西藥藥品優良製造規範
(第二部:原料藥)
PIC/S:Guide to Good Manufacturing Practice for Medicinal Products
Part II
PE009-12 (1 October 2015)
© PIC/S October 2015
第 2 頁,共 97 頁
西藥藥品優良製造規範
(第二部:原料藥)
目 錄
第 1 章 前言……….. P.3 第 2 章 品質管理……….. P.9 第 3 章 人事……….. P.14 第 4 章 建築物與設施……….. P.16 第 5 章 製程設備……….. P.22 第 6 章 文件製作與紀錄……….. P.27 第 7 章 原物料管理……….. P.37 第 8 章 生產與製程中管制……….. P.41 第 9 章 原料藥及中間產物的包裝與識別標示…….. P.47 第 10 章 儲存與運銷……….. P.50 第 11 章 實驗室管制……….. P.52 第 12 章 確效……….. P.59 第 13 章 變更管制……….. P.68 第 14 章 中間產物及原料藥的拒用與再用…………. P.70 第 15 章 申訴與回收……….. P.73 第 16 章 委受託製造廠(含實驗室)………. P.74 第 17 章 代理商、貿易商、經銷商、重分包裝廠及重
標示廠……….. P.75 第 18 章 以細胞培養/醱酵製造之原料藥的特定規範. P.78 第 19 章 臨床試驗用原料藥……….. P.86 第 20 章 術語彙編……….. P.90
第 3 頁,共 97 頁
本規範係採 PIC/S GMP(Part II)(PE 009-12)制訂,考量本國國情及 現況,將不適用之條文刪除,且本規範僅適用人用西藥原料藥。
1. 前言(INTRODUCTION)
1.1 目的(Objective)
本文件意在提供在適當品質管理系統下,原 料藥製造之優良製造準則的指引,以確保其 符合既定品質與純度的要求。
This document (Guide) is intended to provide guidance regarding good manufacturing
practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the requirements for quality and purity that they purport or are represented to possess.
在本規範中,所謂「製造」係指原料藥之原 物料接收、生產、分包裝、重分包裝、標示、
重標示、品質管制、放行、儲存與運銷以及
相關的管制等全部作業。在本規範中,「應」
係指期待其會受適用的建議,除非不適合、
經 GMP 規範之任何相關附則修正或由經證 明可提供至少同等品質保證水準之替代選項 所取代。
In this Guide, “manufacturing” includes all operations of receipt of materials, production, packaging, repackaging, labeling, relabeling, quality control, release, storage and distribution of APIs and the related controls. In this Guide, the term” should” indicates recommendations that are expected to apply unless shown to be inapplicable, modified in any relevant annexes to the GMP Guide, or replaced by an alternative demonstrated to provide at least an equivalent level of quality assurance.
整體而言,本規範不涵蓋與從事製造人員的 安全及與環境之保護相關的層面。此等管制 是製藥廠固有的責任,按國家的法律管理之。
The GMP Guide as a whole does not cover safety aspects for the personnel engaged in the manufacture, nor aspects of protection of the environment. These controls are inherent responsibilities of the manufacturer and are governed by national laws.
第 4 頁,共 97 頁
本規範並非意在界定查驗登記/註冊的要求或 修改藥典的要求,且不影響衛生主管機關在 建立藥品之上市/製造許可申請中,對原料藥 特定查驗登記/註冊之要求的職責。查驗登記/
註冊文件中所做之全部承諾皆須符合。
This Guide is not intended to define registration requirements or modify pharmacopoeial
requirements and does not affect the ability of the responsible competent authority to establish specific registration requirements regarding APIs within the context of
marketing/manufacturing authorizations. All commitments in registration documents must be met.
1.2 範圍(Scope)
本規範適用於人用藥品之原料藥的製造。本 規範適用於無菌原料藥之製造,僅及於原料 藥成為無菌之前,無菌原料藥的滅菌及無菌 作業不包含在本規範中,但應遵循我國西藥 藥品優良製造規範第一部及附則 1 之相關規 定。
This Guide applies to the manufacture of APIs for medicial products for human use. It applies to the manufacture of sterile APIs only up to the point immediately prior to the APIs being rendered sterile. The sterilisation and aseptic processing of sterile APIs are not covered, but should be performed in accordance with the principles and guidelines of GMP as laid down in national legislations and interpreted in the GMP Guide including its Annex 1.
由於 PIC/S GMP 對血液機構訂有關於血液之 收集及測試的詳細要求,本規範不含括全血 及血漿。但包含以血液或血漿為原料所製造 的原料藥。
This Guide excludes whole blood and plasma as the PIC/S GMP Guide for Blood
Establishments lays down the detailed requirements for the collection and testing of blood. However, it does include APIs that are produced using blood or plasma as raw materials.
總之,本規範不適用於大包裝藥品,但適用 於其他所有活性原料。該等活性原料適用於 西藥藥品優良製造規範附則 2、3 及 6 所描述 之任何關於變異規定。可於附則 2、3 及 6 找 到某些原料藥類型之補充規範。
Finally, the Guide does not apply to
bulk-packaged medicinal products. It applies to all other active starting materials subject to any derogations described in the annexes to the GMP Guide, in particular Annexes 2 to 7 where supplementary guidance for certain types of API may be found.
第 5 頁,共 97 頁
「原料藥之起始原料」 係指用於生產原料藥 並且納入該原料藥結構中之一個重要結構部 份的原料、中間產物或原料藥。原料藥之起 始原料可以是依照契約或商業協議從一個或 多個供應商購得的商品,或在廠內所生產的 原料。原料藥之起始原料通常具有界定之化 學性質與結構。
An “API Starting Material” is a raw material, intermediate, or an API that is used in the production of an API and that is incorporated as a significant structural fragment into the
structure of the API. An API Starting Material can be an article of commerce, a material purchased from one or more suppliers under contract or commercial agreement, or produced in-house. API Starting Materials normally have defined chemical properties and structure.
