國立臺灣大學理學院化學系 碩士論文
Department of Chemistry College of Science
National Taiwan University Master Thesis
多牙氮配位基及雙金屬鈀錯合物的合成與其催化應用 Synthesis and Catalytic Application of Dipalladium
Complexes with Multidentate Ligands
楊舒庭 Shu-Ting Yang
指導教授﹕劉緒宗 博士 Advisor: Shiuh-Tzung Liu, Ph.D.
中華民國 106 年 2 月
February, 2017
謝誌
首先最感謝的是指導老師劉緒宗老師,您始終是我最尊敬的老師之一。從我 大三做專題開始就很關心我們,給我們很大的空間,並依照每個人不同的需要給 予教導和協助,在老師身上我真的看到「因材施教」這四個字。進入碩班後實驗 一直不太順利讓我很沮喪,不過在老師的引領下終於找到題目。原本覺得平淡無 奇的反應,在老師的指點下慢慢發覺其中奧妙,體驗到窺見真理的喜悅感。老師 對我說的話我都有放在心上,以後會記取教訓,不論在生活或工作上都要更積極。
我不小心先找到工作,一直對老師感到很抱歉,沒辦法把東西做得更完整再離開,
謝謝老師還是盡心盡力地幫助我完成我的研究,老師對科學的堅持也讓我見識到 一個嚴謹的化學研究是什麼樣子。此外,也要感謝林英智老師和邱靜雯老師對我 的論文指導與提點。
謝謝實驗室的夥伴們:童哥、倍偲、馨雅、Mani、杰哥、萬大、社長、天哥、
洺煒、宏明、雅君、豪哥、泰霖、陶濤、芃、昱皓、米安德和郭宜蓁,都給予我 很多幫助,祝福大家未來都能平安順利。其中特別感謝社長和洪洺煒,你們是讓 B563-1 井井有條的大功臣,有你們在真的很讓人安心,謝謝你們就算是畢業後 也時常關心我,給予我很多實驗上的建議。感謝 STL 之花沈芃贊助我超過 100 mg 的 bpnp,讓我在最後緊要關頭不至於斷炊,還有王昱皓和萬書豪,最後這 半年多有你們三個真的很開心,和你們討論實驗讓我重拾對化學的熱情,謝謝你 們常常陪我到很晚,口試時也幫了我不少忙。
感謝林宣佑,一直是我最好的精神支柱,包容我很忙脾氣不好,雖然在當兵 還是花很多時間幫我修改論文,陪我一起解決很多問題。最後謝謝家人和朋友一 直以來的支持鼓勵與陪伴,不管發生什麼事總是和我站在一起,默默關心我而不 施予我壓力,支持我繼續努力,當我的軍師幫助我順利渡過每個難關,有你們才 有現在的我。
摘要
本篇論文分別合成 2,7-bis(2-pyridyl)-1,8-naphthyridine (bpnp) 和 5-phenyl- 2,8-bis(2,2’-bipyridin-6-yl)-1,9,10-anthyridine (pbbpa) 作為多牙配位基,利用這些 配體與鈀金屬進行錯合反應,探討所生成錯合物的結構和催化應用。多牙氮配位 基 bpnp 與乙酸鈀在甲醇和三氟醋酸下反應生成 Pd2(bpnp)(TFA)3(OH) (9),
pbbpa 與 Pd(MeCN)2Cl2 反應,合成出雙金屬錯合物。由於其溶解度不佳,使用
六氟磷酸鉀置換陰離子,生成 [Pd2(pbbpa)Cl2](PF6)2 (11),以核磁共振光譜和質 譜分析分別鑑定結構。
論文中比較 9 和 11 對於硝基還原反應之催化活性之差異。在先前的研究 工作中發現 [Pd2(pbbpa)Cl2](PF6)2 (11) 經過少量的 NaBH3CN 活化後,能夠在 氫氣下還原硝基苯衍生物,而 Pd2(bpnp)(TFA)3(OH) (9) 則是可以作為催化劑直 接在氫氣下進行該還原反應。另外,對此催化系統的應用範圍做了廣泛性測試,
並藉由動力學和可能的反應中間體的實驗,解析此催化的反應機構。發現 9 的 催化過程是遵循縮合路徑,由 N-苯基羥胺和亞硝基苯縮合成氧化偶氮苯後,藉 由雙金屬的協助進行後續的還原得到苯胺;而 11 的催化過程則和單金屬錯合物 Pd(bpy)(TFA)2 相同,是直接將 N-苯基羥胺還原成產物。
Abstract
The use of bimetallic complexes as catalysts for the catalytic reactions has received much attraction due to the possible synergistic effect between the metal ions.
In this study, a naphthyridine-based multidentate ligand 2,7-bis(2-pyridyl)-1,8- naphthyridine (bpnp) and an anthyridine-based ligand 5-phenyl-2,8-bis(2,2’-bipyridin- 6-yl)-1,9,10-anthyridine (pbbpa) were synthesized. Coordination of bpnp with Pd(OAc)2 in a mixture of MeOH and trifluoroacetic acid yielded the dipalladium complex Pd2(bpnp)(TFA)3(OH) (9). Treatment of pbbpa with Pd(MeCN)2Cl2
followed by anion exchange resulted in the formation of [Pd2(pbbpa)Cl2](PF6)2 (11).
The structures of both complexes were confirmed by 1H-, 13C-NMR and ESI-HRMS.
In a previous work, the resulting complex obtained from treatment of 11 with NaBH3CN was catalytically active toward the reduction of nitroarenes in the presence of H2. In this work, complex 9 showed a similar activity without any pre-treatment.
Comparison of the catalytic activity between these complexes was revealed. This catalytic system is applicable for various nitroarenes. The possible reaction mechanism of this catalytic system is established by the kinetic studies and the reactivity of possible intermediates under the catalytic conditions. 9 catalyzed through the condensation pathway, in which N-phenylhydroxylamine and nitrosobenzene formed azoxybenzene, and gave aniline by following reduction with the assistance of dimetal complex. On the other hand, 11 and Pd(bpy)(TFA)2 catalyzed through direct pathway, in which aniline was obtained by reduction of N-phenylhydroxylamine.
目錄
摘要 ... I
Abstract ... II 目錄 ... III
附圖目錄 ... V
附表目錄 ... VI
流程目錄 ...VII
第一章 緒論 ... 1
1.1 萘啶類多牙基與其雙金屬錯合物 ... 1
1.2 蒽啶類多牙基與其雙金屬錯合物 ... 4
1.3 硝基苯還原反應 ... 7
1.4 研究目的 ... 9
第二章 雙鈀金屬錯合物之合成與鑑定 ...10
2.1 配位基之合成 ... 10
2.2 雙鈀金屬錯合物之合成與鑑定 ... 12
2.3 雙鈀金屬錯合物 9 之配位基置換 ... 17
第三章 雙鈀金屬錯合物之催化應用 ...22
3.1 反應條件最佳化 ... 22
3.2 反應應用範圍 ... 26
3.3 芳基鹵化物之還原反應探討 ... 27
3.4 反應機構探討和與其他鈀催化劑之比較 ... 31
3.5 催化劑重複反應測試 ... 39
