國立臺灣大學生命科學院基因體與系統生物學學程 碩士論文
Genome and Systems Biology Degree Program College of Life Science
National Taiwan University Master Thesis
核黃素激酶調節有絲分裂的意外作用
An Unexpected Role for Riboflavin Kinase in Regulating Mitosis
林依潔 I-CHIEH LIN
指導教授:高承福 博士 Advisor: CHENG-FU KAO, Ph.D.
中華民國 107 年 8 月
August 2018
1
謝辭
真心覺得做研究並不是件容易的事情,正因如此,我更加感謝這段時間幫助 我的人們。
首先,最感謝的是我的父母親,你們給我很大的支持,不論是生活上或精神 上的,真的是非常偉大。再來是高承福老師,感謝您願意收留我,並且很認真的 教導我、指導我跟訓練我,手把手提攜我到這裡,您實在功不可沒,謝謝您總是 不厭其煩的跟我討論實驗結果,也很感謝您建立了這麼溫馨的實驗室,R243 的大 家感情融洽,我覺得實屬可貴,謝謝大家讓我的碩班生活充滿美好回憶,大家開 心吃吃喝喝或是嘻笑,還能一邊做出高品質的研究,可以跟你們一起度過這些日 子我覺得很幸運,幸好有你們,我才能走到今天。奶奶雖然很掉漆但真的給了我 太多研究上和論文上的幫助了(還有營養品,哈);心怡超高效率辦事真的覺得超 棒的,省了超多力;一哥教我做實驗;小宥幫我改論文;馨文負責逗我(?) ;阿 祖雙魚座超懂;小史姊姊很溫暖;祥恩哥哥很罩;太太自身處困境下還跟我討論 實驗細節;謝謝阿姨辛苦幫我們洗瓶子和倒垃圾,覺得阿姨的存在實在太重要,
還有波波和美英,謝謝你們。
也要謝謝鄭淑珍老師和 lab329 的大家,實驗之路就是從此啟蒙的,一起共患 難或開心打鬧的日子也很珍貴。
然後,謝謝我的碩班同學們:佩萱、達錡、劉當、庭蔚,跟你們一起學習、一 起上課真的很棒,因為學程專題和必修的特殊性,我們書報討論的機會超高,再 加上你們都是會動腦的優秀人才,一起討論的過程真的學到很多很多,覺得研究 生的學習我們堪稱典範了,謝謝你們,有你們這些同學令我覺得驕傲。還有學程 的老師們,每一個老師都很厲害、都有值得學習的地方,台大真的不是蓋的,很 強的。
謝謝家鴻學姊,吐苦水不在話下啊,以後請繼續照顧我喔,哈哈。很謝謝孟 澤,各種神 carry,沒你在,我實在不知道該怎麼度過這段日子(也太誇張),只能 以後慢慢還了。
還有謝謝各路朋友,偶爾拉研究生出去放風吃草,真的很重要!最後謝謝我自 己,這麼努力的堅持下來了,這段日子真的很充實,學到很多,辛苦你了!
3
中文摘要
為了確保細胞內基因的穩定性,在有絲分裂期間調節染色體分離是非常重要的一 個過程。有絲分裂主要是由細胞週期蛋白所調控,但合適的營養條件對於細胞週 期的影響也是非常重要。有趣的是,我們發現產生能量的輔因子:黃素單核苷酸
(FMN)在調節有絲分裂中具有重要的角色,黃素單核苷酸 是由核黄素激酶 (Fmn1)
催化核黄素 (維生素 B2) 所形成。在酵母菌中,細胞週期結束時必須要透過 mitotic exit network(MEN) 的訊息調節,而 Lte1 是 MEN 的正調控因子。和組 蛋白修飾有關的基因,SET1,也被報導和 mitotic exit 有關。有趣的是,我們發 現FMN1 和 SET1 還有 LTE1 皆有基因相互作用。我證實了有絲分裂的延遲造成此 基因相互作用,而且,FMN1 的功能與 MEN 是平行的,因為 FMN1 會表現出與 CLA4 和CDC15 有基因的相互作用,而此兩者都是參與 MEN 的調控。另外,lte1Δ fmn1 Δ 的生長缺陷可以通過缺失 BUB2 來解救。而 BUB2 是 MEN 的負調控因子,也就是 說,我發現,很重要地,FMN1 會與細胞週期的調控有關,而且是跟 mitotic exit network 有平行關係。更進一步地,我們發現FMN1 參與在 anaphase promoting complex (APC)中,而且會有metaphase-anaphase 時期轉換的缺陷。Ras2 會抑 制 APC 的功能,而我們發現 fmn1 突變體的生長缺陷可通過缺失 RAS2 而得以挽救。
我們也發現了fmn1 突變體中表現 APC 缺陷的症狀: fmn1 突變體導致染色體分離錯 誤和有絲分裂週期蛋白 Clb2 的異常積累。綜上所述,FMN1 缺陷會導致metaphase
和 anaphase 轉換上出現錯誤。因此,FMN1 可能在調節有絲分裂中扮演重要的角色。
關鍵字:黃素單核苷酸,有絲分裂,染色體分離,MEN,Lte1,APC
英文摘要
The regulation of chromosome separation during the mitotic phase is essential to ensure genome stability. Mitosis is controlled by complex regulation network, including proper nutritional and metabolic status to ensure smooth cell cycle transition. Interestingly, our genetic screening identified that an energy producing cofactor, flavin mononucleotide (FMN) has an important function in regulating mitosis in budding yeast. The Mitotic Exit Network (MEN) is a signaling pathway known to drive cells out of mitosis and to promote the faithful division of cells. We found that FMN1, encoding a Riboflavin kinase, confers a genetic interaction with LTE1, coding for a positive regulator of MEN.
In addition, FMN1 displayed negative interactions with CLA4 and CDC15, both of which are involved in the regulation of MEN. Furthermore, the growth defect of lte1
fmn1 could be rescued by the deletion of BUB2, which is a negative regulator to the
MEN activity. The growth defect of fmn1 could be rescued by the deletion of RAS2,
which inhibits the function of anaphase promoting complex (APC), indicating that the Fmn1 may play a role in regulating APC. Furthermore, the fmn1 mutants exhibited
anaphase entry delay, chromosome segregation error and abnormally accumulation of mitotic cyclin, Clb2. Taken together, ablation of FMN1 leads to defects in
metaphase-anaphase transition. Thus, FMN1 may play a role in regulating mitosis.
5
目錄
謝辭 ...2
中文摘要 ...3
英文摘要 ...4
目錄 ...5
圖目錄 ...6
表目錄 ...7
INTRODUTION ...8
1 The function of riboflavin ... 8
1.1 The function of Flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) ... 9
1.2 The FMN1, Fmn1 and FMN ... 13
2 Cell cycle regulation ... 15
2.1 The cell cycle ... 15
2.2 The mitotic phase and mitotic checkpoints ... 17
3 The mitotic exit ... 23
3.1 The mitotic exit ... 23
3.2 The mitotic exit network ... 24
3.3 The Cdc14 early anaphase release network ... 28
4 Histone protein modification and their function... 32
4.1 H2B ubiquitylation and H3K4 methylation... 32
4.2 The Set1 complex and the regulation of H3K4 methylation ... 34
4.3 The relation between Set1/H3K4me and mitosis ... 37
5 The relationship between SET1, FMN1 and mitosis ... 40
RESULTS ...41
1 FMN1 genetically interacts with SET1 ... 41
2 The synthetic interaction of FMN1 and SET1 leads to defective cell cycle progression ... 45
3 Loss of Fmn1 activity leads to mitosis delay ... 46
4 FMN1 is involved in mitotic exit regulation ... 48
5 Fmn1 activity is required for metaphase-anaphase transition ... 50
DISSCUSION...57
Riboflavin kinase mutants performed mitosis deregulating ... 57
Loss the riboflavin kinase activity leads to chromosome segregation error... 59
METHODS AND MATERIALS ...63
REFERENCE ...93
圖目錄
Figure 1. Mutation of FMN1 displays genetic interaction with SET1 and its upstream regulator H2Bub ... 66 Figure 2. fmn1-105 mutants exhibited slight resistance to Benomyl ... 67 Figure 3. FMN1 displays genetic interaction with LTE1 in three different
backgrounds ... 68 Figure 4. The viability of fmn1-105 set1Δ cells were dependent for Lte1 70 Figure 5. fmn1-105 lte1Δ cells delay in S phase ... 71 Figure 6. The growth rate of fmn1-105 lte1Δ cells is lower than wild-type
and single mutants cells ... 72 Figure 7. fmn1-105 lte1Δ cells are delayed in mitosis ... 73 Figure 8. FMN1 displays genetic interaction with mitotic exit network
regulators ... 75 Figure 9. The growth defect of fmn1-105 lte1Δ is due to MEN restrain .. 77 Figure 10. The genetic interaction between FMN1, FEAR components
and APC regulator ... 79 Figure 11. fmn1-105 cells are later for metaphase-anaphase transition .. 81 Figure 12. fmn1-105 cells exhibit chromosome segregation error ... 83 Figure 13. The spindle elongation defect affects the viability of fmn1-105
cells ... 85
7
表目錄
Table 1. Summary of the result of synthetic genetic array analysis of fmn1-105…86 Table 2. Saccharomyces cerevisiae strains used in this study………...87
INTRODUTION
1 The function of riboflavin
Riboflavin, also called Vitamin B2. The richest natural source is yeast.
