發展早期快速檢測及新的治 療策略,來避免群眾受到腸病毒A71的威脅。 中 文 關 鍵 詞 : 腸病毒A71型、病毒準種、毒力因子、致病機轉
英 文 摘 要 : Enterovirus A71 (EV-A71) infections are significantly increased in the Asia-Pacific region for more than a decade. EV-A71 is a major pathogen causing hand-foot-and-mouth disease, but occasionally causes severe neurological complications. EV-A71 is a hyper-mutagenic virus that exhibits high mutation rate. Therefore, virus population consists mutant spectra of virus rather than identical sequence, termed viral quasispecies. As selection pressure put on the virus population, such as tissue tropism etc., only some viruses will be survival and become dominant haplotype. Recent studies show quasispecies diversity determines viral virulence in vivo, indicating the important roles of mutant spectra for viral virulence. However, no information focused on viral quasispecies of EV-A71 and its contribution to pathogenesis in vivo is available. To understand EV-A71 quasispecies among the hosts will provide new insights to viral virulence and pathogenesis. Utilizing deep sequencing and haplotype
analyses of viruses from various tissues of an autopsy patient, we sought to define the evolutionary pathway by which EV-A71 evolves fitness for invading the central nervous system (CNS) in human. Broad mutant spectra with divergent mutations were observed at the initial infection sites in the respiratory and digestive systems. After viral invasion, we identified a haplotype switch and dominant haplotype, with glycine at VP1 residue 31 in viral
particles disseminated into the integumentary system and CNS. In addition, higher percentage of VP1-31G haplotypes of EV-A71 was found from fatal cases than those from HFMD patients. In vitro viral-growth and fitness analyses indicated that VP1-31G conferred growth and a fitness advantage in human neuronal cells, whereas VP1-31D showed enhanced replication in human colorectal cells. Considering that fatal cases contain enhanced glycine substitution at VP1-31, we suggest that the increased prevalence of VP1-31G may alter viral tropism and aid central nervous system invasion. We further characterized the effect of VP1-31 variants on virulence and pathogenesis in vivo, which provided valuable information for EV-A71 pathogenesis. In summary, EV-A71 poses a persistent global health threat, investigating the virulence determinants and the viral pathogenesis will help to develop early diagnosis of
virulence strain and new therapeutic strategies to prevent children from the threat of EV-A71 infection.
科技部補助專題研究計畫成果報告
(□期中進度報告/■期末報告)
腸病毒 A71 型病毒準種與毒力因子之研究
計畫類別:■個別型計畫 □整合型計畫
計畫編號:
MOST 105-2320-B-006 -032 -MY3
執行期間:2016 年 8 月 1 日至 2019 年 7 月 31 日
執行機構及系所:國立成功大學醫學檢驗生物技術學系
計畫主持人:王貞仁
共同主持人:王世敏
計畫參與人員:黃聖文、黃乙惠
本計畫除繳交成果報告外,另含下列出國報告,共 份:
□執行國際合作與移地研究心得報告
□出席國際學術會議心得報告
□出國參訪及考察心得報告
1目錄
目錄 ... I 中文摘要與關鍵字 ... II 英文摘要與關鍵字 ... III 報告內容 ... 1 Introduction ... 1Background and significance ... 1
Objectives ... 5
Materials and Methods ... 5
Results ... 8 Discussion ... 13 Tables ... 19 Figures ... 20 References ... 26 科技部補助專題研究計畫成果自評表 ... 30 I
中文摘要與關鍵字
腸病毒 A71 型感染在近十幾年內持續地在亞洲環太平洋地區增加,腸病毒 A71 型除了引發輕微的疾病,如:手足口症與疱疹性咽峽炎外,也會伺機造成嚴 重的神經性併發症,如:無菌性腦膜炎、腦炎、急性無力肢體麻痺甚至死亡,目前 並沒有可使用的藥物或疫苗可以使用。腸病毒 A71 型病毒是一個高度突變率的 病毒,因此,在病毒族群的組成中,並非為單一種病毒序列,而為一群帶有不同 突變的病毒所組成,此種現象稱為病毒準種(Quasispecies)。當天擇壓力出現在病 毒族群中,例如:細胞趨性、或需克服免疫反應與抗病毒藥物時,經過篩選後,可 能只有某些病毒能夠存活,並成為主要的群別。近來的研究指出病毒準種的多樣 性,決定了病毒在宿主體內的毒力以及致病機轉,因此,進一步了解在不同宿主 中腸病毒 A71 型病毒準種將可以提供我們了解病毒毒力以及致病機轉一個嶄新 的見解。 我們試圖透過對一位解剖病人各組織的病毒進行次世代定序和病毒突變型 別分析,來定義腸病毒 A71 感染人類後進入中樞神經系統的適應性的進化途徑。 我們發現在呼吸和消化系統等初始感染部位觀察到具有廣泛的突變型別組成,在 病毒進一步入侵後,會發生突變型別的轉換並有一顯性突變型─在 VP1 第 31 個 鹼基處帶有甘氨酸的病毒顆粒為傳播到皮膚和中樞神經系統主要突變型。此外, 在死亡病例中 VP1-31G 比例也比手足口症輕症患者高。我們進一步利用基因重 組病毒探討 VP1-31 突變位點對於病毒的影響,發現在神經細胞中 VP1-31G 的病 毒生長速率較快及有較好的適應性,然而 VP1-31D 的病毒則是在腸道細胞中生 長較好,進一步以老鼠實驗探討 VP1-31 突變點在宿主體內的毒力與致病機轉, 結果顯示 VP1-31 突變位點會影響腸病毒 A71 在宿主體內的毒力。這些研究將提 供關於腸病毒 A71 致病機轉寶貴的資訊。總結,腸病毒 A71 型經常造成全球大 眾健康問題的潛在威脅,了解此病毒的致病機轉將有助於發展早期快速檢測及新 的治療策略,來避免群眾受到腸病毒 A71 的威脅。 關鍵字: 腸病毒 A71 型、病毒準種、毒力因子、致病機轉 II英文摘要與關鍵字
Enterovirus A71 (EV-A71) infections are significantly increased in the Asia-Pacific region for more than a decade. EV-A71 is a major pathogen causing hand-foot-and-mouth disease, but occasionally causes severe neurological complications. EV-A71 is a hyper-mutagenic virus that exhibits high mutation rate. Therefore, virus population consists mutant spectra of virus rather than identical sequence, termed viral quasispecies. As selection pressure put on the virus population, such as tissue tropism etc., only some viruses will be survival and become dominant haplotype. Recent studies show quasispecies diversity determines viral virulence in vivo, indicating the important roles of mutant spectra for viral virulence. However, no information focused on viral quasispecies of EV-A71 and its contribution to pathogenesis in vivo is available. To understand EV-A71 quasispecies among the hosts will provide new insights to viral virulence and pathogenesis. Utilizing deep sequencing and haplotype analyses of viruses from various tissues of an autopsy patient, we sought to define the evolutionary pathway by which EV-A71 evolves fitness for invading the central nervous system (CNS) in human. Broad mutant spectra with divergent mutations were observed at the initial infection sites in the respiratory and digestive systems. After viral invasion, we identified a haplotype switch and dominant haplotype, with glycine at VP1 residue 31 in viral particles disseminated into the integumentary system and CNS. In addition, higher percentage of VP1-31G haplotypes of EV-A71 was found from fatal cases than those from HFMD patients. In vitro viral-growth and fitness analyses indicated that VP1-31G conferred growth and a fitness advantage in human neuronal cells, whereas VP1-31D showed enhanced replication in human colorectal cells. Considering that fatal cases contain enhanced glycine substitution at VP1-31, we suggest that the increased prevalence of VP1-31G may alter viral tropism and aid central nervous system invasion. We further characterized the effect of VP1-31 variants on virulence and pathogenesis
in vivo, which provided valuable information for EV-A71 pathogenesis. In summary,
EV-A71 poses a persistent global health threat, investigating the virulence determinants and the viral pathogenesis will help to develop early diagnosis of virulence strain and new therapeutic strategies to prevent children from the threat of EV-A71 infection.
