୯ҥᆵεᏢғڮࣽᏢଣғϯࣽᏢࣴز܌
ᅺγፕЎ
Graduate Institute of Biochemical Sciences College of Life Science
National Taiwan University Master Thesis
TristetraprolinৎೈқӧλႵѮᏘಒझфૈϐࣴز Functional Characterization of Tristetraprolin Family
Proteins in Mouse Macrophages
Ц߷൛
Kuan-Ting Wang
ࡰᏤ௲Ǻ䐁Ϙ റγ Advisor: Dr. Ching-Jin Chang
!
ύ҇୯ 101 ԃ 6 Д
June, 2012
i
ᇞ
ᇞ!!!!ᖴ!
ӣ२ᅺγٿԃޑВηǴ൩ႽΑფኬǴอኩࠅ҉ғᜤבǶགᖴၡ ᔅշךޑΓॺǴךόૈؒԖգॺǴךঁΓόՉୟǼ!
നགᖴޑࢂךޑࡰᏤԴৣˇ䐁ϘԴৣǶᗋளಃԛ፯Ε O514ǴᆶԴৣय़
ፋਔǴԴৣᒃϪЪಒЈӦᇥܴჴᡍ࠻ޑࣴزБӛޑኬηǹΕჴᡍ࠻ࡕǴԛ
ჴᡍၶډᓍਔǴԴৣᕴ๏ךӭࡰЇǴᡣךճࡋၸᜤᜢǹନΑჴᡍޑࡰ
ᏤǴԴৣΨΜϩᜢЈךޑғࢲǴᡣך७གྕធǶவԴৣيନΑᏢډޕѦǴ
ΨᏢډΑԴৣಒЈǵऐЈǵངЈǵᖰޑғࢲᄊࡋǶགᖴα၂ہǺܿԴৣǵ
էᄪᐥԴৣϷԙ๓ቺԴৣǴᖴᖴԴৣॺୖᆶךޑα၂Ǵ๏ךჴҔޑࡌǴᡣךޑ
ፕЎ׳уֹ๓ǴΨᡣ҂ٰࣴزБӛ׳уమཱǶ!
གᖴεᏢਔයޑᏤৣˇ৪ྷࢪԴৣǴᖴᖴԴৣѤԃٰޑྣ៝аϷࣴز܌ᅩ ਔ๏ךޑᔅշᆶႴᓰǴךωԖᐒډѠεғϯ܌൩᠐Ƕགᖴ୯ፁଣഋⱀԴৣǴ ᖴᖴாሦᚸᔉคޕޑךՉපයᚒࣴزǴᡣךჹჴᡍԖ׳ుቫޑᇡǶ!
གᖴ O514 ޑ܌ԖԋǴૈᇡգॺӳǶགᖴךޑৣРˇۺሺᏢۊǴᖴᖴգ вಒӦךჴᡍǴय़ჹךคЗᅰޑ༿េୢᚒΨόவόགډჇྠǶགᖴךޑӳᎃ
ۚˇٵ✎ᏢۊǴᖴᖴգᕴӧךགډੲᏩਔ๏ךΚໆǴӧךόޕၰჴᡍ၀࡛ሶਔ
๏ךࡌǴᡣךૈኖၸٗ٤ውޑВηǶགᖴךޑЈᡫᏤৣˇࢅউᏢۊǴᖴᖴգ ᕴࢂಒЈӦჸךЈޑಒ༾ᡂϯǴᆶךϩ٦ғࢲύޑזǴΨ໒Ꮴךᡣך׳߿
ඪय़ჹ֚ᜤǶགᖴॕޱᏢۊததӣٰᆶךॺϩ٦ғࢲᗺᅀǶགᖴӵᏢߏᕴࢂᓨ ᓨޑஒჴᡍ࠻ޑख़ނᘜՏǴஒបបޑߡܫӧךॺਫǶགᖴےണᆶᜩהڙ ךޑᎵᆶคᮯᓐǶགᖴکᇳᅴᏢۆǴᖴᖴգॺᄋඉӦගٮךॺ܌ሡǶ!