Riboflavin is involved in many processes in the body and is necessary for normal cell growth and function. Riboflavin deficiency in the body may cause various types of cancer, and for migraine headaches. Taking riboflavin helps for acne, muscle cramps, burning feet syndrome, carpal tunnel syndrome, and blood disorders such as congenital methemoglobinemia and red blood cell aplasia.
Riboflavin is one of eight B vitamins. B vitamins are a class of water-soluble vitamins that play important roles in cell metabolism. Though these vitamins share similar names, they are chemically distinct that often coexist in the same foods. All B vitamins help the body to convert carbohydrates into glucose, which is used to produce energy. These B vitamins also help the body metabolize fats and protein.
Riboflavin functions as a coenzyme, meaning that it is required for enzymes (usually proteins) to perform normal physiological actions. Riboflavin functions for the metabolism of carbohydrates, proteins, and lipids. It plays a crucial role in the
production of hematopoietic stem cell, Nervi nervorum and protein synthesis. It acts as a coenzyme in the intermediate metabolism and participates in the reaction of folate
9
The using forms of riboflavin are riboflavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) in eukaryotes. FMN and FAD bind to proteins to form the flavoproteins. This utilization of FMN and FAD are similar to the distribution across all kingdoms of life (Koch & Macheroux, 2016).
1.1 The function of Flavin mononucleotide (FMN) and flavin adenine dinucleotide
(FAD)
FMN and FAD are important for many metabolic reactions. Riboflavin serves as a precursor, riboflavin kinase (Fmn1) phosphorylate riboflavin to form FMN. FAD synthetase (Fad1) catalyzes adenylylation of the FMN to form FAD. These riboflavin coenzymes play important roles in many metabolic reactions including amino acid carbohydrate and lipid, protein metabolism and in the conversion of folic acid and pyridoxine into their coenzyme forms.
FMN and FAD are the redox cofactor for the prosthetic group of many metabolic proteins. These proteins are called flavoproteins. Flavoproteins contain a
flavin moiety and it is essential for the protein function. Flavoproteins utilize the unique and versatile structure of flavin moieties to catalyze difficult redox reactions. Since flavins have, multiple redox states they can participate in processes that involve the transfer of one or two electrons, hydrogen atoms, or hydronium ions.
One well-known reaction is part of the citric acid cycle (also known as the TCA or Krebs cycle). In the electron transport chain, FMN is one of the components of complex
I while FAD is involved in the activity of complex II (Succinate dehydrogenase). FAD acts as an electron carrier and takes part in both the Kreb’s Cycle and oxidative
phosphorylation. It accepts electrons and is transformed into FADH2 that stored
momentarily of the high-energy electrons from this oxidation. FADH2 then transfers its electrons to complex II of the electron transport chain. For each pair of electrons from FADH2 passed along the electron transport chain, 1.5 units of ATP molecules are formed.
http://web.stanford.edu/group/hopes/cgi-bin/hopes_test/riboflavin/
The genome of Saccharomyces cerevisiae contains 68 genes (~1% of all yeast
11
proteins) which encode for flavin-dependent proteins. Thirty-five flavoproteins require FAD (74%) and fifteen require FMN (26%). A large number of flavoproteins are present in cells suggest their important roles in cells. However, many yeast flavoenzymes are poorly characterized. Biochemical properties such as substrate specificity, kinetic parameters and reaction partners need to be determined to improve our understanding of human orthologs (Koch and Macheroux, 2016).
FMN functions as prosthetic group of various oxidoreductases including NADH dehydrogenase as well as cofactor in biological blue-light photoreceptors (Kerscher, 2000; Joseph-Horne, et al., 2001; Christie, et al., 1999). FMN is the form in which riboflavin is found in cells and tissues. It requires more energy to produce, but is more soluble than riboflavin. FMN is a stronger oxidizing agent than NAD and is particularly useful because it can take part in both one- and two-electron transfers (Casaus, et al., 2002). During the catalytic cycle, a reversible interconversion of the oxidized (FMN), semiquinone (FMNH-) and reduced (FMNH2) forms occurs in the various
oxidoreductases (Jablonski & DeLuca, 1977).
FAD displays important roles in many metabolic processes. FAD is the more complex and abundant form of flavin. FAD is required to convert retinol (vitamin A) to retinoic acid via cytosolic retinal dehydrogenase and tryptophan to niacin (vitamin B3) (Dalfó, 2007; Powers, 1999). FAD is also required for the production of pyridoxic acid
from pyridoxal (vitamin B6) by pyridoxine 5'-phosphate oxidase and the fatty acid
oxidation by fatty acyl CoA dehydrogenase (Powers, 1999). Oxidation of pyruvate, α-ketoglutarate, and branched-chain amino acids requires FAD in the shared E3 portion
of their respective dehydrogenase complexes (Islam, et al., 2009). Synthesis of an active form of folate (5-methyltetrahydrofolate) from 5, 10-methylenetetrahydrofolate by Methylenetetrahydrofolate reductase is FADH2 dependent (Lubran, 1971). Reduction of the oxidized form of glutathione (GSSG) to its reduced form (GSH) by Glutathione reductase is also FAD dependent (Pai & Schulz, 1983).
FAD can exist in four different redox states, which are the flavin-N (5)-oxide, quinone, semiquinone, and hydroquinone (Iyanagi & Mason, 1973; Teufel, et al., 2013).
FAD is converted between these states by accepting or donating electrons. FAD, in its fully oxidized form, or quinone form, accepts two electrons and two protons to become FADH2 (hydroquinone form). The semiquinone (FADH·) can be formed by either reduction of FAD or oxidation of FADH2 by accepting or donating one electron and one proton, respectively. FAD has a more positive reduction potential than NAD+ and is a very strong oxidizing agent (Kumar & Chen, 2007). The cell utilizes this in many energetically difficult oxidation reactions such as dehydrogenation of a C-C bond to an alkene (Büch, 1995). FAD-dependent proteins function in a large variety of metabolic pathways including electron transport, DNA repair, and nucleotide biosynthesis,
13
beta-oxidation of fatty acids, amino acid catabolism, as well as synthesis of other cofactors such as CoA, CoQ and heme groups (Preedy, 2012).
1.2 The FMN1, Fmn1 and FMN
The FMN1 that encodes for yeast riboflavin kinase was found in 2000 (Santos, et al., 2000). The FMN1 is an essential gene and the growth defect shown by the fmn1Δ
null mutants could be rescued by supplementing the medium with externally high concentration of FMN suggests that a diffusion mechanism would be involved in the uptake of FMN by yeast cells (Santos, et al., 2000). Depleted FMN1 causes abnormal mitochondrial morphology (Altmann & Westermann, 2005). Overexpression FMN1 causes slow growth.
The riboflavin kinase (Fmn1) synthesis of FMN is essential for cell viability and that no redundant activities (Santos, et al., 2000). Riboflavin kinase from rat liver shows an activity increase with low concentrations of Zn2+ unlike other ATP-dependent
enzymes that prefer Mg2+, indicating its unusual properties (Merrill and McCormick, et al., 1980). Fmn1 predominantly localizes to the microsomal fraction and also found in
the mitochondrial inner membrane, exposing its COOH terminal domain to the matrix space (Santos, et al., 2000).
Riboflavin kinase of the Schizosaccharomyces pombe represents a novel family
of phosphoryl transferring enzymes (Bauer, et al., 2003). It is a monomer comprising a central β-barrel clasped on one side by two C-terminal helices that display an L-like shape. The opposite side of the β-barrel serves as a platform for substrate binding as
demonstrated by complexes with ADP and FMN (Bauer, et al., 2003).
(Bauer, et al., 2003)
FMN functions as prosthetic group of various oxidoreductases including NADH dehydrogenase as well as cofactor in biological blue-light photoreceptors (Kerscher, 2000; Joseph-Horne, et al., 2001; Christie, et al., 1999). FMN functions as a coenzyme, meaning that it is involved in many biological functions but not specific one. That is, deficiency of FMN makes a global effect in cells due to the defect of energy metabolism (Barile, et al., 1997). However, the characterization of FMN1, FMN has not well
reported until very recently. It is very valuable to investigate the multiple roles of FMN1, Fmn1 and FMN in cells
15
2 Cell cycle regulation
2.1 The cell cycle
It is important for an organism to produce new daughter cells to maintain its structure and inherited material by cell divides into two cells. Cell-division cycle or cell cycle is a well regulating works in cells. The cells go through the cell cycle progression and pass all the necessary checkpoints that rely on specific control mechanisms to ensure it never returns to previous event. Many factors including cyclins,
cyclin-dependent kinases (CDKs), ubiquitin ligases, inhibitors of cyclin-dependent kinases, and reversible phosphorylation to ensure that cell cycle events occur in correct order with least amount of errors (Nigg, 1995; Kaldis, et al., 2013; D'Angiolella, et al., 2003; Fujimitsu, et al., 2016).