Keywords: Enterovirus A71, Quasispecies, Virulence, Pathogenesis
報告內容
IntroductionHuman enterovirus A71 (EV-A71), major causative agent of hand-foot-and-mouth disease (HFMD), belongs to the Enterovirus genus in the Picornaviridae family, Enterovirus A species based on its genome sequence [1, 2]. Several HFMD outbreaks associated with co-circulation of EV-A71 have been reported around Asia-Pacific region including Taiwan, China, Japan, Malaysia, Singapore, Vietnam, Cambodia, Australia, and Thailand [3-7]. According to the comparison of VP1 sequences, EV-A71 can be phylogenetically divided into distinct genotypes: A to G [8]. Until now, it is still not clear whether genotype of EV-A71 associated with disease severity in human. In Taiwan, EV-A71 causes the first large outbreak in 1998 and continuously threaten children in 2000, 2001, 2004, 2005, 2008, and 2012 outbreaks. Our previous investigation reported continuous genotypic change responsible for each new outbreak in Taiwan every 2-5 years from 1998 to 2008; including genotype C2 to B4 (1998-2000), B4 to C4 (2001-2004), C4 to B5 (2005-2008). Most recent outbreak occurs in 2012 which shows a unique scenario in Taiwan EV-A71 epidemiology history that the same genotype B5 results in two large outbreaks in 2008 and 2012 [9].
Background and significance
Most of EV-A71 infection are asymptomatic or caused HFMD, which is self-limited and mild; however, severe complications, including neurological sequelae, such as aseptic meningitis, brainstem encephalitis, poliovirus-like paralysis, neurological pulmonary edema and sudden death, occasionally occur in EV-A71 infected patients especially children [10]. Pathogenesis of EV-A71 is recently better understood based on autopsy case analysis. EV-A71 encephalomyelitis shows intense inflammation in spinal cord, brainstem, hypothalamus and cerebellar dentate nucleus [11]. The inflammation is typical of viral encephalitis and anterior horn cells at all levels of the cord which shows neurotropism with viral antigen or RNA in neuronal bodies. Virus antigen and RNA can also be detected in other inflammation sites including medulla, the fourth ventricle, midbrain, hypothalamus, and subthalamic nucleus [11]. Viral antigens or RNA are detected mostly in neurons with neuronophagia or degeneration, suggesting viral cytolysis is an important mechanism for tissue injury. In addition, other mechanisms of tissue injury, such as immune mediated or bystander effects contribute to pathogenesis because virus antigens or RNA are more focal than expected from the extent and severity of inflammation [12]. Based on human autopsy results supported by findings in mouse model, viruses entry into central nervous system (CNS) through peripheral motor nerves. Because peripheral motor nerves are the first neurons encountered, motor and adjacent neurons in the spinal cord, medulla, pons, and midbrain observe the most severe inflammation. Therefore, it demonstrates that virus spreads from skeletal muscle to motor neuron junctions, up to peripheral motor nerves and motor nuclei in CNS [13].
EV-A71 is a nonenveloped positive-sense single-stranded RNA virus which consists of 60 copies each of four capsid proteins (VP1, VP2, VP3, and VP4) that form a symmetrical icosahedral structure. Among the capsid proteins, VP1, VP2, and VP3 are exposed on the virus surface and the smallest VP4 is arranged inside the icosahedral lattice. A small peptide VPg (encoded by 3B protein coding region) covalently binding to the 5' terminus of genome. Consequently, the external capsid proteins play the roles of receptor binding on the surface of susceptible cells and contain the antigenic determinants. Several receptors or attachment factors have been identified for EV-A71 binding or entry, including human scavenger receptor class B, member 2 (SCARB2) [14], human P-selectin glycoprotein ligand-1 (PSGL1) [15], sialylated glycans [16], annexin II [17], vimentin [18], and nucleolin [19]. To date, receptor expression may not explain neuron tropism as identified receptor molecules or proteins ubiquitously express in various tissues instead of specific expression in CNS, although EV-A71 may use unidentified receptors to entry into CNS. Several capsid protein determinants are identified to change virus virulence in mouse infection model. By using reverse genetics viruses, we previously demonstrated that VP1-145E and VP2-149M increased virus infectivity which contributed to cytotoxicity in neuron cells [20]. Through increasing RNA accumulation, these two mutations cooperatively increased mouse lethality in vivo. In antigenicity analysis, we identified that different subgenotypes of EV-A71 exhibited diverse antigenic properties against human antisera using antigenic cartography [21].
Almost all picornaviruses are able to restrict host-cell translation, which is suggested to be benefit for not only viral replication and translation but protecting virus from the immune response [22]. It is believed that picornaviruses induce apoptosis with various mechanisms. In coxsackievirus, apoptotic cells are observed in viral-infected heart and CNS, and coxsackievirus B3 VP2 protein involves in the pro-apoptotic process with interaction of cellular protein Siva [23-25]. Moreover, expression of picornavirus 2Apro and 3Cpro serine proteases and non-structural 2C protein have been reported to induce cell death in the form of apoptosis [26, 27]. In contrast, picornavirus genome also encodes other viral proteins functionally suppress apoptotic responses. Poliovirus 3A protein can inhibit the tumor-necrosis factor (TNF) receptor on the cell surface and reduce apoptosis [28]. Poliovirus 3Cprotease is also determined to cleave IκBα which causes less cell apoptosis and more viral replication [29]. Therefore, viral proteins have important roles for regulating apoptosis in picornavirus-infected cells. Previous studies have also shown that the CNS injury during paralytic poliomyelitis is associated with apoptosis induced by the virus infection in poliovirus-infected mice [30]. Cell damage involving apoptosis in the CNS following poliovirus infection can be observed [31]. Similar to poliovirus, EV-A71 infection often complicates with severe neurological diseases [32]; however, phylogenetic analysis cannot attribute phenotypic variations to subtype differences so far and the neurotoxicity determinants to CNS pathogenesis of EV-A71 has not been fully clarified [33]. Like poliovirus, either overexpression of 2A and 3C proteases or EV-A71 infection can cleave eIF4G or caspases to induce cell apoptosis in human rhabdomyosarcoma and neuroblastoma [34, 35]. As we analyzed the molecular evolution of intra-genotypic EV-A71, we found that more amino acid
substitutions occurred in non-structural protein coding region instead of structural protein coding region [36], which indicates those mutations potentially play roles in viral fitness in the human hosts. In sum, each viral protein contributes to unique function in viral infection and determines EV-A71 pathogenesis. Mutations at these proteins may result in functional changes of viral infection and influence viral pathogenesis in vitro and in vivo.
In contrast to 5'UTR of poliovirus determining neurovirulence, virulence determinants in viral genome of EV-A71 are not well clarified [37]. Mouse and monkey model studies have been undertaken to investigate the EV-A71 viral genetic determinants that may account for the viral virulence in vivo [38-40]. The 3D polymerase and 3' untranslated region, corresponding to ts determinants of poliovirus Sabin 1, determine distinct temperature sensitivity for EV-A71 [41]. EV-A71 mutant containing mutations in the 5' UTR, 3D and in the 3' UTR shows attenuated viral neurovirulence and limited virus spread in the central nervous system of monkeys. In addition to non-structural protein coding region and untranslated regions, structural proteins corresponding to cellular receptor binding change viral virulence in vivo. By using reverse genetics system, we found that several mutations including those in 3D, 5' UTR, VP2, and VP1 contribute to viral virulence in vivo [20, 40, 42]. As comparing the EV71 sequences between 1998 and 1986 outbreaks in Taiwan, we found that strains from 1998 outbreaks retrieve an amino acid substitution 3D-251I through recombination and this substitution reduces temperature susceptibility at 39℃ in vitro and attenuates viral virulence in mouse model in vivo [42], which may explain the emergent EV-A71 occurrence in 1998 in Taiwan. A single mutation C to U at position 158 of stem loop II in 5' UTR reduces viral translation in vitro and neurovirulence in vivo [40]. C158 substitution contributes to viral replication and protein translation in cells and in mice as well as virus invasion into brain. As mentioned above, two other mutations VP2-149M and VP1-145E were cooperatively responsible for cytotoxicity and RNA accumulation in vitro and mouse lethality in vivo [20]. Also, many studies try to define EV-A71 virulence determinants by analyzing whole genome of EV-A71 strains from severe and mild human patients by using conventional sanger sequencing. As comparing isolates’ sequences from patients with various disease severity in mainland China, genotype C4a isolates containing conservative residues (22H, 249V, and 289A) and the mutated residues (27S, 31S/D, 98K, 145G/Q, 164E, and 240A/S) cause higher proportion of severe disease in EV-A71 infection [43]. Another valine to isoleucine substitution at 3D-263, which locates at the finger domain of the viral RNA-dependent RNA polymerase 3D protein, was identified in virulent strains [44].