第四章 結論 ...40
第五章 實驗部分 ...42
5.1 General part ... 42
5.2 Synthetic procedures and characterization of compounds ... 43
5.2.1 Synthesis of ligand and palladium(II) complexes ... 43
5.2.2 Reduction of Nitroarenes ... 57
5.2.3 Investigation of Reaction Intermediates and Mechanism ... 64
參考文獻 ...69
附錄一 ...73
附圖目錄
Figure 1-1 Naphthyridine-based multidentate ligands ... 1
Figure 1-2 Anthyridine-based multidentate ligands ... 4
Figure 2-1 Partial 1H NMR spectrum of (a)bpnp and (b) 9 ... 12
Figure 2-2 ESI-HRMS spectrum of 9 ... 13
Figure 2-3Partial 1H NMR spectrum of (a) bpnp and (b) 10 ... 14
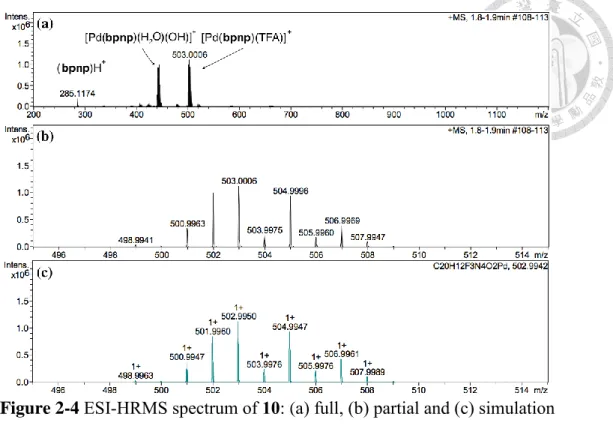
Figure 2-4 ESI-HRMS spectrum of Pd(bpnp)(TFA)2 (10) ... 15
Figure 2-5 Partial 1H NMR spectrum of (a) pbbpa and (b) 11 ... 16
Figure 2-6 ESI-HRMS spectrum of 11 ... 17
Figure 2-7 Partial 1H NMR of (a) 9 and (b) 12 ... 18
Figure 2-8 ESI-HRMS of 12 ... 19
Figure 2-9 Partial 1H NMR of (a) 9 and (b) 13 ... 20
Figure 2-10 ESI-HRMS of 13 ... 21
Figure 3-1 Mono-palladium(II) complexes with N-donating ligands ... 26
Figure 3-2 Kinetic time course of 9-catalyzed reduction of 14b ... 32
Figure 3-3 Kinetic time course of 9-catalyzed reduction of 17a ... 33
Figure 3-4 Kinetic time course of 11-catalyzed reduction of 17a ... 34
Figure 3-5 Kinetic time course of 11-catalyzed reduction of 14b ... 35
Figure 3-6 9-catalyzed reduction of 14b w/ or w/o NaBH3CN ... 36
Figure 3-7 Kinetic time course of Pd(bpy)(TFA)2-catalyzed reduction of 14b ... 37
Figure 3-8 Pd(bpy)(TFA)2-catalyzed reduction of 14b w/ or w/o NaBH3CN ... 38
附表目錄
Table 2-1 The ranges of molar conductivity of complexes with different ratios ... 13
Table 3-1 Preliminary optimization for the reduction of 4-nitrotoluene ... 23
Table 3-2 Reaction optimization for the reduction of 4-nitrotoluene ... 24
Table 3-3 Comparison of various Pd complexes on reduction of 4-nitrotoluene ... 25
Table 3-4 Scope of catalysis with various nitroarenes ... 27
Table 3-5 Influence of bromide in reduction of nitroarenes ... 28
Table 3-6 Reduction of 1-bromo-4-nitrobenzene (14g) ... 29
Table 3-7 Reductive dehalogenation of aryl halides ... 30
Table 3-8 Reduction of 14a, 16a and 18a catalyzed by 9 ... 32
Table 3-9 Reduction of 14a, 16a and 18a catalyzed by 11 ... 34
Table 3-10 Reduction of 14a, 16a and 18a catalyzed by Pd(bpy)(TFA)2 ... 37
流程目錄
Scheme 1-1 Bimetallic complexes [(bpnp)M2Ln] ... 3
Scheme 1-2 Cyclometalation of L1 with dirhodium acetate ... 4
Scheme 1-3 Diruthenium(II) complexes synthesized using photoisomerization ... 5
Scheme 1-4 Dicopper Complexes with Anthyridine-Based Ligands ... 6
Scheme 1-5 Dinickel Complexes with Anthyridine-Based Ligands ... 6
Scheme 1-6 Combined water-gas shift reaction and nitroarene hydrogenation ... 8
Scheme 1-7 Chemoselectivity in hydrogenation with RuCl2(PPh3)3 ... 8
Scheme 2-1 Synthesis of bpnp ... 10
Scheme 2-2 Synthesis of pbbpa ... 11
Scheme 2-3 Synthesis of Pd2(bpnp)(OH)(TFA)3 (9) ... 12
Scheme 2-4 Synthesis of Pd(bpnp)(TFA)2 (10) ... 14
Scheme 2-5 Synthesis of [Pd2(pbbpa)Cl2](PF6)2 (11) ... 15
Scheme 2-6 Synthesis of [Pd2(bpnp)Cl2(OH)](TFA) (12)... 18
Scheme 2-7 Synthesis of [Pd2(bpnp)Br2(OH)](TFA) (13) ... 20
Scheme 3-1 Reduction of p-nitrotoluene ... 22
Scheme 3-2 Reduction of 1-chloro-4-nitrobenzene ... 29
Scheme 3-3 Proposed mechanism of the reduction of nitroarenes ... 31
Scheme 3-4 Multiple rounds of 9-catalyzed reduction of nitroarenes ... 39
Scheme 3-5 High-pressure reaction ... 39
第一章 緒論
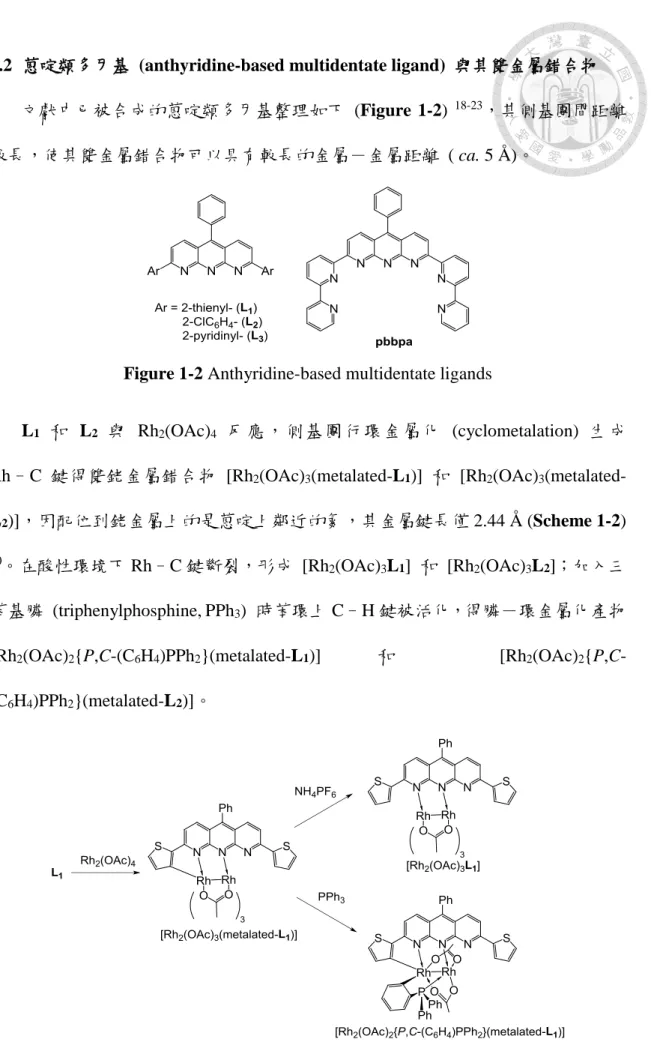
1.1 萘啶類多牙基 (naphthyridine-based multidentate ligand) 與其雙金屬錯合物 在過去的數十年間,在有機合成中使用雙金屬錯合物作為勻相催化劑受到化學 家們高度關注。在雙金屬錯合物中,由於金屬離子間距離短,在催化反應中會展現 協同效應 (synergistic effect) 1,和相對應的單金屬錯合物比較往往具有較好的催化 活性或選擇性。因此,設計出能夠鉗合雙金屬離子的配基,使雙金屬接近就顯得格 外重要。近年來,1,8-萘啶被大量使用作為雙金屬系統中有效的橋接配基,2、7號 位置可修飾出其他具有配位能力的多牙氮配位基,增加與金屬的螯合能力,部分整 理如 Figure 1-1 2-7。