In each turn of cell cycle, cells take place series of events lead to duplicate its DNA (DNA replication) and divide to two daughter cells. According to the cell
morphology, cell cycle can be briefly dividing into three periods: interphase, the mitosis, and cytokinesis (Cooper, 2000). Cells that have temporarily or reversibly stopped
dividing are said to have entered a state of quiescence called G0 phase (Patt & Quastler, 1963). Although the various detailed stages of cell cycle are not usually
morphologically distinguishable, each phase of the cell cycle has a distinct set of
specialized biochemical processes that prepare the cell for initiation of cell division (Wieser & Pines, 2015).
Activation of each phase is dependent on the proper progression and completion of the previous one. The interphase is for cells to grow and prepare their DNA
duplication. The interphase consists of three distinct phases: G1 phase, S (synthesis) phase, G2 phase (Israels & Israels, 2001). During G1 phase, the cells grow and accumulate nutrients needed for mitosis. During S phase, the cells duplicate its DNA.
During G2 phase, the cells prepare for the cell division and chromosome condensation (Sorenson, et al., 1990). Cell’s chromatin condenses into chromosomes during the onset of the mitotic phase. During the mitotic phase, cell separates the chromosomes in its cell nucleus into two identical sets in two nuclei. The final stage, cytokinesis, the
chromosomes and cytoplasm separate into two new daughter cells (Hartwell, 1971).
After cell division, each of the daughter cells begins the interphase of a new cycle.
In budding yeast, the progression through the cell cycle is controlled by a single cyclin-dependent kinase known as CDK1 (Cdc28) (Hartwell, 1973). This kinase is constitutively present through the cell cycle but associated with particular cyclins to differentially modulate its activity at each cell cycle stage. There are three G1 cyclins (Cln1–3) and six B-type cyclins (Clb1–6) that control S phase, G2, and mitosis
(Hadwiger, et al., 1989; Fitch, et al., 1992; Dahmann & Futcher, 1995). G1 cyclins are
17
required for bud emergence, spindle pole body duplication, and the expression of the B-type cyclins. Clb5 and Clb6 are involved in DNA replication and spindle pole body duplication and Clb1–4 promote spindle formation and entry into mitosis
(Cepeda-García, 2017).
2.2 The mitotic phase and mitotic checkpoints
To ensure genome stability, it is very important to well regulate the chromosome separation during the mitotic phase (Hartwell & Weinert, 1989). In addition, the nuclear
division (karyokinesis) during mitotic phase is a relatively short period of the cell cycle, that’s why mitotic phase is complex and highly regulated (Ou & Rattner, 2002). The
sequence of mitotic events is essential to the process of chromosome segregation which is orchestrated by microtubules (a polarized protein polymer of tubulin) as well as
hundreds of other proteins that function together (Walczak & Heald, 2008; Sobel, 1997).
The events divided into several phases: metaphase, anaphase and telophase. To distinguish each phase, it is useful to monitor the morphology of the spindles. The metaphase spindle typically reaches 1.5–2 μm in lengths (Winey, et al., 1995) which present the chromosome attachment and spindle assembly. The proper bipolar
attachments of all chromosomes trigger the signal of cell to enter anaphase, the stage at which chromosome segregation, and then the spindle elongation occurs. The nuclear
envelope of budding yeast does not break down during mitosis; that is to say, it undergoes a closed mitosis (Cavalier-Smith, 2010; Patterson, 1999).
(Marta and Fernando, 2017) The process of the mitosis is to ensure that the mitotic spindle is correctly attached to the kinetochores and correctly positioned or oriented (McIntosh, et al., 2002; Tanaka, 2002). If not, there are two mitotic checkpoints to inhibit mitotic progression in
presence of errors. They are spindle assembly checkpoint (SAC) and spindle position checkpoint (SPOC or SPoC) (Rudner & Murray, 1996; Musacchio & Salmon, 2007;
Yeh, et al., 1995; Bloecher, et al., 2000). The checkpoints and the regulation are essential to stop cell cycle progression in order to maintain genomic stability and cell viability.
The SAC monitors chromosome bi-orientation on the mitotic spindle and
19
chromosomes attachment properly. It is a feedback-control system operates cells halts in mitosis and precludes passage into the final phases of cell division (Musacchio &
Salmon, 2007). The function of SAC is preventing premature chromosome segregation and loss of sister chromatid cohesion to arrest the initiation of anaphase when
unattached or incorrectly attached chromosomes is present (Shonn, et al., 2000). This is an important function for preserves the genome from alterations in chromosome copy number and their viability threats (Musacchio, 2015).
SAC consists of a group of proteins that monitor the attachment of spindle
microtubules and kinetochores (for review see Musacchio & Salmon, 2007). Normally, the duplicated chromosomes properly attached to the mitotic spindle promoter cell to transit to anaphase. However, the SAC delays the metaphase-anaphase transition when duplicated chromosomes have not properly attached to the mitotic spindle (Nicklas, et al., 1995; Pinsky & Biggins, 2005). The highly conserved SAC consists of Mad1, Mad2,
BubR1/Mad3, Bub1, Bub3, and Mps1 kinase (Foley & Kapoor, 2013; Jia et al., 2013).
They associate with kinetochores, and target the anaphase-promoting complex (APC) to inhibit the anaphase progression. The APC is an E3 ubiquitin ligase that requires Cdc20 (Bloom, et al., 2007) to recruit proteins for ubiquitination and subsequent degradation that promote the inactivation of Clb–Cdc28. The SAC proteins bind directly to Cdc20, preventing the function of APC, and then delays the metaphase-anaphase transition (Li,
et al., 1997; Fang & Kirschner, 1998).
(Burgess, et al., 2014)
(Schibler, et al., 2016)
It is also important to silence the SAC when mistakes in chromosome
attachment were resolved (Mao, et al., 2003). However, the SAC silenced has received attention just in recent ten years (Vanoosthuyse and Hardwick, 2009). The
Kinetochore-associated phosphatase protein 1 (PP1) is indicated to display an essential role in SAC silencing (Rosenberg, et al., 2011). PP1 is likely to promote the removal of Mad1/Mad2 from kinetochores through the dephosphorylation of Bub1, and the
dephosphorylation of Bub1 is likely a critical step in SAC silencing. In addition, the kinetochore-localized PP1 may antagonize checkpoint kinases Mps1 to promote anaphase onset and SAC silencing (Bokros and Wang, 2016).
21
(Bokros and Wang, 2016)
For a daughter cell to receive a complete genomic complement, it is essential that the mitotic spindle be positioned accurately within the cell in the budding yeast (Falk, et al., 2011). The spindle position checkpoint (SPOC) prevents cells from becoming
anucleate/ binucleate by arresting cells in late anaphase until the spindle has realigned along the mother–bud axis. The SPOC is a feedback mechanism that delays cell-cycle progression in response to defects in spindle position (Muhua, et al., 1998; Adames, et al., 2001.). After the chromosomes attach properly to kinetochore, one copy of DNA
will be separate into daughter cell. At this time, it is important to make sure that the spindle properly aligned along the mother–bud axis in budding yeast. Some case, the spindle becomes mispositioned in the mother cell compartment, if the (SPOC) are not activate, cells inappropriately exit from mitosis in the mother cell compartment and go on to produce one anucleate cell and one binucleate cell (Falk, et al., 2016).
The SPOC arrest cells from exit the mitosis until the spindle is properly aligned
along the mother-bud axis (Yeh, et al., 1995; Lew & Burke, 2003). The mitotic exit is regulated by mitotic exit network (MEN), which promoters the activation of the phosphatase Cdc14 and causes cell exit from the mitosis (Visintin, et al., 1998). Key SPOC components are the kinase Kin4 and the Bub2–Bfa1 GAP complex that inhibit the key MEN component, Tem1. Kin4 primarily localizes to the mother cell and
associates with spindle pole bodies (SPBs) located to inhibit MEN signaling. In contrast, the Kin4 does not associate with the SPBs in the bud. Thus, only when a MEN bearing SPB leaves the mother cell and the spindle is accurately positioned along the
mother-bud axis can MEN signaling occur and cell division proceed (Falk, et al., 2011).
(Falk, et al., 2011)
23
3 The mitotic exit
3.1 The mitotic exit
When the cell cycle progression goes to division, it is important to ensure the coordinated distribution of cellular material between the mother cell and the daughter cell (Jorgensen & Tyers, 2004; Goranov, et al., 2009). On mitosis completion, mitotic structures disassemble, mitotic regulators are inactivated. This process, termed mitotic exit, starts with the inactivation of Cdk1, the master regulator of mitosis, and the inactivation of its substrates (Murray, et al., 1989; Hershko, et al., 1991; Holloway, et al., 1993). Consequently, the mitotic spindle breaks down, cytokinesis occurs,
chromosomes decondense, and interphase functions resume (Hotz, et al., 2014). Mitotic Exit is an important transition point that signifies the end of mitosis and the onset of new G1 phase for a cell. For a normal eukaryotic cell, the mitotic exit is irreversible.
Moreover, the cell needs to rely on specific control mechanisms to ensure it exits mitosis. The Mitotic Exit Network (MEN) is an extensively studied signaling pathway that promotes mitotic exit, facilitates the control of spindle orientation, and initiates cytokinesis in the budding yeast (Shou, et al., 1999; Visintin, et al., 1999). Another non-essential Cdc Fourteen Early Anaphase Release (FEAR) pathway also promotes this event (Stegmeier, et al., 2002). I will introduce FEAR on next section.