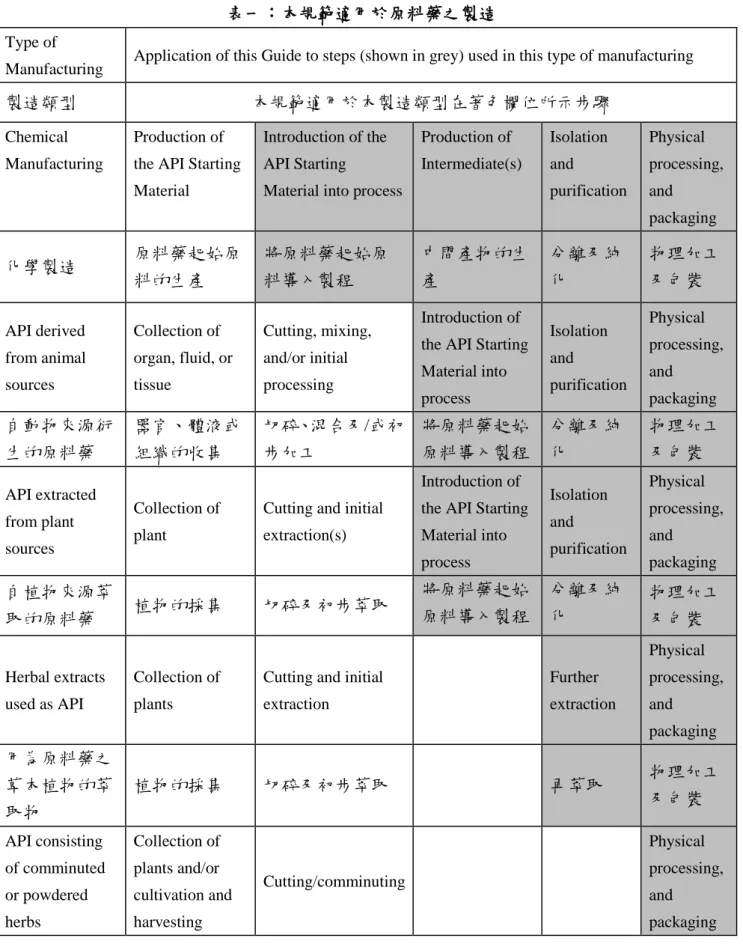
製造者應依理論基礎指定原料藥之生產起始 點並予以文件化。對合成製程而言,該起始 點慣稱為「原料藥起始原料」進入製程之點。
對於其他製程而言(例如醱酵、萃取、純化 等),其理論基礎應依個案建立。表一提供 原料藥之起始原料正常導入製程起始點的指 引。
The manufacturer should designate and document the rationale for the point at which production of the API begins. For synthetic processes, this is known as the point at which
"API Starting Materials" are entered into the process. For other processes (e.g. fermentation, extraction, purification, etc), this rationale should be established on a case-by-case basis.
Table 1 gives guidance on the point at which the API Starting Material is normally
introduced into the process.
從該起始點開始,本規範界定之適當的 GMP 應適用於這些中間產物及/或原 料藥的製造步驟。這當包括經確定會影 響原料藥品質之關鍵製程步驟的確 效。不過,必須注意到的事實是:製造 者選擇確效一個製程步驟,未必需要將 該步驟界定為關鍵步驟。
From this point on, appropriate GMP as defined in this Guide should be applied to these intermediate and/or API
manufacturing steps. This would include the validation of critical process steps determined to impact the quality of the API. However, it should be noted that the fact that a manufacturer chooses to validate a process step does not
necessarily define that step as critical.
第 6 頁,共 97 頁
本規範通常適用於表一灰色區中顯示的步 驟,這不意味以灰色顯示之所有步驟都應完 成。在原料藥的製造中,GMP 的嚴謹性應隨 製程從早期階段原料藥步驟進行到最終步 驟,亦即至純化及包裝,而升高。原料藥的 物理加工,例如製粒、加衣或粒子大小的物 理操作(諸如粉碎、微細化),應至少按本 規範的標準執行。
The guidance in this document would normally be applied to the steps shown in gray in Table 1.
It does not imply that all steps shown should be completed. The stringency of GMP in API manufacturing should increase as the process proceeds from early API steps to final steps, purification, and packaging. Physical
processing of APIs, such as granulation, coating or physical manipulation of particle size (e.g.
milling, micronizing), should be conducted at least to the standards of this Guide.
本規範不適用於界定之「原料藥起始原料」
導入製程之前的步驟。
This GMP Guide does not apply to steps prior to the introduction of the defined "API Starting Material".