Figure 1-1 Naphthyridine-based multidentate ligands
其中,1979 年 Caluwe 以有機方法合成出一具有多氮原子的雜環平面分子,
以1,8-萘啶主體結構,在2、7號位置上用吡啶修飾,稱為 2,7-bis(2-pyridyl)-1,8- naphthyridine (bpnp) 8。此分子可作為金屬錯合物之配位基,利用吡啶和萘啶上的 氮原子和金屬鍵結,形成之金屬錯合物可應用於催化合成反應。多牙配位基與金屬 形成穩定的錯合物,其金屬距離多在 2-3 Å 間,使雙金屬間產生特別的性質和協
同效應。其中 bpnp 之雙金屬錯合物發展彙整於 Scheme 1-1:1980 年代 Kaska 利用 bpnp 與 Ru2(OAc)4Cl 反應,得到 [Ru2(bpnp)(μ-CH3CO2)3]PF6 (Scheme 1-1, structure A) 9,以 X 光繞射光譜鑑定其結構;與二價雙銠金屬 Rh2(OAc)4 反應,
得到類似結構的 [Rh2(bpnp)(μ-CH3CO2)3]PF6 (Scheme 1-1, structure B) 10,11;另外,
取 bpnp 與二價氯化銅反應得到雙銅配位錯合物 [Cu2(bpnp)(Cl)2(μ-Cl)(μ-OH)]
(Scheme 1-1, structure C) 12,X 光繞射光譜鑑定結果顯示其以氯和氫氧根為架橋連 接。這一系列錯合物開啟了 bpnp 雙金屬錯合物的濫觴,但其文獻中並未針對催 化活性作探討。
近年來,本實驗室對於 bpnp 和不同金屬反應形成之錯合物亦有一系列研究。
2012 年 利 用 [Cu2(CF3CO2)4] 與 bpnp 反 應 , 合 成 出 雙 銅 金 屬 錯 合 物 [Cu2(bpnp)(CF3CO2)2(μ-CF3CO2)(μ-OH)] (Scheme 1-1, structure D) 13,雙銅皆為二價,
以氫氧根和三氟醋酸根為架橋基團,其結構類似於 Kaska 於 1984 年所報導之雙 銅錯合物 [Cu2(bpnp)(Cl)2(μ-Cl)(μ-OH)]。該錯合物可催化 2,6-雙取代酚類的氧化 耦合,在反應過程中可觀察到雙銅金屬中心連接兩分子酚類之中間體,利於發生分 子內耦合,展現了雙金屬協同效應。以 Cu2O 在甲酸中和 bpnp 作用,得到另一 結構類似的雙銅錯合物 [Cu2(bpnp)(HCO2)2(μ-HCO2)(μ-OH)] (Scheme 1-1, structure E) 14,可在氧氣下催化醇類及胺類的氧化耦合。使用 [Cu(MeCN)4](PF6) 和 bpnp 錯 合 形 成 [Cu2(bpnp)(MeCN)4](PF6)2, 再 經 過 陰 離 子 置 換 得 一 價 雙 銅 錯 合 物 [Cu2(bpnp)(MeCN)4](BArF4)2 (Scheme 1-1, structure F) 和 [Cu2(bpnp)(μ-Cl)2] (Scheme 1-1, structure G) 15,後者經由 X 光繞射光譜鑑定其為一蝴蝶形結構,銅 金 屬 中 心 和 bpnp 與 氯 呈 正 四 面 體 配 位 , 三 者 皆 能 催 化 2- 溴 苯 甲 酸 和 脒 (amidines) 合成喹唑啉酮 (quinazolinone) 的環化反應。
另外,利用 bpnp 與 Pd(PhCN)2Cl2 和 Pd2(dba)3 錯合,可得一具特殊金屬中 心的水溶性三鈀金屬錯合物 [Pd3(bpnp)2Cl2]Cl2 (Scheme 1-1, structure H) 16。由 X 光繞射分析此錯合物結構,三鈀金屬間夾角為 79.10°,形成一近 L 型結構,兩 bpnp 以正交形式與金屬配位。電子密度分析儀可推測三核金屬的中心鈀為零價,
並以六配位型態存在。若分別加入 NaOH、KBr 和 NaN3,皆可置換金屬上的氯配 位 基 , 再 以 NaPF6 置 換 陰 離 子 即 可 得 到 [Pd3(bpnp)2(OH)2](PF6)2 、 [Pd3(bpnp)2Br2](PF6)2 和 [Pd3(bpnp)2(N3)2](PF6)2。此文獻為目前發表 bpnp 多鈀 金屬錯合物的唯一案例,但尚未發表任何催化應用。
另 外 若 以 Ru3(CO)12 和 bpnp 反 應 , 形 成 一 甲 酸 根 架 橋 之 雙 釕 錯 合 物 [Ru2(bpnp-H3)(μ-HCO2)(CO)4] (Scheme 1-1, structure I),其 bpnp 的萘啶被部分還 原。分別以醋酸和苯甲酸置換配位基,可得到 [Ru2(bpnp-H3)(μ-CH3CO2)(CO)4] (Scheme 1-1, structure J) 和 [Ru2(bpnp-H3)(μ-PhCO2)(CO)4] (Scheme 1-1, structure K) 17。
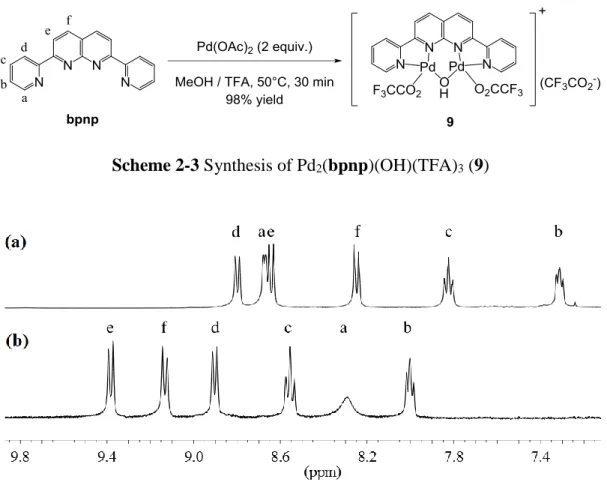
1.2 蒽啶類多牙基 (anthyridine-based multidentate ligand) 與其雙金屬錯合物 文獻中已被合成的蒽啶類多牙基整理如下 (Figure 1-2) 18-23,其側基團間距離 較長,使其雙金屬錯合物可以具有較長的金屬-金屬距離 ( ca. 5 Å )。
Figure 1-2 Anthyridine-based multidentate ligands
L1 和 L2 與 Rh2(OAc)4 反應,側基團行環金屬化 (cyclometalation) 生成 Rh–C 鍵得雙銠金屬錯合物 [Rh2(OAc)3(metalated-L1)] 和 [Rh2(OAc)3(metalated- L2)],因配位到銠金屬上的是蒽啶上鄰近的氮,其金屬鍵長僅 2.44 Å (Scheme 1-2)
20。在酸性環境下 Rh–C 鍵斷裂,形成 [Rh2(OAc)3L1] 和 [Rh2(OAc)3L2];加入三 苯基膦 (triphenylphosphine, PPh3) 時苯環上 C–H 鍵被活化,得膦-環金屬化產物 [Rh2(OAc)2{P,C-(C6H4)PPh2}(metalated-L1)] 和 [Rh2(OAc)2{P,C- (C6H4)PPh2}(metalated-L2)]。
L3 和 [Ru(η6-p-cymene)Cl2]2 作 用 可 得 雙 釕 金 屬 錯 合 物 [Ru2(L3)(p- cymene)2Cl2],能催化三級芳香胺類的氧化氰化23。L3 和 Ru(tpy)Cl3 (tpy = 2,2';6',2"- terpyridine) 反應 形成 Distal-[Ru(tpy)(L3)Cl]2+,加入硝酸銀和氯 離子沉澱得到 Distal-[Ru(tpy)(L3)(OH2)]2+, 經 光 異 構 化 (photoisomerization) 得 Proximal- [Ru(tpy)(L3)(OH2)]2+, 可 再 和 一 分 子 Ru(tpy)Cl3 錯 合 形 成 Proximal,proximal- [Ru2(tpy)2(L3)(μ-Cl)]3+,雙金屬距離為 4.193 Å 。在鹼性水溶液中進行配基取代 (ligand substitution) 得 Proximal,proximal-[Ru2(tpy)2(L3)(μ-OH)]3+ 和 Proximal,proximal-[Ru2(tpy)2(L3)(OH)(H2O)]3+,後者 L3上蒽啶中心的氮和 H2O 上 的氫能夠形成氫鍵,增加額外的穩定度 (Scheme 1-3) 18。
Scheme 1-3 Diruthenium(II) complexes synthesized using photoisomerization
另外,於 L3 吡啶下方增加了額外的吡啶,即為 5-phenyl-2,8-bis(2,2’-bipyridin- 6-yl)-1,9,10-anthyridine (pbbpa)。此三聯吡啶 (terpyridine, tpy) 的平面結構為一鉗 合配基 (pincer ligand) ,可能增強配位基的螯合效應 (chelate effect)。以 Cu(ClO4)2
之六水合物分別和 L3 和 pbbpa 錯合得到 [Cu2(L3)(H2O)4(MeCN)2](ClO4)4 和
[Cu2(pbbpa)(μ-ClO4)2](PF6)2 雙銅錯合物 21,兩者皆可催化苯甲醇的氧化。另外,