The cyclin dependent kinase, Cdc28 must be inactive when cell exit from the mitosis. The Cdc28 was dephosphorylated and inactivated by Cdc14. Cdc14 is a conserved and essential protein phosphatase. Out of anaphase, Cdc14 is sequestered in the nucleolar chromatin bound to its inhibitor Net1, also called Cfi1. Net1 anchors Cdc14, and together with Sir2. They form the RENT (REgulator of Nucleolar silencing and Telophase) complex which inhibits transcription (Baro, et al., 2017). The
interaction is dissolved via the phosphorylation of Cfi/Net1 upon entry into anaphase.
The release of Cdc14 from the nucleolus provides the signal coordinate the chromatin structure and chromosome segregation. The Cdc14 release liberates to dephosphorylate Cdc28 and promote exit from mitosis (Hwang, et al., 2009).
3.2 The mitotic exit network
The Mitotic Exit Network (MEN) is the main signaling pathway that triggers Cdc28 inactivation and the onset of cytokinesis on segregation of the daughter nuclei (Murray, et al., 1989; Hershko, et al., 1991; Holloway, et al., 1993). The MEN from budding yeast is closely related to the Septation Initiation Network (SIN) from the fission yeast Schizosaccharomyces pombe (Bardin & Amon, 2001) and the Hippo pathway from mammals (Hergovich & Hemmings, 2012). The MEN consists of GTPase Tem1, two serine/threonine kinases, Cdc15 and Dbf2- Mob1 (Baro, et al.,
25
2017). The signaling pathway driven by the Tem1, which signals through the Cdc15.
Cdc15, activates Dbf2, which works with the coactivator Mob1 to release the
phosphatase Cdc14 from the nucleolus (Hotz, et al., 2014). Activation of the MEN is controlled through a complex relationship between all components and regulators of this signaling pathway (Bardin, et al., 2000; Pereira, et al., 2000). Changes in the activity of these proteins and in their ability to interact with each other and to localize to different cell structures allow for a tight temporal and spatial regulation of the pathway (Jensen, et al., 2004).
(Caydasi, et al., 2012)
Regulation of the MEN is complex, with multiple, partially redundant pathways.
The most studied regulators are Lte1 and the Bfa1-Bub2 complex, although Cdc5, Kin4, Kel1/2, Gic1 and -2, Ste20, Cla4, Cdc42, and Ras2 are also implicated (Geymonat, et al., 2009). Lte1 (gene name means: low temperature essential) was seen as a positive
mitotic regulator because lte1 mutants undergo a telophase arrest at low temperature
(Shirayama, et al., 1994). Lte1 shares homology with the guanosine nucleotide exchange domain of the Ras-guanosine nucleotide exchange factor (GEF) Cdc25 (Cherfils & Chardin, 1999; Quilliam, et al., 2002). Thus, it was proposed that Lte1 might be a GEF for Tem1. Cla4 kinase is essential for the activity of Lte1. Lte1 depends on Cla4 for its cell cycle-dependent phosphorylation that is essential for Lte1 function (Seshan & Amon, 2005). Bfa1 and Bub2 are negative regulators of the MEN that form a two-component GTPase-activating protein (GAP) for Tem1 in vitro (Geymonat, et al., 2002). The dissociation of Bfa1-Bub2 complex from Tem1 is dependent upon the Polo-like kinase Cdc5 to phosphorylate the Bfa1-Bub2 complex (Hu, et al., 2001). The inactivation of MEN through Bfa1-Bub2 complex belongs to SPOC (Wang, et al., 2000;
Li, 1999; Caydasi & Pereira, 2009). Thus, mitotic exit is prevented when there is spindle damage, spindle misorientation at G2/M phase or DNA damage (Hu and Elledge, 2002).
Lte1 has homology to GEF, and it has long been hypothesized that it could function as a GEF for Tem1 (Bardin, et al., 2000; Seshan and Amon, 2005). However, overexpression of the GEF domains of Lte1 could not suppress the low temperature sensitivity of lte1 (Yoshida, et al., 2003). In contrast, other (central) domain of Lte1 is enough to rescue the defect. Other observations indicated Lte1 cannot stimulate activity of Tem1 in vitro (Geymonat, et al., 2009). Rather than stimulating Tem1’s nucleotide
27
exchange, Lte1 was proposal to be a downstream effector protein of Ras , prevent the small amounts of Kin4 from associating with spindle pole bodies (SPB) in the bud (Bertazzi, et al., 2011; Falk, et al., 2011), or contribute the Bfa1 localization. Recently, it was shown that Lte1’s GEF domain suppressed the lethality of a genetic mitotic exit defect strain by the allele of lte1-ΔEcoRI (Falk, et al., 2016). But still, they could not demonstrate the Lte1’s GEF activity in vitro. Thus, it is likely that Lte1 promotes exit form mitosis in multiple mechanisms (Falk, et al., 2016).
Lte1 localized to the bud cortex is important for Lte1 activity (Bardin, et al., 2000;
Pereira, et al., 2000). Additionally, the bud-localized activating signal is necessary for full MEN activation. The spatial information translate into chemical signal is coordinate by the Lte1 and Kin4. During metaphase, Tem1 and the Bfa1/Bub2 complex localize to both SPBs. During anaphase, they become concentrated at the daughter SPB (dSPB) that migrates into the bud (Fraschini, et al., 1999; Pereira, et al., 2000). At normal condition, SPB function as the sensor to translated the spatial information into chemical signal. The MEN signal will be activated when dSPB escapes the MEN inhibitor Kin4 in the mother cell and moves into the bud where the MEN activator Lte1 presented (Chan and Amon, 2010). In contrast, Bfa1-Bub2 and Kin4 remain on both SPBs when the spindle is misaligned in the mother cell and the SPOC is activated (Pereira, et al., 2000, 2001; Molk, et al., 2004; Pereira and Schiebel, 2005; Fraschini, et al., 2006;
Maekawa, et al., 2007). Thus, the switch from symmetrical to asymmetrical distributions of the MEN regulators precedes MEN activation.
3.3 The Cdc14 early anaphase release network
The Cdc Fourteen Early Anaphase Release (FEAR) network regulates the initiated inactivating of mitotic CDKs by triggering the Cdc14 releasing (Stegmeier, et al., 2002). The mitotic cyclin-CDK inactivation is important for exit from mitosis. The inactivation is initiated at the metaphase-anaphase transition which regulates by the FEAR in the budding yeast (Rahal & Amon, 2008). In addition, the FEAR is critical to ensure accurate anaphase chromosome segregation and the integration of this process with other anaphase events (Rock and Amon, 2009).
While FEAR pathway is not strictly essential for Cdc14 release, it helps coordinate the timing of Cdc14-driven anaphase events (Weiss, 2012). Proposed functions of FEAR include ensuring that the segregation of all chromosomes is initiated
simultaneously, stabilization of anaphase spindles, localization of spindle proteins, positioning of the anaphase nucleus, resolution of the rDNA to allow its segregation, and priming of the MEN so that mitotic exit can occur promptly and efficiently (Rock and Amon 2009; Yellman and Roeder, 2015). That is, the FEAR, although not
essential, has an important biological role in the metaphase-anaphase transition for
29
timely controlling the cell cycle progression.
The FEAR network promotes Cdc14 release by multiple ways. At anaphase onset, the APC be activated by associate with Cdc20, and the FEAR occurs. The Cdc20-APC then degrades the Securin (Pds1), which is an anaphase inhibitor. The Securin inhibits the Seperase (Esp1), which cleavages the cohesion and let the chromosome segregate.
Slk19, Kinetochore-associated protein, forms complex with Seperase. The Separase-Slk19 and the proteins Zds1 and Zds2 are thought to down-regulate PP2A-Cdc55 phosphatase activity (Yasutis, et al., 2010; Wicky, et al., 2011). This down-regulation of PP2A-Cdc55 phosphatase activity allows phosphorylation of
Cfi1/Net1, which sequesters Cdc14 and localizes in the nucleolus (Visintin, et al., 1999).
Another pathway involves Spo12 (Tomson, et al., 2009). Spo12 is a nucleolar
phosphoprotein that binds to Fob1, which binds to Cfi1/Net1 and prevents Cdc14 release. The proteins of Spo12, Fob1 and Net1 regulate for the Cdc14’s sequestration.
CDK phosphorylation on the Spo12 would reduce Fob1’s ability to inhibit Cdc14 release (Stegmeier, et al., 2004). The Spo12 promotes the Cdc14 release by an indirect way. Cdc5 also is thought to be contributing for FEAR-dependent Cdc14 release. Cdc5 phosphorylates Net1 and results in Cdc14 disassociations (Shou, et al., 2002; Yoshida and Toh-e, 2002). However, it has been argued that Cdc5, instead of directly
phosphorylating Net1, primarily stimulating degradation of kinase Swe1, which enables
Clb2-Cdk1 to phosphorylate Net1 (Liang, et al., 2009). Some evidence indicates that the direct phosphorylation of Cdc14 by Cdc5 may facilitate Cdc14 release (Visintin, et al., 2003; Rahal and Amon, 2008). Also, the degradation of Cdc5 protein is essential for
the return of Cdc14 to the nucleolus after exit mitosis (Visintin, et al., 2008). Moreover, Cdc5 also controls the MEN by phosphorylating Bfa1-Bub2 complex which leads to the activation the MEN (Botchkarev and Haber, 2017).