གᖴᅺγӕᏢǺඁනǵΪဖǵӲሺǵᅺةǵгॡǵߪᇬǵठፉǵࡘ఼Ǵᖴ
ii
ᖴգॺӳЈӦॷ๏ךჴᡍ܌ሡǴԵ၂όս༑௲ᏤךǴӧךቪፕЎܣα၂ਔ ᔅךѺݨѺǶགᖴεᏢӕᏢǺۏဖǵܴᑉǵຐԮǵ࠽ֳǵࠄసǵ൛ǵᖴᄨǵ ϡᇅǴঁДԛޑᆫᓓᡣךઢډᚈᓌลภǵΠЃזಥՀǴᡣךב܌Ԗόඍ זǶགᖴଯύӕᏢࢅඁᆶ☰఼Ǵᖴᖴգॺ೭ሶӭԃٰഉךଆᅭᅭᡜᡜǴᆶգॺ ӧଆਔǴਔ໔൩ӳႽӣډךॺଆӧ᐀ܴғࢲޑВηǴߙࡾߓǼ!
གᖴךനᒃངޑৎΓˇவόલৢڂᘶޑྭྭѪѪǴٌधπբޑݿݿ༰
༰ǴٿঁຠЈབઢޑۂۂǴᖴᖴգॺคค৷ӦЍךǵᔅշךǴԛӣৎǴ գॺᕴࢂྗഢᙦޑεᓓעךᗯளႫႫǴஒךޑيЈᡫкᅈૈໆǶৎǴ҉ᇻࢂ
ךനྕཪޑᗉ॥ෝǶ!
നࡕǴགᖴỚᇞ߈ϤԃٰޑഉՔǴᆶךϩ٦ғࢲޑ഻ࡗࠉǴхךޑϪǴ
๏ך٩ǶགᖴգᕴࢂעךܫӧಃՏǴѝाךሡाᔅշਔǴգۓ௨ନ
ᜤǴрӧךޑيᜐǴոΚӦڐշךှ،ୢᚒǴᡣךޕၰך҉ᇻόࢂঁΓǶ ԖգǴӳǼ!
ᜫஒϩ٦ֹԋፕЎޑ഻৹ᆶգॺϩ٦ǴᖴᖴգॺჹךޑбрǴךؒᏁᜤבǶ!
!
!
!
!
!
!
!
!
!
iii
ύ
ύЎᄔा!
ӃϺխࣝϸᔈޑ୷Ӣ߄ၲڙډᝄஏፓǶํচࢲϯೈқᐟ䁙ȐMAPKȑӧӃ ϺխࣝϸᔈύתᄽᜢᗖဌՅǴѬၸᕗለϯΠෞᙯᒵӢηک RNA ่ӝೈқǴёࢲ ϯݹ܄ಒझӢηȐproinflammatory cytokinesȑޑӝԋǶMAPK ᕗለ䁙ȐMKPȑᙖ җѐᕗለϯբҔ٬ MAPK Ѩѐࢲ܄ǶҞࣴزࡰр Mkp-1 ࢂঁख़ाޑॄፓӢ ηǴѬёаᜢഈݹ܄ಒझӢηޑғౢǶךॺςܴ TristetraprolinȐTtpȑёаᙖ җᆶڀԖਸޑӭဏ❥ᩫ-ֿ❣ᾕׇӈȐAU-rich element, AREȑޑ Mkp-1 mRNA ่ ӝǴ٠ၸࡕᙯᒵᐒڋஒ Mkp-1 mRNA फ़ှǶTTP ৎх֖ΟঁЬाԋǴTtpǴ
Zfp36l1ک Zfp36l2ǶҁፕЎޑҞջӧΑှځд TTP ৎೈқӧӃϺխࣝϸᔈύ
תᄽޑفՅǶ२Ӄךॺᢀჸ TTP ৎೈқޑ mRNA ᆶೈқ፦߄ၲǶӧڙډિӭᗐ ȐLipopolyssacharide, LPSȑڈᐟޑԴႵѮᏘಒझ RAW264.7 ύǴTTP ޑ mRNA ک
ೈқ፦ଯࡋᇨᏤǴՠ Zfp36l1 ک Zfp36l2 ޑ mRNA ߄फ़եǴԶೈқ፦߄߾ߥ
ठǶճҔ Zfp36l1 ک Zfp36l2 ޑ knockdown ϩǴךॺวٿঁڙډ Zfp36l1 ک Zfp36l2 ፓޑҞ mRNAǺMkp-1 ک Cox-2Ƕ෧Ͽ Zfp36l1 ک Zfp36l2 ޑ߄Ǵ ёۯߏҞ mRNA ޑъ૰යǴԶ٬Ҟ mRNA ቚуǶಒझϣ Mkp-1 ޑ߄ၲ
ቚуਔǴڋ p38 MAPK ޑࢲ܄Ǵ٬ԴႵѮᏘಒझჹ LPS ڈᐟޑ௵གࡋΠफ़Ƕ ԜѦǴךॺᗋวଯࡋᕗለϯޑ Zfp36l1 ᆶЍࢎೈқ 14-3-3 ่ӝǴ٠٬ Mkp-1
mRNAόफ़ှǶᆕӝॊǴךॺޑࣴز่݀ᡉҢ Zfp36l1 ک Zfp36l2 ޑ߄ᆶᕗ
ለϯঅႬǴёፓԴႵѮᏘಒझ RAW264.7 ڙિӭᗐڈᐟਔǴMkp-1 mRNA ޑ߄
Ǵ೭Ψᇥܴ Zfp36l1 ک Zfp36l2 ࢂӃϺխࣝϸᔈύख़ाޑፓӢηǶ
ᜢᗖӷ
mRNA ᛙۓ܄ǵTristetraprolinǵӃϺխࣝϸᔈǵᕗለϯǵํচࢲϯೈқᐟ䁙
ġ
iv
Abstract
Gene expressions are tightly controlled in the innate immune response.
Mitogen-activated protein kinases (MAPKs) play critical roles in the innate immune
response through phosphorylating downstream transcription factors and RNA binding