[Cu2(pbbpa)(μ-ClO4)2](PF6)2 在雙氧水中能有效地催化一級醇的氧化耦合反應得到 酯類化合物,以質譜觀察到反應中間體為雙銅金屬中心分別連接一分子苯甲醇和 一分子苯甲醛,有利於分子內攻擊 (intramolecular attack),即展現了雙金屬協同效 應。反觀 [Cu2(L3)(H2O)4(MeCN)2](ClO4)4 由於金屬上空配位較多,容易行其他競爭 反應而非分子內反應 (Scheme 1-4)。
Scheme 1-4 Dicopper complexes with anthyridine-based ligands
使 用 Ni(OAc)2 分 別 和 L3 和 pbbpa 反 應 形 成 雙 鎳 錯 合 物 [Ni2(L3)(H2O)6(CF3CO2)2](CF3CO2)2 和 [Ni2(pbbpa)(CF3CO2)4(H2O)] 22,雙金屬距 離為 5.409 和 5.014 Å 。比較兩者對於羧酸還原反應的催化活性,發現後者具有良 好的效果,可能是由於三聯吡啶的螯合效應和較短的金屬距離 (Scheme 1-5)。
Scheme 1-5 Dinickel complexes with anthyridine-based ligands
1.3 硝基苯還原反應
苯胺類化合物是工業上重要的化合物,其合成方式亦成為有機合成上重要的目 標之一。其中最常見的合成路徑是藉由催化加氫反應 (catalytic hydrogenation) 還
原相對應之硝基苯,非勻相催化劑如金屬銅 24、鈷、鈀25和鎳26都常被使用,後
兩者由於活性較高,可能需要加入抑制劑以防止芳香環被氫化27。大多以氫氣或合
成氣 (syngas) 作為還原劑,但也有甲烷28和甲酸29,30的例子。非勻相催化劑可運 用在工業上,以連續攪拌槽反應器 (continuous stirred tank reactors, CSTR) 大量生 產苯胺類。勻相催化劑亦常使用 VIII B 的過渡金屬錯合物如鐵、鈷、鎳、釕、銠 和鈀催化硝基苯還原,雖無法取代非勻相催化劑的工業用途,但因可能具有較佳的
還原選擇性31,32且反應條件溫和,適合用在多官能基的起始物。
Rh(py)3Cl3 (py = pyridine) 在二甲基甲醯胺 (dimethylformamide, DMF) 下和等 當量 NaBH4 作用形成活性物種 Rh(Py)2Cl2(DMF)(BH4) ,在氫氣下可催化還原反 應,若不加入 NaBH4 則無法催化,加入過量 NaBH4 亦會抑制反應33。雙釕錯合 物 [RuL(CO)2Cl]2 (LH = 2-phenylpyridine, benzo[h]quinoline, 1-phenylpyrazole 和 azobenzene 等含氮配基)可選擇性催化硝基苯還原,苯環上的氯取代基不會被氫 解。反應時雙金屬分解成單金屬和溶劑加成,形成催化活性物種 [RuL(CO)Cl·S] (S
= DMF, DMSO),在高壓氫氣和高溫下氫置換氯開啟循環。但此催化系統活性較相
對應之鈀錯合物高,會還原氰基和芳香酮類34。
若結合水煤氣轉化反應 (water-gas shift reaction) 和硝基苯還原,即可以一氧化 碳還原硝基苯,通常使用羰基錯合物作為催化劑 (Scheme 1-6)。1980 年代 Alper 提出三個勻相催化硝基苯還原的系統:Ru3(CO)12 在室溫、一大氣壓一氧化碳下還
原硝基苯,此系統具有良好的選擇性,苯環上的氯和醛均不會被還原 35;[Rh(1,5-
hexadiene)Cl]2 和 Co2(CO)8 組成的雙金屬系統也有類似的效果 36;RuCl2(PPh3)3
在室溫、一大氣壓合成氣下亦可還原立體障礙小的硝基苯,不會還原烯類、羰基和
鹵素,在純氫氣或一氧化碳下產率則較差37。
Scheme 1-6 Combined water-gas shift reaction and nitroarene hydrogenation
除了以氣體作為還原劑以外,使用低碳數脂肪醇和甲酸以催化轉移氫化 (catalytic transfer hydrogenation) 還原硝基苯提供了合成苯胺的另一選擇,且可能得 到 不 同 的 選 擇 性 , 如 RuCl2(PPh3)3 亦 可 以 甲 酸 還 原 4’- 硝 基 苯 乙 酮 (4’- nitroacetophenone) 38,但選擇性不同於氫氣還原 (Scheme 1-7)。
Scheme 1-7 Chemoselectivity in hydrogenation with RuCl2(PPh3)3
2010 年起 Beller 發表了三個催化轉移氫化系統:以 FeBr2-PPh3 催化,
PhSiH3 作為還原劑39、Fe(BF4)2・6H2O-P(CH2CH2PPh2)3 催化,甲酸為還原劑40 和 RuCl2(tpy) 催化,異丙醇為還原劑 41,三者皆具有很好的選擇性,能夠保留反 應物上的鹵基、烯、羰基。一系列的釕、銥錯合物被合成並應用在轉移氫化硝基苯
還原反應上,探討金屬和配基效應對產物選擇性的影響42。
以鈀催化的硝基苯還原反應早期大多是以 Pd/C 催化,近幾年有在反應中以聚 甲基氫矽氧烷 (polymethylhydrosiloxane, PMHS)43,44、三乙基矽烷 (triethylsilane)43 或一氧化碳45等,將Pd(OAc)2 或其他 Pd(II) 還原成 Pd(0) 催化的例子,以氫氣 還原 Pd(II) 的例子目前為止則並未有詳細的報導。
1.4 研究目的
目前為止許多含氮多牙基已被製備並應用於合成雙金屬錯合物,其中 2,7- bis(2-pyridyl)-1,8-naphthyridine (bpnp) 和 5-phenyl-2,8-bis(2,2’-bipyridin-6-yl)- 1,9,10-anthyridine (pbbpa) 引起我們的關注。在文獻中,金屬-金屬距離在約莫 3 Å 的 [M2(bpnp)Ln] 和金屬-金屬距離在 5 Å 左右的 [M2(pbbpa)Ln] 多在催化 反應中展現了協同效應。因此,藉由比較兩配位基 bpnp 和 pbbpa 之雙鈀金屬錯 合物,我們期望能夠探討金屬-金屬距離對硝基還原反應催化活性的影響。
第二章 雙鈀金屬錯合物之合成與鑑定
2.1 配位基之合成
在 1979 年,Caluwe 首次利用 4-aminopyrimidine-5-carboxaldehyde 經過兩次 Friedländer 縮合反應合成出 2,7-bis(2-pyridyl)-1,8-naphthyridine (bpnp) 8 ,然而在 此論文中我們採用另一方法, 經由 Stille 耦合反應連結萘啶中心骨架 2,7-dichloro- 1,8-naphthyridine (3) 和側基團 2-tributylstannylpyridine 46。
首先,取 2,6-diaminopyridine 在酸性環境下和 malic acid 縮合得到 2-amino- 7-hydroxy-l,8-naphthyridine (1) , 經 重 氮 化 並 水 解 取 得 2,7-dihydroxy-1,8- naphthyridine (2)。接著以三氯氧化磷進行氯化得到化合物 3 47,總產率53%。3 和 2-tributylstannylpyridine 在 PdCl2(PPh3)2 催化下進行雙 Stille 耦合即可得 bpnp (Scheme 2-1)。此配位基為已知物,以核磁共振光譜 (NMR spectroscopy) 鑑定其結
構,其氫譜共6 組訊號在芳香環區,積分值為 12 個氫,化學位移和耦合常數均與
文獻相符。
Scheme 2-1 Synthesis of bpnp46,47
Caluwe 在 1977 年以 2,6-diaminopyridine-3,5-dicarboxaldehyde 和兩當量甲 基酮經兩次 Friedländer 縮合反應合成出 2,8-雙取代之蒽啶 48。然而我們使用 Zimmerman 的 改 良 方 法 製 備 5-phenyl-2,8-bis(2,2’-bipyridin-6-yl)-1,9,10-
anthyridine (pbbpa) 49。合成路徑如 Scheme 2-2 所示。trimethyl benzoate 在吡啶 (pyridine) 中 和 malononitrile 反 應 , 加 入 濃 鹽 酸 得 到 2-amino-6-chloro-4- phenylpyridine-3,5-dicarbonitrile (4) 。 接 著 在 濃 氨 水 中 進 行 親 核 芳 香 取 代 (nucleophilic aromatic substitution) 得 到 2,6-diamino-4-phenylpyridine-3,5- dicarbonitrile (5) 。 最 後 在 氫 氣 下 以 Pd/C 催 化 氫 化 得 到 2,6-diamino-4- phenylpyridine-3,5-dicarbaldehyde (6),總產率 5%。