第 7 頁,共 97 頁
Table 1: Application of this Guide to API Manufacturing 表一:本規範適用於原料藥之製造
Type of
Manufacturing Application of this Guide to steps (shown in grey) used in this type of manufacturing
製造類型 本規範適用於本製造類型在著色欄位所示步驟
Chemical Manufacturing
Production of the API Starting Material
Introduction of the API Starting
Material into process
Production of Intermediate(s)
Isolation and
purification
Physical processing, and
packaging
化學製造 原料藥起始原
料的生產
將原料藥起始原 料導入製程
中間產物的生 產
分離及純 化
物理加工 及包裝
API derived from animal sources
Collection of organ, fluid, or tissue
Cutting, mixing, and/or initial processing
Introduction of the API Starting Material into process
Isolation and
purification
Physical processing, and
packaging 自動物來源衍
生的原料藥
器官、體液或 組織的收集
切碎、混合及/或初 步加工
將原料藥起始 原料導入製程
分離及純 化
物理加工 及包裝
API extracted from plant sources
Collection of plant
Cutting and initial extraction(s)
Introduction of the API Starting Material into process
Isolation and
purification
Physical processing, and
packaging 自植物來源萃
取的原料藥 植物的採集 切碎及初步萃取 將原料藥起始
原料導入製程
分離及純 化
物理加工 及包裝
Herbal extracts used as API
Collection of plants
Cutting and initial extraction
Further extraction
Physical processing, and
packaging 用為原料藥之
草本植物的萃 取物
植物的採集 切碎及初步萃取 再萃取 物理加工
及包裝
API consisting of comminuted or powdered herbs
Collection of plants and/or cultivation and harvesting
Cutting/comminuting
Physical processing, and
packaging
第 8 頁,共 97 頁
由磨碎或粉碎 之草本植物所 組成的原料藥
植物的採集及/
或培育與採收 切碎/磨碎 物理加工
及包裝
Biotechnology:
fermentation/cell culture
Establishment of master cell bank and working cell bank
Maintenance of working cell bank
Cell culture and/or
fermentation
Isolation and
purification
Physical processing, and
packaging
生物技術:醱 酵/細胞培養
種細胞庫及工 作細胞庫的建 立
工作細胞庫的維 護
細胞培養及/
或醱酵
分離及純 化
物理加工 及包裝
“Classical”
Fermentation to produce an API
Establishment of cell bank
Maintenance of the cell bank
Introduction of the cells into fermentation
Isolation and
purification
Physical processing, and
packaging 用傳統醱酵以
生產原料藥
細胞庫的建立 細胞庫的維護 細胞導入醱酵 分離及純 化
物理加工 及包裝
GMP 要求遞增
第 9 頁,共 97 頁
2. 品質管理(QUALITY MANAGEMENT)
2.1 原則(Principles)
2.10 品質應為參與製造之所有人員的責 任。
2.10 Quality should be the responsibility of all persons involved in manufacturing.
2.11 每一家藥廠皆應建立及實施有效的品 質管理系統,並予以文件化。該系統包 含管理階層及適當製造人員的主動參 與。
2.11 Each manufacturer should establish, document, and implement an effective system for managing quality that involves the active participation of management and appropriate manufacturing personnel.
2.12 品質管理系統應包含組織架構、程序、
流程及資源,以及必要的作業,以確保 原料藥會符合其品質與純度之預定規 格的信心。所有與品質有關之作業皆應 加以界定並予以文件化。
2.12 The system for managing quality should encompass the organisational structure, procedures, processes and resources, as well as activities necessary to ensure confidence that the API will meet its intended specifications for quality and purity. All quality related activities should be defined and documented.
2.13 應有獨立於生產部門,並擔負品質 保證與品質管制責任的品質單位。品質 單位得為分離之品質保證(QA)部門 及品質管制(QC)部門,或為單一個 人或一組人員的形式,依組織之大小與 架構而定。
2.13 There should be a quality unit(s) that is independent of production and that fulfills both quality assurance (QA) and quality control (QC) responsibilities.
This can be in the form of separate QA and QC units or a single individual or group, depending upon the size and structure of the organization.
2.14 放行中間產物及原料藥的被授權人應 予指定。
2.14 The persons authorised to release intermediates and APIs should be specified.
2.15 所有與品質有關的作業皆應在其執行 時加以記錄。
2.15 All quality related activities should be recorded at the time they are performed.
2.16 與既定程序之任何偏差皆應加以文件 化並予以說明。關鍵性的偏差應加以調 查,且該調查及其結論應予以文件化。
2.16 Any deviation from established procedures should be documented and explained. Critical deviations should be investigated, and the investigation and its conclusions should be documented.
第 10 頁,共 97 頁
2.17 原物料在經品質單位滿意完成評估 前不得放行或使用,除非備有適當的系 統允許其使用(例如,在第 10.20 條所 述的隔離/待驗下放行,或是在原料或 中間產物等待完成評估前使用)。
2.17 No materials should be released or used before the satisfactory completion of evaluation by the quality unit(s) unless there are appropriate systems in place to allow for such use (e.g. release under quarantine as described in Section 10.20 or the use of raw materials or
intermediates pending completion of evaluation).
2.18 就主管機關的檢查、嚴重 GMP 缺失、
產品瑕疵及相關的行動(例如,與品質 有關之申訴、回收及主管機關的管制行 動等),應具備能及時通知負責管理者 之程序。
2.18 Procedures should exist for notifying responsible management in a timely manner of regulatory inspections, serious GMP deficiencies, product defects and related actions (e.g. quality related complaints, recalls, regulatory actions, etc.).
2.19 為可靠達成該品質目標,應有全面設計 並正確實施的品質系統。該系統涵蓋優 良製造規範、品質管制及品質風險管 理。
2.19 To achieve the quality objective reliably there must be a comprehensively
designed and correctly implemented quality system incorporating Good Manufacturing Practice, Quality Control and Quality Risk Management.
2.2 品質風險管理(Quality Risk Management)
2.20 品質風險管理是針對原料藥品質風險 之評價、管制、溝通及檢討的系統過 程。可用前瞻性及回溯性的方式來執 行。
2.20 Quality risk management is a systematic process for the assessment, control, communication and review of risks to the quality of the active substance. It can be applied both proactively and
retrospectively.
2.21 品質風險管理系統應確保下列項目: 2.21 The quality risk management system should ensure that:
- 品質風險的評估是基於科學知
識、製程的經驗,並且最終透過與 原料藥的使用者之溝通連結至病 患之保護。
- the evaluation of the risk to quality is based on scientific knowledge, experience with the process and ultimately links to the protection of the patient through communication with the user of the active
substance.