proteins to elicit the biosynthesis of proinflammatory cytokines. Inactivation of MAPKs
is done by MAPK phosphatases (MKPs) through dephosphorylation. The previous
studies strongly suggested that Mkp-1 was a critical negative regulator for switching off
the production of proinflammatory cytokines. We had demonstrated that Mkp-1 mRNA
containing AU-rich element (ARE) was post-transcriptionally regulated by an
ARE-binding protein Tristetraprolin (Ttp). The TTP family contains three major
members, Ttp, Zfp36l1 and Zfp36l2. To examine whether other family proteins also
play roles in the innate immune response, their expression profiles were determined.
The mRNA and protein of Ttp were highly induced by Lipopolyssacharide (LPS) in
mouse macrophage RAW264.7 cells, whereas the mRNAs of Zfp36l1 and Zfp36l2 were
down-regulated and their proteins were maintained in the consistent levels in the period
of LPS-stimulation. By knockdown analysis, we found that Mkp-1 and
Cyclooxygenase-2 (Cox-2) were the mRNA targets of Zfp36l1 and Zfp36l2 in the
resting condition. Knockdown of Zfp36l1 and Zfp36l2 increased the basal levels of
v
target mRNAs by prolonging their half-lives. Increasing the expression of Mkp-1
repressed the activity of p38 MAPK, and the sensitivity to LPS-stimulation was
decreased. Furthermore, we found that hyper-phosphorylation of Zfp36l1 stabilized
Mkp-1 expression by forming a complex with adapter protein 14-3-3. Our findings
imply the expression and phosphorylation of Zfp36l1 and Zfp36l2 might play roles in
modulating the mRNA level of Mkp-1 to control p38 MAPK activity in LPS-stimulation,
and both Zfp36l1 and Zfp36l2 are important regulators in the innate immune response.