2,6-二溴吡啶加入正丁基鋰和二甲基乙醯胺 (dimethylacetamide, DMAc) 進行 乙醯化得到 1-(6-bromopyridin-2-yl)ethanone (7),和 2-tributylstannylpyridine 經過 Stille 耦合反應得到 1-(2,2′-bipyridin-6-yl)ethanone (8),總產率 32%。6 和 8 在鹼 性 環 境 下 進 行 兩 次 Friedländer 縮 合 反 應 得 到 目 標 產 物 5-phenyl-2,8-bis(2,2’- bipyridin-6-yl)-1,9,10-anthyridine (pbbpa)。此配位基為已知物,以核磁共振光譜鑑
定其結構,其氫譜有11 組訊號在芳香環區,共 23 個氫,化學位移、積分值和耦合
常數均與文獻相符。
Scheme 2-2 Synthesis of pbbpa 49
2.2 雙鈀金屬錯合物之合成與鑑定
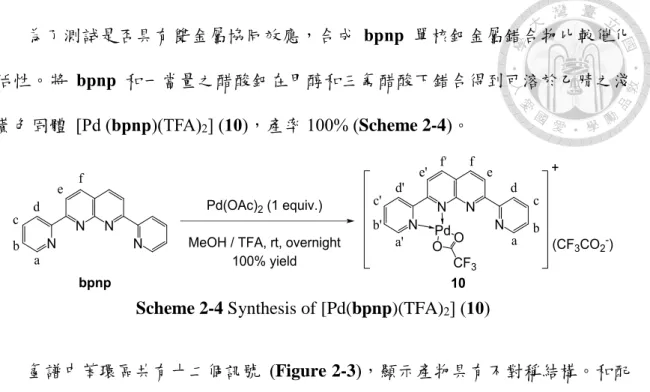
bpnp 和兩當量之醋酸鈀在甲醇和三氟醋酸下錯合得到可溶於乙腈之黃色固 體 Pd2(bpnp)(TFA)3(OH) (9),產率 98% (Scheme 2-3)。氫譜中苯環區共有六個訊號 (Figure 2-1),顯示產物具有對稱結構。和配位基的氫譜比較,最接近金屬的 Ha往
高場移動,而其餘所有訊號皆往低場移動,位移量以Hf最多。氟譜中僅有一組寬
訊號,顯示外圍離子和配位的三氟醋酸根在溶液中可以進行交換。而在電灑式高解 析質譜 (electrospray ionization-high-resolution mass spectrometry, ESI-HRMS) 中,
[Pd2(bpnp)(TFA)2(OH)]+ 理論計算的荷質比 (m/z) 為 740.8869,得到的結果是 740.8892,且同位素模擬結果亦一致 (Figure 2-2);質譜中最高峰之 m/z 為 644.9099,
同 位 素 模 擬 和 [Pd2(bpnp)(TFA)(OH)2]+ 相 符 ( 附 錄 一 ), 可 能 為 [Pd2(bpnp)(TFA)2(OH)]+ 在游離過程中被水氣進行配基置換產生。
Scheme 2-3 Synthesis of Pd2(bpnp)(OH)(TFA)3 (9)
Figure 2-1 Partial 1H NMR spectrum of (a)bpnp in CDCl and (b) 9 in DMSO-d
Figure 2-2 ESI-HRMS spectrum of 9: (a) full, (b) partial and (c) simulation
藉由測量莫耳導電度來判斷錯合物 9 於溶液態中解離的狀況是否與推測結構
吻合。將9 溶於二甲基亞碸 (dimethyl sulfoxide, DMSO) 中,於 0.40 mM 濃度下 測得莫耳導電度為54 (ohm-1 cm2 mol-1),溶於硝基甲烷 (nitromethane, MeNO2) 中,
於0.40 mM 濃度下測得莫耳導電度為 81 (ohm-1 cm2 mol-1)。根據文獻報導 (Table
2-1) 50,當錯合物在溶液中解離成一個陽離子和一個陰離子時,在 DMSO 中莫耳
導電度會在50–90 ohm-1 cm2 mol-1,在MeNO2中莫耳導電度則在75–95 ohm-1 cm2
mol-1,此二實驗數據符合推測結構,主配位基bpnp、兩個三氟醋酸根和一個氫氧
根與雙鈀金屬為陽離子,其外圍有一個三氟醋酸根為陰離子。
Table 2-1 The ranges of molar conductivity of complexes with different ratios Molar conductivity (ohm-1 cm2 mol-1) ranges
Solvent Non-electrolyte 1:1 Electrolyte 2:1 Electrolyte 3:1 Electrolyte
DMSO < 50 50–90 110–195 200–240
MeNO2 < 75 75–95 150–180 220–260
為了測試是否具有雙金屬協同效應,合成 bpnp 單核鈀金屬錯合物比較催化 活性。將 bpnp 和一當量之醋酸鈀在甲醇和三氟醋酸下錯合得到可溶於乙腈之淺 黃色固體 [Pd (bpnp)(TFA)2] (10),產率 100% (Scheme 2-4)。
Scheme 2-4 Synthesis of [Pd(bpnp)(TFA)2] (10)
氫譜中苯環區共有十二個訊號 (Figure 2-3),顯示產物具有不對稱結構。和配 位基的氫譜比較, Ha’、 Hc、 Hd、Hd’和 He’往高場移動,其餘訊號則往低場移動,
位移量以Hf’最多,Ha – Hf 和 Ha’ – Hf’ 相較之下位移量均較小,位移變化最多的 是離金屬最近的Ha 和 Hb。而在電灑式高解析質譜中,[Pd(bpnp)(TFA)]+ 理論計算 的荷質比 (m/z) 為 502.9950,得到的結果是 503.0006,且同位素模擬結果亦一致 (Figure 2-4) 。 質 譜 中 次 高 峰 之 m/z 為 444.9949 , 同 位 素 模 擬 和 [Pd(bpnp)(H2O)2(OH)]+ 相符(附錄一),可能為 [Pd(bpnp)(TFA)]+ 在游離過程中 被水氣進行配基置換產生。
Figure 2-3Partial 1H NMR spectrum of (a) bpnp and (b) 10 in CD3CN
Figure 2-4 ESI-HRMS spectrum of 10: (a) full, (b) partial and (c) simulation
將 10 溶於 DMSO 中,於 0.40 mM 濃度下測得莫耳導電度為 68 (ohm-1 cm2 mol-1);而溶於 MeNO2 中,於 0.40 mM 濃度下則測得莫耳導電度為 96 (ohm-1 cm2
mol-1),此二數據皆符合文獻中陰陽離子比例為 1:1 的電解質,與推測結構相符,
主配位基bpnp、一個三氟醋酸根與雙鈀金屬形成陽離子,其外圍有一個三氟醋酸
根為陰離子。
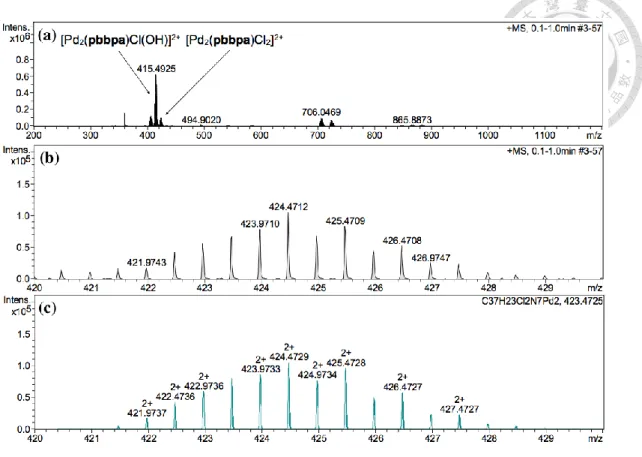
Pd(MeCN)2Cl2 和 pbbpa 錯 合 後 以 KPF6 置 換 陰 離 子 得 到 錯 合 物 [Pd2(pbbpa)Cl2](PF6)2 (11),產率 82% (Scheme 2-5)。
Scheme 2-5 Synthesis of [Pd2(pbbpa)Cl2](PF6)2 (11)
以核磁共振光譜 (Figure 2-5 b) 和電灑式游離高解析質譜 (Figure 2-6) 鑑定 錯合物 11,氫譜顯示錯合物為一對稱結構,和配位基 pbbpa 相比,兩者之氫譜訊 號變化並無明顯的趨勢,Hb、Hc和Hf向高場移動,其餘質子向低場移動,其中以 Hh 位移最多。在電灑式游離高解析質譜中,[Pd2(pbbpa)Cl2]2+ 理論計算的荷質比 (m/z) 為 424.4729,實驗結果為 424.4712,同位素模擬亦相符 (Figure 2-6 c)。質譜 中強度最高峰之 m/z 為 415.4925,同位素模擬和 [Pd2(pbbpa)Cl(OH)]2+ 相符(附 錄一),可能為 [Pd2(pbbpa)Cl2]2+ 在游離過程中被水氣進行配基置換產生。
Figure 2-5 Partial 1H NMR spectrum of (a) pbbpa in CDCl3 and (b) 11 in CD3CN
將 11 溶於DMSO 中,於 0.40 mM 濃度下測得莫耳導電度為 160 (ohm-1 cm2 mol-1),溶於MeNO2中,於1.0 mM 濃度下測得莫耳導電度為 180 (ohm-1 cm2 mol-
1),文獻中陰陽離子比例為 1:2 的電解質在 DMSO 中莫耳導電度範圍會在 110–195 ohm-1 cm2 mol-1,在MeNO2中則在150– 180 ohm-1 cm2 mol-1 (Table 2-1)50,故此數
據符合推測結構,主配位基pbbpa、兩個氯與雙鈀金屬形成陽離子,其外圍有兩個
六氟磷酸根。
Figure 2-6 ESI-HRMS spectrum of 11: (a) full, (b) partial and (c) simulation
2.3 雙鈀金屬錯合物 9 之配位基置換
嘗試將9 上的三氟醋酸根換成其他配位基。在室溫氮氣下將 9 溶於甲醇中,