第 11 頁,共 97 頁
- 品質風險管理過程的努力、正式化
及文件化之程度應與風險程度相 稱。
- the level of effort, formality and documentation of the quality risk management process is
commensurate with the level of risk.
此外,品質風險管理之過程及應用的實 例詳見附則 20。
Examples of the processes and
applications of quality risk management can be found, inter alia, in Annex 20.
2.3 品質單位的職責【Responsibilities of the Quality Unit(s)】
2.30 品質單位應參與所有與品質有關的事 務。
2.30 The quality unit(s) should be involved in all quality-related matters.
2.31 品質單位應審查及核准所有與品質有 關的適當文件。
2.31 The quality unit(s) should review and approve all appropriate quality-related documents.
2.32 獨立的品質單位之主要職責不得委由 其他單位擔任。這些職責應以書面載 明,並應包含,但未必限於下列各項:
2.32 The main responsibilities of the
independent quality unit(s) should not be delegated. These responsibilities should be described in writing and should include, but not necessarily be limited to:
1. 放行或拒用/拒收所有原料藥。放 行或拒用/拒收中間產物供在製造 者管制之外的使用;
1. Releasing or rejecting all APIs.
Releasing or rejecting intermediates for use outside the control of the manufacturing company;
2. 建立放行或拒用/拒收原料、中間 產物、包裝與標示材料的系統;
2. Establishing a system to release or reject raw materials, intermediates, packaging, and labeling materials;
3. 在原料藥放行運銷之前,審查已完 成的關鍵製程步驟之批次製造及 實驗室管制紀錄;
3. Reviewing completed batch production and laboratory control records of critical process steps before release of the API for distribution;
4. 確保關鍵性偏差業經調查並解決; 4. Making sure that critical deviations are investigated and resolved;
5. 核准所有規格及製造管制標準書; 5. Approving all specifications and master production instruction;
6. 核准會影響中間產物或原料藥品 質的所有程序;
6. Approving all procedures impacting the quality of intermediates or APIs;
第 12 頁,共 97 頁
7. 確保已執行內部稽查(自我查 核);
7. Making sure that internal audits (self-inspections) are performed;
8. 核准中間產物及原料藥之受託製 造者;
8. Approving intermediate and API contract manufacturers;
9. 核准可能衝擊中間產物或原料藥 品質的變更;
9. Approving changes that potentially impact intermediate or API quality;
10. 審查與核准確效計畫書及報告; 10. Reviewing and approving validation protocols and reports;
11. 確保與品質相關之申訴經過調查 並解決;
11. Making sure that quality related complaints are investigated and resolved;
12. 確保使用有效系統以維護與校正 關鍵性設備;
12. Making sure that effective systems are used for maintaining and calibrating critical equipment;
13. 確保原物料經過適當測試並提報 其結果;
13. Making sure that materials are appropriately tested and the results are reported;
14. 確保對原料藥及/或合適時對中間 產物有安定性資料支持其再驗日 期或末效日期及儲存條件;
14. Making sure that there is stability data to support retest or expiry dates and storage conditions on APIs and/or intermediates where appropriate; and
15. 執行產品品質檢討(如第 2.6 節所 界定)。
15. Performing product quality reviews (as defined in Section 2.6).
2.4 生產作業的責任(Responsibility for Production Activities)
對生產作業的責任應以書面說明,並應 包括,但未必限於下列各項:
The responsibility for production activities should be described in writing and should include, but not necessarily be limited to
1. 依照書面程序擬訂、審查、核准及 發佈中間產物或原料藥的生產指 令;
1. Preparing, reviewing, approving, and distributing the instructions for the production of intermediates or APIs according to written
procedure;
2. 依照預先核准之指令,生產原料藥 及合適時生產中間產物;
2. Producing APIs and, when
appropriate, intermediates according to pre-approved instructions;
第 13 頁,共 97 頁
3. 審查所有批次製造紀錄,並確保這 些紀錄已經完成與簽章;
3. Reviewing all production batch records and ensuring that these are completed and signed;
4. 確保所有生產偏差已經提報與評 估,且關鍵性偏差經過調查並記錄 其結論;
4. Making sure that all production deviations are reported and evaluated and that critical
deviations are investigated and the conclusions are recorded;
5. 確保生產設施/設備是潔淨的,並 經消毒(合適時);
5. Making sure that production facilities are clean and, when appropriate, disinfected;
6. 確保必要之校正已經執行並保存 其紀錄;
6. Making sure that the necessary calibrations are performed and records kept;
7. 確保廠房設施與設備經維護保養 並保存其紀錄;
7. Making sure that the premises and equipment are maintained and records kept;
8. 確保確效計畫書與報告經審查及 核准;
8. Making sure that validation protocols and reports are reviewed and approved;
9. 評估產品、製程或設備上所提議的 變更;以及
9. Evaluating proposed changes in product, process or equipment; and 10. 確保新增,及合適時經修改之設施
及設備經過驗證。
10. Making sure that new and, when appropriate, modified facilities and equipment are qualified.
2.5 內部稽查(自我查核)【Internal Audits (Self Inspection)】
2.50 為證實遵從原料藥 GMP 之原則,應依 照核定的時程表執行定期的內部稽查。
2.50 In order to verify compliance with the principles of GMP for APIs, regular internal audits should be performed in accordance with an approved schedule.
2.51 稽查所見與改正措施應予以文件化,並 呈報公司的負責管理人。同意之改正措 施應以適時且有效的方式完成。
2.51 Audit findings and corrective actions should be documented and brought to the attention of responsible management of the firm. Agreed corrective actions should be completed in a timely and effective manner.
