光異構性膽固醇高分子膜之製備及光學特性探討(3/3)
Synthesis and Characterization of Photo-isomerizable Cholesteric Liquid Crystalline Polymer Films (3/3):
(Thermal Recordable Novel Cholesteric Liquid Crystalline Polyacrylates Containing Various Chiral Moieties)
Jui-Hsiang Liu(劉瑞祥)
Department of Chemical Engineering, National Cheng Kung University, Tainan 70101, Taiwan
NSC 96-2221-E-006 -009
ABSTRACT:
In order to investigate the effect of spacer length and linkages between the rigid mesogenic core and the terminal group on the molecular interaction and physical properties of polymers, two series of novel side chain liquid crystalline polyacrylates were synthesized. These were composed of liquid crystalline monomers with six or eleven methylene segments as spacers, and chiral monomers end capped with menthyl or cholesteryl groups. Liquid crystalline phases of the polymers were investigated using differential scanning calorimetry and polarized optical microscopy, and confirmed with X-ray diffractometry. Color image recording of the synthesized polymer films was achieved using a thermal treatment, and then fixed by quenching. This investigation demonstrates that the introduction of carbonate linking groups between the rigid mesogenic core and terminal group decreases both the lateral molecular interaction and thermal stability of the liquid crystalline polymers.
Keywords: Cholesteric Liquid Crystals, Chiral, Copolymers, Reflection, Photo-isomerization, Thermochromic 摘要: 為了探討在液晶元及末端基間,分子間隙長度以及結合基種類對分子間作用力,以 及高分子物性之相關性,本研究中合成了兩不同系列之側鏈型液晶壓克力高分子。該 系列高分子包含有6 碳及 11 碳亞甲基之分子鏈,以及末端含有 menthyl 或 cholesteryl 光學活性基之單體。所合成高分子,以掃描式熱分析儀(DSC),偏光顯微鏡(POM),以 及X-光散亂儀分析其相變化圖以及液晶相。所合成之膽固醇型高分子膜,以加熱方式 及淬冷方式,改變膽固醇型液晶之螺旋距及反射顏色,而達到以溫度控制顏色改變特 性之高分子膜設計。本研究中發現,在液晶元與末端基間導入柔軟基,可降低分子間 側向作用力,以及降低液晶性高分子之熱穩定性。 關鍵字:膽固醇液晶,光學活性,共聚物,反射光,光異構化,熱變色性
INTRODUCTION
Systematic investigations of the synthesis, characterization and applications of side chain liquid crystalline polymers began only after Ringsdorf1 and others proposed that a flexible spacer should be inserted between the polymeric main chain and the mesogenic side groups to decouple the motions of the main chain and side groups in the liquid crystalline state. Ringsdorf et al. developed the first side chain liquid crystal polymers in 1978. Since then, research in polymer liquid crystalline science and its technological applications has dramatically increased.2-7
Cholesteric polymeric materials continue to warrant a great deal of attention, as their unique helical structures give rise to the selective reflection of incident light. The helical structure of cholesteric materials is induced by the presence of chiral components in nematic liquid crystals.8 A cholesteric structure is characterized by its handedness and pitch. Equal numbers of enantiomeric “guests” of equal enantiomeric purity induce helical structures with identical pitch and opposite handedness.9 The reflection of colors raised by the interference of light, due to the periodic molecular arrangement, is related to the helical pitch. The helical pitch is very sensitive to the influence of external conditions. Cholesteric liquid crystals have found applications in twisted nematic displays, medical thermography and imaging, linear and non-linear optics, sensors, and in novel electro- and magneto-optic devices and detectors.10 The helical structure shows constructive interference effects that lead to spectrally selective light reflections.11 Using this property, cholesteric liquid crystals have been used as media for optical data recording and storage12 and for photonic band gap (PBG) materials for display technology, telecommunications and fiber optics.13 Different substances show different capacities for twisting a nematic phase. The ability of a chiral dopant to induce helical pitch is quantitatively defined as helical twisting power, β. The helical twisting power of chiral compounds can be defined as the slope of the plot of 1/p versus the concentration of the chiral compound.14-16 In theory, many factors could influence the pitch of the cholesteric liquid crystals, for example the molecular chirality, the chemical structure of the chiral moiety, and the molecular interaction between host and guest molecules, among others.
Recently there has been much development in the utilization of this property of cholesteric liquid crystals in the field of information technology, mainly in reflective displays, reflective polarizers, diffuse reflectors, and optical filters.17 Tamaoki et al. showed that cholesteric liquid crystals can be used to develop full color rewritable recording devices that could operate in the thermal and photon modes.18
Marcelis et al. investigated the influence of the linking group between the spacer and mesogenic core on the alternating properties of dimer liquid crystals.19 It was found that the phase behaviors of the cholesterol-containing dimer liquid crystals with ether and ester linkages are very similar, showing
odd-even effects for the phase transition temperatures, isotropization entropies and selective reflection wavelengths. In a series of studies, chiral monomers and chiral dopants were synthesized and their applications in the induction of cholesteric liquid crystal phases were also studied.20-25 The linkages between the mesogenic core and the terminal group were designed with ester segments. This article presents the synthesis and characterization of two series of novel side chain liquid crystalline polyacrylates, incorporating liquid crystalline monomers with six or eleven methylene segments as spacers, and chiral monomers end capped with menthyl or cholesteryl groups. These syntheses were carried out in order to investigate the effect of methylene spacer length and linking group, between the rigid mesogenic core and terminal group, on the molecular interaction and physical properties of polymers. This allowed us to investigate the effect of linkages on the liquid crystalline properties of polymers. The liquid crystalline phases and thermal stability of chiral polymers were also examined.
EXPERIMENTAL Materials
4-Hydroxybenzoic acid (99.0 %), 6-chloro-1-hexanol (95.0 %), 11-bromo-1-undecanol (97.0 %), acryloyl chloride (98.0 %), 4-butoxyphenol (99.0 %), L(-)-menthol (99.7 %), (-)-cholesteryl chloroformate (97.0 %; [α]D= –28o, c=2 in CHCl3), hydroquinone (99.5 %), 4,4'-biphenol (97.0 %),
triethylamine (Et3N; 99.0 %), N,N’-dicyclohexyl-carbodiimide (DCC; 99.0 %),
and 2,2’-azobisisobutyronitrile (AIBN; 98.0 %) were purchased from Acros Chemical Co. 4-Dimethylaminopyridine (DMAP; 99.0 %) and (-)-menthyl chloroformate (97.0 %; [α]D= –82o, c=1 in CHCl3) were purchased from
Lancaster Chemical Co. All organic solvents were purchased from Aldrich Chemical Co. Dichloromethane (CH2Cl2) was distilled over calcium hydride
under argon immediately before use, and other solvents were purified by standard methods. Analytical thin-layer chromatography was conducted on Merck aluminum plates with 0.2 mm of silica gel 60F-254. Anhydrous sodium sulfate was used to dry all organic extracts. AIBN was freshly recrystallized from methanol.