KeywordsǺǺ
mRNA stabilityˣ Tristetraprolin ˣ Innate immune response ˣ Phosphorylation ˣ
Mitogen-activated protein kinase
ġ
ġ
ġ
ġ
ġ
vi
Contents
ᇞᖴ ... i
ύЎᄔा ... iii
Abstract... iv
Contents ... vi
Abbreviations ... ix
1. Introduction ... 1
1.1 TTP Family Proteins... 1
1.1.1 Identification of TTP Family Proteins ... 1
1.1.2 Function of TTP Family Proteins ... 2
1.1.3 TTP Family Proteins Mediate ARE-containing mRNA Decay ... 3
1.1.4 Ttp in the Transcriptional Regulation ... 4
1.1.5 The Phosphorylation Modification of TTP Family Proteins ... 5
1.1.6 TTP family Proteins in Cancers... 6
1.2 Innate Immune Response... 7
1.2.1 Mitogen-activated Protein Kinase (MAPK) in the Innate Immune Response... 7
1.2.2 Mitogen-activated Protein Kinase Phosphatases (MKPs) in the Innate Immune Response ... 7
1.2.3 TTP Family Proteins in the Innate Immune Response ... 8
2. Materials and Methods ... 10
2.1 Plasmid Constructs ... 10
2.2 Cell Culture ...11
2.3 Preparation Whole Cell Extracts and Cytoplasmic/Nuclear Extracts ... 12
2.4 Alkaline Phosphatase, Calf Intestinal (CIP) Treatment ... 13
vii
2.5 Western Blot Assay and Antibodies... 13
2.6 RNA Extraction and Reverse-transcription ... 14
2.7 Real-time PCR... 15
2.8 RNA Pull-down Assay... 16
2.9. Dual Luciferase Reporter Assay ... 17
2.10 Short-hairpin RNA (shRNA) ... 17
2.11. Lentivirus Knockdown ... 18
2.12. Co-immunoprecipitaion (Co-IP) ... 19
2.13. GST Fusion Protein Production and GST Pull-down Assay ... 19
2.14 Statistical Analysis... 21
3. Specific Aims... 22
4. Results ... 23
4.1 The Consistent Expression and Protein Phosphorylation of Zfp36l1 and Zfp36l2 in the Period of LPS-stimulation in RAW264.7 Cells... 23
4.2 Zfp36l1 and Zfp36l2 Destabilize MAPK Phosphotase-1 (Mkp-1) and Cyclooxygenase-2 (Cox-2) mRNAs in Resting RAW264.7 Cells. ... 24
4.3 Zfp36l1 and Zfp36l2 Down-regulate the Mkp-1 and Cox-2 3’UTR-mediated Luciferase Reporter Activity and Interact with Deadenylase Caf1a. ... 25
4.4 Regulation of Mkp-1 mRNA Stability by Phosphorylation of Zfp36l1... 27
4.5 The Induction of Mkp-1 mRNA in Early LPS-stimulation is Post-transcriptionally Modulated by Zfp36l1 and Zfp36l2. ... 29
4.6 p38 MAPK Activity is Regulated by Zfp36l1 and Zfp36l2 Through Mkp-1... 30
5. Discussion... 32
6. Figures ... 39 Figure 1. The consistent expression and protein phosphorylation of Zfp36l1 and
viii
Zfp36l2 during the period of LPS-stimulation in RAW264.7 cells... 39
Figure 2. Zfp36l1 and Zfp36l2 destabilize MAPK phosphotase-1 (Mkp-1) and cyclooxygenase-2 (Cox-2) mRNAs in resting RAW264.7 cells. ... 42
Figure 3. Zfp36l1 and Zfp36l2 down-regulate the Mkp-1 and Cox-2 3’UTR -mediated luciferase reporter activity and interact with deadenylase Caf1a. ... 46
Figure 4. Regulation of Mkp-1 mRNA stability by phosphorylation of Zfp36l1. .. 49
Figure 5. The induction of Mkp-1 mRNA in early LPS-stimulation is post-transcriptionally modulated by Zfp36l1 and Zfp36l2... 52