2.6 產品品質檢討(Product Quality Review)
第 14 頁,共 97 頁
2.60 應以證實製程的一致性為目標,執行原 料藥之定期的品質檢討。該等檢討通常 應每年執行一次,並予以文件化,且至 少應包含下列各項:
2.60 Regular quality-reviews of APIs should be conducted with the objective of verifying the consistency of the process.
Such reviews should normally be
conducted and documented annually and should include at least:
關鍵製程中管制及關鍵原料藥試 驗結果之檢討;
A review of critical in-process control and critical API test results;
不符合既定規格之所有批次的檢 討;
A review of all batches that failed to meet established specification(s);
所有關鍵偏差或不符合及相關調 查的檢討;
A review of all critical deviations or non-conformances and related investigations;
對製程或分析方法所執行之任何 變更的檢討;
A review of any changes carried out to the processes or analytical
methods;
安定性監測計畫之結果的檢討; A review of results of the stability monitoring program;
所有與品質有關之退回、申訴及回 收的檢討;以及
A review of all quality-related returns, complaints and recalls; and
改正措施之適當性的檢討。 A review of adequacy of corrective actions.
2.61 本檢討之結果應進行評估,並評估是否 應採取改正措施或任何再確效。對該改 正措施的理由應予以文件化。同意之改 正措施應以適時且有效的方式完成。
2.61 The results of this review should be evaluated and an assessment made of whether corrective action or any revalidation should be undertaken.
Reasons for such corrective action should be documented. Agreed
corrective actions should be completed in a timely and effective manner.
3. 人事(PERSONNEL)
3.1 人員資格檢核(Personnel Qualifications)
3.10 應有適當教育、訓練及/或經驗並經檢 核符合資格的足夠人員,以執行與監督 中間產物及原料藥的製造。
3.10 There should be an adequate number of personnel qualified by appropriate education, training, and/or experience to perform and supervise the manufacture of intermediates and APIs.
第 15 頁,共 97 頁
3.11 參與中間產物及原料藥之製造的所有 人員之責任,應以書面規定之。
3.11 The responsibilities of all personnel engaged in the manufacture of intermediates and APIs should be specified in writing.
3.12 訓練應由符合資格的人員定期執行,且 至少應涵蓋作業人員執行之特定作業 及與該作業人員的職能有關之 GMP。
訓練紀錄應予保存。訓練應定期評估。
3.12 Training should be regularly conducted by qualified individuals and should cover, at a minimum, the particular operations that the employee performs and GMP as it relates to the employee's functions. Records of training should be maintained. Training should be
periodically assessed.
3.2 個人衛生(Personnel Hygiene)
3.20 作業人員應力行優良的衛生及健康習 慣。
3.20 Personnel should practice good sanitation and health habits.
3.21 作業人員應穿戴適合其參與之製造作 業的潔淨衣服,且合適時,該衣服應予 更換。必要時,應穿戴附加的保護性裝 備,例如頭、臉、手及臂膀的覆罩,以 防止中間產物及原料藥受到污染。
3.21 Personnel should wear clean clothing suitable for the manufacturing activity with which they are involved and this clothing should be changed, when appropriate. Additional protective apparel, such as head, face, hand, and arm coverings, should be worn, when necessary, to protect intermediates and APIs from contamination.
3.22 作業人員應避免直接接觸中間產物或 原料藥。
3.22 Personnel should avoid direct contact with intermediates or APIs.
3.23 吸菸、飲食、咀嚼及食物的存放,應限 制在與製造區分離之某特定場所。
3.23 Smoking, eating, drinking, chewing and the storage of food should be restricted to certain designated areas separate from the manufacturing areas.
第 16 頁,共 97 頁
3.24 罹患傳染性疾病或身體之暴露表面有 開放性傷口的人員,不得參與可能導致 損及原料藥之品質結果的作業。任何人 員在任何時刻(經由體檢或監督者的觀 察)顯現有明顯疾病或開放性傷口,且 該健康狀態對原料藥之品質可能會有 不良影響時,應排除在生產作業外,直 到該病況已治癒或合格的醫療人員認 定該員之加入不會損害該原料藥的安 全性或品質為止。
3.24 Personnel suffering from an infectious disease or having open lesions on the exposed surface of the body should not engage in activities that could result in compromising the quality of APIs. Any person shown at any time (either by medical examination or supervisory observation) to have an apparent illness or open lesions should be excluded from activities where the health condition could adversely affect the quality of the APIs until the condition is corrected or qualified medical personnel determine that the person's inclusion would not jeopardize the safety or quality of the APIs.
3.3 顧問(Consultants)
3.30 指導中間產物或原料藥之製造及管制 的顧問,應有充分的教育、訓練及經 驗,或其中之任何組合,以指導其受聘 指導的主題。
3.30 Consultants advising on the manufacture and control of intermediates or APIs should have sufficient education, training, and experience, or any combination thereof, to advise on the subject for which they are retained.
3.31 載明姓名、地址、資格以及這些顧問提 供之服務類型的紀錄應予保存。
3.31 Records should be maintained stating the name, address, qualifications, and type of service provided by these consultants.
4. 建築物與設施(BUILDINGS AND FACILITIES)
4.1 設計與建造(Design and Construction)
第 17 頁,共 97 頁
4.10 使用於製造中間產物及原料藥之建築 物及設施應予配置、設計及建造,以適 合該製造類型及階段並便於清潔、維護 保養及操作。設施也應予設計,以將潛 在的污染減到最低。對中間產物或原料 藥已建立其微生物學上的規格者,合適 時,其設施也應予設計,以限制其暴露 於不合宜之微生物學上的污染物。
4.10 Buildings and facilities used in the manufacture of intermediates and APIs should be located, designed, and constructed to facilitate cleaning, maintenance, and operations as appropriate to the type and stage of manufacture. Facilities should also be designed to minimize potential
contamination. Where microbiological specifications have been established for the intermediate or API, facilities should also be designed to limit exposure to objectionable microbiological contaminants, as appropriate.
