國立臺灣大學藥學專業學院藥學研究所 碩士論文
Graduate Institute of Pharmaceutical Sciences School of Pharmacy
National Taiwan University Master Thesis
探討 Sildenafil 增強化療藥物 Doxorubicin 在人類荷爾蒙 不依賴型前列腺癌細胞之抗癌作用機轉
Mechanism Study of Sildenafil as a Chemosensitizer of Doxorubicin against Hormone-Refractory Prostate
Cancers
張若凡 Jo-Fan Chang
指導教授:顧記華 博士 Advisor:Jih-Hwa Guh, Ph. D.
中華民國 105 年 7 月 July 2016
致謝
時光飛逝,想當初剛進臺大藥學所時,其實滿是慌張的情緒,身邊的朋友都 到了不同學校不同系所就學,瞬間覺得自己好像少了什麼支柱似的。但是幸運的 是,顧老師讓我有機會來到這個充滿溫馨關懷的實驗室進行研究,開啟了我充實 又愉快的碩班生涯。
很感謝顧老師總能在繁忙的事務當中仍不忘關心我的實驗、選課、助教工作 和生涯規劃等等,並給予我許多指導與建議。每每看著老師忙碌的背影,都總是 很敬佩您那樣認真盡責的做事態度,這也是我在老師身上學到的寶貴精神,希望 我之後也能保有一顆熱忱不滅的心去完成所有的挑戰。
感謝口試委員蕭哲志老師、黃聰龍老師、許麗卿老師以及指導教授顧記華老 師,謝謝您們撥冗審閱論文,並細心的指導給予建議,讓本論文變得更加完整。
也很感謝實驗室成員一直以來的照顧,像是我的師父瑞苓學姊,各種實驗疑 難雜症都最喜歡跟妳一起討論,妳做事快又準且思路清楚根本是偶像;佳純學姊 總是觀察力入微,教我實用的實驗小技巧並耐心地跟我分享選課經驗;還有婉禎,
我大概忘不了我剛進實驗室時,妳還陪我一起參加迎新,讓我不那麼害怕新環境,
妳總是那麼貼心; 還有考前幫忙提及重點之記憶力超好的美玲學姊;和開朗活潑的 社紅學姐與老師間有趣的對答,讓實驗室常常歡笑不斷;還有學妹們敬婷、庭卉、
緒昶,跟妳們一起參加研討會、聊天談心真的很開心。
很感謝我的摯友依玟,總在我最煩躁無助的時候給我幫助、鼓勵,讓我再度 充滿正能量繼續努力;還有順美、欣樂,雖然我們各自在不同地方努力著,但都 用訊息、明信片相互聯繫並加油打氣。
最要感謝的是我的父母、家人,謝謝你們讓我無憂無慮的念書、做研究,讓 我沒有負擔的完成各階段的學業。很感動的是,畢業典禮時大家揪團參加(感謝東 東姊夫開車載親友團;美育姊姊犧牲假日來看我),尤其是媽媽胃不舒服,卻為了參 加我的畢典而沒有住院,讓我超級心疼。還有總是支持我興趣要我放手去做的老 爸,幫我拍了許多照片並做成影片送我當畢業禮物;以及寫論文時,不斷精神喊話 的大姊、二姊和妹妹。
太多感謝無法一一表達,只能誠摯地感謝一路上所有幫助我、支持我、激勵 我的家人、師長、朋友們,謝謝您們!
List of Abbreviations
ABC transporters ATP-binding cassette transporters
ABCB1 ATP binding cassette subfamily B member 1 ABCC10 ATP binding cassette subfamily C member 10 ABCG2 ATP-binding cassette subfamily G member 2
ADT Androgen deprivation therapy
AJCC American Joint Committee on Cancer ATM Ataxia telangiectasia mutated
ATR Ataxia telangiectasia and Rad3-related protein Bak Bcl-2 homologous antagonist/killer
Bax Bcl-2 associated X
Bcl-2 B-cell lymphoma 2
Bid BH3 interacting-domain death agonist
BSA Bovine serum albumin
Caspase Cysteine-aspartate-specific proteases CD95 Cluster of differentiation 95
Chk1 Checkpoint kinase 1
Chk2 Checkpoint kinase 2
DAPI 4',6-diamidino-2-phenylindole
DCFH-DA 2',7'-dichlorodihydrofluorescein diacetate
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
DNA-PKcs DNA-dependent protein kinase catalytic subunit
DR3 Death receptor 3
DR4 Death receptor 4
DR5 Death receptor 5
DRE Digital rectal examination
DSB DNA double-strand break
ELISA Enzyme-linked immunosorbent assay FAP-1 Fas associated phosphatase-1
FBS Fetal bovine serum
FDA Food and Drug Administration
FITC Fluoresceine isothiocyanate
FLIP Fas-associated death domain (FADD) interleukin-1-converting enzyme (FLICE)-like inhibitory protein
H2A.X Histone H2A.X
HR Homologous recombination
HRP Horseradish peroxidase
HRPC Hormone-refractory prostate cancer LHRH Luteinizing hormone-releasing hormone
NAC N-acetylcysteine
NHEJ Non-homologous end joining
PARP-1 Poly [ADP-ribose] polymerase 1
PBS Phosphate buffered saline
PBST Phosphate buffered saline with 0.1% Tween 20
PDE5 Phosphodiesterase-5
PI Propidium iodide
PI-3K Phosphoinositide 3-kinase
PIKKs Phosphatidylinositol 3-kinase-related kinases
PSA Prostate-specific antigen
RNA Ribonucleic acid
ROC Reactive oxygen species
RPA14 Replication protein A 14 kDa subunit RPA32 Replication protein A 32 kDa subunit RPA70 Replication protein A 70 kDa subunit
SD Standard deviation
siRNA Small interfering RNA
Smac/DIABLO Second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI
TNFR1 Tumor necrosis factor receptor 1 Topo IIα Topoisomerase IIα
TRUS Transrectal ultrasound
中文摘要
近年來,老藥新用在藥物開發上的發展潛力逐漸被受重視,像是威而鋼其學名
sildenafil (Phosphodiesterase-5 抑制劑),為治療男性勃起功能障礙的藥物,但在最 近研究發現,其在活體內外實驗中都具有加強 doxorubicin 毒殺賀爾蒙不依賴型前 列腺癌細胞的能力,但其相關分子機制仍不清楚。在我們的研究結果中發現,
sildenafil 本身並不會影響賀爾蒙不依賴型前列腺癌細胞 (PC-3 及 DU145) 的存活 率,但是當其與 doxorubicin 同時作用下,sildenafil 能協同性地增強 doxorubicin 所 誘導的前列腺癌細胞之細胞凋亡現象,包括增加核小體 DNA 斷裂、sub-G1 細胞數 量、內因性與外因性細胞凋亡訊號,並減少抗凋亡 Bcl-2 蛋白質家族的表現量 (Mcl-1, Bcl-xL 及 Bcl-2)。此外,值得注意的是,雖然合併使用 sildenafil 與 doxorubicin 能增加 PC-3 細胞中的氧化壓力,但當我們利用抗氧化劑 (NAC 及 trolox) 來去除 該氧化壓力後,卻無法阻止細胞凋亡的發生,意味著氧化壓力並非造成藥物合併 後細胞凋亡增加之原因。有趣的是,我們也發現 sildenafil 可以加劇其他第二型拓 譜異構酶 (etoposide 及 mitoxantrone) 所產生之細胞凋亡情況,然而 sildenafil 對於 第 一 型 拓 譜 異 構 酶 (camptothecin) 所 導 致 的 細 胞 凋 亡 則 沒 有 影 響 。 已 知 doxorubicin 毒殺癌細胞的其一機制是增加 DNA 雙骨斷裂的產生,我們進一步探討 sildenafil 對於 doxorubicin 所誘導的 DNA 損傷及其修補機制之影響。實驗結果發 現,doxorubicin 造成的 DNA 雙骨斷裂之兩種修復機制 (homologous recombination, HR; non-homologous DNA end joining, NHEJ) 都會被 sildenafil 所抑制,例如:合併 使用 sildenafil 與 doxorubicin 會減少 HR 修補機制中 RPA32 的過量磷酸化、降低 Rad51 之表現量及其在細胞核內形成 foci 的能力;並在 NHEJ 修補機制中,減少 DNA-PKcs (Thr2609)的磷酸化、抑制 Ku80 鍵結到 DNA 斷裂尾端之能力。另外,
已知 sildenafil 為 phosphodiesterase-5 (PDE5) 之抑制劑,我們也進一步探討抑制 PDE5 對於 sildenafil 增強 doxorubicin 毒殺效果的重要性。實驗結果顯示,不論是 利用其他 PDE5 活性抑制劑 (vardenafil 或 tadalafil),或是使用 siRNA 去抑制 PDE5 的表現,都可以加強 doxorubicin 之細胞毒殺效果,然而我們卻發現只有 PDE5 活 性抑制劑才能有效減少 doxorubicin 所誘導的 DNA 雙骨斷裂 HR 修補機制。總結來 說,sildenafil 可經由抑制 HR 與 NHEJ 途徑來減少 doxorubicin 所誘導的 DNA 雙骨 斷裂之修補,導致核小體 DNA 片段化與內外因性細胞凋亡信號增強,藉此增加
doxorubicin 毒殺賀爾蒙不依賴型前列腺癌細胞之能力。而目前我們不排除 PDE5 在此機制中可能扮演部分腳色,但這仍須要更進一步的研究去驗證。
關鍵字:Doxorubicin; Sildenafil; Hormone-refractory prostate cancer; HR; NHEJ;
PDE5; Apoptosis
Abstract
Drug repositioning is a potential strategy for drug development. Recently, sildenafil, a phosphodiesterase-5 (PDE5) inhibitor, has been repurposed as a chemosensitizer to synergistically potentiate doxorubicin-induced cell killing in hormone-refractory prostate cancer (HRPC) both in vitro and in vivo. However the synergistic anticancer mechanism has not been well identified. In the present study, the data demonstrated that sildenafil by itself did not affect cell survival of PC-3 and DU145 (two HRPC cell lines), but significantly enhanced cell apoptosis induced by doxorubicin, as evidenced by the synergistic increase of nucleosomal DNA fragments and sub-G1 (apoptosis) population, and the activation of both intrinsic and extrinsic apoptotic pathways. Moreover, the anti-apoptotic Bcl-2 family proteins, Mcl-1, Bcl-xL and Bcl-2, were significantly downregulated by the combinatorial treatment with sildenafil and doxorubicin. It is noteworthy that an increase in cellular ROS at early combinatorial treatment was noted; however, both ROS scavengers, NAC and trolox, dramatically abolished the ROS production but failed to inhibit cell apoptosis, indicating the sensitization mechanism beyond the oxidative stress. Interestingly, sildenafil also enhanced cell apoptosis induced by other topoisomerase II inhibitors (e.g., etoposide and mitoxantrone) but not topoisomerase I inhibitor (e.g., camptothecin).