Instrumentations
All new compounds were identified by 1H-NMR, FT-IR spectra, and elemental analysis (EA). FTIR spectra were recorded as a KBr disk on a Jasco VALOR III (Tokyo, Japan) FTIR spectrophotometer. 1H-NMR (400 MHz) and
13C-NMR (100.6 MHz) spectra were obtained on a Bruker AMX-400
(Darmstadt, Germany) high-resolution NMR spectrometer, and chemical shifts were reported in ppm with tetramethylsilane (TMS) as an internal standard. Optical rotations were measured at 30oC in chloroformusing a Jasco DIP-370 polarimeter with the D-line of sodium (λ= 589 nm) with a precision of ±0.001o.
The measurements were performed using 1 % solutions of substances in chloroform. Elemental analyses were carried out on a Heraeus CHN-O (Darmstadt, Germany) rapid elemental analyzer. Gel permeation chromatography (GPC) measurements were carried out at 40 oC on a Hitachi L-4200 (Osaka, Japan) instrument equipped with TSK gel GMH and G2000H columns. CHCl3 was used as an eluent in these columns, and the rate of
elution was 1.0 mlmin-1. The instrument was calibrated with a polystyrene standard. Differential scanning calorimetry (DSC) was conducted with a Perkin Elmer DSC 7 at a heating and cooling rate of 10 Kmin-1 under a nitrogen atmosphere. The phase transitions were investigated with an Olympus BH-2 polarized light microscope (POM) equipped with Mettler hot stage FP-82, and the temperature scanning rate was determined to be 10 Kmin-1. Thermal decomposition temperature data were recorded under a nitrogen atmosphere at a heating rate of 20 Kmin-1 with a thermogravimetric analyzer (TGA) Perkin Elmer TGA 7. UV/Vis absorption spectra were measured with a Jasco V-550 spectrophotometer. The X-ray diffraction data was recorded on a Rigaku RINT 2500 series instrument with Ni-filtered CuKα radiation. The sample was held in a quartz capillary inside a temperature-controlled cell (Rigaku LC high- temperature controller). Scanning electron microscope (SEM) microphotographs were measured with a JEOL HR-FESEM JSM-6700F (Osaka, Japan) instrument.
Synthesis of Monomers (Schemes 1 and 2)
The chemical structures of chiral and liquid crystalline monomers used in this investigation are illustrated in Scheme 1. Scheme 2 shows the synthetic routes for the target chiral monomers M1-M3. 4-(6-Acryloyloxyhexyloxy) benzoic acid was synthesized in our laboratory according to the literature.23 Chiral monomers M4-M6 were synthesized following processes similar to those reported in our previous reports. 20,23-25 Liquid crystalline monomers M7 and M8 were synthesized according to the procedures described in the literature26,27 and our previous reports.20,23 The obtained products were purified and then identified using FTIR, 1H-NMR, and elemental analysis.
O C O O CH2 6 CH2 CH C O O C O O H H H M2 O ** * O C O O CH2 6 CH2 CH C O O C O O O ** * O C O O CH2 6 CH2 CH C O O C O O O M1 M3 O C O O CH2 6 CH2 CH C O O C O H H H M5 O ** * O C O O CH2 6 CH2 CH C O O C O O ** * O C O O CH2 6 CH2 CH C O O C O O M4 M6 (a) Chiral monomers
(b) Liquid Crystalline monomers
O C O O CH2 n CH2 CH C O O O M7 n= 6 M8 n= 11 C4H9 Scheme 1.
Monomer n R1 M1 1 menthyl 2 menthyl M3 1 cholesteryl O C O O CH2 6 CH2 CH C O O C O R1 O ** * HO OH Et3N CH2Cl2 + DCC DMAP CH2Cl2 R1= menthyl cholesteryl R C O Cl O n HO O n C O R1 n= 1 or 2 O C O O CH2 6 CH2 CH C O OH + 1, 2, 3 1-3 4, 5, 6 (M1-M3) O H H H or M2 n Scheme 2.
(-)-Menthyl 4-Hydroxyphenyl Carbonate (1)
Hydroquinone (2.64 g, 0.024 mol), triethylamine (2.63 g, 0.026 mol), and dry dichloromethane (50 ml) were put inside of a 150 ml two-necked flask equipped with a magnetic stirrer. Then, menthyl chloroformate (4.37 g, 0.02 mol) was added dropwise under vigorous stirring of the resulting solution. After the reaction mixture was stirred under a nitrogen atmosphere for 24 h at 30 oC, the solution was poured into water and extracted with chloroform. The organic layer was washed several times with water, dried over anhydrous MgSO4, and evaporated to dryness. The crude product was purified by column
chromatography (silica gel, ethyl acetate/hexane= 1/5). Yield: 3.16 g (45.1 %). FTIR (KBr, νmax/cm-1): 3362 (OH), 2961, 2895 (CH2), 1693 (C=O in Ar-COO-),
1605, 1503 (C-C in Ar). 1H-NMR (acetone-d6, δ in ppm): 0.81-0.90 (m, 9H,
CH3), 1.05-1.88 (m, 9H, CH2), 4.83 (m, 1H, OCHCH2), 6.51 (d, 2H, Ar-H),
7.05 (d, 2H, Ar-H).
(-)-Menthyl 4-Hydroxybiphenyl-4'-Carbonate (2)
The synthesized procedure of 2 and 3 was analogous to 1.
Yield: 42.4 %. FTIR (KBr, νmax/cm-1): 3346 (OH), 2960, 2893 (CH2), 1692
(C=O in Ar-COO-), 1604, 1505 (C-C in Ar). 1H-NMR (acetone-d6, δ in ppm):
6.91-7.49 (d, 8H, Ar-H).
(-)-Cholesteryl 4-Hydroxyphenyl Carbonate (3)
Yield: 39.6 %. FT-IR (KBr, νmax/cm-1): 3352 (OH), 2949, 2866 (CH2), 1715
(C=O in Ar-COO-), 1597, 1502 (C-C in Ar). 1H-NMR (acetone-d6, δ in ppm):
0.68 (m, 3H, CH3), 0.86-1.02 (m, 12H, CH3), 1.10-2.25 (m, 28H, CH2),
4.55-4.63 (m, 1H, OCH), 5.40 (t, 1H, CHCH2), 6.51-6.54 (d, 2H, Ar-H),
7.03-7.07 (d, 2H, Ar-H).
(-)-Menthyl 4-(6-Acryloyloxyhexyloxy) Phenyl-4’-Phenyl Carbonate (4) (M1)
4-(6-Acryloyloxyhexyloxy) benzoic acid (2.92 g, 10.0 mmol), compound 1 (3.50 g, 12.0 mmol), and 50 ml dry dichloromethane were added to a two-necked glass reactor. N,N’-dicyclohexylcarbodiimide (DCC) (3.09 g, 15.0 mmol) and 4-dimethylaminopyridine (DMAP) (0.12 g, 1 mmol) were dissolved in CH2Cl2 (30 ml), and then they were added to the solution under a nitrogen
atmosphere. The reaction mixture was stirred for 24 h at 30 oC. A solid, N,N’-dicyclohexyl urea, was precipitated and filtered off. The resulting solution was washed with water, dried with anhydrous MgSO4, and evaporated
to dryness. The crude product was purified by column chromatography (silica gel, ethyl acetate/hexane= 1/5).