4.11 建築物及設施應有為整齊放置設備及 原物料之適當空間,以防止混雜及污 染。
4.11 Buildings and facilities should have adequate space for the orderly placement of equipment and materials to prevent mix-ups and contamination.
4.12 設備本身(例如,密閉性或圍堵性系統)
對原物料提供適合之保護者,該設備得 座落於室外。
4.12 Where the equipment itself (e.g., closed or contained systems) provides adequate protection of the material, such
equipment can be located outdoors.
4.13 通過建築物或設施之物流及人流應予 設計,以防止混雜或污染。
4.13 The flow of materials and personnel through the building or facilities should be designed to prevent mix-ups or contamination.
4.14 對於下列作業,應有經界定之區域或其 他管制系統:
4.14 There should be defined areas or other control systems for the following activities:
等候放行或拒用之進廠原物料的 接收、識別、抽樣及隔離/待驗;
Receipt, identification, sampling, and quarantine of incoming materials, pending release or rejection;
中間產物及原料藥在放行或拒用 前之隔離/待驗;
Quarantine before release or
rejection of intermediates and APIs;
中間產物及原料藥的抽樣; Sampling of intermediates and APIs
第 18 頁,共 97 頁
拒用的原物料在進一步處置(例 如,退回、重處理或銷毀)前之保 存;
Holding rejected materials before further disposition (e.g., return, reprocessing or destruction);
已放行之原物料的儲存; Storage of released materials;
生產作業; Production operations;
分裝或包裝及標示作業;以及 Packaging and labeling operations;
and
實驗室作業。 Laboratory operations.
4.15 應對於人員提供足夠且潔淨之盥洗設 施。該設施應提供冷水與熱水,合適時 並提供肥皂或清潔劑、烘乾機,或單次 使用的紙巾。盥洗設施應與製造區分 離,但便於使用。合適時,並應提供足 夠之淋浴及/或更衣的設施。
4.15 Adequate and clean washing and toilet facilities should be provided for personnel. These facilities should be equipped with hot and cold water, as appropriate, soap or detergent, air dryers, or single service towels. The washing and toilet facilities should be separate from, but easily accessible to,
manufacturing areas. Adequate facilities for showering and/or changing clothes should be provided, when appropriate.
4.16 實驗室(區)通常應與生產區隔離。若 生產作業對實驗室量測之準確性無不 良影響,且實驗室及其作業對生產作業 或中間產物或原料藥無不良影響者,則 有些實驗室(區)得座落在生產區中,
特別是使用於製程中管制的實驗室
(區)。
4.16 Laboratory areas/operations should normally be separated from production areas. Some laboratory areas, in
particular those used for in-process controls, can be located in production areas, provided the operations of the production process do not adversely affect the accuracy of the laboratory measurements, and the laboratory and its operations do not adversely affect the production process or intermediate or API.
4.2 公用設施(Utilities)
第 19 頁,共 97 頁
4.20 會影響產品品質之所有公用設施(例 如,蒸汽、氣體、壓縮空氣及空調系統)
應予驗證並適當地監測,且當超過限值 時,應採取行動。應有這些公用設施系 統之建構圖。
4.20 All utilities that could impact on product quality (e.g. steam, gases, compressed air, and heating, ventilation and air conditioning) should be qualified and appropriately monitored and action should be taken when limits are exceeded. Drawings for these utility systems should be available.
4.21 合適時,應提供適當的通風、空氣過濾 及排氣系統。這些系統應經設計及建 造,以將污染及交叉污染的風險降到最 低,並應包含適合該製造階段之空氣壓 力、微生物(合適時)、粉塵、濕度以 及溫度的控制設備。對於原料藥暴露於 環境的區域,應給予特別注意。
4.21 Adequate ventilation, air filtration and exhaust systems should be provided, where appropriate. These systems should be designed and constructed to minimise risks of contamination and
cross-contamination and should include equipment for control of air pressure, microorganisms (if appropriate), dust, humidity, and temperature, as
appropriate to the stage of manufacture.
Particular attention should be given to areas where APIs are exposed to the environment.
4.22 空氣再循環至生產區者,應採取適當措 施,以管制污染及交叉污染的風險。
4.22 If air is recirculated to production areas, appropriate measures should be taken to control risks of contamination and cross-contamination.
4.23 永久性安裝的管線應適當地識別。這可 利用辨識個別管線、文件製作、電腦管 制系統,或其他替代方法達成之。管線 應裝設於適當位置,以避免中間產物或 原料藥之污染的風險。
4.23 Permanently installed pipework should be appropriately identified. This can be accomplished by identifying individual lines, documentation, computer control systems, or alternative means. Pipework should be located to avoid risks of contamination of the intermediate or API.
4.24 排水管應有足夠的尺寸,且配置空氣阻 斷裝置,或在合適時配置適當裝置,以 防止虹吸回流。
4.24 Drains should be of adequate size and should be provided with an air break or a suitable device to prevent
back-siphonage, when appropriate.
4.3 水(Water)
第 20 頁,共 97 頁
4.30 原料藥之製造用水應證明適合其預定 之用途。
4.30 Water used in the manufacture of APIs should be demonstrated to be suitable for its intended use.
4.31 除另有正當理由外,製程用水應至少符 合世界衛生組織對飲用水品質之指引。
4.31 Unless otherwise justified, process water should, at a minimum, meet World Health Organization (WHO) guidelines for drinking (potable) water quality.
4.32 飲用水不足以確保原料藥之品質,且要 求更嚴格之化學及/或微生物學上的水 質規格者,應另訂其物理/化學屬性、
總生菌數、不合宜的微生物及/或內毒 素的適當規格。
4.32 If drinking (potable) water is insufficient to ensure API quality and tighter
chemical and/or microbiological water quality specifications are called for, appropriate specifications for physical/chemical attributes, total microbial counts, objectionable
organisms, and/or endotoxins should be established.