Due to DNA-damaging properties of doxorubicin, the regulators and signaling of DNA double-strand break (DSB) and repair pathways were studied. As a result, the combinatorial treatment reduced the protein expression of hyperphosphorylated RPA32 and phosphorylated DNA-PKcs (Thr2609), which were involved in DSB repair pathways, homologous recombination (HR) and non-homologous DNA end joining (NHEJ), respectively. The defects in HR and NHEJ pathways were further substantiated by the reduced levels of nuclear Rad51 foci formation and DNA end-binding activity of
nuclear Ku80. The role of PDE5 in the sensitization mechanism was examined as well.
The data revealed that inhibition of PDE5 activity by two other inhibitors, vardenafil or tadalafil, or PDE5 knockdown by siRNA potentiated the cell-killing effect of doxorubicin. However, only PDE5 inhibitors but not PDE5 knockdown reduced the HR-mediated DSB repair in response to doxorubicin. In conclusion, the data suggest that sildenafil enhances doxorubicin-induced apoptosis in HRPC through the impairment of HR and NHEJ-mediated DSB repair systems, leading to synergistic increase of nucleosomal DNA fragments and activation of both intrinsic and extrinsic apoptotic pathways. Inhibition of PDE5 is, at least partly, responsible for the sensitization mechanism.
Key words: Doxorubicin; Sildenafil; Hormone-refractory prostate cancer; HR; NHEJ;
PDE5; Apoptosis
Contents
口試委員會審定書 ... i
致謝... ii
List of Abbreviations ... iii
中文摘要 ... v
Abstract.. ... vii
Contents. ... ix
Aim of the study ... 1
Chapter 1: Introduction ... 3
1.1. Prostate ... 3
1.2. Prostate cancer ... 3
1.3. Human Prostate cancer cell lines ... 6
1.4. Doxorubicin ... 7
1.5. Sildenafil ... 7
1.6. Cell death ... 10
1.7. Apoptosis ... 11
1.8. Oxidative stress ... 14
1.9. DNA double-strand break signaling and repair ... 15
Chapter 2: Materials and Methods... 19
Chapter 3: Results ... 26
3.1. Effect of doxorubicin and sildenafil on cell cycle progression in PC-3 and DU145 cells... 26 3.2. Validation of sildenafil-mediated sensitization of doxorubicin-induced
apoptosis... 26
3.3. Effect of doxorubicin or/and sildenafil on the expression of Bcl-2 family proteins ... 27
3.4. Effect of doxorubicin or/and sildenafil on ROS production ... 28
3.5. Effect of sildenafil on doxorubicin-induced DNA double-strand break signaling and repair system ... 28
3.6. Effect of combinatorial treatment on DNA end-binding capacity and protein expression of Ku80 ... 30
3.7. Effect of combinatorial treatment on nuclear foci formation and expression of Rad51 ... 30
3.8. Effect of other PDE5 inhibitors on doxorubicin-induced cell apoptosis ... 31
3.9. Effect of other PDE5 inhibitors on HR-mediated repair of doxorubicin-induced DSB ... 31
3.10. Effect of PDE5 knockdown on doxorubicin-induced cell death and DSB signaling and repair ... 32
3.11. Effect of sildenafil on the sensitization of apoptosis induced by other topoisomerase inhibitors ... 33
Chapter 4: Discussions ... 34
4.1. Effect of sildenafil on apoptosis induced by doxorubicin in PC-3 cells ... 34
4.2. The role of ROS production in the sensitization mechanism ... 35
4.3. Effect of sildenafil on doxorubicin-induced DNA double-strand break signaling and repair ... 36
4.3.1. DNA double-strand break signaling ... 37
4.3.2. DNA double-strand break repair ... 38 4.4. Effect of PDE5 inhibitors or PDE5 knockdown on doxorubicin-induced cell
death and DSB signaling and repair ... 41 4.5. Effect of other topoisomerase inhibitors or/and sildenafil on cell apoptosis. 42 Chapter 5: Conclusion ... 44 Tables
Table 1. The stages and estimated 5-year survival rates for prostate cancers ... 5 Table 2. The most commonly used human prostate cancer cell lines for research . 6 Table 3. PDE5 inhibitors approved by FDA. ... 8 Table 4. Proposed mechanisms for sildenafil as a chemosensitizer in cancers... 9 Figures
Figure 1. Effect of doxorubicin and/or sildenafil on cell cycle distribution in PC-3 cells. ... 46 Figure 2. Effect of doxorubicin and/or sildenafil on cell cycle distribution in
DU145 cells. ... 47 Figure 3. Effect of sildenafil on doxorubicin-induced apoptosis in PC-3 cells. ... 49 Figure 4. Effect of doxorubicin and/or sildenafil on the expression of Bcl-2 family in PC-3 cells. ... 51 Figure 5. Effect of doxorubicin and/or sildenafil on ROS production in PC-3 cells.
... 52 Figure 6. Effect of ROS scavengers NAC and trolox on cell apoptosis induced by
doxorubicin and sildenafil. ... 53 Figure 7. Effect of doxorubicin and/or sildenafil on the integrity of chromosomal
DNA in PC-3 cells. ... 54 Figure 8. Effect of sildenafil on doxorubicin-induced DNA double-strand break
(DSB) signaling and repair in PC-3 cells. ... 57 Figure 9. Effect of sildenafil on doxorubicin-induced DNA double-strand break
(DSB) signaling and repair in DU145 cells. ... 58 Figure 10. Effect of doxorubicin and/or sildenafil on DNA end-binding capacity,
total and nuclear expression of Ku80 in PC-3 cells. ... 59 Figure 11. Effect of doxorubicin and/or sildenafil on the nuclear foci formation
and expression of Rad51 in PC-3 cells. ... 61 Figure 12. Effect of other PDE5 inhibitors, vardenafil and tadalafil, on
doxorubicin-induced cell apoptosis in PC-3cells. ... 63 Figure 13. Effect of other PDE5 inhibitors, vardenafil and tadalafil, on
doxorubicin-induced cell apoptosis in DU145 cells. ... 64 Figure 14. Effect of PDE5 inhibitors, vardenafil and tadalafil, on HR-mediated
repair of DSB induced by doxorubicin in PC-3 cells. ... 65 Figure 15. Correlation between Rad51 expression and the level of nucleosomal
DNA fragments (apoptosis) in PC-3 cells treated with doxorubicin or in combination with different PDE5 inhibitors. ... 66 Figure 16. Effect of PDE5 knockdown on doxorubicin-induced cell death in
PC-3cells. ... 67 Figure 17. Effect of PDE5 knockdown on doxorubicin-induced DSB signalling
and repair in PC-3 cells. ... 68 Figure 18. Effect of other topoisomerase inhibitors and/or sildenafil on cell death
in PC-3 cells. ... 70 Figure 19. A schematic of how sildenafil sensitizes HRPC cells to chemotherapy
drug doxorubicin. ... 71 Appendixes ... 72 References ... 76
Aim of the study
According to the statistics from the World Health Organization in 2012, prostate cancer is the second most frequently diagnosed cancer and the fifth-leading cause of cancer death in men worldwide1, 2. Prevalence of prostate cancer is generally higher in developed countries than in developing countries. Based on the American Cancer Society’s estimates for 2016, prostate cancer becomes the most common cancer among American men, and it is the second-leading cause of cancer death in men3. In Taiwan, cause of death statistics reported by the Ministry of Health and Welfare in 2014 show that prostate cancer is the seven-leading cause of cancer death in men4. Besides, the incidence and mortality rate of prostate cancer in Asian populations have gradually increased these years5. Moreover, since prostate cancer mainly occurs in men aged 65 or older, the incidence rate of prostate cancer in the aging society may be continuously rising.