(Caydasi and Pereira, 2012)
While the FEAR and the MEN both control the Cdc14 release and mitotic exit occurrence, they still have distinct contribution. The MEN drives export of Cdc14
31
from nucleus to the cytoplasm (Mohl, et al., 2009). Export from the nucleus would allow Cdc14 access to mitotic exit regulators in the cytoplasm. It has been suggested that Cdc14 release by the FEAR pathway might be limited to the nucleus (Lu and Cross, 2009; Yellman and Roeder, 2015), or alternatively that there could be some export to the cytoplasm (Rock and Amon, 2009).
4 Histone protein modification and their function
Eukaryotic genomic DNA wraps around histones to form the nucleosome units of chromatin. Nucleosomes allow for regulated access of nuclear factors to DNA and provide a platform for recruitment of factors that do not interact directly with nucleic acids. Posttranslational modifications of histone tails mediate selective recruitment of factors with roles in gene transcription, DNA damage response, and other nuclear processes. Combinatorial and interdependent patterns of histone modifications create a multilayered system of chromatin domains and gene expression control (Suganuma and Workman, 2011).
Ubiquitination of histone H2B (H2Bub) are the most abundant ubiquitin
conjugates in yeast corresponding to ~10% of H2B. Monoubiquitination of H2B is by far the most abundant form (Robzyk, et al., 2000). The H2Bub as a landmark is important for gene activation. In addition, H2Bub has histone crosstalk to promote histone H3 lysine 4 di- and trimethylation (H3K4me2 and H3K4me3) at actively transcribing genes.
4.1 H2B ubiquitylation and H3K4 methylation
Monoubiquitination of histone H2B has emerged as an important chromatin modification with roles not only in transcription but also in cell differentiation. The
33
primary role for H2Bub is modifying nucleosome accessibility or recruiting the multicomponent histone methyltransferase. Additionally, H2Bub was indicated to be involving in many biological functions, such as transcription, RNA processing, DNA repair, cell differentiation, regulation of chromosome segregation by the kinetochore machinery (Soares & Buratowski, 2013).
Histone H2B is monoubituitinated (H2Bub) on lysine 123 in yeast or the corresponding lysine 120 in mammals (Osley, 2006). In Saccharomyces cerevisiae, Rad6 was identified as the only H2B ubiquitinconjugating enzyme (E2) and Bre1 as the only H2B ubiquitin ligase (E3) (Wood, et al., 2003). H2Bub also regulates the activity of Dot1 and activates of the Set1 complex (COMPASS) who mediated H3 lysine 79 and H3 lysine 4 methylation (Nakanishi, et al., 2009).
H2Bub regulates the methylation of histone H3 lysine 4 (H3K4me) by Set1 methyltransferase complex. This signal is linked to gene activation from yeast to humans. The requirement of H2Bub to regulate the H3K4 methylatation by Set1 complex is explained by at least three nonexclusive models. The crosstalk between H2Bub and H3K4 methylation (Soares and Buratowski, 2013) is explained for the followings. First, H2Bub may change the configuration of the nucleosome H3 tail to make it more accessible to the histone methyltransferase (HMT). A second simple model is that H2Bub may bind one of the HMT subunits, tethering the complex to
promote higher-level methylation by increasing the residence time. Alternatively, H2Bub binding to the HMT may trigger a conformation change that exposes or activates the catalytic site. Some experimental evidence exists for each of these
possibilities, and all three mechanisms may contribute (Soares and Buratowski, 2013).
(Soares and Buratowski, 2013)
4.2 The Set1 complex and the regulation of H3K4 methylation
The H3K4 methylation (H3K4me) is important for gene transcription. Moreover, the different levels of H3K4me still have different functions. There are three levels of H3K4me: mono-, di-, and trimethylation; yet they have different functions and location (Soares and Buratowski, 2013). H3K4me1 is furthest from transcriptional start sites (TSSs) and is linked to nucleosome dynamics and chromatin remodeling at
stress-responsive genes in yeast (Nadal-Ribelles, et al., 2015). H3K4me2 peaks slightly further downstream from TSSs and recruits a histone deacetylase. And it is linked to
35
ongoing transcription as well as gene repression in yeast (Margaritis, et al., 2012).
H3K4me3 is highest near active promoters or around TSSs and recruits multiple factors that promote transcription, including TFIID, histone acetyltransferases, and chromatin remodelers (De Santa, et al., 2010; Zhang, et al., 2016). In general, H3K4me3 is considered a universal hallmark of active transcription, as genome-wide studies from yeast to humans have shown a strong correlation between active transcriptions (Weiner, et al., 2015).
All the status of H3K4 methylation is only performed by Set1 complex in
Saccharomyces cerevisiae. The SET1 encodes H3K4 methyltransferase (Set1) and the
single H3K4 methyltransferase is found in a complex (ySet1C/COMPASS) with seven other subunits (Swd1, Swd3, Bre2, Sdc1, Spp1, Swd2, and Shg1) (Dehe and Geli, 2006).
Set1 is homologous to human MLL1 (Roguev, et al., 2001). Six of COMPASS subunits conserved in the metazoan SET1 complexes, with four also conserved in the metazoan MLL complexes.
Set1 domain and COMPASS (Kim et al., 2013)
The Set1 methylated histone H3K4 is regulated by H2Bub. Set1, Swd1, Swd3,
Bre2, and Sdc1 are the minimal Set1 complex subunits required for H2B
ubiquitylation-dependent H3K4 methylation in the in vitro system. The same group also reveals that the n-SET Domain of Set1 is essential for H2B ubiquitylation-dependent H3K4 methylation (Kim, et al., 2013). In contrast, the independent of the
requirement for H2Bub regulation is linked by the Set1C/COMPASS subunit, Swd2.
Previous reports (Vitaliano-Prunier, et al., 2008) showed that Swd2 is required for H2B ubiquitylation-dependent H3K4 methylation in vivo. Histone H2B promotes
ubiquitylation at Lys 68 and Lys 69 of Swd2, and mutation of the two sites reduced H3K4 trimethylation. Additionally, Swd2 ubiquitylation controls the recruitment of Spp1, which is specific necessary for H3K4 trimethylation.
Each subunits of Set1C/COMPASS differentially affects Set1 stability, complex integrity, global H3K4methylation, and distribution of H3K4 methylation along active genes (Dehe and Geli, 2006). The effects of them were analyzed by the loss of
individual Set1C/COMPASS subunits. The complex requires Set1, Swd1, and Swd3 for integrity, and the Swd1-Swd3 heterodimer present is important for amount of Set1.
Bre2 and Sdc1 also form a heteromeric subunit, which requires the SET domain of Set1 for interaction with the complex. Sdc1 strongly interacts with itself. Inactivation of either Bre2 or Sdc1 has very similar effects. The presence of Spp1 is important for the amount of Set1 and retains trimethylated H3K4, whereas, Shg1 slightly reduce levels of
37
both di- and trimethylation (Dehé, et al., 2006).
4.3 The relation between Set1/H3K4me and mitosis
H3K4 methylation by Set1 complex is important in regulating cell cycle beside of the function in regulating gene expression. H3K4me alters chromatin structure and serves to recruit or exclude binding of nonhistone proteins to chromatin. H3K4me is known to be associated with gene expression; however, H3K4me also contributes to cell cycle, such as DNA damage repair, timely DNA replication (Sollier, et al., 2004), and mitosis (Schibler, et al., 2016). Although the mechanism of Set1 complex regulating mitosis had not well studied, there still have some puzzle pieces
The Set1C complex has been involved in the regulation of chromosome segregation during mitosis (Zhang, et al., 2005). Proper chromosome segregation requires that centromeres of sister chromatids be attached to microtubules. The attachments are mediated by kinetochores. Set1 is required for the methylation of conserved lysines in a kinetochore protein, Dam1 (Zhang, et al.,2005). Dam1 is phosphorylated by Ipl1 in response to inappropriate microtubule–kinetochore interactions (Keating, et al., 2009). The Dam1 phosphorylation is inhibited by Set1 methylation of flanking amino acid. Set1 opposes the functions of Ipl1 in regulating chromosome segregation, and this suppression is not linked to methylation of H3.
The Ipl1 phosphorylate Dam1 lead to triggering of the spindle assembly
checkpoint (SAC) (Biggins and Murray, 2001), which inhibited the anaphase promoting processes. The fact that Set1 is opposite of the Ipl1 function suggest that loss of SET1 may bypass the SAC. Indeed, deletion of SET1 leads to resist Benomyl, which interferes mitotic spindle polymerizing and induces cell cycle arrest at metaphase (Schibler, et al., 2016; Beilharz, et al., 2017). The resistance was proposed that Set1/H3K4 stable the tubulin formation by delaying the G1/S or resolving the SAC. Another explanation is that the SAC component, Mad2, binds to H3K4me and leads to the relief from the SAC.
The highly conserved HORMA domain in Mad2 is indicated to be a novel H3K4 methyl reader (Schibler, et al., 2016). The interaction of Mad2 and H3K4 limits the capability of Mad2 to inhibit Cdc20 function, and then promote the relief of the SAC.