As a RNA virus, EV-A71 exhibits dynamic evolutionary. Due to the error-prone of viral RdRp, EV-A71 has high mutation rates and increases its genetic diversity [45], resulting mutational robustness in quasispecies (reviewed in [46]). To date, many investigation clarify that RNA viruses generate 10-4 to 10-6 mutations per nucleotide, average a mutation per genome in each replication cycle [47]. Such large population diversity can be observed both in experimental and natural infections with RNA viruses, any combinations of point or double mutations are potentially generated during each cycle of replication. Even a virus derived from molecular clones can be quickly transformed into a population with relative sequences. Previous studies proposed that mutational robustness provides
advantages for virus to adapt in a new environment under various selective pressures [48]. For example, reduction of mutation rate of poliovirus without changing the replication rate causes virulence decrease in mice [49]. In addition, a pre-existing minor drug-resistant variants of HIV-1 was reported to facilitate viral adaptation, which causes the failure of anti-viral therapy [50]. Similar to HIV-1, a novel high-fidelity variant of EV-A71 with 3D-L123F and 3D-G64S mutations attenuate viral virulence and pathogenesis in vivo [51], indicating the error-prone property of the RdRp of EV-A71 viruses helps infection of virus populations. Therefore, low replication fidelity is of crucial benefit to RNA virus survival in host replication. In the life of virus, the viruses are challenged by selective pressures all the time, such as infecting a new host with distinct immune pressure [52], infecting different types of cells, or exposing to different chemical agents or antiviral drugs [53]. Nonetheless, anything is a double-edged sword, as well as RNA replicative fidelity. High replicative fidelity has deleterious effects on viral fitness and can attenuate virus in vivo. According to vesicular stomatitis virus investigation, most of random single mutation reduces virus replication and 40% are lethal mutations [54]. Other viruses such as tobacco etch virus and the phages show similar scenario [55]. In the experimental infection, increasing mutation rate by mutagen agents such as nucleoside drugs leads to viral extinction. Taken together, mutation rate in the RNA virus populations is close to the maximum tolerable error rate, which determines the various “haplotypes” (genomes containing various mutations in structural and non-structural protein coding regions) composition to extend virus genetic diversity maintaining in the population. Both of genetic diversity and mutation tolerance drive virus evolution and adaption in hosts. In the complex environment like virus spread within the population, the ability to generate a highly diverse mutant spectra will allow virus to adapt and survive in various intra-host environment, such as selection pressures generated by host adaptation and host immune response [49, 56, 57]. In the course of viral infection and spread among hosts, virus population faces frequent selection events that the haplotypes preferred under selection pressure will become dominant in the viral population. Since high genome diversity can be a fitness advantage for virus to adapt a new environment or to escape from immune responses, it raises the question whether potential virulence factors hidden in quasispecies or haplotype diversity of quasispecies contribute to EV-A71 virulence and affect viral pathogenesis. Thus, knowledge of the evolutionary dynamics under selective pressures within hosts is the key to understand EV-A71 virulence and pathogenesis, which are of direct relevance of improvement for clinical therapy and disease management [58]. In order to accurately determine an intra-host EV-A71 evolutionary dynamics, sensitive sequencing method will be necessary to detect and quantify viral rare variants. Recently, next-generation sequencing becomes a powerful tool for reconstructing genomic diversity of viral quasispecies [59]. This technique offers a deep coverage sequence data from more than millions of reads, providing exceptional resolution for investigating mutant spectra of complex virus population [60]. Using next-generation sequencing to monitor and quantify EV-A71 population variant network will aid to dissect evolution landscape and viral fitness within human hosts.
Objectives
Although various mutations have been identified according to sequence comparison among circulating strains by using conventional sanger sequencing, none of substitution is well-defined as virulence factors in human in vitro and in vivo. In our plan, we determined EV-A71 quasispecies of clinical isolates from patients with different disease severity from 1998 to 2012 and define virulence factors of EV-A71 among mutant spectra. By utilizing next-generation sequencing technique, we examined substitution frequency of variants and predict haplotype structure of EV-A71 quasispecies. Clinical manifestations of patients associated with substitution frequency and haplotype composition to determine potential virulence factors of EV-A71 were examined. The effects of identified potential virulence factors on virus characteristics were examined by reverse genetics system. We characterized viral growth and fitness among mutant viruses containing various potential virulence factors. Also, we investigated the mechanism of virulence factors affecting viral growth and fitness, including virion stability, virus binding and uncoating, RNA accumulation in vitro. In addition to in vitro experiments, we analyzed viral virulence and pathogenesis in mouse model in vivo. Mutant viruses containing potential viral virulence factors were inoculated into neonatal mice and examined their effects on clinical scores, body weight, and lethality in EV-A71 infection. Through these novel investigations by using next-generation sequencing, we defined novel virulence factors which increase disease severity in EV-A71 infection. The results lead us to better understanding of EV-A71 pathogenesis in human hosts.
Materials and Methods
Clinical EV-A71 isolates
A retrospective study of 22 clinical isolates from inpatients and outpatients from 1998 were obtained from collections of the NCKU Virology Laboratory, as approved by the Institutional Review Board of National Cheng Kung University Hospital (A-ER-104-365). Fifteen virus strains isolated from throat swab specimens were randomly selected for quasispecies analysis: 7 from fatal cases, and 8 from HFMD cases. In addition, 7 viral strains isolated from various tissues or specimens of 1 autopsy case were analyzed. RD (rhabdomyosarcoma, Bioresource Collection and Research Center, Taiwan), DLD-1 (human colorectal adenocarcinoma, Bioresource Collection and Research Center, Taiwan), and SK-N-SH (human neuroblastoma, American Type Culture Collection, USA) cells were maintained, respectively, in Dulbecco’s modified Eagle’s medium (DMEM) (RD cells), Roswell Park Memorial Institute 1640 medium (DLD-1 cells), or minimum essential medium (SK-N-SH cells) containing 10% fetal bovine serum plus 2 mM L-glutamine, 100 IU penicillin, and 100 μg streptomycin per ml at 37 ℃. The cells were tested and found to be free of mycoplasma contamination. To prepare EV-A71 stocks, viruses were propagated for one passage and titrated by performing plaque assays in RD cells.
Virus cDNA amplification and next-generation sequencing
The cDNA amplicons of EV-A71 isolates from various patients will be prepared as previous described (28). Briefly, RNA purification of EV-A71 isolates was performed by using QIAmp Viral RNA kit (QIAGEN). Viral cultures (140 ul) will be collected, 560 ul of buffer AVL was added and placed at room temperature for 10 minutes; a further 560 ul of pure alcohol will be added for complete vortexing. The above mix was placed through a QIAmp spin column, washed twice with the buffer AW1; RNA was dissolved with 80℃ RNase-free water. After extraction of virus RNA, cDNA was synthesized using EV-A71 specific primers and SuperScript II (Life Technologies) as recommended by the manufacturer. RNA (5 ul) was added to SuperScript II RT reaction mix containing 400 nM of the primer. The RT was performed at 55℃ for 60 minutes. Synthesized cDNA will be treated by RNase H (Life Technologies) and RNase T1 and stored at −20 °C. To amplify cDNA amplicons of different EV-A71 isolates, synthesized cDNA served as template for PCR, using primer pairs by high fidelity KOD plus DNA polymerase (TOYOBO). KOD DNA polymerase was activated at 95℃ for 5 minutes and PCR was performed for 25 cycles for denaturing at 95℃ for 1 second, and annealing-extension at 68℃ for 8 minutes. PCR products was analyzed and extracted from 1% agarose gel for complete genome sequencing of these cDNA amplicons by using shotgun sequencing with Illumina Miseq System (Illumina Inc.) After retrieved shotgun reads at various depth, we used bowtie and CLC Bio genomic Workbench software to assemble the combined reads into contigs/scaffolds and generate full-genome alignment between assembly and target genome. Both mutation and indels were counted as variants (mismatches). Genetic variations of viruses were determined by the ratio of mismatch mutations vs. the total reads in each genomic site. Haplotypes of each isolate were predicted by QuasiRecomb and ShoRah program. Results of variants and haplotype for isolates were statistically analyzed and correlated with disease severity of patients.
Construction of infectious cDNA clones
After extracting viral RNA, cDNA was synthesized using EV-A71 specific primers and SuperScript III (Life Technologies) as recommended by the manufacturer. The synthesized cDNA was treated with RNase H and RNase T1, and stored at −20℃. To construct infectious cDNA clones of EV-A71 isolates, synthesized cDNA was used as templates for PCR using KOD Plus Polymerase (TOYOBO). PCR amplicons were purified by phenol/chloroform extraction and cloned using a TOPO XL PCR Cloning Kit (Invitrogen). Specific primers designed based on the published EV-A71 sequences were used for complete genome sequencing of these cDNA constructs.