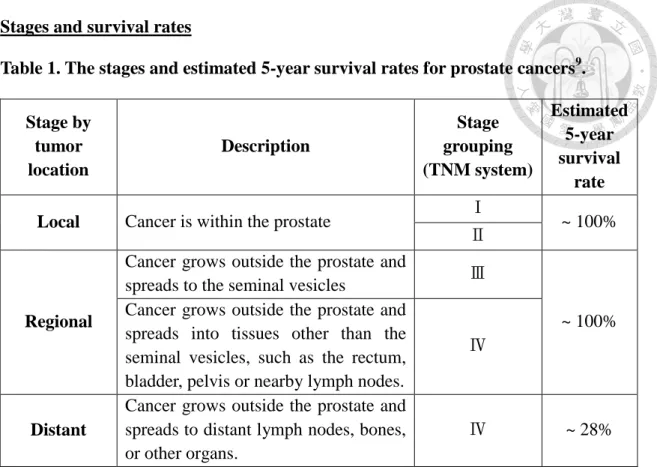
Based on results of prostate biopsy graded with American Joint Committee on Cancer (AJCC) TNM system, prostate cancer can be categorized into four stages. For stage І and Ⅱ,patients have no symptoms and cancer has not spread outside of the prostate. Treatment options usually include active surveillance, radical prostatectomy, and radiation therapy. 5-year survival rate of the early stages is almost 100%. For stage
Ⅲ in which tumor has grown outside the prostate and may have spread to the seminal vesicles, the radiation plus hormone therapy is necessary. After treatment, the 5-year survival rate of this stage is still nearly 100%. However, patients with stage Ⅳ prostate cancer, which may has spread to nearby lymph nodes and bones, the 5-year survival rate decreases to 28%. In general, different types of hormone therapies such as luteinizing hormone-releasing hormone (LHRH) analog, LHRH antagonist, anti-androgen and
estrogens are used initially to treat advanced prostate cancer. Unfortunately, nearly all metastatic prostate cancers treated with hormone therapies become resistant after a period of months or years. When prostate cancer no longer responses to hormone therapy, it is referred as hormone-refractory prostate cancer (HRPC). Chemotherapy is a key therapy for HRPC treatment. Among the chemotherapeutic agents, doxorubicin is one of the treatment options6. However, the major side effect of the drug is cumulative dose-dependent cardiotoxicity7. To minimize the side effect, combination of doxorubicin with other drugs may provide a good regimen by lowering the dosage while retaining the therapeutic function of doxorubicin6. Recently, sildenafil, an FDA-approved PDE5 inhibitor, has been repurposed to potentiate doxorubicin-induced killing in HRPC both in vitro and in vivo8. Nevertheless, the synergistic anticancer mechanism of doxorubicin combined with sildenafil has not yet been well identified.
Therefore, the aim of this study is to delineate the sensitization mechanism of sildenafil in HRPC.
Chapter 1: Introduction
1.1. Prostate
The prostate is a gland found only in male reproductive and urinary systems. It lies below the bladder and in front of the rectum. The function of the prostate gland is to produce some of weak alkaline fluid that is part of semen, which neutralize the acidity of the vaginal tract and prolong the lifespan of sperm. The growth and function of the prostate gland relies on androgens (male hormones) produced by the testes 9, 10.
1.2. Prostate cancer
Prostate cancer is a malignant tumor that starts when cells in prostate grow uncontrollably. Almost all prostate cancers (95%) belong to adenocarcinomas, which develop from the gland cells in the prostate. Although some of prostate cancers can grow and spread rapidly, most prostate cancers usually grow slowly 9, 10.
Incidence and mortality rates
According to the global cancer fact in 2012 estimated by the World Health Organization, prostate cancer is the second most frequently diagnosed cancer and the fifth-leading cause of cancer death in men worldwide1, 2. Based on the American Cancer Society’s estimates for prostate cancer for 2016, prostate cancer is the most common cancer in American men, aside from skin cancer, and it is the second leading cause of cancer death3. In Taiwan, cause of death statistics reported by the Ministry of Health and Welfare in 2014 show that prostate cancer is the sixth-leading cause of cancer death in men4.
Causes and risk factors
The exactly causes that lead to prostate cancer have not been well-identified. But some risk factors can be referred, such as age over 50, African American ethnicity,
developed countries in the western world, inherited gene mutations (RNASEL, BRCA1/2, MSH2 and MLH1 or HOXB13), having higher levels of androgens, diet, obesity, etc9. Symptoms
Early stage of prostate cancer usually has no symptoms. More advanced prostate cancers may have problems urinating, blood in the urine or semen, erectile dysfunction, bone pain, leg pain and foot pain, etc9, 10.
Diagnostic tests
1. Medical history and physical examination: Doctors diagnose prostate cancer through asking patients some questions about family history of prostate cancer and any changes in bladder habits, then using digital rectal examination (DRE) to check prostate for any lumps or changes in size and shape.
2. Prostate-specific antigen (PSA) test: PSA is a protein made by prostate gland.
When the PSA level becomes abnormal in the blood, it may indicate the presence of prostate cancer. The higher the PSA level, the more possibly that prostate cancer occurs.
3. Transrectal ultrasound (TRUS): Image the prostate by using an ultrasound probe placed into the rectum. It is used to measure the size of prostate and check for abnormal areas.
4. Biopsy: During a biopsy, 6-12 biopsies were taken from different areas of prostate using a core needle biopsy. The collected tissues are tested in a laboratory to confirm whether cancer cells are present. If cancer cells exist, Gleason score is used to describe how aggressive cancer cells are.
Stages and survival rates
Table 1. The stages and estimated 5-year survival rates for prostate cancers9. Stage by
tumor location
Description
Stage grouping (TNM system)
Estimated 5-year survival
rate Local Cancer is within the prostate Ⅰ
~ 100%
Ⅱ
Regional
Cancer grows outside the prostate and
spreads to the seminal vesicles Ⅲ
~ 100%
Cancer grows outside the prostate and spreads into tissues other than the seminal vesicles, such as the rectum, bladder, pelvis or nearby lymph nodes.
Ⅳ
Distant
Cancer grows outside the prostate and spreads to distant lymph nodes, bones, or other organs.
Ⅳ ~ 28%
Prostate cancer treatments
Depends on the different stages of prostate cancer, ages and health states of patients, treatment options for men with prostate cancer may include9-11:
1. Watchful waiting or active surveillance: For men whose cancer is small and slow-growing, treatments such as surgery or radiation may not be suitable, because the side effects of these treatments may outweigh the benefits for the early stage prostate cancer. Therefore, active monitoring the prostate cancer using DRE and PSA blood test, or less intensive follow-up may be adopted.
2. Surgery: Radical prostatectomy is the main type of surgery to completely remove the localized prostate cancer plus some of the tissue nearby it, including the seminal vesicles.
3. Radiation therapy: Radiation therapy uses high-energy rays to kill cancer cells. The types of radiation therapy for prostate cancer include external beam radiation and
brachytherapy (internal radiation therapy). They are usually combined with hormone therapy for treatment of more advanced prostate cancer.
4. Hormonal therapy: It is also known as androgen deprivation therapy (ADT), which slows the growth of prostate cancer by lowering the level of male hormones (androgens) in the body, or blocking the action of androgen from affecting prostate cancer cells. There are several types of hormone therapy can be used, including luteinizing hormone–releasing hormone (LHRH) agonists, LHRH antagonists, anti-androgens, CYP17 inhibitor or removal of the testicles (orchiectomy).
5. Chemotherapy: It is used to treat metastatic hormone-refractory prostate cancer.
Some of chemotherapy drugs used to treat prostate cancer include:
First-line chemotherapy combination: Docetaxel and a steroid drug prednisone Second-line chemotherapy combination: Cabazitaxel and prednisone
Palliative chemotherapy: mitoxantrone and prednisone Others:Estramustine, epirubicin, paclitaxel, etc.
1.3. Human Prostate cancer cell lines
Table 2. The most commonly used human prostate cancer cell lines for research12,
13:
Cell lines Source Androgen
sensitivity p53 PTEN
PC-3 Lumbar
metastasis Insensitive Deletion mutation
Deletion mutation
DU145 Brain
metastasis Insensitive Missense
mutation PTEN
+/−
LNCaP Lymph node
metastasis Sensitive Silent mutation
Nonsense mutation
1.4. Doxorubicin
Doxorubicin is a chemotherapy drug belonging to a class of compounds with similar structures, named anthracyclines. Doxorubicin was first isolated from Streptomyces peucetius var. caesius, a soil bacterium, in the 1970’s14, and has shown great efficacy to kill both solid and liquid tumors. It has been routinely used for the treatment of several cancers including breast, lung, gastric, ovarian, thyroid, non-Hodgkin’s and Hodgkin’s lymphoma, multiple myeloma, sarcoma and pediatric cancers15. Despite the common use of doxorubicin in the clinics, the mechanisms of doxorubicin are still not fully understood. So far, there are two proposed anticancer mechanisms of doxorubicin that are widely accepted. First, acting like almost all anthracycline drugs, doxorubicin intercalates into DNA and disrupts the topoisomerase-II (Topo II)-mediated DNA repair, leading to DNA double strand break.
Second, redox-dependent metabolism of doxorubicin generates free radicals which damage the cellular membranes, DNA and proteins16. In addition to these two mechanisms, other models have also been proposed to explain the doxorubicin-mediated cell death, such as Topo-II-independent DNA adduct formation, ceramide overproduction and intercalation-induced DNA torsional stress and nucleosome destabilization17. Although doxorubicin exerts excellent killing effects in many cancers, its side effect of cumulative and dose-dependent cardiotoxicity has limited its usage7.
1.5. Sildenafil
Sildenafil, also known as Viagra, is a cGMP-specific phosphodiesterase type 5 (PDE5) inhibitor. It has been approved by FDA for the indications of erectile dysfunction and pulmonary arterial hypertension since 1998 and 2005 respectively18, 19.