Set1/H3K4 also is indicated to regulate Cdc14 release which is a critical event during mitotic exit in the budding yeast (Hwang & Madhani, 2009). The Cdc14 is a phosphatase, which is requiring for proteins degradation during anaphase, leads to mitotic exit and restart the cell cycle (Hwang, 2009). The cross talk between
39
chromosome and mitosis progression is important. The soluble proteins, Cdc14 is one of them, tightly chromatin-bound factors on chromosomes, is part of the regulatory complexity involves the requirement that a variety of communications. In addition, the Set1 is the factor that involved in the important control to ensure the timely and correct inheritance of sister chromatids followed by cytokinesis (Hwang & Madhani, 2009).
5 The relationship between SET1, FMN1 and mitosis
In summary, properly mitosis is important for genome stability. Previous studies indicate that the mitosis is critically regulated by chromatin factors (Zlotorynski, 2016;
Aguilera, et al., 2000; Kadauke & Blobel, 2013; Chen, et al., 2005; Black, et al., 2016).
In addition, the mitosis progression is dependent on its nutrition condition. However, how the metabolism affects mitosis is not well defined. Our unpublished data have revealed a genetic interaction between SET1 and FMN1. The SET1 is important in regulating mitosis, thus, I aimed to study if the FMN1 functions in mitosis, and the role of FMN1 in regulating mitosis.
41
RESULTS
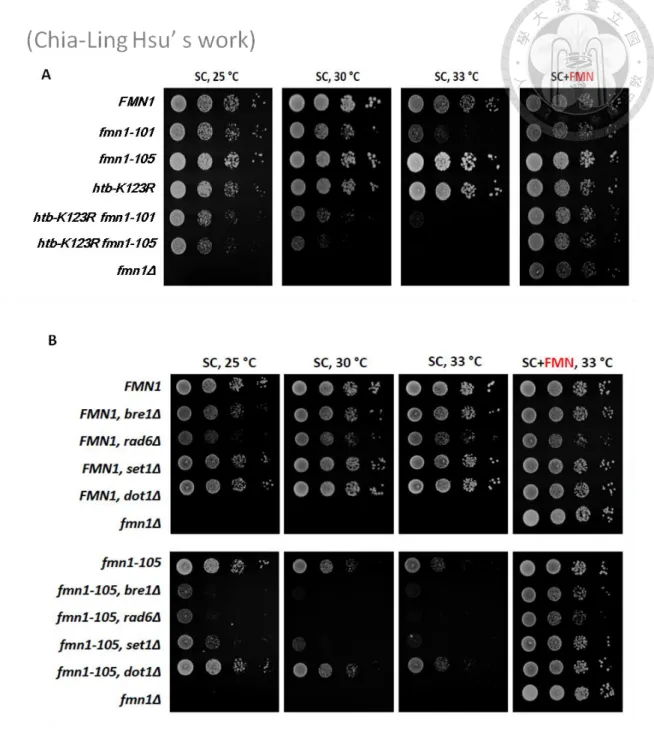
1 FMN1 genetically interacts with SET1
Our genetic screen recovered the htb-K123R mutation, which abolishes the H2B ubiquitylation, displays synthetic lethality with a point mutation resides in FMN1 which encodes Riboflavin kinase, produces riboflavin monophosphate (FMN); FMN is a necessary cofactor for many enzymes (Figure 1A). To explore a potential link between H2Bub and FMN1, we created temperature sensitive alleles of FMN1 by random
mutagenesis. In total, we identified nine fmn1 alleles and all of them showed synthetic sickness with htb-K123R, and they could be rescued when supplied with the addition of FMN in the culture medium. This observation may indicate that the mutation of FMN1 may reduce the Fmn1 protein kinase activity and the production of FMN. In addition, the synthetic phenotype between FMN1 and htb-K123R could be rescued with the addition of FMN in the culture medium. The result of this screening suggested that the FMN synthesis is likely the only function of Fmn1. And the interaction with FMN1 suggests H2Bub may carry an unappreciated role in regulating cellular metabolism.
Alternatively, FMN may have an unknown function that regulates a cellular process downstream of H2Bub pathway. Among those FMN1 alleles, the fmn1-105 mutation showed relatively minor temperature sensitivity but still displayed significant sickness than wild-type when combined with htb-K123R. The fmn1-105 allele bears a point
mutation at Phenylalanine 95, replaced by Serine. This site is near V91, which is
homological to human flavin binding site. Its temperature sensitivity rises around 30~33
C depending strain backgrounds. The fmn1-105 allele was thus selected to further
characterization of the genetic interaction between H2Bub and FMN1.
Monoubiquitination of histone H2B is catalyzed by Rad6-Bre1 (Wood, et al., 2003). To investigate the requirement of FMN in htb-K123R cells is due to the H2B ubiquitination abolished, we confirmed the synthetic interaction and observed the viability of the bre1Δ fmn1-105 and rad6Δ fmn1-105 cells (Figure 1B). The
fmn1-105 mutant displayed strong genetic interaction with BRE1 and RAD6, suggesting
that the FMN1 is important for cellular functions that regulated by H2B ubiquitination.
H2Bub regulates Set1-mediated H3K4 methylation and Dot1-mediated H3K79
methylation. We found that FMN1 does not have genetic interaction with DOT1 while the fmn1-105 mutant specifically displayed genetic interaction with SET1, suggesting that the FMN1 is important for regulating some of the function of SET1 (Figure 1B).
It has been proposed that set1Δ promotes microtubule stability as the mutation was deficient in spindle elongation (Reijo, et al., 1994). Benomyl is a microtubule destabilizing drug, which contains the active compound methyl
benzimidazol-2-yl-carbamate (MBC). MBC is known to cause the depolymerization of microtubules in vivo (Jacobs et al. 1988) and in vitro (Kilmartin 1981), most likely by
43
directly binding to tubulin (Neff et al. 1983). I treated cells with 30 μg/ml Benomyl and in consistent with published results, set1Δ mutant cells were highly resistant to high levels of Benomyl than wild-type (Figure 2). The fmn1-105 cells also display Benomyl resistance; suggest that FMN1 may play a role in mitosis by a microtubule stability dependent manner. Interestingly, the double mutants of fmn1-105 set1Δ resisted to Benomyl at the same level as fmn1-105 but less than the set1Δ single mutant. The loss of fmn1-105 activity lead to reduce the Benomyl resistance of set1Δ, suggesting that the function of SET1 in mitosis is partially dependent on FMN1. To determine whether the Benomyl resistance complementary defect of set1Δ is specifically to fmn1-105 cells, we examined a microtubule-irrelevant gene, ACS2, is coding for an acetyl-coA synthetase, the nuclear source of acetyl-coA for histone acetylation (Frenkel & Kitchens, 1977).
The ACS2 display genetic interaction with SET1, however, the acs2-ts1 could not resist to Benomyl, in addition, the loss of Acs2 activity did not lead to reduce the Benomyl resistance of set1Δ. These observations indicate that the Benomyl resistance of set1Δ is specifically dependent on the function of Fmn1.
FMN is involved in many metabolic pathways. To further explore the role of FMN1 in mitosis, we used synthetic genetic array (SGA) analysis (Tong, 2001) to
screen for genes that may cause synthetic sickness/lethality with fmn1-105. We mated the single mutant library of MAT strains with, MATa fmn1-105. After selection, the
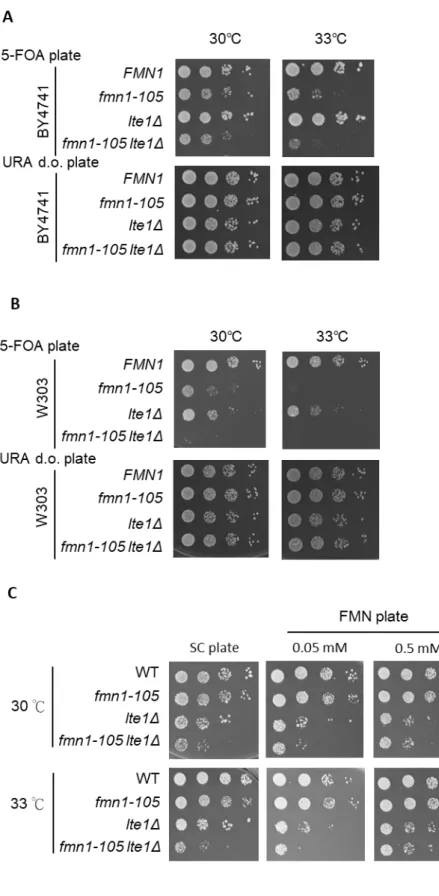
double mutation strains were picked up if they were significantly slow growing. A list of candidate genes is found (Table 1), 74% are involved in metabolic process, 45% are involved in gene expression and 21% are involved in regulation of transcription by RNA polymerase II. These three biological processes are large scale to analyze, however, there were ~15% of candidates were related ―mitotic cell cycle process‖. It supports that FMN1 could be involved in mitosis. Remarkably, LTE1 was one of the candidates, which was reported to cause synthetic lethality when combine with SET1
deletion. To ensure the genetic interaction between LTE1 and SET1, I deleted the lte1 in set1Δ cells. The set1Δ lte1Δ cells indeed showed synthetic lethal, in consistent with the
previous study (Hwang & Madhani, 2009). To further confirm the genetic interaction between FMN1 and LTE1, I created the double deletion of FMN1 and LTE1 in three different genetic backgrounds, null deletion of FMN1 covered by a fmn1-105 or wild-type plasmid (URA marker) in BY4741 or W303 (Figure 3A. B) and W303 RAD5+ with fmn1-105 mutation residing on the genome (Figure 3C). Whichever
background, the fmn1-105 lte1Δ exhibited strongly genetic interaction at 33℃.