4.33 製程用水係由藥廠自行處理,以達界定 之品質者,該處理程序應予確效,並按 適當的行動限值監測之。
4.33 Where water used in the process is treated by the manufacturer to achieve a defined quality, the treatment process should be validated and monitored with appropriate action limits.
4.34 非無菌原料藥之製造廠意欲或宣稱其 非無菌原料藥適合進一步加工,以生產 無菌藥品者,其最終分離與純化步驟之 用水的總生菌數、不合宜微生物以及內 毒素應加以監測與管制。
4.34 Where the manufacturer of a nonsterile API either intends or claims that it is suitable for use in further processing to produce a sterile drug (medicinal) product, water used in the final isolation and purification steps should be
monitored and controlled for total microbial counts, objectionable organisms, and endotoxins.
4.4 圍堵(Containment)
4.40 在高致敏性物質,例如,青黴素或頭孢 子菌素的生產上,應使用專用生產區,
該區可包括設施、空氣處理設備及/或 製程設備。
4.40 Dedicated production areas, which can include facilities, air handling equipment and/or process equipment, should be employed in the production of highly sensitizing materials, such as penicillins or cephalosporins.
第 21 頁,共 97 頁
4.41 除建立並維持經確效之去活化及/或清 潔程序者外,當涉及具感染本質或高藥 理活性或高毒性的物質時(例如,某些 類固醇或細胞毒性的抗癌劑),也應考 慮專用生產區。
4.41 Dedicated production areas should also be considered when material of an infectious nature or high
pharmacological activity or toxicity is involved (e.g., certain steroids or cytotoxic anti-cancer agents) unless validated inactivation and/or cleaning procedures are established and
maintained.
4.42 應制訂並執行適當的措施,以防止源自 人員及原物料等從一個專用區移動到 另外一個專用區的交叉污染。
4.42 Appropriate measures should be
established and implemented to prevent cross-contamination from personnel, materials, etc. moving from one dedicated area to another.
4.43 高毒性非藥用原料,例如,除草劑與殺 蟲劑之任何生產作業(包含秤重、粉碎 或分裝或包裝),不得使用原料藥之生 產的建築物及/或設備。高毒性非藥用 原料的處理與儲存,應與原料藥隔離。
4.43 Any production activities (including weighing, milling, or packaging) of highly toxic non-pharmaceutical materials such as herbicides and
pesticides should not be conducted using the buildings and/or equipment being used for the production of APIs.
Handling and storage of these highly toxic non-pharmaceutical materials should be separate from APIs.
4.5 照明(Lighting)
4.50 在所有區域皆應提供足夠的照明,使便 於清潔、維護保養,以及正確作業。
4.50 Adequate lighting should be provided in all areas to facilitate cleaning,
maintenance, and proper operations.
4.6 污水與廢料(Sewage and Refuse)
4.60 源自廠房內及其緊鄰之周圍區域的污 水、廢料及其他廢棄物(例如,源自製 造之固體、液體或氣體的副產物)應以 安全、適時且衛生的方式處置。廢棄物 的容器及/或管線應清楚地識別。
4.60 Sewage, refuse, and other waste (e.g., solids, liquids, or gaseous by-products from manufacturing) in and from
buildings and the immediate surrounding area should be disposed of in a safe, timely, and sanitary manner. Containers and/or pipes for waste material should be clearly identified.
4.7 衛生措施與維護保養(Sanitation and Maintenance)
第 22 頁,共 97 頁
4.70 中間產物及原料藥之製造使用的建築 物,應適當地維護保養及維修,並保持 在潔淨狀態中。
4.70 Buildings used in the manufacture of intermediates and APIs should be properly maintained and repaired and kept in a clean condition.
4.71 應制訂書面程序,指定衛生處理之職責 及規定在建築物及設施之清潔上使用 的清潔時程表、方法、設備,以及材料。
4.71 Written procedures should be established assigning responsibility for sanitation and describing the cleaning schedules, methods, equipment, and materials to be used in cleaning buildings and facilities.
4.72 必要時,對適當的滅鼠劑、殺蟲劑、殺 黴菌劑、燻蒸劑,以及清潔與減菌劑的 使用,也應制訂書面程序,以防止設 備、原料、包裝/標示材料、中間產物,
以及原料藥受污染。
4.72 When necessary, written procedures should also be established for the use of suitable rodenticides, insecticides, fungicides, fumigating agents, and cleaning and sanitizing agents to prevent the contamination of equipment, raw materials, packaging/labeling materials, intermediates, and APIs.
5. 製程設備(PROCESS EQUIPMENT)
5.1 設計與建造(Design and Construction)
5.10 中間產物及原料藥之製造設備,應有適 當的設計及足夠的大小,並安裝在適當 的位置,以適合預定用途、清潔、合適 時之減菌處理及維護保養。
5.10 Equipment used in the manufacture of intermediates and APIs should be of appropriate design and adequate size, and suitably located for its intended use, cleaning, sanitization (where
appropriate), and maintenance.
5.11 設備應適當建造,以使其接觸原料、中 間產物或原料藥的表面,不會改變中間 產物及原料藥的品質超出法定或其他 既定規格。
5.11 Equipment should be constructed so that surfaces that contact raw materials, intermediates, or APIs do not alter the quality of the intermediates and APIs beyond the official or other established specifications.