The selective vasodilatory mechanism of PDE5 inhibitors in penis and lung has been well identified. PDE5 is an enzyme that hydrolyzes second messenger molecule cGMP into biologically inactive 5’GMP. Therefore, sildenafil can increase levels of cGMP by inhibiting the degradation of cGMP via PDE5. PDE5 is found in particularly high concentrations in the corpus cavernosum and lung vasculature. The elevated cGMP in these tissues activates the downstream cGMP-dependent protein kinase (PKG) and cGMP-gated ion channels, finally resulting in the reduction in the intracellular calcium level. The decreased calcium level in cells leads to the relaxation of vascular smooth muscle, thereby increasing the blood flow in penis and lung. Of note, the activation of the NO/cGMP system by sexual stimulation or alveolar distension of lung is the prerequisite for sildenafil to exert its functions through cGMP/PKG signaling in penis or lung20.
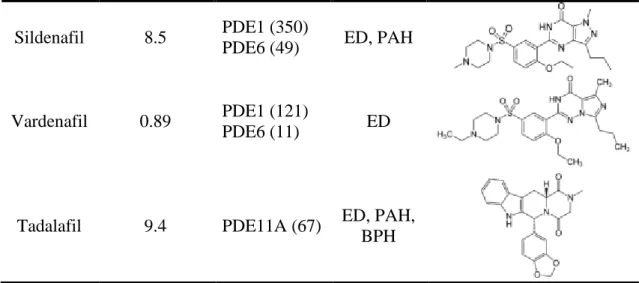
Other PDE5 inhibitors
Table 3. PDE5 inhibitors approved by FDA21
PDE5 inhibitor
IC50 for human recombinant PDE5A (nM)
Selectivity
(nM) Indication Structure
Sildenafil 8.5 PDE1 (350)
PDE6 (49) ED, PAH
Vardenafil 0.89 PDE1 (121)
PDE6 (11) ED
Tadalafil 9.4 PDE11A (67) ED, PAH, BPH
* ED: Erectile dysfunction; PAH: Pulmonary arterial hypertension; BPH: Benign prostatic hyperplasia
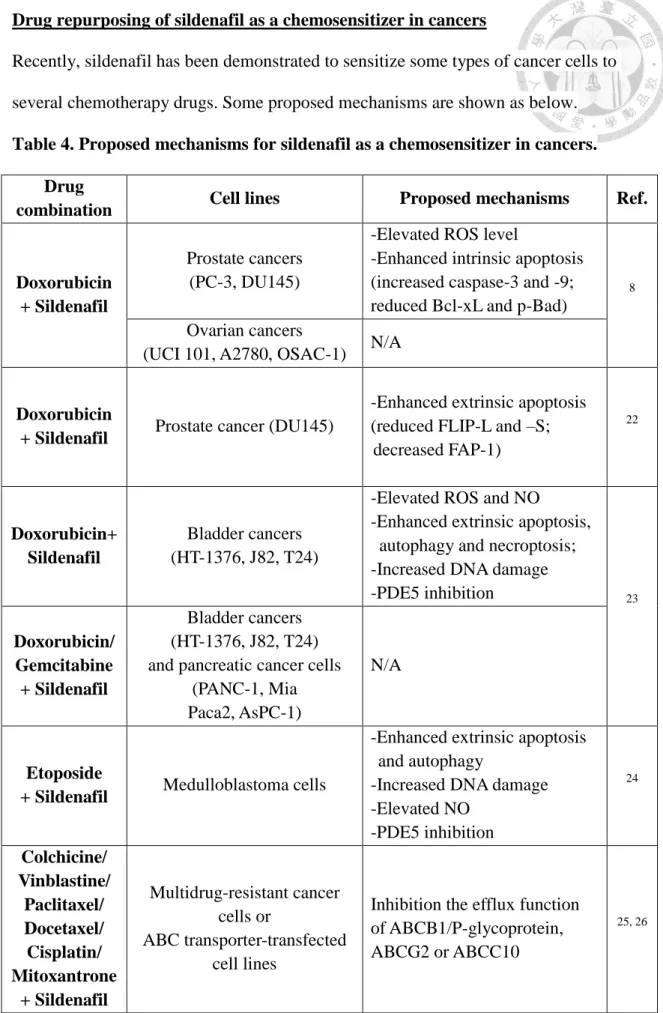
Drug repurposing of sildenafil as a chemosensitizer in cancers
Recently, sildenafil has been demonstrated to sensitize some types of cancer cells to several chemotherapy drugs. Some proposed mechanisms are shown as below.
Table 4. Proposed mechanisms for sildenafil as a chemosensitizer in cancers.
Drug
combination Cell lines Proposed mechanisms Ref.
Doxorubicin + Sildenafil
Prostate cancers (PC-3, DU145)
-Elevated ROS level
-Enhanced intrinsic apoptosis (increased caspase-3 and -9;
reduced Bcl-xL and p-Bad)
8
Ovarian cancers
(UCI 101, A2780, OSAC-1) N/A
Doxorubicin
+ Sildenafil Prostate cancer (DU145)
-Enhanced extrinsic apoptosis (reduced FLIP-L and –S;
decreased FAP-1)
22
Doxorubicin+
Sildenafil
Bladder cancers (HT-1376, J82, T24)
-Elevated ROS and NO
-Enhanced extrinsic apoptosis, autophagy and necroptosis;
-Increased DNA damage
-PDE5 inhibition 23
Doxorubicin/
Gemcitabine + Sildenafil
Bladder cancers (HT-1376, J82, T24) and pancreatic cancer cells
(PANC-1, Mia Paca2, AsPC-1)
N/A
Etoposide
+ Sildenafil Medulloblastoma cells
-Enhanced extrinsic apoptosis and autophagy
-Increased DNA damage -Elevated NO
-PDE5 inhibition
24
Colchicine/
Vinblastine/
Paclitaxel/
Docetaxel/
Cisplatin/
Mitoxantrone + Sildenafil
Multidrug-resistant cancer cells or
ABC transporter-transfected cell lines
Inhibition the efflux function of ABCB1/P-glycoprotein, ABCG2 or ABCC10
25, 26
1.6. Cell death
There are many types of cell death which can be classified according to its morphological appearance (which may be apoptotic, necrotic or autophagic), enzymological preference (the involvement of nucleases or of specific classes of proteases, such as caspases, calpains or cathepsins), functional aspects (programmed or accidental) or immunological characteristics (immunogenic or non-immunogenic)27. Among all types of cell death, programmed cell death has been regarded as a barrier to restrict cancer cells from keep surviving. Apoptosis, autophagy and necroptosis are the most common programmed cell death targeted by anticancer therapy nowadays28. Apoptosis
Apoptosis was first described by Kerr et al. in 1972, and is characterized with respect to the morphological changes in dying cells, including cell shrinkage, nuclear condensation and fragmentation, membrane blebbing, loss of adhesion to the neighboring cells and detachment from the extracellular matrix29. Biochemical changes include cleavage of high molecular weight of chromosomal DNA into internucleosomal fragments, phosphatidylserine externalization and numerous cleavages of intracellular substrate through specific proteolytic process. Of note, since apoptosis does not cause localized inflammatory response and damage to surrounding tissues, it has been considered as a useful target in cancer therapy30.
Autophagy
Autophagy, is a catabolic process in which autophagosomes, double membrane-bound structures, encapsulate cytoplasmic macromolecules and organelles and destine these cellular components for degradation and renewal. Autophagy is an evolutionarily conserved strategy for cell to survive under stress conditions, such as nutrient deprivation, ROS, hypoxia, drug stimuli, endoplasmic reticulum stress, etc.
However, when coping with excessive stress or being dysregulated, autophagy may lead to non-apoptotic cell death. In cancers, autophagy plays dual roles, either tumor suppressor or protector31. The regulation of autophagy in cancers is complicated and depends on different cell conditions28.
Necroptosis
Necroptosis, different from necrosis, is a form of programmed necrosis, which can be executed by regulated mechanisms like apoptosis, but in a caspase-independent manner. Necroptosis can be induced by the activation of the TNF receptor superfamily, Toll-like receptors (TLRs), cellular metabolic and genotoxic stresses or various anticancer agents. One of the critical characteristics of necroptosis is to form necrosome by receptor-interacting protein kinase 1 (RIP1) and RIP3. The formation of the necrosome can be inhibited by chemical compounds such as necrostatin-1, which is the most widely used agent to detect necroptosis in cells. The advantage of the necroptosis is that it can target apoptosis-resistant forms of cancers28, 32.
1.7. Apoptosis
Apoptosis is the major type of programmed cell death and can be divided into two core pathways, the extrinsic death receptor-mediated apoptosis and intrinsic mitochondria-dependent apoptosis (Appendix 1).
Extrinsic death receptor-mediated apoptosis
Type 1 TNF receptor (TNFR1) and Fas (CD95) are two most well-known death receptors (DR), and their corresponding ligands are TNF and Fas ligand (FasL). The intracellular portion of death receptor is known as the death domain (DD). Once three or more DR-ligands bind to DR, they initiate trimerization of DR in which DD were brought together to form a binding site for the adaptor protein. The specific adaptor
proteins respectively for TNFR1 and Fas are called TNF receptor-associated DD (TRADD) and Fas-associated DD (FADD). The function of TRADD or FADD is to recruits pro-caspase-8 to form a complex of ligand-receptor-adaptor protein-pro-caspase-8, named the death-inducing signaling complex (DISC), which is responsible for the cleavage of pro-caspase-8 to caspase-8. Activated caspase-8 can then stimulate apoptosis through activation of caspase-3, or promote crosstalk signaling to enhance intrinsic apoptosis by truncated Bid (tBid)33.