加入 3 當量的 PPh3,氫譜訊號變寬,顯示配基正在進行可逆交換,但無法得到單 一產物;在室溫下加入 3 當量的醋酸鈉,溶液顏色和氫譜皆沒有改變,在甲醇下 迴流半小時則溶液顏色由黃色轉棕色,氫譜訊號變寬,但位置仍和原本相同;若改 以大量醋酸置換,則氫譜為 9 和自由配基之疊加,顯示醋酸將 bpnp 置換下來。
將 9 溶於 DMSO 以質譜鑑定,顯示 DMSO 並未配位;溶於 DMF,質譜同樣沒 有變化。
在室溫 CO/N2下將 9 溶於甲醇中,溶液由黃轉黑,可知 9 被 CO 還原為 Pd(0),
氫譜訊號變寬。在室溫氮氣環境下將 9 和過量 KI 溶於甲醇中,溶液初呈黃色,
攪拌後呈暗紅色且出現黑色固體,氫譜上苯環區僅看到自由配基的訊號,顯示碘將
bpnp 置換下來了。接著選用 Na2S 進行配基置換,為了避免過量的硫置換 bpnp,
僅使用一當量的 Na2S 和 9 在氮氣下溶於無水乙腈,溶液迅速由黃轉黑棕色,但氫
譜十分雜亂。
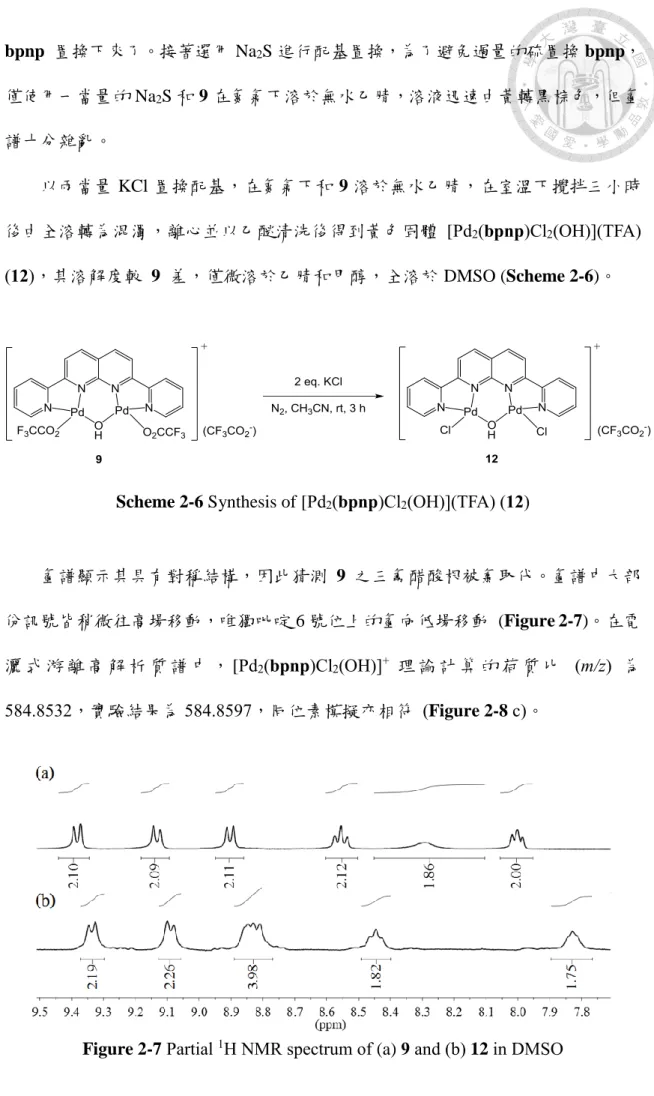
以兩當量 KCl 置換配基,在氮氣下和 9 溶於無水乙腈,在室溫下攪拌三小時 後由全溶轉為混濁,離心並以乙醚清洗後得到黃色固體 [Pd2(bpnp)Cl2(OH)](TFA) (12),其溶解度較 9 差,僅微溶於乙腈和甲醇,全溶於 DMSO (Scheme 2-6)。
Scheme 2-6 Synthesis of [Pd2(bpnp)Cl2(OH)](TFA) (12)
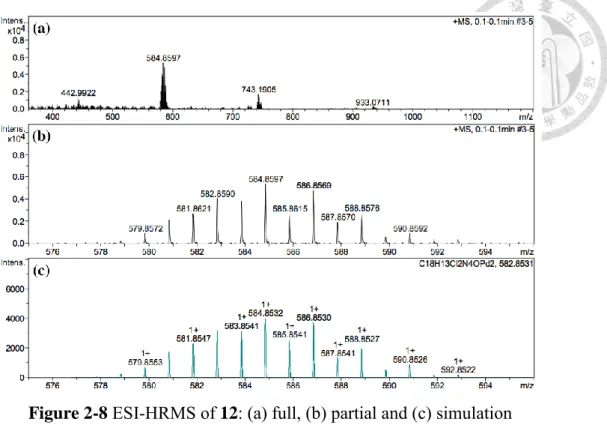
氫譜顯示其具有對稱結構,因此猜測 9 之三氟醋酸根被氯取代。氫譜中大部 份訊號皆稍微往高場移動,唯獨吡啶 6 號位上的氫向低場移動 (Figure 2-7)。在電 灑 式 游 離 高 解 析 質 譜 中 , [Pd2(bpnp)Cl2(OH)]+ 理 論 計 算 的 荷 質 比 (m/z) 為 584.8532,實驗結果為 584.8597,同位素模擬亦相符 (Figure 2-8 c)。
Figure 2-7 Partial 1H NMR spectrum of (a) 9 and (b) 12 in DMSO
Figure 2-8 ESI-HRMS of 12: (a) full, (b) partial and (c) simulation
將 12 溶於 DMSO 中,於 0.40 mM 濃度下測得莫耳導電度為 54 (ohm-1 cm2 mol-1),同濃度下 MeNO2 中莫耳導電度為 100 (ohm-1 cm2 mol-1),符合文獻中陰陽 離子比例為 1:1 的電解質。推測結構為主配位基 bpnp、一個氫氧根和兩個氯與雙 鈀金屬形成陽離子,其外圍有一個三氟醋酸根為陰離子。
欲以氯同時置換氫氧根和三氟醋酸根,取四當量 KCl 在氮氣下和 9 溶於無水 乙腈,在室溫下攪拌三小時後溶液同樣由全溶轉為混濁,離心以 DMSO 測譜,但 其氫譜變寬,顯示氯離子會和配基進行可逆交換。
以兩當量 KBr 置換配基,在氮氣下和 9 溶於無水乙腈,在室溫下攪三小時後 溶液由全溶轉混濁,離心並以少量乙醚清洗得黃色固體 [Pd2(bpnp)Br2(OH)](TFA) (13),其溶解度較 9 差,微溶於乙腈和甲醇,全溶於 DMSO (Scheme 2-7)。
Scheme 2-7 Synthesis of [Pd2(bpnp)Br2(OH)](TFA) (13)
氫譜顯示其具有對稱結構,因此猜測其結構應為 9 上的三氟醋酸根被溴取代 。 氫譜中吡啶 6 號位上的氫向低場移動,離溴較遠的萘啶上的氫位移幾乎不變,其 餘 氫 則 稍 微 向 高 場 移 動 (Figure 2-9) 。 在 電 灑 式 游 離 高 解 析 質 譜 中 , [Pd2(bpnp)Br2(OH)]+ 理論計算的荷質比 (m/z) 為 674.7513,實驗結果為 674.7503,
同位素模擬亦相符 (Figure 2-10 c)。質譜中強度次高峰之 m/z 為 630.8002,同位素 模擬和 [Pd2(bpnp)Br(H2O)(OH)2]+ 相符(附錄一),可能為 [Pd2(bpnp)Br2(OH)]+ 在 游離過程中被水氣進行配基置換產生。
將 13 溶於 DMSO 中,於 0.40 mM 濃度下測得莫耳導電度為 56 (ohm-1 cm2 mol-1),同樣濃度在 MeNO2 中莫耳導電度為86 (ohm-1 cm2 mol-1),此數據符合文
獻中陰陽離子比例為1:1 的電解質,與推測結構相符,主配位基 bpnp、一個氫氧
根和兩個溴與雙鈀金屬形成陽離子,其外圍有一個三氟醋酸根為陰離子。
Figure 2-9 Partial 1H NMR of (a) 9 and (b) 13 in DMSO
Figure 2-10 ESI-HRMS of 13: (a) full, (b) partial and (c) simulation
第三章 雙鈀金屬錯合物之催化應用
許多鈀催化反應已被報導,如耦合反應、氧化反應、還原反應、異構化反應等
51-53。因此,在此節我們探討雙鈀金屬錯合物 Pd2(bpnp)(OH)(TFA)3 (9) 對於硝基
苯還原之催化活性。
3.1 反應條件最佳化
選擇 4-硝基甲苯 (4-nitrotoluene) 作為起始物進行硝基苯還原反應最佳化測 試 (Scheme 3-1),結果如 Table 3-1。
Scheme 3-1 Reduction of p-nitrotoluene
由於文獻中鈀催化硝基苯還原中的活性物種皆為 Pd(0) 29,30,43-45,54,55,若使用 Pd(II) 則需搭配還原劑如聚甲基氫矽氧烷 (polymethylhydrosiloxane, PMHS) 43,44、 三乙基矽烷 (triethylsilane) 43 或一氧化碳 45等,將Pd(II) 還原成 Pd(0) 後才能開 啟催化循環。在本實驗室先前的研究工作中探討 [Pd2(pbbpa)Cl2](PF6)2 (11) 對硝 基苯還原的催化效果,測試了多種氫陰離子 (hydride) 來源(NaBH3CN、Et4NBH4、 NaBH4、NaH、HCO2Na 和 HCO2NH4),發現NaBH3CN 活化 11 的效果最好,因 此一開始選用 NaBH3CN 作為活化劑。
加入 2.5 mol% 之 NaBH3CN 還原鈀金屬,但得到的產率並不佳 (entry 1);反 應加熱至迴流溫度,則產率稍微上升 (entry 2);增加 NaBH3CN 量至 10 mol% , 則產率大幅上升 (entry 3);但使用 50 mol% 之 NaBH3CN 得到的產率不佳 (entry 4),推測是由於金屬錯合物分解成為鈀黑 (palladium black)。為了減少鈀黑的生成,
參考文獻加入了 DMSO 或對苯醌 (p-benzoquinone)56,但效果並不顯著(entries 5 和 6)。然而另外嘗試不加入額外的活化劑,發現產率亦相當高,表示預催化劑 (precatalyst) Pd2(bpnp)(OH)(TFA)3 (9) 能夠直接被氫氣活化 (entry 7),於是接下來 即不加入額外的活化劑進行反應最佳化。
Table 3-1 Preliminary optimization for the reduction of 4-nitrotoluene a
Entry Additives Conv.
(%)b
Yield (%)b 16b 17b 20b 1 NaBH3CN 2.5 mol% < 5 < 5 0 < 5
2c NaBH3CN 2.5 mol% 35 < 5 0 32
3 NaBH3CN 10 mol% 76 < 5 0 69
4 NaBH3CN 50 mol% 6 < 5 0 < 5 5 NaBH3CN 50 mol%, DMSO 10 mol% 6 < 5 0 < 5 6 NaBH3CN 50 mol%, benzoquinone 10 mol% < 5 0 0 0
7 None 100 0 0 100
a Standard conditions: a mixture of 4-nitrotoluene (0.50 mmol), additives and 9 (0.0025 mmol) was stirred in MeOH (0.5 mL) at 50°C under H2 for 6 h. b Conversion and yields were determined by
1H NMR spectroscopy using CH2Br2 as the internal standard. c under refluxing temperature.