Figure 6. p38 MAPK activity is regulated by Zfp36l1 and Zfp36l2 through Mkp-1. ... 54
Figure 7. Hypothesized regulatory networks between Zfp36l1, Zfp36l2, Mkp-1, and p38 MAPK in RAW264.7 cells. ... 56
7. Tables... 57
Table 1. Primers for PCR... 57
Table 2. Primers for real-time PCR ... 58
8. Appendix... 59
Appendix 1. mRNA targets of human TTP family proteins. ... 59
Appendix 2. The pathway of ARE-mediated mRNA decay. ... 60
Appendix 3. Mammalian MAP kinase pathways. ... 60
Appendix 4. Classification of MAPK phosphatases ... 61
Appendix 5. The three types of gene expression profiles in Tnf-α activated genes. ... 61
9. References ... 62
ix
Abbreviations
3’UTR 3’-untranslated region
ARE Adenylate/uridylate-rich element CCL-2 Chemokine (C-C motif) ligand 2 CCR4 Carbohydrate catabolism repression 4 cIAP2 Cellular inhibitor of apoptosis 2 CIP Calf intestinal alkaline phosphatase
COX-2 Cyclooxygenase-2
DCP Decapping enzyme
DEPC Diethyl pyrocarbonate
DTT Dithiothreitol
EDC3 Enhancer of mRNA decapping 3 EDTA Ethylenediaminetetraacetic acid EGTA Ethylene glycol tetraacetic acid ERK Extracellular signal-regulated kinase
FBS Fetal bovine serum
GST Glutathione S-transferase HDAC Histone deacetylases
Hedls Human enhancer of decapping large subunit
HEPES 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid hnRNP Heterogeneous nuclear ribonucleoprotein
HNSCC Human head and neck squamous cell carcinoma
HuR Hu antigen R
I-cam1 Intercellular adhesion molecule 1
IEG Immediate early gene
IL Interleukin
IPTG Isopropyl β-D-1-thiogalactopyranoside JNK c-Jun amino-terminal kinase
LPS Lipopolysaccharide
MAPK Mitogen-activated protein kinase
MK2 Mitogen-activated protein kinase-activated protein kinase 2
MKP MAPK phosphatase
NF-κB Nuclear factor of kappa light polypeptide gene enhancer in B cells PARN Poly(A)-specific ribonuclease
P-body Processing body
PBS Phosphate buffered saline
x
SDS-PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis shRNA Short hairpin RNA
T-ALL T-cell acute lymphoblastic leukemia Tnf-α Tumor necrosis factor-α
TPA 12-O-tetradecannoylphorbol-13-acetate
TTP Tristetraprolin
TZF Tandem zinc finger
Xrn1 5'-3' exoribonuclease 1
Zfp36l1 Zinc finger protein 36, C3H type-like 1 Zfp36l2 Zinc finger protein 36, C3H type-like 2 Zfp36l3 Zinc finger protein 36, C3H type-like 3
ġġ
1
1. Introduction
1.1 TTP Family Proteins
1.1.1 Identification of TTP Family Proteins
The sequence of Tristetraprolin (Ttp; also known as Zfp36, Tis11) was first
confirmed from the cDNA of TPA-treated (12-O-tetradecannoylphorbol-13-acetate)
Swiss 3T3 cells [1], and the expression of Ttp was transiently activated by insulin or
growth factor [2,3]. The characteristics of amino acid sequence of Ttp are three
Pro-Pro-Pro-Pro repeats [2] and two tandem zinc finger (TZF; also known as CCCH
domain) domains which are required for RNA binding [1].