In vitro transcription and transfection
All cDNA constructs were digested with MluI to produce DNA templates, purified by phenol extraction and ethanol precipitation, and then dissolved in RNase-free water. For in vitro RNA-transcription reactions, 5 μg linear DNA template was added to a buffer containing 80 mM HEPES (pH 7.5), 12 mM MgCl2, 2 mM spermidine, 40 mM dithiothreitol, 3.125 mM each NTP, 50 U RNasin RNase inhibitor, and 30 U T7 polymerase (Promega) in a total volume of 50 μl. Transcription reactions were terminated by the addition of 6 U RNase-free DNase (Promega) and incubation at 37℃ for 30
min. The RNA concentration was determined by measurement of the optical density at 260 nm, and checked by denaturing agarose gel electrophoresis. Then, 2 μg RNA was transfected into RD cells to produce recombinant viruses, as previously described [47].
Thermostability, virus growth, and fitness assays
To assess thermostability, 107 plaque-forming units (pfu) of EV-A71 VP1-31D and VP1-31G viruses were incubated at the indicated temperatures. After a 1–3 day incubation, the numbers of infectious viruses were titrated by performing plaque assays. For viral growth, SK-N-SH, RD, and DLD-1 cell monolayers were infected with various EV-A71 recombinants at a multiplicity of infection (moi) of 0.1, as described above. Infected cells were harvested at different intervals and whole-cell lysates were prepared for plaque assays to determine virus titers after a single freeze-thaw. For viral fitness, equal quantities of wild-type and mutant EV-A71 (106 pfu) virus were used to co-infect the indicated cells, and viruses were harvested after >75% cells appeared to exhibit cytopathic effects. Virus mixtures were continuously passaged in cells, and the cDNAs of viruses after the indicated passages were sequenced to determine the dominant virus in the viral mixtures that defined the viral fitness of the compared viruses.
Virus-binding assay
To examine virus-binding ability by ELISA, 2 × 104 cells/well were seeded in 96-well plates and incubated for 2 days. The cells were washed with phosphate-buffered saline (PBS) and various recombinant EV-A71 strains with mutations were added to cells at an MOI of 20 for a 1h incubation at 4°C. Unbound virus was washed away from the cells with ice-cold PBS. Cells were fixed using 80% acetone and stained with a monoclonal antibody against EV-A71 (MAb 979; Millipore) at a 1:2,000 dilution in 1% bovine serum albumin for 1 h. This was followed by washing with PBS-T (0.05% Tween 20 in PBS) and staining with a peroxidase-conjugated, goat-anti-mouse IgG polyclonal antibody (Cell Signaling Technology) at a 1:2,000 dilution in 1% bovine serum albumin for 1 h. After washing with PBS-T, freshly prepared substrate, 3,3',5,5'-tetramethylbenzidine (100μl; Invitrogen), was added to each well and the absorbance was measured at 450 nm.
Virus RNA release assay
Neutral red (NR)-labeled viruses were prepared by infecting RD cells with the mutant viruses indicated on the figure in the presence of 10μg/ml neutral red. At 3 days postinfection in the dark, cell lysates were harvested and subjected to three rounds of freeze-thawing. Cell debris was removed by low-speed centrifugation, and virus titers were determined by performing plaque assays. To analyze viral RNA release of mutant viruses, confluent cells were infected with the indicated mutant viruses at an MOI of 1. Following virus absorption to the indicated cells at 37°C for 2 h, unbound viruses were washed away using DMEM. After the postinfection time indicated on the figure, NR-labeled viruses that did not uncoat their capsids were inactivated by light exposure for 30 min. To determine the numbers of viruses that had released their RNA, infected cells were suspended using trypsin, and 104 cells were added to plates with confluent RD cells for plaque counts. After 4 days, the plaques in 104 infected cells were counted to obtain the numbers of viruses that had released RNA into the cells.
Viral infection of ICR mice
The 3-day-old ICR mice were infected with EV-A71 VP1-31D or VP1-31G virus (106 TCID50/ml,
100μl/mice) by intraperitoneal injection. Control mice were given with viral medium (DMEM, 2% P/S, 2% FBS). Animals were observed daily until 21 days for clinical score, weight gain or loss, and mortality. Clinical score were followed: 0, healthy; 1, ruffled fur and hunchbacked appearance; 2, wasting; 3, limb weakness; 4, limb paralysis; and 5, moribund and death.
Results
Dynamic VP1-31 haplotype changes driven by a selective bottleneck among diverse tissues We first investigated viral quasispecies and EV-A71 dissemination across diverse specimens and tissues obtained in 1998 at National Cheng Kung University Hospital from a child with fatal disease. EV-A71 isolates were collected from 7 autopsy specimens or tissues from the (1) respiratory and digestive systems (orogastric tube swab, throat swab, and intestinal mucosa); (2) integumentary system (skin lesion); and (3) CNS: (cervical vertebrae, medulla oblongata, and basal ganglia) (Fig 1). We amplified the whole genomes of isolates and analyzed their variants in quasispecies via deep sequencing.
Diverse traits contributing to virus infectivity might also be conferred by combinations of various mutations present in individual virus genomes, referred to as ‘haplotypes’. Haplotype reconstruction is used to identify each haplotype sequence and its frequency distribution among mutant spectra, which can reveal dynamic changes of haplotypes within hosts. To precisely quantify the genetic diversity of EV-A71 quasispecies in various tissues, we reconstructed the haplotypes of collected isolates, using QuasiRecomb software [61].
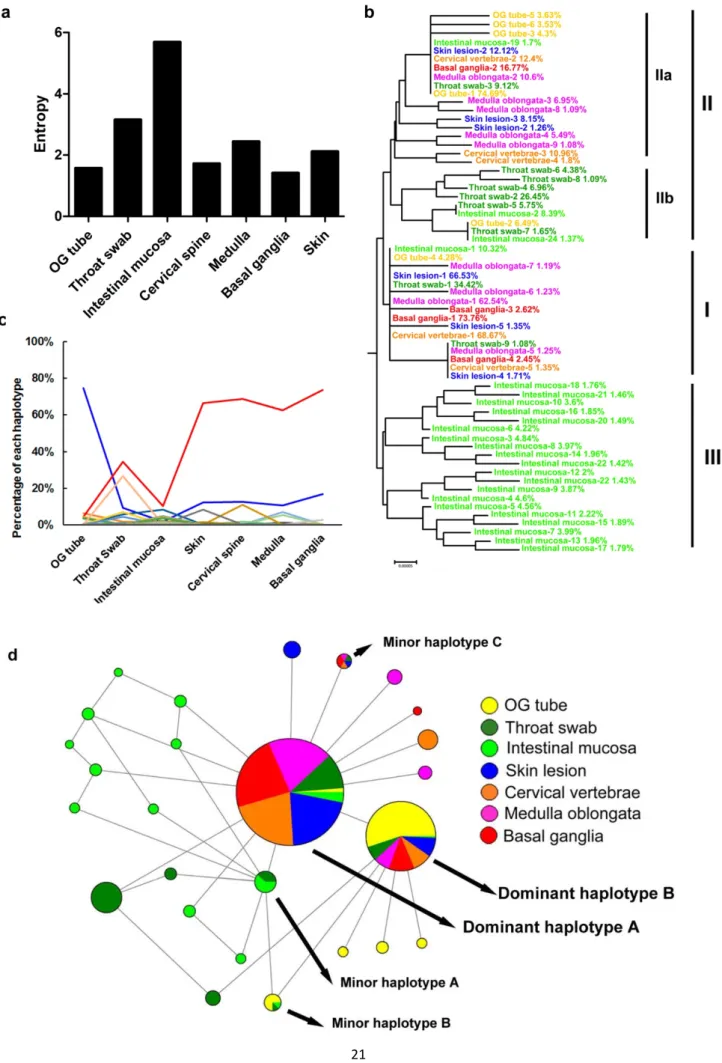
According to the estimated haplotype distribution, we first defined intra-host virus diversity by measuring the Shannon entropy, H. Consistent with the single-nucleotide variation (SNV) results, the highest diversity was found in samples from the throat swab (H = 3.15) and intestinal mucosa (H = 5.69) (Fig 2a). As EV-A71 transmits through the oral–fecal or oral–oral routes between human hosts, the nasopharyngeal and gastric-intestinal tissues are thought to be primary infection sites [62]. Together, these results indicated that EV-A71 exhibited broad mutant spectra, with the highest numbers of both SNVs and haplotypes at the primary infection sites in human dissemination routes. This phenomenon may facilitate viral adaptation to different hosts, tissues, or environments. However, as viruses invaded the integumentary system and CNS, the genetic diversity decreased. The reduction of mutant spectra implied that selective pressure potentially arose during viral dissemination within the host, forming a bottleneck with the tissues around the external lumens to specific sites in the integumentary system and CNS. Thus, whereas diverse variants infected other tissues, variants with high viral fitness in these tissues were maintained in the viral populations.