Yield: 38.8 %. Tm= 39.7 oC. [α]D= –32.5o. FT-IR (KBr, νmax/cm-1): 2922,
2850 (CH2), 1725 (C=O in Ar-COO-), 1590, 1500 (C-C in Ar), 1200, 1130
(COC), 1631 (C=C). 1H-NMR (CDCl3, δ in ppm): 0.82-0.95 (m, 9H, CH3), 1.11-2.04 (m, 17H, CH2), 4.12-4.17 (t, 4H, CH2O), 4.57-4.64 (m, 1H, OCHCH2), 5.85 (dd, 1H, CH2=CH), 6.13 (dd, 1H, CH2=CH), 6.33 (dd, 1H, CH2=CH), 7.07-7.11 (d, 2H, aromatic), 7.27-7.33 (d, 4H, aromatic), 8.09-8.12(d, 2H, aromatic). 13C-NMR (100.6 MHz, CDCl3): 26.34, 27.28 (CH3); 23.61, 24.81, 25.69, 28.78, 29.23, 34.20, 38.65 (CH2); 62.99, 68.16 (OCH2); 22.66, 27.65, 49.13, 76.54 (CH); 113.15, 121.20, 123.35, 129.51 (aromatic); 128.81, 131.85 (C=C); 120.57, 145.93, 149.89, 159.72 (aromatic quaternary); 154.06, 165.89, 166.30 (C=O). Anal: calcd for C33H42O8,C 69.94,
H 7.47; found C 69.96, H 7.47 %.
(-)-Menthyl 4-{4-[4-(Acryloyloxyhexyloxy)-Phenyl Carbonyloxy] Biphenyl} Carbonate (5) (M2)
The synthesized procedure of M2 and M3 was analogous to M1.
Yield: 41.3 %. Tm= 87.1 oC. [α]D= –28.4o. FT-IR (KBr, νmax/cm-1): 2932,
2852 (CH2), 1730 (C=O in Ar-COO-), 1600, 1512 (C-C in Ar), 1252, 1220
(COC), 1635 (C=C). 1H-NMR (CDCl3, δ in ppm): 0.84-0.96 (m, 9H, CH3), 1.47-2.16 (m, 17H, CH2), 4.14-4.17 (t, 4H, CH2O), 4.60-4.65 (m, 1H, OCHCH2), 5.87 (dd, 1H, CH2=CH), 6.14 (dd, 1H, CH2=CH), 6.33 (dd, 1H, CH2=CH), 7.10-7.13 (d, 2H, Ar-H), 7.30-7.37 (d, 4H, Ar-H), 7.72-7.75 (d, 4H, Ar-H), 8.12-8.15 (d, 2H, Ar-H). 13C-NMR (100.6 MHz, CDCl3): 26.33, 27.28 (CH3); 23.61, 24.80, 25.68, 28.78, 29.22, 34.21, 38.65 (CH2); 62.99, 68.16 (OCH2); 22.65, 27.66, 49.13, 76.53 (CH); 113.15, 119.70, 119.87, 127.04,
129.05, 129.52 (aromatic); 128.81, 131.85 (C=C); 120.56, 134.21, 134.64, 150.31, 153.25, 159.71 (aromatic quaternary); 154.07, 165.88, 166.30 (C=O). Anal: calcd for C39H46O8,C 72.87, H 7.21; found C 72.50, H 7.17 %.
(-)-Cholesteryl 4-(6-Acryloyloxyhexyloxy) Phenyl-4’-Phenyl Carbonate (6) (M3)
Yield: 36.7%. K 110.3 oC N* 204.9 oC I. [α]D= –20.7o. FT-IR (KBr, νmax/cm-1):
2937, 2864 (CH2), 1720 (C=O in Ar-COO-), 1603, 1511 (C-C in Ar), 1253,
1179 (COC), 1632 (C=C). 1H-NMR (CDCl3, δ in ppm): 0.69 (m, 3H, CH3),
0.85-1.02 (m, 12H, CH3), 1.10-2.50 (m, 28H, CH2), 4.03-4.20 (t, 4H, OCH2),
4.53-4.62 (m, 1H, OCHCH2), 5.43 (t, 1H, CHCH2), 5.80-5.84 (dd, 1H,
CH2=CH), 6.07-6.17 (dd, 1H, CH2=CH), 6.37-6.43 (dd, 1H, CH2=CH),
6.95-6.97 (d, 2H, Ar-H), 7.19-7.26 (d, 4H, Ar-H), 8.11-8.14(d, 2H, Ar-H).
13C-NMR (100.6 MHz, CDCl 3): 12.01, 18.90, 19.25, 22.54, 22.80 (CH3); 21.02, 23.80, 24.25, 25.68, 27.60, 28.51, 28.95, 31.88, 36.52, 37.90, 39.48, 39.68 (CH2); 64.42, 68.05 (OCH2); 42.28 (quaternary); 27.98, 31.81, 35.76, 49.95, 56.10, 56.65, 78.94, 122.61, 139.08 (CH); 114.27, 121.94, 123.19, 132.27 (aromatic); 128.52 130.54 (C=C); 121.35, 148.44, 152.83, 164.71 (aromatic quaternary); 163.47, 166.27 (C=O). Anal: calcd for C50H68O8,C 75.34, H 8.60;
found C 75.20, H 8.63 %.
(-)-Menthyl 4-(6-Acryloyloxyhexyloxy) Phenyl-4’-Benzoate (M4), (-)-Menthyl 4-{4-[4-(Acryloyloxyhexyloxy) Phenyl Carbonyloxy] Biphenyl} Carbonylate (M5), (-)-4-Cholesteryl 4-(6-Acryloyloxyhexyl- oxy) Phenyl-4’-Benzoate (M6), 4-Butoxyphenyl-4’-(6-Acryloyloxy- hexyloxy) Benzoate (M7), 4-Butoxyphenyl-4’-(6-Acryloyloxyundecyloxy) Benzoate (M8)
were synthesized following the processes described in our previous reports20,23-25 and literature. 26-27
Polymerization (Scheme 3)
Homopolymers and copolymers were obtained by the polymerization of monomers in benzene in presence of 3 mol % of 2,2’-azobisisobutyronitrile (AIBN) at 60 oC for 24 hours. A feed molar ratio of 83/17 of the comonomers was used to prepare binary copolymers. The typical synthetic procedures for the polymers are described as follows: To a solution of predetermined amount monomers in benzene was added with 3 mol % of 2,2’-azobisisobutyronitrile (AIBN). These monomers were poured into a glass polymerization tube equipped with a sealing cap, which was degassed in a vacuum using a freeze-thaw technique and then sealed. After completion of polymerization, the polymers were precipitated in a large amount of methanol solution, and then the crude polymers were purified by dissolution in benzene and reprecipitation in methanol. Polymers were then dried in vacuum.