TheW303 RAD5+ background exhibited a growth defect in double mutation but not fmn1-105 only at 33℃. Moreover, it exhibited the phenotype only when growing in SC medium, but not in rich YPD medium. Taking into account the convenience of
operation, I chose the fmn1-105 mutated on the genome background for subsequent
45
experiments.
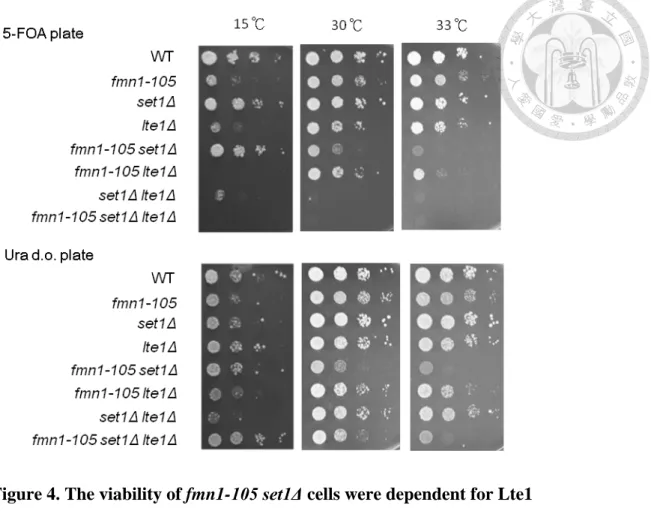
To further confirm that the growth defect of fmn1-105 lte1Δ was due to the loss the Fmn1 activity, I supplied the cells with 0.05 mM and 0.5 mM of FMN in the media. I found the supplement of 0.5 mM FMN recuses the growth defect of fmn1-105 lte1Δ to the level of lte1Δ (Figure 3C). However, supplied cells with 0.05 mM FMN could not rescue the growth defect of fmn1-105 lte1Δ to the level of lte1Δ. These observations indicated that FMN producing activity is required the proper growth of fmn1-105 lte1Δ cells. Furthermore, I found that the 0.05mM of FMN also rendered the lte1Δ cells sicker than growing on medium without supplement. These observations indicated that cells require proper concentration of cellular FMN when loss of Lte1 activity. In addition, LTE1 was deleted in set1Δ fmn1-105 cells. I found that deletion of LTE1 causes the lethality of set1Δ fmn1-105 cells (Figure 4). Collectively, these data indicated that SET1 and FMN1 both display strong interactions with LTE1, indicating that a role for FMN1 in mitosis.
2 The synthetic interaction of FMN1 and SET1 leads to defective cell cycle
progression
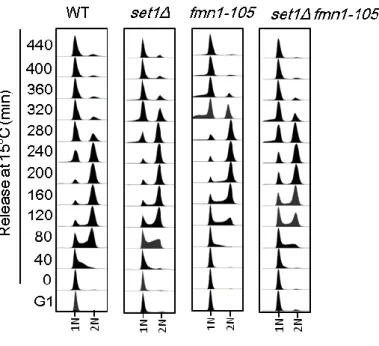
To study the role of FMN1 and SET1 during cell cycle, I monitored the DNA content in mutant cells throughout a complete cell cycle progression. To clearly reveal the pace of cell cycle, cells were arrested in G1 with 2 mg/ml α factor and released into
pheromone-free media at 15 degrees to slow down the cell cycle. After 160 min, 4 mg/ml α factor was added to prevent entry into the subsequent cell cycle. Cells were
collected every 40 min and stained with SYBR GREEN for analysis DNA content by flow cytometry (Figure 5). The fmn1-105, set1Δ and fmn1-105 set1Δ cells were progressing from G1 into S phase slower (80min) than the wild-type (40min). The duration of G2/M was no significant difference between the fmn1-105, set1Δ and fmn1-105 set1Δ cells. However, a subset of the fmn1-105 set1Δ cells accumulated cells
at G1 without migrating into cell cycle. This observation indicated that the genetic interaction between FMN1 and SET1 may lead to the G1-S transition defect. Indeed, we found the evidences of the defect in S phase transition of fmn1-105 set1Δ cells by another group in our lab (data not show). The current work only focuses on the role of FMN1 in regulating the mitosis.
3 Loss of Fmn1 activity leads to mitosis delay
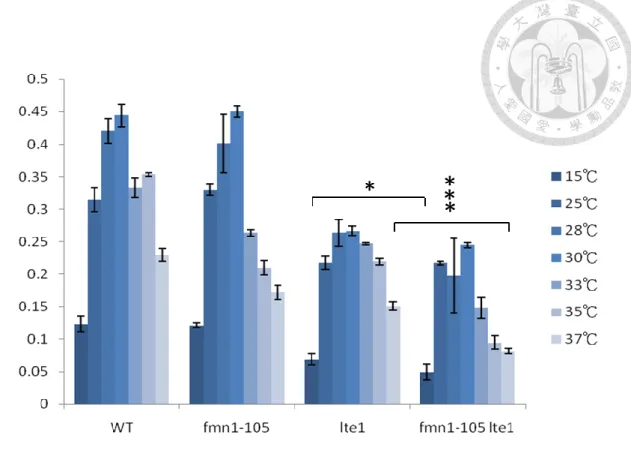
To investigate the role of FMN1 in mitosis, I characterized the role of FMN1 in mitotic defective cells by deleting LTE1. The accurate determination of cell growth and viability is pivotal to monitoring the cells bioprocess, so I first determined the growth rate of mutant cells. I cultured cells in flasks in constant temperature rooms, and on shakers drums to provide sufficient aeration. At intervals of every one hour, a sample would be sterilely removed and measured the cell density by spectrophotometer. OD600
47
values between 0.1 and 1 at the temperature from 15 -37 degree were used to calculate the growth rate. I found that the growth rate of fmn1-105 lte1Δ cells was slowest in all the temperature (Figure 6) but compare to lte1Δ cells, the growth defect was strongest at 37C.
Next, I analyzed the distribution of cells in the cell cycle to determine whether a
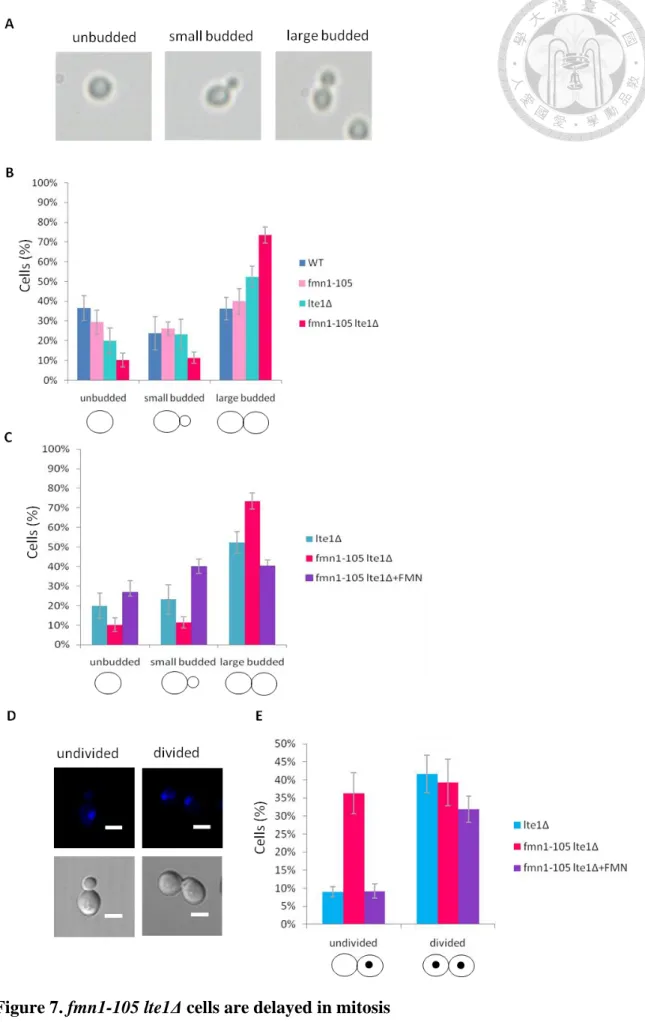
loss of FMN1 function altered cell cycle progression. The fmn1-105, fmn1-105 lte1Δ, lte1Δ and W303 (wild-type) cells were grown asynchronously at 37℃ for 8 hours and
budding morphology was scored (Figure 7A, B). 74% of fmn1-105 lte1Δ cells were large budded compared to 52% in lte1Δ, 40% of fmn1-105 cells and 36% in WT (wild-type) cells. These data suggested that the majority of fmn1-105 lte1Δ cells was arrested at mitosis. Moreover, supplied fmn1-105 lte1Δ cells with high concentration of FMN in the medium, the proportion of large budded cells was reduce to 40%, similar to the level of lte1Δ single mutant (52%) (Figure 7A, C). The increased proportion of mitotic cell in fmn1-105 lte1Δ could be rescued to as lte1Δ by the addition of FMN, suggesting that the effect in mitosis of fmn1-10 5lte1Δ is caused by the insufficient cellular concentration of FMN.