Intrinsic mitochondria-dependent apoptosis
The intrinsic pathway is initiated by the cellular stimuli such as DNA damage, oxidative stress, growth-factor deprivation, hypoxia, etc. Regardless of the stimuli, this pathway eventually increases mitochondrial permeability and releases several pro-apoptotic molecules such as cytochrome-c, Smac/DIABLO, apoptosis-inducing factor (AIF) and endonuclease-G into the cytoplasm. Among these released molecules, cytochrome c bind to cytosolic apoptotic protease activating factor 1 (Apaf-1), procaspase-9 and ATP to form apoptosome, which subsequently activates downstream caspases to induce apoptosis. The intrinsic apoptosis induced by this pathway is closely regulated by the Bcl-2 family33, 34.
Bcl-2 family
Proteins in Bcl-2 family are subdivided into three groups based on structural and functional similarities. Group I is multi-domain (BH1-BH4) anti-apoptotic proteins including Bcl-2, Bcl-xL, Mcl-1, Bcl-w, Bcl-b and A1. Groups Ⅱ is multi-domain (BH1-BH4) pro-apoptotic proteins, including Bax, Bak and Bok. Groups Ⅲ is BH-3 only proteins, containing Bim, Bad, tBid, PUMA, NOXA, Bik, Bmf and Hrk35 (Appendix 2).
In general, anti-apoptotic Bcl-2 family members can promote ADP/ATP exchange
and prevent the opening of permeability transition pores, thus maintaining the mitochondrial membrane permeability. In contrast, both pro-apoptotic Bak and Bax can oligomerize to form a pore on the outer mitochondrial membranes, leading to release of pro-apoptotic substances from the mitochondria. Several models have been proposed to understand the consequence of the interactions between these three subgroups of Bcl-2 protein family. In the “displacement” or called “indirect activation” model, anti-apoptotic Bcl-2 family members such as Bcl-2, Bcl-xL and Mcl-1 directly bind to pro-apoptotic Bak or Bax to prevent them from forming oligomeric pore on the outer membrane of mitochondria. In another model called “direct activation”, BH3-only proteins with high affinity to Bax and Bak are termed as “activators,” while those that only bind the anti-apoptotic proteins are named “sensitizers.” The activator BH3 such as Bim and puma directly interact with and activate Bax, Bak and tBid to promote mitochondrial outer membrane permeabilization (MOMP). While the anti-apoptotic proteins such as Bcl-2 and Bcl-xL indirectly inhibit MOMP by binding to BH3 activators. However, the BH3 sensitizer proteins such as Bmf and Bad can further interact with anti-apoptotic proteins, thus releasing the BH3 activator for promoting MOMP by activation and oligomerization of Bax and Bak36, 37.
Caspase
Caspases, cysteine-aspartate-specific proteases, are a family of protease enzymes playing important roles in apoptosis. Caspase has a cysteine in its active site that hydrolyzes target protein at the C-terminal of an aspartic acid amino acid. Caspases have been generally classified by their known roles in apoptosis (caspase-2, -3, -6, -7, -8, -9 and -10), and in inflammation (caspase-1, -4, -5, -12). Caspases involved in apoptosis can be further subclassified by their mechanism of actions as either initiator caspases (caspase-8 and -9) or effector caspases (caspase-3, -6, and -7). Caspases are initially
produced as inactive monomeric pro-caspases, which can be cleaved and activated by either exposure to another activated caspase, autocatalysis or association with an activator protein complex, for instance apoptosome38.
PARP cleavage
PARP-1, also known as NAD+ ADP-ribosyltransferase 1, is a nuclear enzyme that catalyzes the transfer of poly(ADP-ribose) from its substrate β-NAD+ onto itself and other nuclear proteins in response to DNA single-strand breaks in the base excision repair pathway. During apoptosis, activated caspases-3 and -7 have been shown to cleave PARP-1 into fragments of 89 and 24 kDa. The cleaved PARP-1 becomes incapable of responding to DNA damage. Therefore, PARP cleavage with fragments of 89 or 24 kDa has become a useful hallmark of apoptosis39, 40.
1.8. Oxidative stress
Oxidative stress occurs in the biological systems when the levels of oxidants or reactive oxygen species (ROS) overwhelm the antioxidants or radical scavenging mechanisms, leading to damages of cellular components including proteins, lipids and DNA. ROS are molecules derived from oxygen that have accepted extra electrons and can oxidize other molecules. ROS includes hydrogen peroxide (H2O2), singlet oxygen (1O2), superoxide (O2−∙), hydroxyl radical (OH∙), etc. Mitochondrial respiratory chain and various intracellular enzymes such as NADPH oxidase, xanthine oxidase and lipooxygenases are all endogenous ROS generators. In normal cellular metabolism, the optimal amount of ROS plays an important role in signal transduction. On the other hand, ROS can also be produced from exogenous sources such as radiation and chemotherapies, which has been one kind of mechanism to kill cancer cells. To reduce excessive ROS, cells are equipped with different enzymatic antioxidant defenses such as
superoxide dismutases and catalase, and nonenzymatic antioxidants including glutathione and thioredoxin41.
1.9. DNA double-strand break signaling and repair Formation of DNA double-strand breaks (DSBs)42
DSBs can be generated in various routes. In addition to “programmed” DSB formation during V(D)J recombination, class switch recombination and meiosis, DSB can also be formed by the “accidental” events, such as ionizing radiation, treatment of radiomimetic drugs like topoisomerase II poisons, or topoisomerase I poisons/crosslinking agents-mediated replication fork collapse (one-ended DSB).
DSB-induced DNA damage response43-45
The DNA damage response (DDR) is a network of cellular pathways that sense DNA lesions, activate cell cycle checkpoints and allow DNA repair during cell cycle arrest to avoid the generation of deleterious mutations. When the amount of DNA damage exceeds the repair capacity, DDR signaling will eliminate damaged cells by triggering apoptosis or senescence. In general, three main roles are involved in the DDR—DNA damage sensors, signal transducers and effectors.
When DSBs occur, the DSB sensor, MRE11/RAD50/NBS1 (MRN) complex, will bind to and recruit partially autoactivated ATM, one of phosphoinositide 3-kinase (PI3K)-like protein kinases, to the DSB site, then inducing fully activation of ATM46. Activated ATM phosphorylates numerous local substrates including histone variant H2A.X (γ-H2A.X) around DSB. γ-H2A.X can extend megabase pair distances from the DSB and trigger histone modifications around the DSB to increase DNA accessibility for downstream protein assembly.
DNA damage sensors transmit signals to transducers, which then amplify and
transduce signals to downstream effectors to result in cell cycle arrest. Chk2 and Chk1 are important transducers of ATM and ATR in response to DSB and DNA single-strand break (SSB) respectively. ATM/Chk2 and ATR/Chk1 pathways were historically thought to act in parallel with overlapping functions. However, more recently studies found these two pathways can be an upstream-downstream relationship, which explains why both ATM/Chk2 and ATR/Chk1 are activated and responsible for DSB-induced cell cycle arrest47-50. The proposed model45 (Appendix 3) suggests that upon DSBs, MRN complex recognizes the DSB and leads to recruitment and full activation of ATM. The activated ATM then can phosphorylate several effector kinases including Chk2, which results in G1 arrest through ATM-Chk2-p53-p21 pathway. Activated ATM can also promote the enrollment of CtIP to the site of DSB, where CtIP interacts with and stimulates the nuclease activity of MRE11 of MRN complex to start the end resection of DSB and generate short tracts of ssDNA. Other nucleases and helicases, such as Exo1 and BLM, further resect the ssDNA to form the more extensive regions for RPA to bind and initiate the homologous recombination-mediated DSB repair. Importantly, it has been noted that the exposed ssDNA regions act like SSBs to activate ATR/Chk1 pathway. Activated Chk1 can induce p53-independent S phase arrest via phosphorylation of Cdc25A for degradation51, or it can turn on G2/M checkpoint by phosphorylating and promoting Cdc25C for association with 14-3-3 proteins, preventing Cdc25C from activating mitotic Cdk1/cyclin B complex52, 53. Cell cycle arrest modulated by these transducer and effector kinases is important for the additional replication checkpoint responses (fork stabilization, inhibition of origin firing and S/M checkpoint) and the following DSB repair.
DNA double-strand break repair (Appendix 4)
Except for one-ended DSB which can only be repaired by homologous recombination (HR), two-ended DSB, such as IR or topoisomerase II-mediated DNA damage, can be repaired by either HR or non-homologous DNA end joining (NHEJ)54.
Homologous recombination (HR)
HR is an error-free DSB repair pathway. It is restricted to the late S and G2 phases of the cell cycle, where the homologous sequence located on the sister chromatid is available to serve as a donor template for repair of the damaged strand. The repair process of HR can be divided into three phases55, 56.
PhaseⅠ: Presynapsis
The ends of DSB are initially resected by MRN complex (Mre11-Rad50-Nbs1) and endonuclease CtIP complexed with BRCA1 in a 5’ to 3’ direction to generate short 3’-overhangs of single-strand DNAs (ssDNA). Further end resection is subsequently extended by Exo1, Dna2 and BLM to ensure the maintained resection. The resected ssDNA-ends are then coated by replication protein A (RPA) filaments, which keep ssDNAs unwound. Later, Rad51 together with other mediator proteins, such as BRCA2, Rad52 and Rad51 paralogs, replace RPA to form helical nucleoprotein filament on DNA.
Phase Ⅱ: Synapsis
Rad51 nucleofilaments promote the searching for homologous DNA sequences (sister chromatid in mitosis) similar to that of the 3’-overhangs, and catalyze strand invasion with the formation of displacement loop (D-loop).