C O O CH2 O C O O m CH2 CH x C O O CH2 O 6 CH2 CH y C O O O C O C4H9 n O ** * R1= menthyl cholesteryl O or O R1 m= 6 or 11 n = 1 or 2 Series A CP1-CP6 C O O CH2 O C O O m CH2 CH x C O O CH2 O 6 CH2 CH y C O O O C4H9 n Series B CP7-CP12 C O R1 CH2 CH n C O O CH2 O C O O m O C4H9 m= 6 for P1, m=11 for P2 Scheme 3.
P1: M7 (0.2 g, 0.455 mmol), AIBN (2.24 mg, 0.0136 mmol), and benzene (2 ml) were used. Yield: 76.0 %. FTIR (KBr, νmax/cm-1): 2940, 2863 (CH2), 1730
(C=O in Ar-COO-), 1606, 1509 (C-C in Ar), 1252, 1197 (COC). 1H-NMR (CDCl3, δ in ppm): 0.97 (m, 3H, CH3), 1.26-2.28 (m, 15H, CH2), 3.94 (m, 6H,
CH2O), 6.88 (m, 4H, Ar-H), 7.05 (m, 2H, Ar-H), 8.07 (m, 2H, Ar-H). Anal:
calcd for (C26H32O6)n,C 70.91, H 7.27; found C 71.34, H 7.05 %.
P2 was prepared by a procedure similar to that for P1, using M8 instead of M7. Yield: 75.1 %. FTIR (KBr, νmax/cm-1): 2934, 2856 (CH2), 1726 (C=O in
Ar-COO-), 1613, 1516 (C-C in Ar), 1256, 1203 (COC). 1H-NMR (CDCl3, δ
in ppm): 0.96 (m, 3H, CH3), 1.12-2.27 (m, 25H, CH2), 3.95 (m, 6H, CH2O),
6.90 (m, 4H, Ar-H), 7.07 (m, 2H, Ar-H), 8.08 (m, 2H, Ar-H). Anal: calcd for (C31H42O6)n,C 72.94, H 8.24; found C 72.35, H 8.38 %.
CP1 to CP12 were synthesized by a procedure similar to that for hopolymerization. The synthesized copolymers were identified using FT-IR, and 1H-NMR.
Fabrication of Liquid Crystalline Polymer Films
Glass plates were cleaned using a detergent solution and then were washed with water and acetone using ultrasonic equipment for 20 and 60 minutes, respectively. After completion of the cleaning process, the plates were dried in vacuum. The surface of the glass was coated with polyvinyl alcohol (Mw=20,000), dried, and then rubbed. A solution of polymer was made by dissolving roughly 0.2 g of the sample in 2 ml of benzene. The solution was coated onto a glass substrate and further dried in a vacuum for 24 hours. The cholesteric polymer film was achieved by thermal annealing under the temperature of liquid crystal phase for 40-60 minutes. The obtained films were 80-100 μm in thickness and were examined for their optical properties.
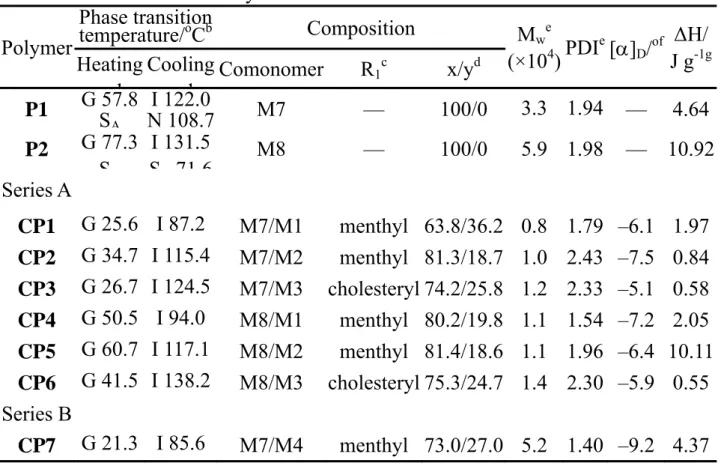
RESULTS AND DISCUSSION Synthesis and Characterization
In order to investigate the effect of spacer length and linking group between the rigid mesogenic core and terminal group on molecular interaction and physical properties of polymers, as shown in Scheme 1, a series of both chiral monomers end capped with various chiral segments and liquid crystalline monomers with six or eleven methylene segments were synthesized. The synthetic routes of the chiral monomers M1 to M3 are shown in Scheme 2. Elemental analysis, FTIR, 1H, and 13C-NMR spectroscopic techniques were used to identify the structures and constitutional composition of target monomers, as shown in the experimental section. A reasonable quantification of all protons of the synthesized monomers was also measured by peak area integrals in the 1H-NMR spectra. 13C-NMR and DEPT135 measurements were adapted to confirm the structure of M3. Assignments of each carbon and proton were assisted by the two-dimensional C-H HMQC spectrum (Figure 1), and the spectrum agreed well with the proposed molecular structure of M3. A full assignment of the resonances of the aromatic protons and carbons was assisted by the HMQC experiments, shown in the signals e, f, and g of Figure 1. The aromatic protons at the positions (Hf) appeared at the most downfield (8.12
ppm) as a doublet. The other 13C signals were well connected with the corresponding protons, as shown in the C-H HMQC spectrum (Figure 1). The structures of monomers were also confirmed by elemental analysis.
Figure 1.
The free radical polymerization of the monomers initiated by AIBN in dry benzene generates the corresponding polymers in good yields. The chemical structures of homopolymers P1, P2 and copolymers CP1 to CP12 are shown in Scheme 3. The synthesized polymers are soluble in common organic solvents such as chloroform, toluene, and THF. The molecular weight and specific rotation of the polymers are summarized in Table 1. After polymerization, the corresponding signals of the vinyl group at 5.85-6.33 ppm in 1H-NMR spectra disappeared completely. The peaks of the chemical shifts corresponding to the polymer were quite broad and consistent with the expected polymer structure. The final molar ratio composition of the copolymers, incorporating chiral and liquid crystalline monomers as co-monomers, was assessed through 1H-NMR spectrometry by comparing the integrated signals in the aliphatic regions (menthyl or cholesteryl moiety) of the chiral unit, located at 4.53-4.97 ppm, and in the aromatic regions of these two co-monomers. The results are summarized in Table 1, which shows that the molar content of co-monomers in the copolymers does not reflect the feed composition. As compared with the
a a b c d e f g h i j kl m no C O O O O C O O h i m l o n j k O O C CH2 CH a b c d e f g g
feed molar ratio, the content of M1 to M6 in copolymer shown in Table 1 is higher than that in feed composition. The results suggest that the monomer reactivity ratios may affect the copolymerization leading to the variations of the copolymer composition.28
Scheme 3 shows the molecular structure of the polymers synthesized in this investigation. As seen in Table 1, the molecular weight (Mw) of copolymers in series B is higher than that in series A. This might be due to the difference in the molecular structure, polarity, and molecular interaction between monomers, leading to the variation of the polymerization rates. Theoretically, specific rotation reveals the effect of the net vector of the polar bonds of the chiral molecules on plane polarized light. The specific rotation of menthyl chloroformate and cholesteryl chloroformate are -82o and -28o, respectively. Accordingly, the specific rotation of the chiral monomers derived from these compounds revealed negative values. As seen in Table 2, chiral monomers end capped with a menthyl group revealed a higher specific rotation than those end capped with cholesteryl group. As seen in Table 1, the specific rotation of the copolymers was also estimated. Cleavage of the double bond and the binding of other monomers together did not seem to significantly affect the chirality of the compounds. The results also indicate that the specific rotation depends not only on the content of chiral units in polymers but also the coherence of polarity due to chemical bonding after polymerization.