We next verified the genetic association of haplotypes in various tissues. First, we performed phylogenetic analysis of all identified haplotypes (>1%), based on the complete genome sequences
(Fig 2b). Phylogeny demonstrated that all haplotypes could be divided into 3 lineages according to their sequences. Linage I contained the most prevalent haplotype in most tissues (except for the orogastric tube), which we designated as dominant haplotype A. This haplotype also represented the common ancestor of several sporadic haplotypes (occurrence frequency <3%) found in throat swab, skin lesion, cervical vertebrae, medulla oblongata, and basal ganglia samples. Lineage II was divided into sublineages, IIa and IIb. Sublineage IIa was the most prevalent haplotype occurring in the orogastric tube and we designated it as dominant haplotype B, although it ranked second (cervical spine, medulla oblongata, basal ganglia and skin), third (throat swab), or eighteenth (intestinal mucosa) in terms of prevalence in other tissues. Several sporadic haplotypes from other tissues (frequency <11%) were also identified in sublineage IIa. The common haplotypes from the respiratory and digestive systems clustered in sublineage IIb, indicating that closely related variants coexisted in these 2 systems. The haplotypes from the intestinal mucosa clustered in another independent lineage, namely lineage III. To combine the frequency and genetic evolution results, we performed Network analysis [63] to cluster haplotypes from all tissues based on their frequency in mutant spectra by the size of the associated circles. Clearly, dominant haplotypes A and B accounted for a rather high proportion of haplotypes among the mutant spectra (Fig 2d). Network results also revealed sporadic haplotypes in the respiratory and digestive systems that clustered in lineage III and sublineage IIb. Taken together, these data showed that dominant haplotypes A and B respectively clustered in lineage I and II (sublineage IIa) among various tissues with high frequency, and variants in the respiratory system, digestive system, and intestinal mucosa clustered in lineage IIb and lineage III. We suggest that the diverse mutant spectra of viral variants containing dominant haplotypes A and B evolved from the respiratory and digestive tracts to the CNS in the host. The haplotypes in the orogastric tube and throat swab were closely related, and those colonizing the intestinal mucosa showed broadened, evolving mutant spectra, which might be advantageous for adapting in the gut.
To determine dynamic changes in the haplotypes, we next analyzed the proportion of each haplotype appearing in all specimens along the EV-A71-infection route to identify dynamic haplotype changes within the host. A major B-to-A haplotype switch from peripheral to CNS specimens was clearly apparent (Fig 2c). Dominant haplotype A increased in prevalence from the orogastric tube (4.3%) to the basal ganglia (73.7%), whereas the dominant haplotype B was reduced (74.7% to 16.7%). Comparing the haplotype frequencies revealed that the 2 dominant haplotypes (A and B) switched in the specimens from the initial infection sites to the CNS (Fig 2c). This haplotype switch demonstrated an evolutionary force acting on EV-A71. To identify which region of the EV-A71 genome that the evolutionary force acted upon, we compared the sequences of the 2 dominant haplotypes (A and B). Unexpectedly, only 1 non-synonymous mutation was identified: an aspartic acid (D) to glycine (G) mutation at VP1 residue 31. Thus, we speculated that VP1 residue 31 was altered by evolutionary pressure and that the VP1-31D-to-G mutation may increase viral adaption and fitness as the virus spreads from the respiratory and digestive systems to the CNS in humans.
The frequency switch between dominant haplotypes A and B, which contained the VP1-31G and 9
D variants, respectively, suggest that viruses replicating in the initial infection sites (including the respiratory and gastrointestinal tracts and their lumens) contained distinct haplotypes relative to other human tissues. Comparison of the dominant haplotype sequences to identify genomic regions of EV-A71 susceptible to evolutionary pressure revealed only the VP1-31 non-synonymous mutation from aspartic acid (dominant haplotype B) to glycine (dominant haplotype A). However, the VP1-31G prevalence increased from the orogastric tube to the CNS (Fig 3), revealing that the dominant B-to-A haplotype switch was mediated by differing tissue tropisms, caused by VP1 residue 31. Therefore, we considered that a selective bottleneck shaped the evolutionary pathway and drove a haplotype switch after the virus invaded the respiratory and digestive systems. The VP1-31 substitution might act under selective pressure to facilitate EV-A71 dissemination to specific sites, including the integumentary system and CNS, which potentially contributed to EV-A71 infectivity and dissemination in the human body.
The selection bottleneck appearing in viral quasispecies from different tissues involved VP1 residue 31.
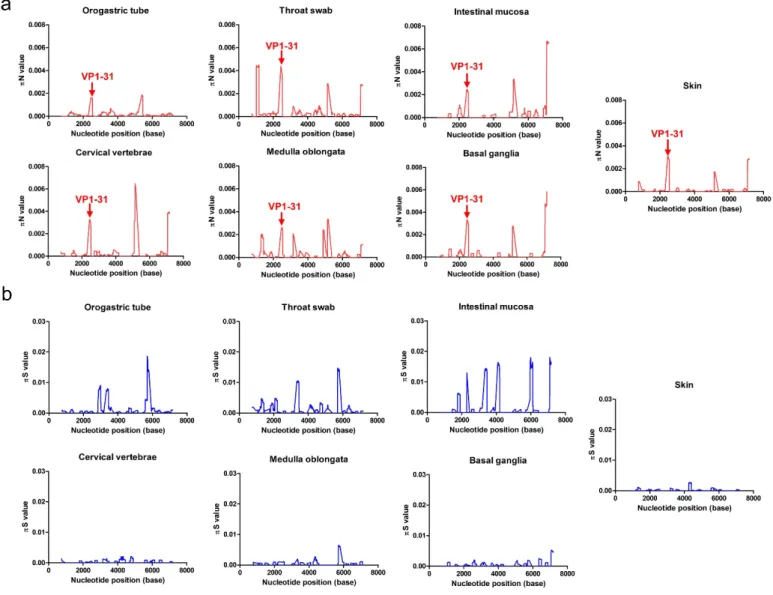
To identify the selective pressure that acted on specific viral gene(s) to help the virus break through the tissue-tropism bottleneck, we quantified the frequency of synonymous (πS) and non-synonymous (πN) mutations in each protein. In general, πN > πS or πS > πN indicates that positive (diversifying) or negative (purifying) selection is acting on the viral genome to introduce new mutations or remove deleterious mutations, respectively. Most tissues except for the orogastric tube showed πN > πS in the VP1 and 3A proteins (Table 1). Thus, the selection bottleneck positively selected EV-A71 mutations at these 2 proteins as the virus colonized the host tissues.
To further define the genetic sites where selection occurred, we estimated the πN and πS values with a sliding window of 50 codons and a step of 1 codon. Divergent πN and πS peak distributions indicated that various amino acid residues were positively selected in different tissues (Fig 3). In the VP1 protein, the non-synonymous VP1-31 substitution between aspartic acid (D) and glycine (G) appeared in all πN plots from different tissues (Fig 3a). Although the presence of many πS peaks (Fig 3b) among the various specimens and tissues tested indicated potential purifying selection sites in the mutant spectra, no πS peak consistently appeared in all tissues (Fig 3b). Thus, the VP1-31 SNV was the only residue subjected to consistent and positive selection pressure in all tissues through which the virus spread, suggesting that EV-A71 might maintain a dynamic, balanced distribution of VP1-31 variants from the initial host infection and that positive selection pressure might fine-tune the relative proportions of VP1-31 polymorphisms in viral quasispecies among various tissues. Two other residues with high πN values were frequently identified among these mutant spectra: the 3A-S73C mutation was commonly found in all isolates except the orogastric tube, while the 3D-W423R mutation was identified in all isolates other than the orogastric tube and medulla oblongata (Fig 3a). The results indicated that these residues broadly accumulated non-synonymous mutations during viral dissemination in humans.
In addition to the πN peaks of amino acid residues under positive selection pressure, many πS 10
peaks (Fig 3b), which displayed potential purifying-selection sites, occurred in these mutant spectra. Among the various πS peaks observed in various tissues, we defined 2 common purifying selection sites at the 2A-42 and 3C-152 residues. The πS peak at the 2A-42 residue was found in all respiratory and digestive system samples (orogastric tube, throat swab, and intestinal mucosa), whereas the peak at 3C residue 152 was found in the orogastric tube, throat swab, medulla oblongata, and basal ganglia. Among these positively and negatively selected SNVs, VP1 residue 31 was the only one consistently selected in all tissues during viral dissemination.