5.12 生產設備應只在其驗證過的作業範圍 內使用。
5.12 Production equipment should only be used within its qualified operating range.
5.13 在中間產物或原料藥之生產中使用的 主要設備(例如,反應器、儲存容器)
及永久性安裝的作業線,應適當地識 別。
5.13 Major equipment (e.g., reactors, storage containers) and permanently installed processing lines used during the production of an intermediate or API should be appropriately identified.
第 23 頁,共 97 頁
5.14 與設備之操作關聯的任何物質,例如,
潤滑劑、熱媒或冷媒,不得接觸中間產 物或原料藥,以免導致其品質改變而超 出法定或其他既定規格。有異於此之任 何偏差,應加以評估,以確保其對該中 間產物或原料藥之預定用途的適用性 無有害的影響。可能時,應使用食品級 的潤滑劑及油品。
5.14 Any substances associated with the operation of equipment, such as lubricants, heating fluids or coolants, should not contact intermediates or APIs so as to alter their quality beyond the official or other established
specifications. Any deviations from this should be evaluated to ensure that there are no detrimental effects upon the fitness for purpose of the material.
Wherever possible, food grade lubricants and oils should be used.
5.15 合適時,應使用密閉性或圍堵性的設 備。當使用開放性的設備,或設備打開 時,應採取適當的防範措施,以使污染 的風險降到最低。
5.15 Closed or contained equipment should be used whenever appropriate. Where open equipment is used, or equipment is opened, appropriate precautions should be taken to minimize the risk of
contamination.
5.16 設備及關鍵的裝置(例如,儀表裝置及 公用設施系統),應保存一套其最新的 建構圖。
5.16 A set of current drawings should be maintained for equipment and critical installations (e.g., instrumentation and utility systems).
5.2 設備維護保養及清潔(Equipment Maintenance and Cleaning)
5.20 應建立設備之預防性維護保養的時程 表及程序(包含責任的指派)。
5.20 Schedules and procedures (including assignment of responsibility) should be established for the preventative
maintenance of equipment.
5.21 設備之清潔及其隨後放行供中間產物 及原料藥之製造使用,應建立書面程 序。清潔程序應包含充分的細節,以使 操作者能以可再現且有效的方式清潔 每一型式的設備。這些程序應包括:
5.21 Written procedures should be established for cleaning of equipment and its
subsequent release for use in the
manufacture of intermediates and APIs.
Cleaning procedures should contain sufficient details to enable operators to clean each type of equipment in a reproducible and effective manner.
These procedures should include:
設備之清潔責任的指派; Assignment of responsibility for cleaning of equipment;
第 24 頁,共 97 頁
清潔時程,合適時,包含減菌處理 時程表;
Cleaning schedules, including, where appropriate, sanitizing schedules;
清潔方法及材料之完整說明,包含 清潔設備使用之清潔劑的稀釋方 法;
A complete description of the methods and materials, including dilution of cleaning agents used to clean equipment;
合適時,拆解及組裝設備之每一物 件的指令,以確保正確之清潔;
When appropriate, instructions for disassembling and reassembling each article of equipment to ensure proper cleaning;
先前批次標識之移除或塗消的指 令;
Instructions for the removal or obliteration of previous batch identification;
保護潔淨設備在使用前免於污染 的指令;
Instructions for the protection of clean equipment from
contamination prior to use;
可行時,使用前檢查設備之潔淨 度;以及
Inspection of equipment for
cleanliness immediately before use, if practical; and
合適時,建立在作業完成後與設備 清潔前最長的時間間隔。
Establishing the maximum time that may elapse between the completion of processing and equipment cleaning, when appropriate.
5.22 設備及器具應予清潔、儲存,以及可行 時,減菌處理或滅菌,以防止污染或殘 留物的移轉,導致改變中間產物或原料 藥的品質超出法定或既定之規格。
5.22 Equipment and utensils should be cleaned, stored, and, where appropriate, sanitized or sterilized to prevent
contamination or carry-over of a material that would alter the quality of the
intermediate or API beyond the official or other established specifications.
第 25 頁,共 97 頁
5.23 當設備用於連續或時段切換生產同一 中間產物或原料藥時,設備應在適當間 隔時間予以清潔,以防止污染物的積集 及移轉(例如,分解產物或過量的微生 物)。
5.23 Where equipment is assigned to continuous production or campaign production of successive batches of the same intermediate or API, equipment should be cleaned at appropriate intervals to prevent build-up and carry-over of contaminants (e.g., degradants or objectionable levels of microorganisms).
5.24 非專用設備在不同物質的生產間應加 以清潔,以防止交叉污染。
5.24 Non-dedicated equipment should be cleaned between productions of different materials to prevent
cross-contamination.
5.25 殘留物之允收標準及清潔程序與清潔 劑的選擇,應予界定並證明其合理。
5.25 Acceptance criteria for residues and the choice of cleaning procedures and cleaning agents should be defined and justified.
5.26 設備之內容物及其潔淨度狀態應以適 當方法予以識別。
5.26 Equipment should be identified as to its contents and its cleanliness status by appropriate means.
5.3 校正(Calibration)
5.30 為確保中間產物或原料藥品質,其關鍵 性之管制、秤重、量測、監測,以及測 試的設備,應依書面程序及既定時程表 予以校正。
5.30 Control, weighing, measuring, monitoring and test equipment that is critical for assuring the quality of intermediates or APIs should be calibrated according to written
procedures and an established schedule.
5.31 如有可追溯到公認標準的標準件,則應 使用該標準件執行設備校正。
5.31 Equipment calibrations should be performed using standards traceable to certified standards, if existing.
5.32 校正紀錄應予保存。 5.32 Records of these calibrations should be maintained.
5.33 應知悉關鍵設備之最近校正狀態並可 證實。
5.33 The current calibration status of critical equipment should be known and
verifiable.
5.34 不得使用未符合校正標準的儀器。 5.34 Instruments that do not meet calibration criteria should not be used.