Table 1. Results of Polymerizationa Polymer Phase transition temperature/oCb Composition Mwe (×104)PDI e [α] D/of ΔH/ J g-1g Heating l Cooling l Comonomer R1 c x/yd P1 G 57.8 S A I 122.0 N 108.7 M7 — 100/0 3.3 1.94 — 4.64 P2 G 77.3 S I 131.5 S 71 6 M8 — 100/0 5.9 1.98 — 10.92 Series A CP1 G 25.6 I 87.2 M7/M1 menthyl 63.8/36.2 0.8 1.79 –6.1 1.97 CP2 G 34.7 I 115.4 M7/M2 menthyl 81.3/18.7 1.0 2.43 –7.5 0.84 CP3 G 26.7 I 124.5 M7/M3 cholesteryl 74.2/25.8 1.2 2.33 –5.1 0.58 CP4 G 50.5 I 94.0 M8/M1 menthyl 80.2/19.8 1.1 1.54 –7.2 2.05 CP5 G 60.7 I 117.1 M8/M2 menthyl 81.4/18.6 1.1 1.96 –6.4 10.11 CP6 G 41.5 I 138.2 M8/M3 cholesteryl 75.3/24.7 1.4 2.30 –5.9 0.55 Series B CP7 G 21.3 I 85.6 M7/M4 menthyl 73.0/27.0 5.2 1.40 –9.2 4.37
CP8 G 41.8 I 125.5 M7/M5 menthyl 80.9/19.1 5.6 1.36 –10.1 6.61 CP9 G 68.5 I 160.1 M7/M6 cholesteryl 72.3/27.7 4.2 1.32 –2.8 3.01 CP10 G 58.7 I 107.6 M8/M4 menthyl 79.8/20.2 1.7 1.82 –4.1 4.13 CP11 G 49.0 I 133.3 M8/M5 menthyl 80.2/19.8 3.6 2.41 –7.7 10.03 CP12 G 45.5 I 152.0 M8/M6 cholesteryl 78.6/21.4 3.0 2.44 –1.2 6.76
a In benzene at 60 oC for 24 hours in the presence of 3 mol % AIBN, feed molar ratio x/y=
83/17.
b G, glassy; N, nematic; S
A, smectic A; N*, chiral nematic (cholesteric); SA*, chiral smectic
A; I, isotropic phase.
c Terminal substituents of chiral monomers.
d Final molar ratio, estimated by 1H-NMR spectroscopy.
e Mw and PDI of the polymers were determined by gel permeation chromatography using
polystyrene standards in chloroform.
f Specific rotation of compounds, 0.1 g in 10 mL chloroform.
g Determined by DSC thermograms on the second heating cycle at a scanning rate of 10
Kmin-1; ΔH =enthalpy change on clearing.
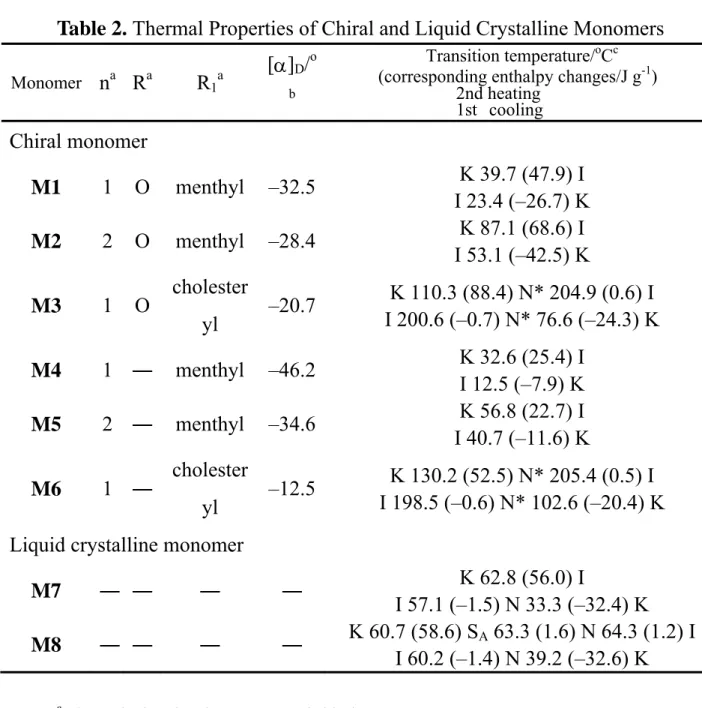
Liquid Crystalline Phases and Thermal Stabilities
The thermal properties and the phases of the synthesized monomers and polymers are summarized in Tables 1 and 2. As shown in Table 2, the chiral monomers M1, M2, M4, and M5, which incorporate a terminal menthyl group, did not reveal any mesophase. In this case, the formation of mesophases may be limited by aspect ratio, steric, and polarity of the monomers.29 The results suggest that incoherence of monomers’ aspect ratio, steric, and polarity might influence the molecular interaction between monomers leading to the absence of mesophases. However, the introduction of cholesteryl groups in chiral monomers M3 and M6 resulted in the exhibition of mesomorphic phases. Both M3 and M6 exhibited enantiotropic mesophases and revealed chiral nematic phases (N*; oily streaky texture) during heating and cooling cycles. The liquid crystalline phases were confirmed using DSC and X-ray diffraction analyses and were compared with the polarized optical microscopic (POM) textures reported in the literature.30 Figure 2 shows DSC thermograms and the POM texture of monomer M3 at a heating rate of 10 Kmin-1. During the heating cycle of M3, a chiral nematic mesophase was observed between 110.3 and 204.9 oC before isotropization. During the cooling scan, an isotropic-to-chiral nematic phase transition at 200.6 oC and crystallization at 76.6 oC were observed. As seen in Table 2, monomer M7, with an electron releasing –OC4H9
segment as the terminal group, exhibited a monotropic mesophase and revealed a nematic phase (N; schlieren texture) at the temperature range of 57.1 to 33.3
(SA; fan-shape texture) during the heating cycle. This result suggests that the
relatively longer methylene in M8 might increase the lateral molecular interaction.