Phase Ⅲ: Postsynapsis
After the successful strand invasion, DNA synthesis of the invading strand is carried out
by DNA polymerase using the donor sequence serving as a template. Depending on the different types of HR, D-loop can be resolved by dissociation of one of the invading strands (synthesis-dependent strand annealing pathway, SDSA), or through migrating double Holliday junction intermediate that is dissolved by BLM–RMI–TOP3 or cleaved by resolvases.
Non-homologous end joining (NHEJ)
NHEJ is an error-prone DNA double-strand break (DSB) repair pathway that is active throughout all cell cycle phases. Compared to HR pathway, NHEJ is a much faster repair process because it simply joins the DSB ends without ensuring the restoration of the original DNA sequence around the DSB site. It has been known that NHEJ is the predominant pathway to repair IR or topoisomerase Ⅱ inhibitors-induced DSBs in mammalian cells.
The repair process of NHEJ 55, 56:
The initiation of NHEJ begins from the binding of the Ku70/Ku80 heterodimer (Ku) to the exposed ends of DSB. Upon binding to DNA, Ku-DNA complex recruits and activates DNA-PKcs to the site of DSB. Activated DNA-PKcs has two important functions. It first thethers two opposing ends of DSB closly, and then recruits end-processing factors (for example Artemis, polynucleotide kinase/phosphatase (PNKP), AP endonuclease 1 (APE1) and tyrosyl–DNA phosphodiesterase 1 (TDP1)) to process the ends of DSB, which allows for religation by the XRCC4- XLF- LIG4 complex together with the polymerases λ and µ.
Chapter 2: Materials and Methods
2.1. MaterialsHuman prostate adenocarcinoma cell lines, PC-3 and DU-145, were purchased from American Type Culture Collection (Rockville, MD, USA). RPMI 1640 medium, fetal bovine serum (FBS), penicillin, streptomycin and 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased from GIBCO/BRL Life Technologies (Grand Island, NY). Control siRNA, antibodies of PARP-1, Bax (6A7), Bcl-2 (C-2), Bcl-xL, Bak (G-23), Mcl-1 (22), Rad51, α -tubulin (B-7), DNA-PKcs (H-163), anti-mouse and anti-rabbit IgGs were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Antibodies of γ-H2A.X (Ser139), Bid, caspase-8, cleaved caspase-9 and Ku80 were from Cell Signaling Technologies (Boston, MA). Caspase-3 was from Imgenex, Corp.
(San Diego, CA). Antibodies of p-Chk2 (Thr68) and p-DNA-PKcs (Thr2609) (10B1) were from Abcam PLC, Inc. (Massachusetts, US). Antibody of RPA32 (12F3.3) was from GeneTex Inc. Antibody of PDE5 was from OriGene Technologies, Inc. (Rockville, MD, USA). PDE5 siRNA was from GE Healthcare Dharmacon Inc. (Chicago, USA).
Doxorubicin, camptothecin, etoposide, sildenafil, vardenafil, tadalafil, propidium iodide (PI) and all other chemical compounds were purchased from Sigma–Aldrich (St. Louis, MO, USA).
2.2. Methods
2.2.1. Cell culture
HRPC cell lines, PC-3 and DU-145, were cultured in RPMI 1640 medium with 5%
FBS (v/v), penicillin (100 units/ml) and streptomycin (100 g/ml). Cultures were maintained in a 37℃ incubator with 5% CO 2. When cells were 90%-100% confluent, cells were detached by using 0.05 % trypsin-EDTA for passaging.
2.2.2. PI staining and flow cytometric analysis
Cells with the indicated treatments were harvested by trypsinization, fixed with 70% (v/v) alcohol at -20°C for at least 30 minutes and washed with PBS. After centrifugation, cells were resuspended with 0.5 ml PI solution containing Triton X-100 (0.1% v/v), RNase (100 μg/ml) and PI (80 μg/ml). DNA content was analyzed with FACScan and CellQuest software (Becton Dickinson, Mountain View, CA).
2.2.3. Western blotting Sample preparation
After the indicated treatment, the cells were trypsinized and lysed with 60 l ice-cold lysis buffer (20 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 10 g/ml leupeptin, 1mM Na3VO4, 1mM NaF and
1mM dithiothreitol)
.
The lysate was incubated on ice for 10 minutes and then clarified by centrifugation at 14,000 rpm (13,000 g) at 4°C for 20 minutes. After the centrifugation, supernatant (cell extract) was collected to determine protein concentration using Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). To prepare sample for loading into gels, 5 sample buffer (0.3 M Tris pH 6.8, 10 % SDS, 50 % glycerol, 10 % β -mercaptoethanol, 0.02 % bromophenol blue) was added to the cell extract in a ratio of 1:4, and samples were mixed thoroughly then being heated at 95℃ for 5 minutes. Samples can be stored at -20°C for future use.Protein separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
The polyacrylamide gel used in a single electrophoresis run can be divided into stacking gel and separating gel. The acrylamide percentage (6~14%, pH 8.8) of the
separating gel depends on the sizes of the target proteins in the sample. After the gelation of the separating gel in the casting frame, stacking gel (acrylamide 5%, pH 6.8) was added on top of the separating gel and a gel comb was inserted into the stacking gel.
The fully gelated polyacrylamide gels were then set up into the vertical gel electrophoresis tank, and the running buffer (25 mM Tris-base, 192 mM glycine, 3.5 mM SDS, pH 8.3) was poured into the inner and outer chamber of tank. The protein marker and the equal amount of proteins (30 g) from each prepared sample were loaded into wells, and the gel was ready to be run at 60 V (for protein stacking) ~ 90 V (for protein separation) for about 2.5 hours.
Transferring the protein from the gel to the membrane
When performing a wet transfer, the separating gel was put into the a “transfer sandwich” (from cathode to anode pad: sponge-filter paper-gel-PVDF membrane-filter paper-sponge), which was placed in a tray with transfer buffer (25 mM Tris-base, 192 mM glycine, 20 % methanol, pH 8.3). Transferring the protein from the gel to the membrane at 65~75 V (about 200~250 mA) for 2~2.5 hours.
Immunoblotting
After an overnight incubation in PBST/5% nonfat milk at 4 ℃, the membrane was washed with PBS/0.1% Tween 20 (PBST) for three times and immuno-reacted with the indicated first antibody (1:1000~1:3000 dilutions) for 2 h at room temperature. After four washings with PBST, the anti-mouse or anti-rabbit IgG-HRP secondary antibody (1:5000 dilution) was applied to the membranes for 1 h at room temperature. The membranes were washed with PBST for 1 h and the detection of signal was performed with an enhanced chemiluminescence detection kit (Amersham Biosciences). The light signal was captured by X-ray films. The relative protein levels were quantified using the Bio-Rad Quantity One software (Hercules, CA, USA).
2.2.4. Measurement of reactive oxygen species (ROS)
Cells pretreated with or without the antioxidant NAC (1 mM) or trolox (0.3 mM) were then incubated with 0.1 % DMSO (control), doxorubicin or/and sildenafil for the indicated times. Thirty minutes before the termination of the incubation period, DCFH-DA (final concentration of 10 mM) was added to the cells and incubated for the last 30 min at 37 ℃ . Cells with different drug treatments were then harvested respectively for the detection of ROS production (%) by measuring the percentage of DCF fluorescence-positive cells in the collected total cells (10000 events) using FACScan flow cytometric analysis.
2.2.5. Comet assay
After treatment, the cells were pelleted and resuspended in ice-cold PBS. The resuspended cells were mixed with 1.5% low melting point agarose. This mixture was loaded onto a fully frosted slide that had been pre-coated with 0.7% agarose and a coverslip was then applied to the slide. After the gelation of the cell mixture, the coverslip was removed. The slides were then submerged in pre-chilled lysis solution (1% Triton X-100, 2.5 M NaCl, and 10 mM EDTA, pH 10.5) for 30 minutes at 4°C.
After soaking with pre-chilled unwinding and electrophoresis buffer (0.3 N NaOH and 1 mM EDTA) for 30 minutes, the slides were subjected to electrophoresis for 15 minutes at 0.5 V/cm (25 mA). After electrophoresis, slides were stained with 1X SYBR Gold (Molecular Probes) and nuclei images were visualized and captured at 200X magnifications with an Zeiss AxioImager A1 fluorescent microscope (Zeiss, Germany) equipped with a CCD camera (Optronics, Goleta, CA). Over one hundred of cells in each sample were scored to calculate the average of comet tail moment (Tail moment =
%DNAtail × Lengthtail) using TriTek CometScoreTM software.
2.2.6. Nuclear extraction
Nuclear extracts were prepared by sequential cell lysis and nuclear lysis. Cell pellet was suspended in 200 l buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol and 0.2 mM PMSF. The cells were subjected to vigorous vortex for 20 seconds, ice-cold incubation for 10 min to disrupt the cell membrane and centrifuged at 2000 rpm for 2 min. The pelleted nuclei were washed twice with 100 l buffer containing 10 mM HEPES, 50 mM NaCl, 25% glycerol and 0.1 mM EDTA without resuspension the pellet. After removal of the washing buffer by centrifugation at 2000 rpm for 5 min, the pelleted nuclei were lysed with 30 l buffer containing 20 mM HEPES (pH 7.9), 25% glycerol, 1.5 mM MgCl2 , 420 mM NaCl, 0.2 M EDTA, 0.5 mM dithiothreitol and 0.2 mM PMSF. After 20 minutes on ice, the lysates were centrifuged at 14,000 rpm for 5 min. The supernatants containing the solubilized nuclear proteins were used for Western blotting.
2.2.7. DNA fragmentation assay
The DNA fragmentation was determined using the Cell Death Detection ELISAPLUS kit (Roche, Mannheim, Germany). The assay was based on the quantitative in vitro determination of cytoplasmic histone-associated DNA fragments (mono- and oligo-nucleosomes) in cells after the induction of cell death. After treated with the indicated agents, cells were lysed and centrifuged, and the supernatant was used for the detection of nucleosomal DNA fragments according to the manufacturer’s protocol.