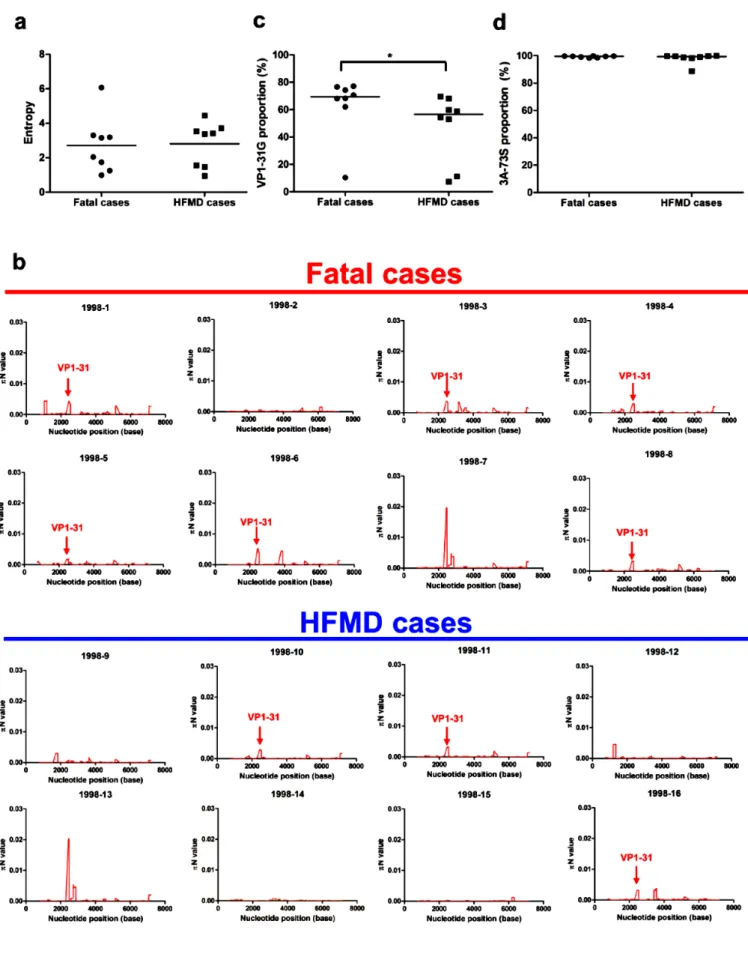
The proportion of VP1-31G correlated with disease severity in patients with EV-A71 infection. To determine whether evolutionary selection commonly acts on the mutant spectra of other EV-A71 strains while infecting human hosts, we collected 15 additional EV-EV-A71 strains, 7 of which were isolated from fatal cases and 8 of which were from HFMD cases. To rule out genetic diversity resulting from different genotypes or specimen types, the collected strains were consistently isolated from throat swabs and belonged to genotype C2, which is the same as the strain from the first fatal autopsy case. We first analyzed the mutant spectra of these strains by determining the frequency of the haplotypes involved. Analysis of the reconstructed haplotypes in the mutant spectra showed that the haplotype entropy did not correlate with patient disease severity (Fig 4a). We further analyzed the selection pressure that acted on specific viral protein(s) in these strains by quantifying πN and πS. Similar to the findings with the autopsy, 68.75% (11/16) and 62.5% (10/16) strains evolved under positive selection at the VP1 and 3A proteins, respectively. Further cross-matching of strain mutations with πN > πS values indicated the 2 SNVs were found in the autopsy cases: the VP1-31 SNV was found in 6 of 8 viruses from fatal cases and in 4 of 8 HFMD cases; 3A-S73C SNV was found in 5 of 8 viruses from either fatal or HFMD cases (Fig 4b). To determine whether the VP1-31G or 3A-73C mutations correlated with EV-A71 disease severity, we compared the frequency of the 2 mutations in viruses from fatal cases and HFMD cases. Analysis of virus isolates from patients with differing disease severities indicated that the frequency of VP1-31G (but not 3A-73C) was significantly higher among patients with fatal disease, compared to those with HFMD (Fig 4c, d). These data implied that a selective bottleneck, which shaped haplotype evolution acting at VP1 residue 31, existed as the virus penetrated the respiratory and digestive tracts to other tissues including the CNS. We speculate that VP1-31G might confer higher replication and a fitness advantage for adapting to CNS tissues. As more haplotypes with VP1-31G appear at the initial infection sites, this increases the potential for EV-A71 to invade the CNS and cause severe neurological disease. Thus, VP1-31G may be a potential viral determinant that contributes to intra-host EV-A71 spread, as well as disease severity in different hosts. VP1-31 determined thermostability, viral replication, and fitness in vitro.
The VP1 protein determines virus binding and uncoating by interacting with cellular receptors and serves as the major target protein of neutralizing antibodies. As the VP1-D31G substitution may constitute a potential viral mutation to facilitate EV-A71 bottleneck breakthrough and invasion into CNS tissues, we next examined the effect of the VP1-D31G mutation on viral replication in vitro. We generated recombinant viruses according to the haplotype sequences of dominant haplotype A
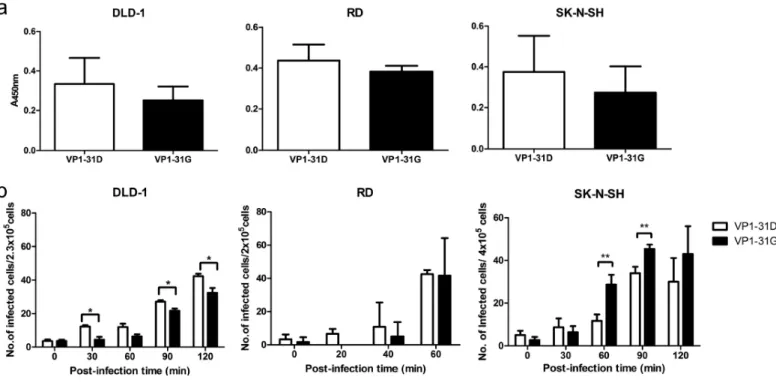
31G) and B (VP1-31D). As capsid protein VP1 is critical for virion structure stability and virus entry, we first examined virus thermostability. The thermostability of infectious VP1-31G virus particles decreased faster than VP1-31D particles at all tested temperatures (Fig 5a).
We next examined the effect of the VP1-31 substitution on viral infection. Because the VP1-31G haplotype (dominant haplotype A) frequency increased from the respiratory and digestive systems to the CNS in infected individuals, we utilized human colorectal adenocarcinoma DLD-1 cells (digestive system), human rhabdomyosarcoma RD cells (muscle), and human neuroblastoma SK-N-SH cells (CNS) to simulate VP1-31G/D viral infection in various tissues. The VP1-31G virus exhibited lower titers in DLD-1 cells than did the VP1-31D virus (Fig 5b). In the RD cells, no significant difference in viral replication was observed between strains; however, the VP1-31G virus exhibited significantly increased growth compared with the VP1-31D virus in SK-N-SH cells.
To assess viral fitness, we performed co-infections with both VP1-31 haplotype viruses in the different cell lines. Virus mixtures were continuously passaged in the respective cells, and the dominant virus in the viral population was determined by cDNA sequencing, targeting the VP1-31 residue. Similar to the viral replication results, virus competition demonstrated that the VP1-31G virus exhibited low viral fitness in colorectal cells, but high viral fitness in muscle and neuronal cells (Fig 5c–e). In the colorectal cells (Fig 5c), both viruses were mixed in the population in the first 5 passages but VP1-31D emerged as the only haplotype in the 6th passage. This change was not due to sequential passaging, as 10 passages of each virus individually did not alter the VP1-31 residue, but rather reflected the higher viral fitness of the VP1-31D virus in cells originating from the digestive system. Conversely, VP1-31G viruses showed high viral fitness in RD muscle cells (Fig 5d) and SK-N-SH neuronal cells (Fig 5e). In these cell types, only VP1-31G could be detected after 6 passages, suggesting that the VP1-31G virus exhibited higher viral fitness in cells derived from the muscular system and CNS. Together, the viral growth and fitness results were consistent with the observed haplotype distribution in the mutant spectra (Fig 2c and 2d), implying that VP1-31G conferred the most fitness for CNS infection whereas VP1-31D conferred more fitness for respiratory and digestive tract tissue infection.
To further define the viral replication step affected by the VP1-D31G mutation, we examined the contribution to virus binding and uncoating. No significant difference in virus binding was observed between virus variants in enzyme-linked immunosorbent assays (ELISAs) (Fig. 6a), consistent with the unlikelihood of changes in the inaccessible internal VP1 protein surface impacting the ability of virions to bind cellular receptors. Next, neutral red-labeled viruses were utilized to determine the uncoating rates of the 31D and 31G viruses. Similar to the viral replication results, the VP1-31D and VP1-31G viruses released their RNA faster in DLD-1 and SK-N-SH cells, respectively (Fig. 6b), with no obvious difference observed in RD cells. Even though the RNA release results (Fig. 6b) displayed approximately 2-fold changes in various tissues, these effects would be obviously amplified after multiple replication cycles. Our viral fitness results (Fig. 5b to d) also demonstrated that the VP1-31 mutation contributed to viral fitness, which was obvious after three to five passages in vitro. Taken
together, our data suggest that the VP1-31D and VP1-31G viruses exhibited different viral replication and fitness in vitro. The accordance with the relative variant frequencies across tissues indicates that EV-A71 adapted to different tissues by changing its RNA release ability through the VP1-D31G substitution.