Table 2. Thermal Properties of Chiral and Liquid Crystalline Monomers Monomer na Ra R1a
[α]D/o b
Transition temperature/oCc (corresponding enthalpy changes/J g-1) Chiral monomer M1 1 O menthyl –32.5 K 39.7 (47.9) I I 23.4 (–26.7) K M2 2 O menthyl –28.4 K 87.1 (68.6) I I 53.1 (–42.5) K M3 1 O cholester yl –20.7 K 110.3 (88.4) N* 204.9 (0.6) I I 200.6 (–0.7) N* 76.6 (–24.3) K M4 1 ― menthyl –46.2 K 32.6 (25.4) I I 12.5 (–7.9) K M5 2 ― menthyl –34.6 K 56.8 (22.7) I I 40.7 (–11.6) K M6 1 ― cholester yl –12.5 K 130.2 (52.5) N* 205.4 (0.5) I I 198.5 (–0.6) N* 102.6 (–20.4) K Liquid crystalline monomer
M7 ― ― ― ― K 62.8 (56.0) I I 57.1 (–1.5) N 33.3 (–32.4) K M8 ― ― ― ― K 60.7 (58.6) SA 63.3 (1.6) N 64.3 (1.2) I I 60.2 (–1.4) N 39.2 (–32.6) K 2nd heating 1st cooling
a The typical molecular structure of chiral monomers:
O C O O CH2 6 CH2 CH C O O C O R1 R n
b Specific rotation of compounds, 0.1 g in 10 mL chloroform.
c Phase transition temperature during heating and cooling scans at a rate of 10 Kmin-1; K,
40 60 80 100 120 140 160 180 200 220 0 10 20 30 40 50 T = 76.6 o C H = -24.3 J/g T = 200.6 o C H = -0.7 J/g T = 204.9 oC H = 0.6 J/g T = 110.3 oC H = 88.4 J/g H e at flow (mw ) Temperature (oC) M3 (a) (b) Figure 2.
As shown in Scheme 3, a series of rod-like side chains with polar pendant group polymers were synthesized. As shown in Table 1, all polymers were revealed to have a high interaction between side chain units leading to the generation of liquid crystalline phases. As shown in Scheme 3 and Table 1, homopolymers P1 and P2, containing terminal electron releasing butoxy (–OC4H9) groups, exhibited a smectic A phase (SA; fan-shaped texture). The
additional ordering on polymerization caused mesophases to be more ordered than for the monomeric nanlogue and transition temperatures and clearing points were higher. This result also demonstrates that the introduction of a butoxy group at the terminal end of the monomers dramatically increases the molecular interaction and polymerization rate, and stabilizes the side chain segment orientation of the polymer, leading to the generation of high molecular weight and thermally stable polymers. In series A, chiral polymers CP1-CP6, comprising a pendant carbonate group as a linkage in the chiral monomer, exhibited chiral nematic mesophases (N*), with the exception of CP5. The introduction of a carbonate group as a linkage might relatively decrease the lateral molecular interaction, leading to the variation of smectic A (SA*) to
chiral nematic mesophases (N*), and a decrease in the phase transition temperatures of the polymers. Contrary to that, for series B, chiral polymers CP7-CP12, comprising a pendant ester group as a linkage in the chiral monomer, showed chiral smectic A (SA*; fan-shape texture) mesophases.
Furthermore, as shown in Table 1, the enthalpy changes between the chiral smectic phase and the isotropic transition in the range of 3.01-10.92 Jg-1 were larger than those (about 0.55-2.05 Jg-1) between the chiral nematic phase and the isotropic transition, which were consistent with the order of the molecular arrangement. Figure 3 presents the DSC curves for the representative polymers P2, CP4, CP5, CP10, and CP11. Detailed phase transition temperatures are summarized in Table 1. Figure 4 shows the crossed POM textures of polymers
CP5, CP6, and CP10. The POM textures in Figure 4 (a) and (c), exhibited the presence of chiral smectic A phases (SA*; fan-shaped texture) in CP5 and
CP10, while chiral nematic characteristics (N*; oily streaky texture) were seen in CP6. As compared with CP3, CP6 containing longer methylene unit in side chain exhibited higher phase transition temperature. The result indicates that the variation in spacer length might affect both the freedom of the mesogenic unit from the polymer backbone and the overall length of the side chain unit.29
40 60 80 100 120 140 en do I CP11 CP11 CP10 CP10 CP5 CP5 CP4 CP4 P2 P2 II H eat flow Temperature (OC) Figure 3. (a) (b) (c) Figure 4.
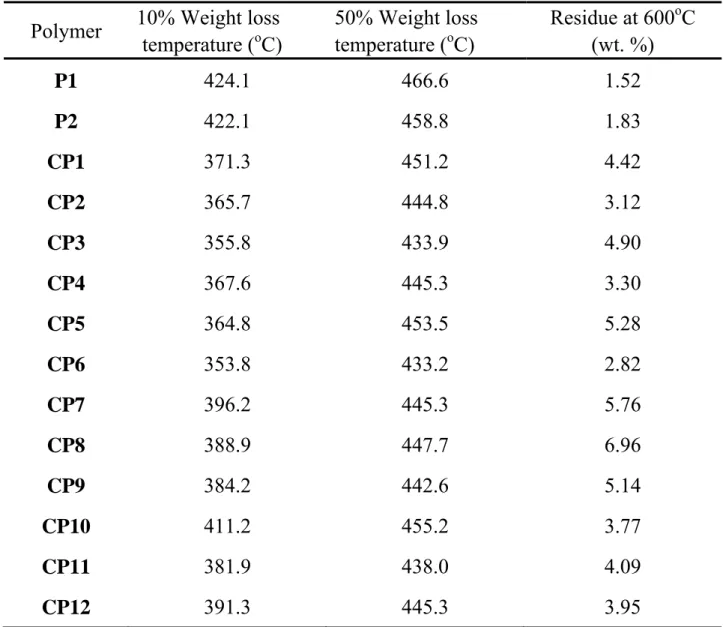
As shown in Table 3, liquid crystalline homopolymers P1 and P2, synthesized from M7 and M8, revealed much higher thermal resistance than the corresponding copolymers. Series A copolymers revealed relatively lower thermal resistance, as compared to that of series B copolymers. Based on molecular structure, the existence of the carbonate group as a linkage might reduce the lateral molecular interaction, leading to the decrease of molecular arrangement order from a smectic to a nematic phase, as shown in Table 1, and the decrease of thermal resistance, as shown in Table 3. Theoretically, the thermal resistance of polymers reveals polymer bond strength. However, the
mesophase stability reveals the strength of intermolecular forces.
Table 3. Thermal Stability of Polymersa Polymer 10% Weight loss
temperature (oC) 50% Weight loss temperature (oC) Residue at 600oC (wt. %) P1 424.1 466.6 1.52 P2 422.1 458.8 1.83 CP1 371.3 451.2 4.42 CP2 365.7 444.8 3.12 CP3 355.8 433.9 4.90 CP4 367.6 445.3 3.30 CP5 364.8 453.5 5.28 CP6 353.8 433.2 2.82 CP7 396.2 445.3 5.76 CP8 388.9 447.7 6.96 CP9 384.2 442.6 5.14 CP10 411.2 455.2 3.77 CP11 381.9 438.0 4.09 CP12 391.3 445.3 3.95
a Estimated using TGA at a heating rate of 20 Kmin-1 under a nitrogen
atmosphere.