2.2.8. Small interfering RNA (siRNA) transfection
PC-3 cells were seeded into a 6-well plate with 30% confluence for each well and grown for 24 hours to 50% confluence. Each well was washed twice with PBS and 1
mL of serum-free Opti-MEM (Life Technologies, Ground Island, NY) was added.
Aliquots containing control or PDE5 siRNA in serum-free Opti-MEM were transfected into cells using Lipofectamine 2000 according to the instructions. The sequences of PDE5 siRNA are GAAGACAGCUCCAAUGACA, GAAAUCAGGUGCUGCUUGA,
GAUGACAGCUUGUGAUCUU and GGAAACGGUGGGACAUUUA. After
transfection for 5 hours, cells were washed twice with PBS and incubated in 10%
FBS-containing RPMI-1640 medium for 48 hours. The cells were then treated with or without doxorubicin for 48 hours, and the level of protein of interest was detected using Western bot analysis.
2.2.9. Immunofluorescence staining of nuclear Rad51 foci
PC-3 cells were grown on coverslips placed in a 6-well plate (1.8 cells/well).
All procedures for immunofluorescence staining were conducted at room temperature.
Following treatment with 1 μ M doxorubicin or/and 10 μ M sildenafil for 24 hours and 8 hours of cell recovery in drug-free medium, PC-3 cells were washed twice with PBS and fixed with 4 % paraformaldehyde in PBS for 20~30 min. After fixation, cells were washed three times with PBS, permeabilized with 0.1% Triton X-100 for 10 minutes followed by three times of wash with PBS and then blocked with 5% BSA/PBS for 1 hour. To examine the nuclear Rad51 foci, cells were subsequently stained with the anti-Rad51 antibody (1:200 dilution in 2.5% BSA/PBS) for 1 hour with gentle agitation and washed three times with PBS. Cells were next incubated with the FITC-conjugated secondary antibody for 1 hour (1:100 dilution in 2.5% BSA/PBS) with gentle agitation.
After washing cells three times with PBS, nuclear staining was performed using 0.15 μg/ml DAPI for 5~10 minutes. Cells on coverslip were finally washed three times with PBS. The air-dried coverslips were next mounted onto glass slides using prolong®
diamond antifade mountant (Thermo Fisher Scientific inc., Waltham, MA, USA). The slides were then kept in the dark at 4℃ for at least one day to dry the antifade mountant.
The immunofluorescent images of nuclear Rad51 foci and nuclei were captured at 630X magnifications (63x/1.4 NA oil immersion objective lens) using Zeiss AxioImager A1 fluorescent microscope (Zeiss, Germany) equipped with a CCD camera (Optronics, Goleta, CA). At least 100 cells were examined in each sample, and the percentage of cells containing over five Rad51 foci in each sample was estimated.
2.2.10. DNA end-binding activity of Ku80 protein
Assessment of DNA end-binding activity of Ku80 was carried out by using a Ku70/Ku80 DNA Repair kit (Active Motif). Briefly, equivalent amounts of nuclear proteins (4 g) were loaded into an oligonucleotide coated 96-well plate. Then, Ku80 proteins contained in nuclear extract specifically bound to the oligonucleotide.
Anti-Ku80 antibody provided by this kit detected DNA bound-Ku80. Addition of the secondary HRP-conjugated antibodies and developing solution provided a colorimetric readout (λ = 450 nm) quantified by spectrophotometry.
2.2.11. Data Analysis
Data are presented as mean standard deviation (SD) for the indicated number of separate experiments. Statistical analysis of data was performed with one-way analysis of variance followed by Bonferroni t-test and p-values < 0.05 were considered significant.
Chapter 3: Results
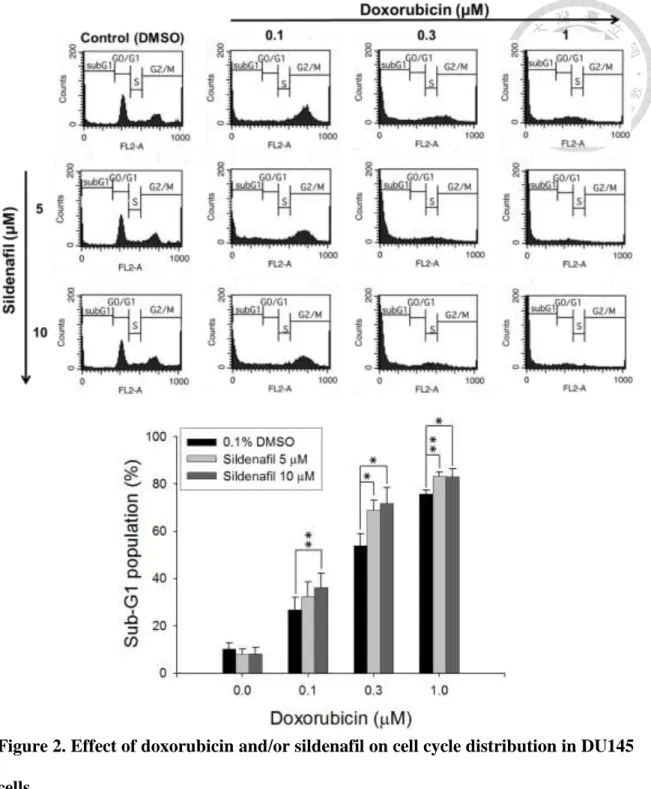
3.1. Effect of doxorubicin and sildenafil on cell cycle progression in PC-3 and DU145 cells
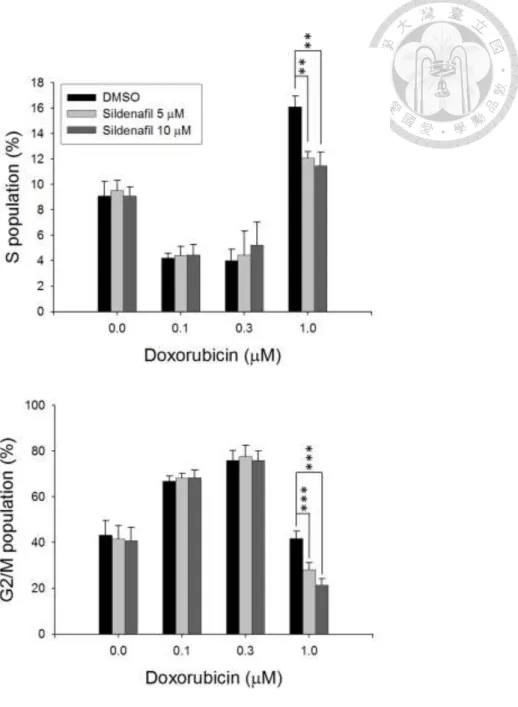
The cell population of the cell cycle in both PC-3 and DU145 cells treated with doxorubicin and sildenafil was determined by PI staining and analyzed using FACScan flow cytometry. Results showed that sildenafil alone did not affect the cell cycle distribution (Fig. 1). However, it significantly potentiated doxorubicin-induced increase of sub-G1 phase population, while decreased both S and G2/M cell population in PC-3 cells (Fig. 1). Similar effects in increasing sub-G1 population were observed in DU145 cells (Fig. 2). Since sub-G1 cell population was considered as apoptotic cells with DNA fragmentation followed by loss of DNA content, the validation of apoptotic cell death was conducted.
3.2. Validation of sildenafil-mediated sensitization of doxorubicin-induced apoptosis
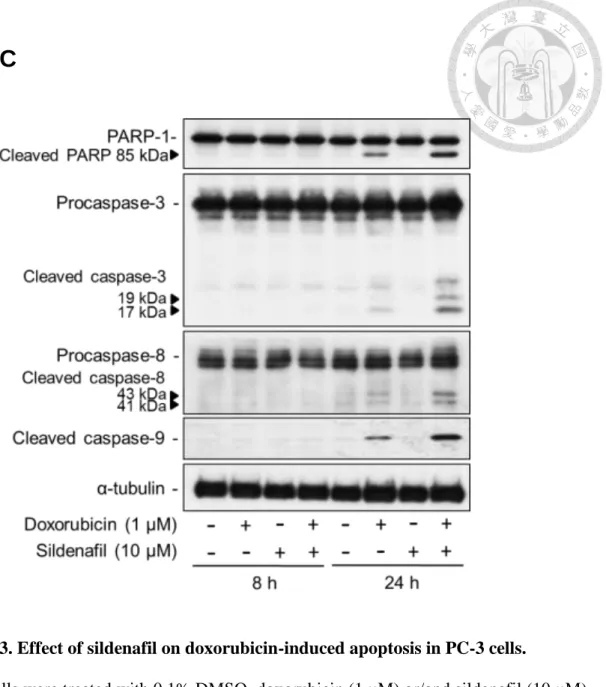
Several assays were performed to confirm whether sildenafil could synergistically enhance apoptotic cell death induced by doxorubicin. The results of microscopic examination showed that, in contrast to doxorubicin alone, the combinatory treatment of doxorubicin and sildenafil for 24 h caused a profound increase of cell shrinkage and apoptotic bodies in PC-3 cells (Fig. 3A). It was worth noting that although sildenafil was able to sensitize PC-3 cells to doxorubicin, sildenafil itself did not have any cytotoxic effect on PC-3 cells. Besides, cell death detection ELISAPLUS kit which detects nucleosomal DNA fragments in the cytoplasm of apoptotic cells was employed to validate cell apoptosis. As a consequence, sildenafil alone did not induce an increase of
nucleosomal DNA fragments in PC-3 cells, but significantly increased doxorubicin-induced effects by 1.67 times (doxorubicin, 1.68-fold; combination, 2.80-fold) (Fig. 3B). Moreover, the expression of apoptotic markers in intrinsic and extrinsic pathway was monitored by Western blot in PC-3 cells. The apoptotic markers including cleaved caspase-9 and -8 which served as initiator caspases in intrinsic and extrinsic pathways, respectively, and their common downstream substrates, effector caspase-3 and PARP-1, were examined. The results demonstrated that the combinatorial treatment for 24 hours significantly increased the expression of cleaved caspase-3, 8, 9 and PARP-1 as compared to doxorubicin alone (Fig. 3C). These results suggested that sildenafil was able to potentiate doxorubicin-induced cell apoptosis by activation of both intrinsic and extrinsic apoptotic pathways.