進一步的實驗參數最佳化結果整理在 Table 3-2。低溫時反應的轉化率並不理 想,但隨溫度上升而有所提升 (entries 2–4),在 50°C 時能夠達到 100% (entry 1),
但在 60°C 時轉化率和產率反而下降,可能是 9 在高溫下分解 (entry 5)。另外室 溫反應的氫譜中可以觀察到 17% 的 N-hydroxy-p-toluidine (16b) 的生成,可能為 反應中間體 (entry 3)。至於不同溶劑的影響,在極性溶劑如甲醇、乙腈和丙酮等中 都能夠得到很好的產率(entries 1, 7 和 8),但在水中由於反應物溶解度差轉化率 並不佳 (entry 6)。另一方面,催化反應在低極性溶劑 (less polar solvent) 如二氯甲 烷、四氫呋喃和甲苯中則不易進行 (entries 9–11)。因此,以甲醇作為此鈀催化劑對
硝基苯還原的最佳溶劑。在不同的預催化劑 9 使用量,發現需要 0.5 mol% 的催 化劑才能得到最佳效益 (entries 12–14)。值得注意的是,在 0.1 mol%催化劑量的實 驗中,觀察到微量的 4,4’-azoxytoluene (17b) 伴隨生成,猜測 17b 可能為催化中間 體,將於 3.4 節作進一步的探討。
Table 3-2 Reaction optimization for the reduction of 4-nitrotoluene a Entry Temp. Solvent Cat. Loading Conv.
(%)b
Yield (%)b
16b 17b 20b
1 50°C MeOH 0.5 mol% 100 0 0 100
2 5°C MeOH 0.5 mol% < 5 0 0 0
3 25°C MeOH 0.5 mol% 25 17 0 8
4 40°C MeOH 0.5 mol% 76 0 0 68
5 64°C MeOH 0.5 mol% 87 0 0 87
6c 50°C H2O 0.5 mol% 45 < 5 < 5 32 7 50°C MeCN 0.5 mol% 100 < 5 < 5 87 8 50°C Acetone 0.5 mol% 75 < 5 7 41 9d 40°C CH2Cl2 0.5 mol% 32 < 5 < 5 23 10 50°C THF 0.5 mol% 41 10 < 5 0 11 50°C Toluene 0.5 mol% 17 < 5 < 5 8 12 50°C MeOH 0.1 mol% 59 0 < 5 37
13 50°C MeOH 0.2 mol% 95 0 0 89
14 50°C MeOH 1.0 mol% 100 0 0 100
a Standard conditions: a mixture of 4-nitrotoluene (0.50 mmol) and 9 (0.0025 mmol) in MeOH (0.5 mL) was stirred at 50°C under H2 (1 atm) for 6 h. b Conversion and yields were determined by 1H NMR integration using CH2Br2 as the internal standard. c Using CH3NO2 as the internal standard for determination. d under refluxing temperature.
為了展現催化劑 9 的獨特之處,比較了一系列鈀錯合物在上述最佳化條件下 對 4-nitrotoluene 還原反應的催化活性,結果如 Table 3-3。與 9 相似的雙金屬鈀
錯合物 11 在相同條件下產率不佳,但加入 NaBH3CN 活化金屬中心後產率大幅提 升(entries 2 和 3)。若直接以 NaBH3CN 而非氫氣作為還原劑 ,則 11 的催化反 應僅能得到微量的 20b,顯示 NaBH3CN 在反應中的角色是作為催化劑活化劑,而 非還原劑 (entry 4)。錯合物 13 的催化效果不佳,僅能得到 16% 的 20b (entry 5)。
[Pd(tpy)Cl](PF6) 57 和含膦配基鈀錯合物 PdCl2(PPh3)2 在此條件下皆無法催化此還 原反應(entries 6 和 11),而其他含氮配基鈀單金屬催化劑 Pd(bpy)(TFA)2 58、 Pd(bpy)Cl259、Pd(bpnp)(TFA)2 (10) 和 [Pd(dien)Cl]Cl 60 (Figure 3-1)亦具有良好的 催化效果 (entries 7–10)。Pd(MeCN)2Cl2 和 Pd(OAc)2 雖然也展現出不錯的催化活 性,但其產率和選擇性均不如 9 (entries 12–13)。
Table 3-3 Comparison of various Pd complexes on reduction of 4-nitrotoluene a Entry Pd catalyst (mol%) Conv.
(%)b
Yield (%)b
16b 17b 20b
1 9 (0.5) 100 0 0 100
2 11 (0.5) 20 0 0 19
3c 11 (0.5) 100 0 0 100
4d 11 (0.5) < 5 0 0 < 5
5 13 (0.5) 27 < 5 6 16
6 [Pd(tpy)Cl](PF6) (1.0) 0 0 0 0
7 Pd(bpy)(TFA)2 (1.0) 100 0 0 100
8 Pd(bpy)Cl2 (1.0) 92 0 0 92
9 10 (1.0) 100 0 0 100
10 [Pd(dien)Cl]Cl (1.0) 100 < 5 0 100 11 PdCl2(PPh3)2 (1.0) 0 0 0 0 12 Pd(MeCN)2Cl2 (1.0) 57 0 < 5 50 13 Pd(OAc)2 (1.0) 89 < 5 < 5 65
a Reaction conditions: a mixture of 4-nitrotoluene (0.50 mmol) and Pd catalyst was stirred in MeOH (0.5 mL) under H2 for 6 h. b Conversion and yields were determined by 1H NMR spectroscopy using CH2Br2 as the internal standard. c NaBH3CN (2.5 mol%) was added. d 2.0 eq.
of NaBH3CN was used under N2.
Figure 3-1 Mono-palladium(II) complexes with N-donating ligands
3.2 反應應用範圍
得到反應最佳化條件後,進一步測試該催化系統的應用範圍。Table 3-4 中列 出各種取代硝基苯類的反應結果,該反應容許許多官能基如烷基 (alkyl groups, 20b)、羥基 (hydroxy groups, 20c)、胺基 (amino groups, 20d)、芳基氟化物 (aryl fluoride, 20e)、氰基 (cyano group, 20h)、酮基 (keto group, 20i) 和羧基 (carboxyl group, 20j),皆能夠在催化系統中保留,此外具有立體障礙的反應物如 1,3-二甲基 -2-硝基苯 (1,3-dimethyl-2-nitrobenzene, 14k) 和 1-硝基萘 (1-nitronaphthalene, 14l) 也能夠成功轉換成相對應的苯胺類。具有多個取代基的硝基苯,如 2,3-二甲基-6- 硝 基 苯 胺 (2,3-dimethyl-6-nitroaniline, 14m) , 和 多 硝 基 苯 如 間 二 硝 基 苯 (o- dinitrobenzene, 14n) 均有很好的產率。然而,含氯或溴之苯化物如 1-氯-4-硝基苯 (1-chloro-4-nitrobenzene, 14f) 和 1-溴-4-硝基苯 (1-bromo-4-nitrobenzene) 反應性較 差,或甚至不反應;含有芳香雜環噻吩 (thiophene) 和吡啶 (pyridine) 的 2-硝基噻 吩 (2-nitrothiophene) 和 3-羥基-2-硝基吡啶 (3-hydroxy-2-nitropyridine) 毫無反應;
脂 肪 類 (aliphatic) 反 應 物 如 2,3- 二 甲 基 -2,3- 二 硝 基 丁 烷 (2,3-dimethyl-2,3- dinitrobutane) 亦無法還原得到對應的產物。
Table 3-4 Scope of catalysis with various nitroarenes a
Product (Yield, %)b
a Reaction conditions: a mixture of nitroarene (0.5 mmol) and 9 (0.0025 mmol) was stirred in MeOH (0.5 mL) at 50°C under H2 for 12 h. b Isolated yield. c 1 mol% of 9 was used. d 28% of 20f and 30% of aniline was observed. e p- nitroaniline was used.
3.3 芳基鹵化物之還原反應探討
儘管多數反應物能夠順利進行催化還原得到高產率預期產物,但 1-chloro-4- nitrobenzene (14f) 和 1-bromo-4-nitrobenzene (14g) 卻不然。為了探究溴基對催化 反應的影響,在一已知可行的硝基苯還原反應中加入了溴化鉀和 1,3,5-三溴苯,可 以發現溴離子對反應沒有影響,而芳溴化物則會減低催化活性,但拉長反應時間仍 然能夠反應完全 (Table 3-5)。
Table 3-5 Influence of bromide in reduction of nitroarenes a
Entry Additive Time (h) Yield of 20a (%) b
1 None 6 100
2 KBr 6 100
3 1,3,5-tribromobenzene 6 39
4 1,3,5-tribromobenzene 16 100
a Reaction conditions: a mixture of bromide source (10 mol%) and 9 (0.5 mol%) was stirred in 0.5 mL MeOH at 50°C under H2 for 2 h, and then nitrobenzene (0.50 mmol) was added and stirred for specific time. b Conversion and yields were determined by 1H NMR spectroscopy using CH2Br2 as the internal standard.