X-ray Diffraction of Polymers
X-ray diffraction measurements of the polymers were carried out to further elucidate the structures of the mesophases. Samples were heated to the temperature ranges of the mesophases and then quenched. Figures 5 (a) and (b) show the representative X-ray diffraction curves of CP5 and CP3/CP6, respectively. The X-ray pattern of CP5 showed the presence of a sharp and strong peak at a small angle region around 2θ = 2.92o, corresponding to the layer d-spacing value of 30.2 Å at 91.5 oC under cooling. Furthermore, a fan-shaped texture could be clearly observed by POM, as shown in Figure 4 (a), which is a characteristic texture of the smectic A phase. According to the
molecular modeling calculation using CS Chem3DPro, employing MM2 energy parameters, the estimated all-trans molecular length l of the most extended conformation of chiral monomer M3 is around 37.5 Å (the layer d-spacing value is ca. 30.2 Å by XRD patterns; l/d= 0.81). Therefore, a possible layer structure of CP5 is suggested to exhibit intercalated packing of the side chains. However, for the copolymers with nematic characteristics, no peak appeared in the small angle region, and the broad peak in the range of 2θ = 15-25o was examined, classically due to the average lateral distance between the neighboring chains with d-spacing of 3-5 Å at mesophases, as shown in Figures 5 (b).31 0 5 10 15 20 25 30 35 40 0 500 1000 1500 2000 2500 d = 30.2 Å 2θ Intensit y 10 15 20 25 30 2θ CP6 CP3 In te ns ity ( a .u .) (a) (b) Figure 5.
Selective Light Reflection of Cholesteric Polymer
Due to the presence of molecular chirality, the cholesterically ordered material has regions that selectively reflect circularly polarized electromagnetic radiation of a band of wavelengths. The central wavelength (λo) of the band of
reflected wavelengths is determined by the pitch (p) of the molecular helix, according to the following formula of Bragg reflection
λo= npcosθ (1)
where n is the average refractive index of the cholesterically ordered material
and θ is the angle of incidence of the beam. The bandwidth ∆λ is given by ∆λ= p×∆n (2)
where ∆n is the birefringence of the uniaxially oriented phase corresponding to the cholesterically ordered phase. In the visible range, the regions selectively reflect circularly polarized light of a particular color.18,32,33 The influence of the copolymer composition on the pitch of the cholesteric helix, and therefore the reflection wavelength, is described by the helical twisting power (HTP) which is defined as the initial slope of the inverse helical pitch p-1 vs. the mole fraction x of the chiral dopant.34
Figure 6 (a) shows the dependence of temperature on the reflective bands of the cholesteric liquid crystalline polymer CP3. Real color images of the cholesteric liquid crystalline polymer CP3 recorded at various temperatures were also given. Cholesteric liquid crystalline polymer CP3 showed a red iridescence in the range of 30-80 oC, green at 100 oC, green-yellow at 120 oC, and blue at 130 oC. It was found that the whole visible range is accessible by varying the temperature. An increase in the film temperature induced blue shifts of the reflection bands, which were shifted from the infrared to a visible light region. Additionally, the bandwidth of the reflected light peak was narrow. The reflective wavelength and helical twisting power in CP3 and CP6 as a function of temperature are shown in Figure 6 (b). The HTP value of CP3 at 30
oC is calculated as 7.81μm-1. An increase in temperature led to an increase in
the HTP value of CP3, indicating that heat treatment might alter the polarity and the molecular interaction of the side chains, leading to a variation in the twisting pitches.35-36 As seen in Figure 6, both HTP of CP3 and CP6 are not dependent upon temperature from 50 to 70 oC. Beyond this range, the HTP of CP6 is depends less upon temperature, as compared with the HTP of CP3. The effect of temperature on the reflective wavelengths might also be attributed to the formation of smectic clusters in the cholesteric phase, or differences in the elastic constants.37 The results also suggest that the shortening of the spacer between the mesogenic phenylbenzoate group and the backbone from eleven to six methylene units hinders the rotation of side groups, and thus promotes a larger twisting of the mesogenic groups in the copolymer.38 Figure 7 presents the side view of the SEM image of the helical structure after thermal annealing. The pitch length (about 570 nm) of the CP3 cholesteric polymer film was estimated. The undulated texture in the helical structure might be due to the unsophisticated process of cutting and thermal annealing treatments. Figure 8 shows sample films synthesized in this investigation patterned at various mesophase temperatures by contact with a flat hot or cool plate. After heat treatment, the polymer films were quenched to room temperature to fix the molecular arrangement and patterns. The reflected color of the fabricated cholesteric cells varied with applied voltage. A detailed investigation of the voltage dependence of the colorful cells is now in progress.
HTP = dp -1 dλ o-1 = n T T dx
( ) ( )
dx (3)400 600 800 1000 0 20 40 60 80 100 T ra n sm ittanc e (%) Wavelength (nm) 30 o C 40 o C 60 o C 80 o C 100 o C 120 o C 130 o C Temperature (oC) 20 40 60 80 100 120 140 HT P ( μm -1) 7 8 9 10 11 12 13 Refl ecti ve wavel ength (nm ) 500 550 600 650 700 750 800 (a) (b) Figure 6. Patterned Quenching Heating Polymer in packed glass plates Heating to isotropic phase Patterning with various temperatures Cooling to cholesteric phase Figure 7. (a) Figure 8. (b)
CONCLUSIONS
A series of both chiral monomers end capped with various chiral segments, and liquid crystalline monomers with six or eleven methylene segments were synthesized. Introduction of the carbonate group as a linkage in side chains might decrease both the molecular arrangement order and thermal stability of these liquid crystalline polymers. We prepared novel cholesteric polymer films whose iridescent colors could be recorded and erased repeatedly in order to generate color images through thermal treatment. The results of this investigation showed that RGB reflected colors could be obtained through a thermal annealing under various mesophase temperatures.
REFERENCES AND NOTES
1. (a) Finkelmann, H.; Ringsdorf, H.; Wendorff, J. H.; Makromol Chem 1978, 179, 273-276; (b) Ichimura, K. Chem Rev 2000, 100, 1847-1874.
2. Bobrovsky, A. Y.; Boiko, N. I.; Shibaev, V. P.; Springer, J. Adv Mater 2000, 12, 1180-1183.
3. Natansohn, A.; Rochon, P.; Meng, X.; Barret, C.; Buffeteau, T.; Bonenfant, S. Macromolecules 1998, 31, 1155-1161.
4. Meyer, J. G.; Ruhmann, R.; Sumpe, J. Macromolecules 2000, 33, 843-850. 5. Suzuki, K.; Saito, H.; Tokita, M.; Watanabe, J. Polymer 2005, 46,
8313-8320.