3.3. Effect of doxorubicin or/and sildenafil on the expression of Bcl-2 family proteins
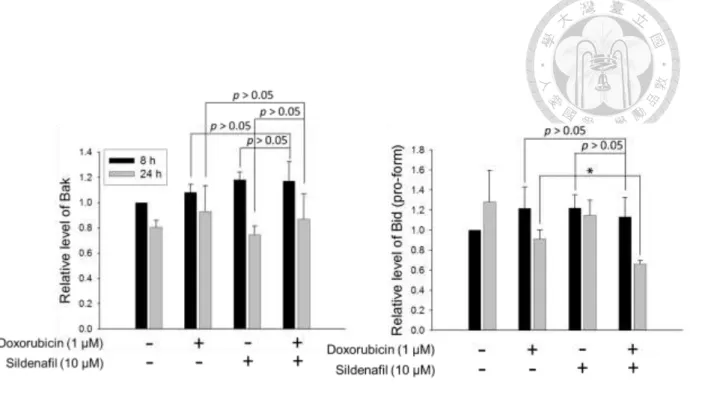
Bcl-2 family proteins, consisting of anti-apoptotic and pro-apoptotic members, are gatekeepers of mitochondria and crucial regulators of apoptosis particularly in the intrinsic pathway to govern the mitochondrial outer membrane permeability. Since the combinatorial treatment enhanced the activation of caspase-9, a key initiator caspase in the mitochondria-involved intrinsic apoptotic pathway, the effect of the combinatorial treatment on the expression of Bcl-2 family proteins was further examined by Western blot. Consequently, the combinatorial treatment for 24 h could further reduce doxorubicin-mediated decease of anti-apoptotic Bcl-2 family proteins, including Mcl-1, Bcl-xL and Bcl-2, but not those of pro-apoptotic members, such as Bax and Bak, in PC-3 cells (Fig. 4A and 4B). Moreover, combinatorial treatment further downregulated the protein level of Bid pro-form (Fig. 4A and 4B). The decreased Bid pro-form was
indicative of the caspase-8 activation since Bid served as a downstream substrate of caspase-8. Taken together, the data suggest that sildenafil may augment the mitochondria-dependent apoptosis induced by doxorubicin through modification of specific members of Bcl-2 family.
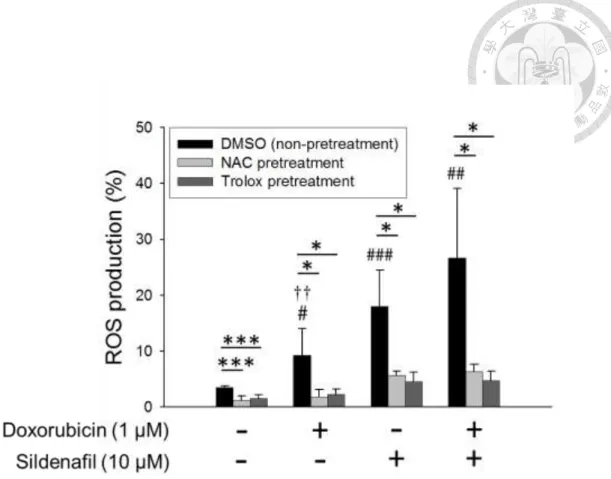
3.4. Effect of doxorubicin or/and sildenafil on ROS production
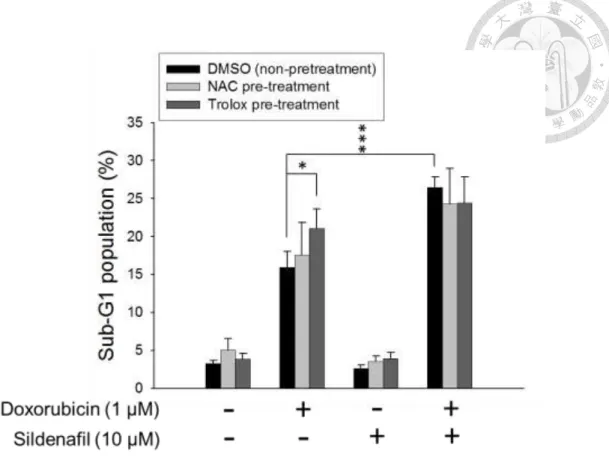
Since mitochondrial dysfunction is often associated with an increase in ROS production, the intracellular ROS levels in PC-3 cells after combinatorial treatment were determined by DCFH-DA assay. The data revealed that the short-term (e.g., 3 h) exposure to doxorubicin or sildenafil alone or combinatorial treatment significantly elevated cellular ROS levels. All the increased levels of ROS production were dramatically abolished in the presence of ROS scavengers, NAC and trolox (Fig. 5). To further determine whether the oxidative stress contributed to the apoptotic sensitization, the flow cytometric analysis of PI staining was performed to determine apoptotic sub-G1 population. As a result, neither NAC nor trolox significantly blunted the synergistic cell apoptosis caused by the combinatorial treatment (Fig. 6). The data suggest that the increase of oxidative stress is not responsible for the apoptotic sensitization mechanism.
3.5. Effect of sildenafil on doxorubicin-induced DNA double-strand break signaling and repair system
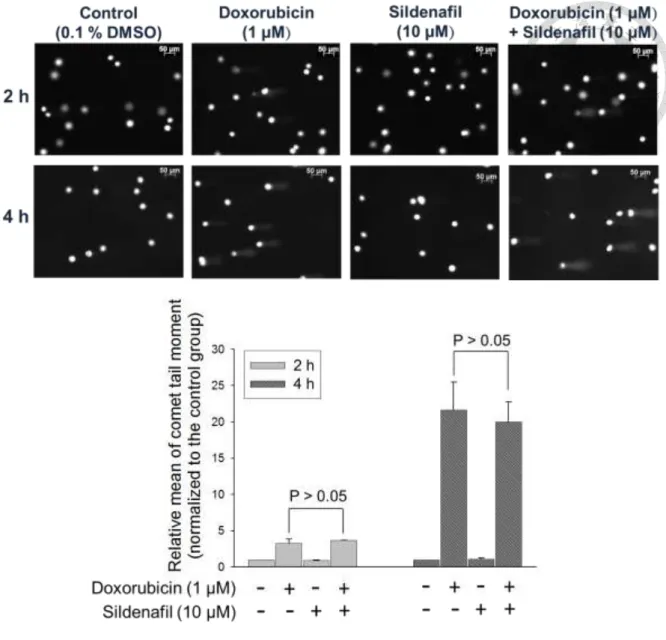
Doxorubicin is a DNA damaging drug known to cause DNA double-strand breaks (DSB). The effects of sildenafil on doxorubicin-mediated DSB signaling and repair system were examined. We first employed the alkaline comet assay to monitor the chromosomal DNA integrity. Comet tail moment was utilized as a scoring parameter to
assess the DNA damage level in individual cells. The data revealed that although doxorubicin alone induced a rapid and profound increase of comet tail moment, sildenafil did not augment the DNA damage levels caused by doxorubicin (Fig. 7). The results of Western blot also showed that sildenafil did not increase doxorubicin-induced phosphorylation of histone H2A.X at Ser139 (γ-H2A.X) and Chk2 phosphorylation at Thr68 (the hallmarker and transducer kinase of DSB, respectively) at an early exposure time (e.g., 3 hours) (Fig. 8A, B). Of note, sildenafil significantly increased the levels of doxorubicin-induced γ-H2A.X formation at a longer exposure time (e.g., 24 hours) (Fig.
8A). The results indicate that sildenafil is unable to potentiate direct DNA damage induced by doxorubicin, but can ultimately sensitize the DNA damaging effect and apoptosis through certain programmed mechanism, such as impairment of DNA repair systems. Accordingly, several markers involved in DNA repair were examined. The data demonstrated that sildenafil blunted the initial RPA32 hyperphosphorylation induced by doxorubicin (Fig. 8C and 8D) and lowered doxorubicin-elicited DNA-PKcs phosphorylation (Thr2609) (Fig. 8F and 8G). In addition, sildenafil further decreased doxorucibin-induced down-regulation of Rad51 (Fig. 8C and 8E). Similar effects were observed in DU145 cells with combinatorial treatment (Fig. 9). It has been well recognized that RPA32 binds to ssDNA during the initial phase of homologous recombination (HR) pathway and its hyperphosphorylation plays a critical role in promoting DSB repair to maintain genome stability in response to DNA damage57, 58. Rad51 plays a major role in homologous recombination (HR) for repairing DSB59. Differently, DNA-PKcs is required for non-homologous end joining (NHEJ) pathway of DNA repair, which rejoins DSBs60. Altogether, the data suggest that sildenafil may impair both HR and NHEJ pathways of DNA repair during doxorubicin-induced DNA damage effect.