另外,添加鹼或銀鹽以進一步觀察反應,結果如 Table 3-6。選用無機鹼 Cs2CO3
時能夠觀察到鈀黑出現,且反應毫無變化,可能是因為銫能夠和配位基配位61-63,
大量的銫將鈀置換掉成為鈀黑而失去反應活性;無機鹼 K2CO3 能夠還原去鹵化
(reductive dehalogenation) 後再進一步還原部分硝基苯,由產物分佈可知還原去鹵
化發生在硝基還原之前 64。若在反應中加入非親核鹼如 2,2,6,6-四甲基哌啶
(2,2,6,6-tetramethyl-piperidine, TMP)、三乙基胺 (triethylamine, NEt3) 和 1,4-二氮雜 二環[2.2.2]辛烷 (1,4-diazabicyclo[2.2.2]octane, DABCO),能夠大幅提升轉化率 (entries 4–6),然而由於還原去鹵化較容易發生,主產物為還原去溴—硝基還原的 產物苯胺 20a。雖然 NEt3 和 DABCO 都具有很好的效果,但考量毒性以及操作
的方便性,在後續的實驗中選用DABCO 進行測試。接著進一步降低 DABCO 的
用量,發現使用 0.25 當量亦可達到同樣的效果 (entries 6–9)。在原反應系統中芳 基鹵化物能夠氧化加成到金屬上,金屬催化劑被佔據而無法催化硝基苯還原,當其 和鈀上的氫陰離子還原脫去時生成一分子芳香族和一分子溴化氫,由於鹼能夠中
和反應中產生的溴化氫,加入鹼有利於還原脫去,反應因而能夠順利完成65,66。加
入銀鹽的目的為使溴沉澱,效果不佳可能是因為銀離子為強氧化劑,會氧化催化劑 Pd(0) 使反應無法進行(entries 10 和 11)。我們將改良後的反應條件運用到 1-
chloro-4-nitrobenzene (14f),亦可得到預期的產物苯胺 (Scheme 3-2)。
Table 3-6 Reduction of 1-bromo-4-nitrobenzene (14g) a
Entry Additive Conv.
(%)b
Yield (%)b
Nitrobenzene Aniline
1 None 0 0 0
2 Cs2CO3 1 eq. 0 0 0
3 K2CO3 1 eq. 100 62 19
4 TMP 2 eq. 100 33 57
5 NEt3 2 eq. 100 0 90
6 DABCO 2 eq. 100 0 83
7 DABCO 1 eq. 100 0 87
8 DABCO 0.5 eq. 100 0 85
9 DABCO 0.25 eq. 100 0 83
10 AgNO31 eq. 0 0 0
11 AgBF4 1 eq. 0 0 0
a Reaction conditions: a mixture of 1-bromo-4-nitrotoluene (0.50 mmol), additive and 9 (0.0025 mmol) was stirred in 0.5 mL MeOH at 50°C under H2 for 12 h. b Conversion and yields were determined by 1H NMR spectroscopy using CH2Br2 as the internal standard.
Scheme 3-2 Reduction of 1-chloro-4-nitrobenzene
將此條件應用到芳鹵化物,如溴苯、氯苯上,我們發現芳鹵化物會進行還原去 鹵化並得到苯 (Table 3-7, entries 1–2);苯環上有其他取代基的芳鹵化物,如 4- bromo-N,N-dimethylaniline 亦 可 順 利 去 鹵 化 (entry 3) ; 選 用 2,4′- dibromoacetophenone 進行測試,發現苯環上和 位的溴均會被氫取代,且乙醯基 更進一步還原為醇類 (entry 4);多溴取代的芳香化合物如 1,3,5-三溴苯 (1,3,5- tribromobenzene) 能夠完全被還原成苯。此外,比較在氮氣下和氫氣下的反應結果 可以確認氫氣在此作為還原劑(entries 1 和 6)。
Table 3-7 Reductive dehalogenation of aryl halides a
Entry Aryl halide Atmosphere Product (yield)b
1 Bromobenzene H2 Benzene (95)
2 Chlorobenzene H2 Benzene (97)
3 4-Bromo-N,N-dimethylaniline H2 N,N-dimethylaniline (96) 4 2,4′-Dibromoacetophenone H2 Acetophenone (16)
1-Phenylethanol (79) 5c 1,3,5-Tribromobenzene H2 Benzene (97)
6 Bromobenzene N2 Benzene (0)
a Reaction conditions: a mixture of aryl halide (0.50 mmol), DABCO (1.0 mmol) and 9 (0.0025 mmol) was stirred in 0.5 mL MeOH at 50°C under certain atmosphere for 12 h. b Yields were determined by 1H NMR spectroscopy using dimethyl sulfone as the internal standard. c 1.5 mmol of DABCO was used.
3.4 反應機構探討和與其他鈀催化劑之比較
文 獻 報 導 的 硝 基 苯 還 原 成 苯 胺 路 徑 有 二 42: 硝 基 苯 還 原 成 亞 硝 基 苯 (nitrosobenzene) 後,進一步加氫還原成 N-苯基羥胺 (N-phenylhydroxylamine),接 著 N-苯基羥胺可直接加氫脫水得到苯胺 (Scheme 3-3 a),或亦可和亞硝基縮合 (condensation) 形成氧化偶氮苯 (azoxybenzene)再加氫脫水得偶氮苯 (azobenzene),
氫化得到二苯胼 (hydrazobenzene),最後加氫分解得苯胺 (Scheme 3-3 b)。
Scheme 3-3 Proposed mechanism of the reduction of nitroarenes
為了決定主要的反應路徑,以較溫和的反應條件用 9 為催化劑,分別以 N-苯 基羥胺 16a 和偶氮苯 18a 為起始物測試其反應性42,67,結果整理於 Table 3-8。在 40°C 下,14a 的轉化率有 80%,且觀察到中間體 18a 與 19a (entry 1)。遂以相同條 件探討 16a 和 18a 的性質。僅以 16a 為起始物時,雖然反應的轉化率高達 97%,
卻只得到 38% 的目標產物 20a,遠低於在相同條件下以 14a 為起始物的 73%,
另外還產生較多量的 18a 與 19a。這顯示在 9 催化下 16a 不容易直接還原成 20a,
而需先氧化成 15a 後,縮合得到 17a,接著才會還原成 18a、19a 和 20a。而以 18a
為起始物時,則能夠得到 81% 的目標產物,因此合理推測反應較可能遵循路徑 (b)。為確認此推論,更進一步做了反應的產物與時間的動力探討。
Table 3-8 Reduction of 14a, 16a and 18a catalyzed by 9
Entry Starting material Conv. (%)b 17a (%)b 18a (%)b 19a (%)b 20a (%)b
1a 14a (0.50 mmol) 80 0 < 5 < 5 73
2a 16a (0.50 mmol) 97 < 5 30 25 38
3a 18a (0.25 mmol) 99 0 -- 18 81
a Reaction conditions: a mixture of starting material and 9 (0.0025 mmol) was stirred in 0.5 mL MeOH at 40°C under H2 for 6 h. b Yields were determined by 1H NMR spectroscopy using CH2Br2 as the internal standard.
追蹤以 9 催化硝基苯還原的反應,其反應動力學曲線圖如 Figure 3-2。在反應 前 120 分鐘中苯胺 20b 的產率和起始物 14b 的轉化率僅不到 10%,亞硝基苯 15b 開始緩慢地生成,這段誘導期 (induction period) 主要是 Pd(II)被還原成 Pd(0) 的步驟。之後,15b 開始和已被還原的 N-苯基羥胺 16a 縮合產生氧化偶氮苯 17b。
在開始反應 180 分鐘後 17b 的量遂達到穩定態 (steady state),即濃度保持固定,
此結果表示其是此催化反應中的一個中間體,和前述的反應路徑推論符合。這區間 伴隨產物 20b 逐漸生成,在 300 分鐘時反應幾乎已完成。
a Reaction conditions: a mixture of 14b (0.5 mmol) and 9 (0.0025 mmol, 0.5 mol%) was stirred in MeOH (0.5 mL) at 50°C under H2. b Conversion and yields were determined by 1H NMR spectroscopy using dimethyl sulfone as the internal standard.
Figure 3-2 Kinetic time course of 9-catalyzed reduction of 14b a, b
為了進一步確認反應路徑,將 15a 和 16a 混合在反應中生成 17a,觀察各成分 的變化 (Figure 3-3)。由圖中可發現誘導期仍然存在,隨後 17a 始開始反應生成 18a 和 19a,且 19a 持續累積。20a 在氧化偶氮苯 17a 幾乎消耗後才開始生成,
表示氫化偶氮苯 18a 的速率要比氫解二苯胼 19a 快很多,這可說明由 17a 還原至 20a 的速率決定步驟應為 N–N 鍵的氫解。
a Reaction conditions: a mixture of 15a (0.25 mmol), 16a (0.25 mmol) and 9 (0.0025 mmol) was stirred in MeOH (0.5 mL) at 50°C under H2. b Conversion and yields were determined by 1H NMR spectroscopy using dimethyl sulfone as the internal standard.
Figure 3-3 Kinetic time course of 9-catalyzed reduction of 17a a, b
以同樣的方式來決定 11 催化硝基苯還原的反應路徑,結果如表 Table 3-9。在 相同條件下 (40°C),以 16a 為起始物時,得到 53% 的苯胺 20a,而 14a 與 18a 為起始物,則分別得到 36% 與 27% 的苯胺 20a,這與催化劑 9 的性質截然不同。
由於反應過程中僅有極少量 18a 與 19a 伴隨生成,同時對於 18a 的活性相對較低,
所以推論以 11 為催化劑的催化系統是遵循路徑 (a)。