To define the potential structural effects of this substitution on the virion, we utilized the EV-A71 crystal structure (PDB code 4AED) to localize VP1-31 on the capsid protein pentamer and to predict the possible changes by protein modeling. According to the pentamer structure (Fig. 7a), the VP1-31 residue is exposed on the virion interior rather than at the exterior surface. Thus, this residue cannot be accessed by cellular receptors or neutralizing antibodies, indicating that the VP1-D31G substitution might not change receptor binding ability to cells or antibody recognition by humoral immunity in the host. The VP1 N terminus forms a loop-like structure to stabilize the mature virion [64]. Upon virus uncoating, the proximal end of the VP1 N-terminal extension rearranges to exit through the capsid at the quasi-3-fold axis near the base of the canyon, thereby loosening the capsid structure on the virion. The N-terminal VP1 region then migrates across the top of the VP2 protein and binds the membrane. This connection may be involved in viral RNA transfer into cells. Our protein-modeling results indicated that the VP1-31 residue was predicted to interact with asparagine at VP1-71, both of which are within the N-terminal region of VP1 (Fig. 7b); however, VP1-D31G exhibited reduced salt bridge formation (Fig. 7c). Since N-terminal extension is essential for virus binding and RNA release, we believe that the potential decrease in the VP1-31 and VP1-71 interaction strength upon VP1-D31G substitution might reduce thermostability and conversely increase the release of viral RNA.
The virulence of VP1-31 substitution in ICR mice
To further evaluate the effect of VP1-31 variations on virulence and pathogenesis in vivo, ICR mice were infected with viruses and the survival rates, body weights, and clinical scores were measured (Fig. 8a-c). The 3-day-old ICR mice were observed daily for 21 days after intraperitoneal injection of EV-A71 VP1-31D or VP1-31G. Results showed that all mice survived in both VP1-31D and VP1-31G groups (Fig. 8a). In addition, no significant difference in body weight between the mice of the two virus-infected groups were observed (Fig. 8b). Mice in the VP1-31D group were similar to control group, which displayed no clinical signs. However, mice of the VP1-31G group displayed a hunched back appearance at 5 days post infection, but recovered gradually after 10 dpi. (Fig. 8c). Those results showed that VP1-31 substitution might influence the virulence of EV-A71 in ICR mice which will provide valuable information for EV-A71 pathogenesis.
Discussion
RNA viruses generally contain highly diverse quasispecies due to the low fidelity of their RNA polymerases. When facing selection pressures with various strengths, such as tissue tropism, environment stresses, or host immune responses, the viruses displayed changing haplotypes as an evolutionary response. A broad mutant spectrum may increase the chance of successfully adapting to
the new selective pressures, such as when establishing infection and colonization at initial infection sites. Our results defined a selection bottleneck existing when EV-A71 invades from the respiratory and digestive systems into other tissues, including the CNS. When EV-A71 infects human hosts through fecal–oral or oral–oral routes, virus initially enters the body through the lumen of the respiratory and digestive tracts and colonizes the epithelial tissues of either the tonsillar crypt [62] or intestinal mucosa in the respiratory and digestive systems, followed by spreading to other tissues. Virus collected from the orogastric tube that spans from the mouth to the stomach represents the mutant spectrum in the lumen of the alimentary canal. This mutant spectrum comprises dominant haplotype B with the 31D substitution (Fig 2c–d). Our in vitro experiment results indicated that the VP1-31D variant increases virion stability (Fig 5a) that potentially facilitates the maintenance of virus infectivity in cell-free environments, such as the lumen of the alimentary canal. This substitution also increased viral infectivity and fitness (Fig 5b–c) in the cells originating from the digestive system. These advantages may aid viral adaptation to environmental changes and resistance to high body temperatures due to a fever following viral transmission to other tissues. These advantages may also explain why the VP1-31D haplotype was still present at a low proportion among tissues from the respiratory and digestive tracts to the brain after selection. The VP1-31D variant also helps viruses infect epithelial cells and then colonize the alimentary canal. Following virus colonization of initial infection sites in the alimentary canal, we observed a founder effect wherein the dominant haplotype switched from VP1-31D to VP1-31G as EV-A71 broke through the selection bottleneck and disseminated to tissues of the integumentary system and CNS. In contrast to the replication advantage of VP1-31D in cells of the digestive system, VP1-31G exhibits higher growth and fitness in neuronal cells of the CNS in vitro (Fig 5b–c). We speculate that the differing tissue tropism of EV-A71 between the respiratory and digestive systems versus the integumentary system or CNS forms a selection bottleneck for viral populations with mutant spectra, resulting in the adaptive VP1-31G haplotype becoming dominant in neuronal tissues. Once EV-A71 establishes infection in the respiratory and intestinal tracts, the VP1-31G mutation may facilitate bottleneck breakthrough and invasion into the skin and CNS. Together, these data indicate that EV-A71 quasispecies utilize the dynamic balance of the diverse haplotype population, especially the VP1-31D and VP1-31G haplotypes, to cooperatively maintain population plasticity and facilitate viral adaptation and spread among various tissues.
Despite the 2 dominant haplotypes, the minor haplotypes in the intestinal mucosa displayed a series of clustering variants as a clade, compared with other haplotypes (Fig 2d). To survive in a complex environment involving mucosal immunity and microbes, complex haplotypes can occur in the intestines to facilitate virus adaption. According to phylogenetic and network-analysis results, we observed 3 minor haplotypes (A to C) co-existing in various tissues. Two haplotypes co-existed in collected isolates from the respiratory and digestive systems (intestinal mucosa, throat swab, and orogastric tube): minor haplotype A was identified in both the intestinal mucosa and throat swab, and minor haplotype B was identified in all 3 respiratory and digestive system specimens. As determining the genetic sequences of viral haplotypes, like the footprints of viral evolution, can help us identify
variants to trace their transmission between different tissues, the observed co-existence of minor haplotypes A and B among the respiratory and digestive systems indicated that viruses can be transmitted between the orogastric tube, throat swab, and intestinal mucosa. Interestingly, we also identified the minor haplotype C, which appeared in the throat swab, skin lesion, cervical vertebrae, medulla oblongata, and basal ganglia, but not in the intestinal mucosa, which is suspected as the initial replication site of enteroviruses. Because this haplotype was only present in the throat swab, but not in the intestinal mucosa, this haplotype potentially reflects another dissemination route, whereby EV-A71 directly invades the host after replication in the upper respiratory tract and spreads to other tissues, including the CNS.
In addition, we examined whether selective pressure commonly existed at VP1 residue 31 in EV-A71. We analyzed the mutant spectra of an additional 15 EV-A71 strains from patients with various disease severities. All these circulating strains were isolated from throat swabs and belonged to the same genotype (C2) circulating in the 1998 outbreak in Taiwan. The πN and πS values provided an indication that approximately 60% of circulating EV-A71 strains face bottleneck selection at VP1 residue 31 (Fig. 3), and more viruses showed high πN values in fatal cases. Our data showed that VP1 residue 31 is common at positive-selection sites among circulating EV-A71. Studying the VP1-31G proportion among these strains further indicated that a higher proportion of VP1-31G appeared among the mutant spectra of EV-A71 in throat swabs in fatal cases (Fig. 4c). Since the EV-A71 VP1-31G haplotype increased in the mutant, this raised the possibility that EV-A71 could break through the bottleneck and invade CNS tissues, promoting fatality in patients with EV-A71 infection. To further determine if 31 selection occurred across genotypes, we also examined the properties of VP1-31G virus isolates belonging to genotypes B4 and B5, but no VP1-31 variation was identified in these mutant spectra (data not shown). Thus, we suggest that our identified selection bottleneck (and even unidentified bottlenecks for other genotypes) might shape unique evolutionary pathways and act at a different genetic site(s) for each genotype. Although we may not have defined all virulence determinants causing severe neurological diseases among various EV-A71 strains or genotypes in this study, our novel findings demonstrated that the viral quasispecies utilized the dynamic balance of the diverse population to cooperatively maintain population plasticity and facilitate viral adaptation and spread when facing a selection bottleneck between the respiratory and digestive systems and the CNS, which may be associated with the viral pathogenesis of EV-A71.