To further investigate if loss of FMN1 function altered the specific progression of mitosis, large budded cells’ nuclear DNA was stained by the addition
4,6-diamidino-2-phenylindole (DAPI) and inspected under the microscope. I found that
36% of fmn1-105 lte1Δ cells were large budded with undivided nuclei compared to 9%
in lte1Δ cells (Figure 7D, E). This observation may suggest that fmn1-105 lte1Δ cells have delayed mitosis prior to anaphase. In addition, the undivided nuclei cells of fmn1-105 lte1Δ decreased when supplying high concentration of FMN in the medium,
indicating that the proper FMN concentration is important for mitosis in fmn1-105 lte1Δ cells.
4 FMN1 is involved in mitotic exit regulation
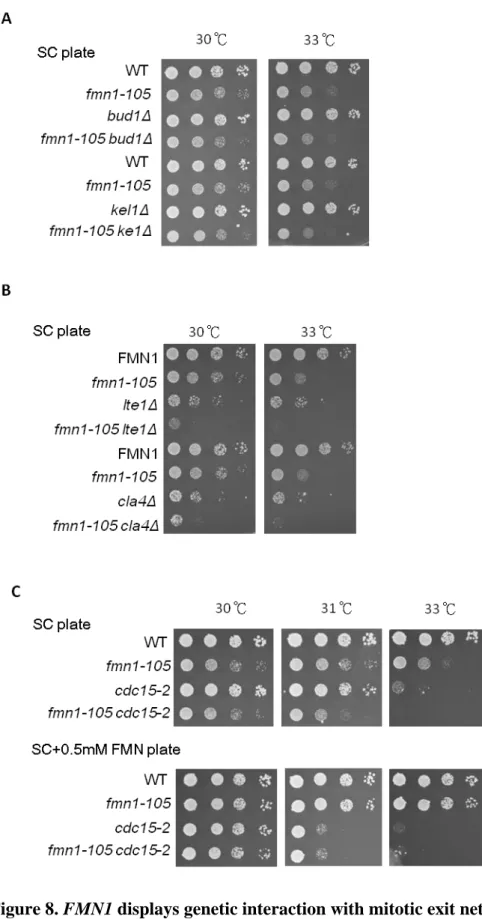
A previous study indicates that beside the function of mitotic exit network regulation (Geymonat, et al., 2010), lte1 mutants also displays altered bud morphology during mitotic arrest. Lte1 localizes specifically at the bud cortex, where it interacts with Ras2 and the polarity cap component, Kel1. Kel1 is required for bud site selection.
Lte1 also can interact with and inhibit Bud1 to avoid untimely bud polarization. I therefore asked if fmn1-105 activity affected the process of bud morphology. I examined the viability of fmn1-105 cells with BUD1 or KEL1 deletion (Figure 8A), compare to single mutant, there was no growth defect in the double mutant cells, indicating that interfering Fmn1 activity did not influence the process of bud morphology.
The above observations narrowed down the impact of that the genetic interaction between FMN1 and LTE1 to the co-regulation mitotic exit events through the mitotic
49
exit network. To determine if fmn1-105 allele affected mitotic exit network (MEN) and the MEN activation. I examined the viability when mitotic exit network was ablated or the MEN were deactivated in fmn1-105 cells. I deleted CLA4, which has an epistatic effect in Lte1 phosphorylation and found that fmn1-105 mutants with CLA4 deletion exhibited synthetic growth defects (Figure 8B), and the viability profile is similar to fmn1-105 lte1Δ cells. Next, to deactivate the MEN, I introduced a hypomorphic allele of
CDC15, a MEN component, into cells harboring the fmn1-105 allele. The double
mutants exhibited enhanced temperature sensitivity than fmn1-105 at 31~33°C, the temperature is lower than the cdc15-2 cells deactivation temperature (Figure 8C). To further confirm that the MEN deactivation was due to loss the Fmn1 activity, I supply the cells with high concentration of FMN in the media. The supplement of FMN recused the growth defect of cdc15-2 fmn1-105 to cdc15-2 level, however, the high concentration of FMN also rendered the cdc15-2 cells sicker than growing on medium without supplement. These observations indicated that the MEN activation requires proper concentration of cellular FMN.
The function of Bub2-Bfa1 antagonizes Lte1 to inhibit the MEN activity. Deletion of BUB2 completely suppressed the proliferation defect of the lte1Δ mutant (Chan and Amon, 2010). This observation indicated that BUB2 deletion in lte1Δ cells restored the MEN activity. I deleted BUB2 in cells harboring the set1Δ lteΔ, and found the deletion
of BUB2 rescued the lethality of set1Δ lteΔ, consistent to the previous study (Chan and Amon, 2010; Hwang, et al., 2009) (Figure 9A). To shed light on whether the MEN activity could be restrained in fmn1-105 lte1Δ cells, I then deleted BUB2 in cells harboring the fmn1-105 lteΔ. The deletion of BUB2 also rescued the growth defect of fmn1-105 lteΔ mutant, indicating that the sickness of the fmn1-105 lte1Δ was due to the
restrained MEN activity (Figure 9B). Interestingly, the deletion had no effect on the viability of fmn1-105 set1Δ, suggesting that the interaction between FMN1 and SET1 may not lie at the process of mitosis (Figure 9C). In summary, FMN1 and SET1 may contribute independent roles to MEN which regulating mitotic exit and the genetic interaction between FMN1 and LTE1 resides at the regulation MEN activity.
5 Fmn1 activity is required for metaphase-anaphase transition
The mitotic exit regulation is an important function for anaphase progression (Queralt and Uhlmann, 2008). The anaphase progression is critically regulated by Cdc14 (Mohl, et al., 2009). The Cdc14 phosphatase is essential for both CDK down regulation and reversal of the phosphorylation events mediated by CDK (Stegmeier and Amon, 2004). In addition to MEN, the FEAR (CdcFourteen Early Anaphase Release) network is prior to control the Cdc14 activity during early anaphase (Stegmeier, et al., 2009). The previous result suggested that Fmn1 functions in parallel to MEN, to
understand whether Fmn1 functions in the FEAR, I tested whether the deletion of Slk19,
51
a component of FEAR would lead to cells growth defect in fmn1-105 mutants. The interactions of slk19Δ and fmn1-105 (Figure 10A) were relatively neutral, suggesting that FMN1 may be epistatic to the FEAR pathway.
Another FEAR component, Spo12, contributes to FEAR by promoting
dissociation of Cdc14 from the RENT (REgulator of Nucleolar silencing and Telophase) complex. I also found that the viability of spo12Δ fmn1-105 double mutants were the same as the fmn1-105 single mutant (Figure 10B). To ensure the previous reported that the spo12Δ lte1Δ double mutant exhibited lethality to cells, I deleted both of them and kept the cells alive by a centromeric URA3-based plasmid expressing LTE1. I treated cells with 5-FOA, leading the cells to abandon the plasmid. The result confirmed that the spo12Δ lte1Δ was inviable (Figure 10C). These observations indicate that the function of FMN1 is epistatic to FEAR and in parallel to MEN.
At the metaphase-to-anaphase transition the anaphase promoting complex or cyclosome (APC/C) primes Pds1 for ubiquitin mediated degradation and promotes the FEAR (Rock and Amon, 2009). Ras2 signal is important for the metaphase-anaphase transition. A previous study indicated that APC/C defective mutants are suppressed by reduced Ras signaling activity, by a deletion of the RAS2 gene (Irniger, et al., 1999). If the Fmn1 is epistatic to regulates the APC/C, deletion of RAS2 should rescue the viability of fmn1-105. Indeed, fmn1-105 ras2Δ cells exhibited significant improvement
of growth at 33°C relative to the fmn1-105 single mutants (Figure 10D). The viability of fmn1-105 cells could be partially suppressed by the perturbation of Ras2 function,
suggesting that Ras2 may inhibit Fmn1 protein kinase activity.
The spindle assembly checkpoint (SAC) inhibits the APC/C functions through the mitotic checkpoint complex and arrests cell at metaphase when present not proper attachment of the spindle. BUB3 is a non-essential gene whose product is involved in SAC (Hoyt, et al., 1991;Roberts, et al., 1994). Like other spindle checkpoint mutants, bub3 loss-of-function mutants are sensitive to Benomyl and cannot delay cell division
in response to spindle depolymerization. To further define the role of Fmn1 in metaphase-anaphase transition, I deleted the gene encoding SAC component in the fmn1-105 strain. Deletion of BUB3 enhanced the sensitivity of fmn1-105 cells to
Benomyl at 33 degrees (Figure 11A). The bub3Δ cells display more resistance to Benomyl at 33 degrees than 30 degrees may due to high temperature accelerate the break down rate of Benomyl (Mallat, et al., 1977). This data indicated that fmn1-105 mutant required intact SAC to grow when the proper chromosomes attachment is lost.
To determine whether the dependence of fmn1-105 on the SAC defect in metaphase-anaphase transition, I treated SAC defective cells with Nocodazole.
Nocodazole is a drug that induces microtubules disassembly; therefore, it destroys the mitotic spindle and activates the spindle assembly checkpoint (Künkel, 1980).