6. Hu, J. S.; Zhang, B. Y.; Wang, Y.; Meng, F. B. J Polym Sci Part A: Polym Chem 2004, 42, 3870-3878.
7. Callau, L.; Giamberini, M.; Reina, J. A.; Mantecón, A. J Polym Sci Part A: Polym Chem 2006, 44, 1187-1889.
8. De Jeu, A. W.; Verteugen, G. Thermotropic Liquid Crystals; Fundamental, Springer-Verlag, Berlin, 1977.
9. C. Ruslim, K. Ichimura. J Phys Chem B 2000, 104, 6529-6535.
10. Demus, D.; Goodby, J.; Gray, G. W.; Spiess, H. W.; Vill, V. Handbook of Liquid Crystals; Weinheim; Wiley-VCH, 1998; Vol. 2A.
11. (a) Barmatov, E. B.; Bobrovsky, A. Y.; Pebalk, D. A.; Barmatova, M. V.; Shibaev, V. P. J Polym Sci Part A: Polym Chem 1999, 37, 3215-3225; (b) Lee, H. K.; Doi, K.; Harada, H.; Tsutsumi, O.; Kanazawa, A.; Shiono, T.; Ikeda, T. J Phys Chem B 2000, 104, 7023-7028.
12. (a) Bobrovsky, A. Y.; Boyko, N. I.; Shibaev, V. P. Chem Mater 2001, 13, 1998-2001; (b) Bobrovsky, A. Y.; Boyko, N. I.; Shibaev, V. P. Liq Cryst 2001, 28, 919-931; (c) Bobrovsky, A. Y.; Boyko, N. I.; Shibaev, V. P. Liq Cryst 1999, 26, 1749-1765; (d) Mena, E.; Van de witte, P.; Lub, J. Liq Cryst 2000, 27, 929-933; (e) Van de witte, P.; Neuteboom, E. E; Brehmer, M.; Lub. J. J Appl Phys 1999, 85, 7517-7521; (f) Van de witte, P.; Galan, J. C.; Lub, J. Liq Cryst 1998, 24, 819-827.
6961-6964; (b) Takeda, H.; Yoshino, K. Phys Rev E 2003, 67, 56607-5; (c) Yang, Y. C.; Kee, C. S.; Kim, J. E.; Park, H. Y. Phys Rev E 1999, 60, 6852-4; (d) Huang, C. Y.; Stott, J. J.; Petschek, R. Phys Rev Lett 1998, 80, 5603-5606.
14.(a) Pfeuffer, T.; Kürschner, K.; Strohriegl, P. Macromol Chem Phys 1999, 200, 2480-2486; (b) Andreas, S.; Strohriegl, P. Macromol Chem Phys 1998, 199, 751-756.
15.Hoshino, N.; Matsuoka, Y.; Okamoto, K.; Yamagishi, A. J Am Chem Soc 2003, 125, 1718-1719.
16.Cook, M. J.; Wilson, M. R. J Chem Phys 2000, 112, 1560-1564.
17. (a) Schwarz, G.; Kricheldorf, H. R. J Polym Sci Part A: Polym Chem 1996, 34, 603-611; (b) Chen, S. H.; Shi, H.; Conger, B. M.; Mastrangelo, J. C.; Tsutsui, T. Adv Mater 1996, 8, 998-1001; (c) Chen, C. T.; Chou, Y. T. J Am Chem Soc 2000, 122, 7662-7672.
18. (a) Tamaoki, N.; Parfenov, A. V.; Masaki, A.; Matsuda, H. Adv Mater 1997, 9, 1102-1104; (b) Tamaoki, N.; Kruk, G.; Matsuda, H. J Mater Chem 1999, 9, 2381-2384; (c) Tamaoki, N. Adv Mater 2001, 13, 1135-1147; (d) Mallia, V. A.; Tamaoki, N. Chem Soc Rev 2004, 33, 76-84.
19. Marcelis, A. T. M.; Koudijs, A.; Karczmarzyk, Z.; Sudholter, E. J. R. Liq Cryst 2003, 30, 1357-1364.
20. Liu, J. H.; Hsieh, C. D.; Wang, H. Y. J Polym Sci Part A: Polym Chem 2004, 42, 1075-1092.
21. Liu, J. H.; Yang, P. C.; Lin, T. H.; Chen, Y. J.; Wu, C. H.; Fuh, Y. G. Appl Phys Lett 2005, 86, 161120-3.
22. Liu, J. H.; Yang, P. C.; Wang, Y. K.; Wang, C. C. Liq Cryst 2006, 33, 237-248.
23. Liu, J. H.; Yang, P. C. Polymer 2006, 47, 4925-4935.
24. Liu, J. H.; Yang, P. C.; Chiu, Y. H.; Suda, Y. J Polym Sci Part A: Polym Chem 2007, 45, 2026-2037.
25. Yang, P. C.; Liu, J. H. J Polym Sci Part A: Polym Chem 2008, 46, 1289-1303.
26. Portugall, M.; Ringsdorf, H.; Zentel, R. Makromol Chem 1982, 183, 2311-2321.
27. Bobrovsky, A. Y.; Boiko, N. I.; Shibaev, V. P. Liq Cryst 1998, 24, 489-500.
28. Odian, G. Principles of Polymerization; 3rd ed., John Wiley & Sons: New York, 1991; Chapter 6, pp 452-496.
29. Collings, P. J.; Hird, M. Introduction to Liquid Crystals Chemistry and Physics; Taylor and Francis, London, 1997; Chapter 3.
30. (a) Dierking, I. Textures of Liquid Crystals; Wiley-VCH: Weinheim, 2003; (b) Hans, K.; Rolf, H. Handbook of liquid crystals; Verlag Chemie: Weinheim, 1980; (c) Http://bly.colorado.edu/lcphysics/textures/.
NY, 1989; Chapter 6.
32. Brehmer, M; Lub, J; Van de witte, P. Adv Mater 1998, 10, 1438-1441. 33. Demus, D.; Goodby, J.; Grey, G.; Spiess, H.; Vill, V. editors. Handbook of
liquid crystals; Wiley: New York, 1998.
34. Shibaev, V.; Bobrovsky, A.; Boiko, N. Prog Polym Sci 2003, 28, 729-836. 35. Chilaya, G.; Oestreicher, F.; Scherowsky, G. Mol Mater 1998, 9, 291.
36. (a) Chen, S. W.; Shi, H.; Mastrangelo, J. C.; Ou, J. J. Prog Polym Sci 1996, 21, 1211-1233; (b) Kutulya, L.; Vashchenko, V.; Semenkova, G.; Shkolnikova, N. Mol Cryst Liq Cryst 1999, 331, 583-591; (c) Pieraccini, S.; Donnoli, M. I.; Ferrarini, A.; Gottarelli, G.; Licini, G.; Rosini, C.; Superchi, S.; Spada, G. P. J Org Chem 2003, 68, 519-526.
37. (a) Mallia, V. A.; Tamaoki, N. J Mater Chem 2003, 13, 219-224; (b) Blatch, A. E.; Fletcher, I. D.; Luckhurst, G. R. J Mater Chem 1997, 7, 9-17.
38. (a) Finkelmann, H.; Rehage, G. Makromol Chem Rapid Commun1980, 1, 733-740; (b) McARDLE, C. B. Side Chain Liquid Crystal Polymers; Blackie: New York, NY, 1989; Chapter 9.