Several animal models, including neonatal mice, transgenic mice, and monkeys, have been used to investigate EV-A71 pathogenesis. Although EV-A71 caused neurological symptoms in these models, the routes of infection were mostly intraspinal, intracerebral, intratracheal, intraperitoneal, intramuscular, and subcutaneous routes. Only rare models successfully and consistently established infection in animals through natural fecal-oral and oral-oral routes. In addition, in the limited animal models that employed the natural oral routes, the virus was not detected in the intestinal mucosa, which differs from our observations in humans. We suggested that the different tissue tropisms observed between humans and other available animal models will result in distinct selection pressure for virus
quasispecies during dissemination in humans and animals. To investigate enterovirus dissemination in humans, we directly analyzed EV-A71 quasispecies among various tissues in autopsy patient specimens. To further evaluate the important role of the VP1-31 substitution identified in the autopsy patient, we analyzed additional EV-A71 isolates from other patients. Both the results from the autopsy case and various isolates from patients indicated that the VP1-31 substitution was associated with the ability of the virus to invade the CNS to cause severe neurological infection, resulting in fatalities. Thus, continuously analyzing the mutant spectra of circulating strains is necessary to survey virus evolution and to understand viral pathogenesis.
In addition to VP1-31G, other unidentified virulence determinants in viral quasispecies may also contribute to disease severity [20, 39-42], although few studies have been conducted to examine the correlation between viral variation and disease outcome. Data from a genetic-variation study focused on human EV-A71 strains by comparing sequences available in the GenBank database suggested that the substitution of N31D in genotype C, as well as E145G/Q, E164D/K, and VP1-T292N/K in genotype B, was associated with neurological symptoms [65]. Data from another study, using viral strains from various specimens and collection dates, also revealed that VP1-L97R contributes to neuronal cell tropism [66]. All of these virus substitutions represent useful tools for predicting the disease severity of EV-A71.
In conclusion, we identified a novel selection bottleneck appearing between the respiratory and digestive systems and the CNS during EV-A71 dissemination in humans. This bottleneck shaped the mutant spectra of EV-A71 in these tissues and drove a dominant haplotype switch from VP1-31D to VP1-31G. The proportion of VP1-31G virus also increased among fatal cases of EV-A71 infection. The VP1-31G virus, with the dominant haplotype in the CNS, exhibited high viral growth and fitness in neuronal cells, indicating that VP1-31G might contribute to CNS invasion and cause neurological disease in patients. Together, the process of haplotype selection might not only associate with viral pathogenesis in A71-infected patients but also correlate with disease severity among different EV-A71-infected individuals with various disease severities, especially for those haplotypes isolated from fatal cases. VP1-31G represents a potential viral factor that resides among quasispecies of EV-A71 isolates and contributes to viral dissemination and disease severity within humans.
Figure legends
Fig 1. Divergent EV-A71 mutant spectra among various tissues in human. Pie charts displaying the proportion of each haplotype among the indicated specimens or tissues. Among all 7 pie plots, each haplotype is presented with a unique color, which displays the distribution of each haplotype across each specimen or tissue.
Fig 2. Dominant haplotype switching between the respiratory tract and the CNS. (a) The Shannon entropy of each viral quasispecies isolated from the indicated tissues was calculated based on the frequency distribution of all haplotypes detected in the indicated specimens or tissues. (b)
Phylogenetic analyses of each haplotype from different tissues are displayed. Haplotypes with a frequency >1% were included in the analysis. The haplotypes were colored according the collected tissues. The rank and observed frequency of each haplotype were annotated for each tissue studied. (c) The plot presents the frequency distributions of each haplotype in different tissues. Each line with a unique color indicates the proportion of changes of a haplotype among specimens or tissues in the respiratory and digestive systems, integumentary system, and central nervous system. OG tube: orogastric tube. (d) Phylogeny networks and appearance frequency of each haplotype from different tissues. The sizes of the solid circles indicate the total abundance of each haplotype among all specimens or tissues. Different colors displayed in each solid cycle show the abundance contributed by the indicated specimens or tissues.
Fig 3. Genetic-diversity contributions of non-synonymous and synonymous mutations of viruses isolated from various tissues. The πN (a) and πS (b) values were calculated throughout each viral genome using a 50-codon sliding window and a 1-codon step. High πN values indicate cases where the SNVs evolved under positive selection, whereas high πS values indicate SNVs that evolved under purifying selection. Amino acid residues for the major peaks of πN and πS are noted in the plots. The arrows indicate the πN peak of the VP1-31 residue among different specimens or tissues.
Fig 4. Higher proportion of VP1-31G viruses from fatal cases than HFMD cases. (a) The Shannon entropies of quasispecies isolated from patients with fatal disease or HFMD were compared based on the frequency distribution of all haplotypes. The 1998-1 virus was isolated from a throat swab of an autopsy case. The center values represent the Shannon entropies among the selected fatal and HFMD cases. The proportion of VP1-31G (b) and 3A-73S (c) among virus isolates from patients with fatal disease or HFMD was determined based on the SNV-calling results from deep-sequencing reads. The center values represent mean proportions among the selected fatal and HFMD cases. The Mann– Whitney U test was used to calculate p values. * p < 0.05. (d) The πN values for EV-A71 from fatal cases and HFMD cases were calculated for each viral genome using a 50-codon sliding window and a 1-codon step.
Fig 5. Effects of VP1-31 substitution on thermostability, viral replication, and viral fitness. Plots (a) and (b) display, respectively, the thermostability and viral replication of a virus with VP1-31 substitutions. (a) A total of 107 pfu of the VP1-31G (green) or VP1-31D (blue) virus was incubated at different temperatures. Viruses were harvested after the indicated number of days and titrated in plaque assays (n = 6). The center values and error bars represent the mean and ± 1 standard deviation, respectively. A paired Student’s t test was used to calculate p values. (b) DLD-1, RD, and SK-N-SH cells were infected with VP1-31G (red) or VP1-31D (blue) at an moi of 0.1, as indicated. Viral titers were determined at the indicated days post-infection. The center values and error bars represent the mean ± 1 standard deviation, respectively. A 2-tailed paired Student’s t test was used to calculated p values. (c) DLD-1, RD, and SK-N-SH cells were individually inoculated or co-inoculated with the VP1-31G and VP1-31D viruses, using the same amount of each virus. Viruses were harvested once obvious cytopathic effects were observed. Harvested viruses were sequentially passaged in the same
cell line. The dominant haplotype appearing in various passages of viruses were defined by Sanger sequencing of the VP1-31 residue.
Fig 6. Effects of VP1-31 substitution on virus binding and RNA release. (a) VP1-31G and VP1-31D viruses were absorbed to DLD-1, RD, and SK-N-SH cells, as indicated. Bound viruses were quantified by ELISA. Values and error bars are the mean ± 1 standard deviation, respectively. (b) DLD-1, RD, and SK-N-SH cells were infected with neutral red-labeled VP1-31G and VP1-31D viruses, as indicated. Based on the characteristics of a neutral red-labeled virus, which can be inactivated under light exposure unless the virus releases its RNA, the numbers of viruses that released their RNA genome into the cells were determined using the infectious center assay. Values and error bars are the mean ± 1 standard deviation, as indicated. A paired Student’s t test was used to calculate P values. The error bars represent 1 standard deviation from triplicate experimental results.
Fig 7. Localization of the VP1-31 residue and potential protein structure changes at the VP1-31 substitution site. (a) The external and internal surfaces of the EV-A71 capsid pentamer are shown, as generated using the UCSF Chimera program, version 1.9. The red residue indicates the location of the VP1-31 residue in the outer (left panel) or internal (right panel) surface of the pentamer structure (PDB code 4AED). (b) The VP1-31 residue (red) is displayed on the VP1 protein ribbon structure. N and C indicate the N- and C-terminal ends of the VP1 protein. The red box region is enlarged in panel c of this figure. (c) Diagram displaying protein modeling of the structural consequences of the VP1-31 D-to-G mutation. Blue and red sticks expanding from the backbone structure represent the side chains of VP1-71D (in both panels), VP1-31D (left panel), and VP1-31G (right panel) residues. Solid yellow lines indicate potential interactions between atoms of VP1-71D and VP1-31D (left panel) or VP1-31G (right panel).
Fig.8 Survival rate, body weight and clinical score of ICR mice. The 3-day-old ICR mice were infected with 105 TCID50 of EV-A71 VP1-31D and VP1-31G virus as well as a mock control by
intraperitoneal injection. (a) Survival rates, (b) body weight (c) and clinical scores were monitored daily for 21 days after infection. Clinical score were followed: 0, healthy; 1, ruffled fur and
hunchbacked appearance; 2, wasting; 3, limb weakness; 4, limb paralysis; and 5, moribund and death.