Zfp36l1 (Tis11b, Brf1, cMG1, Erf1, and Berg36) and Zfp36l2 (Tis11d, Brf2, and
Erf1) which belong to TTP family proteins were also identified by Dr. Blackshear group
[1] . It was reported that the TZF domains of Zfp36l1 and Zfp36l2 were more than 70%
identical to Ttp. Although their sequences are similar, their responses to TPA stimulation
are different. Zfp36l1 and Zfp36l2 are detectable without TPA stimulation; however, Ttp
is undetectable in Swiss 3T3 cells without TPA stimulation [1].
The fourth protein of TTP family, Zfp36l3, is located on the X chromosome of
mouse and rat, but it is not found in human [4]. It suggests that Zfp36l3 may play a
unique role in rodent, but the function of Zfp36l3 in non-rodent may be replaced by
2
other TTP family proteins.
1.1.2 Function of TTP Family Proteins
The Ttp knockout mice develop complex syndromes of inflammatory arthritis,
dermatitis, autoimmunity, cachexia, and myeloid hyperplasia. Those symptoms can be
recovered by using Tumor necrosis factor-α (Tnf-α) antibodies [5]. The amounts of
Tnf-α which secreted by bone marrow-derived macrophages from the Ttp knockout
mice are about 5 times than control cells [6]. Those studies indicate that Ttp is involved
in the regulation of Tnf-α in vivo. Ttp binds to mRNA 3’-untranslated region (3’ UTR)
of Tnf-α containing adenylate/uridylate rich element (ARE) and down-regulates the
expression of Tnf-α in the post-transcriptional level [7]. Ttp has been confirmed to
modulate the expressions of mRNAs containing ARE [8]. As a result, Ttp participates in
many cellular processes, including immune response, cell cycle, carcinogenesis,
angiogenesis, development, and protein glycosylation (Appendix 1) [8].
The Zfp36l1 knockout mice are failed to pass chorioallantoic fusion and die at
embryonic day 10.5 [9]. The Zfp36l2 knockout mice die after 14 days of birth because
the formation of hematopoietic stem cells are failed [10]. The ova of female mice with
N-terminal truncated 29 amino acids (the first exon) of Zfp36l2 can be fertilized, but the
3
embryos die in two-cell stage of development [11]. The conditional knockout mice of
Zfp36l1 and Zfp36l2 will both bother the development of thymus and develop T-cell
acute lymphoblastic leukemia (T-ALL) [12]. The RNA binding abilities of Zfp36l1 and
Zfp36l2 are similar to Ttp due to the conserved TZF domains of those proteins. Tnf-α
mRNA are also destabilized by the overexpression of Zfp36l1 and Zfp36l2 [13], and the
other mRNA targets of Zfp36l1 and Zfp36l2 are listed in Table 1.
1.1.3 TTP Family Proteins Mediate ARE-containing mRNA Decay
Ttp associates with ARE-containing mRNAs and results in mRNAs decay. The
sequences of ARE motifs are highly conserved in those mRNA. In the statistics, 96% of
ARE motifs are AUUUA (pentamers), and 44% are UUAUUUAUU (nonamers) [14].
There are three major deadenylation complexes: Ccr4/Caf1/Not, poly-A specific
ribonuclease (Parn), and Pan2/Pan3. They initiate mRNA degradation through removing
the poly-A tail of mRNA. Using co-immunoprecipitation experiments, Ttp and Zfp36l1
interacte with Ccr4 [15], and Ttp can also associate with Caf1 through Not1 [16].
However, Ttp cannot interact with Parn [15].
After the deadenylation of mRNA, there are two pathways of mRNA degradation.
The first one is mRNA decapping followed by 5’ɦ3’!exonucleolytic decay in p-bodies,
4
and the second is 3’ɦ5’!exonucleolytic decay. Ttp existes in mRNA decapping
complexes, involving Edc3 and Hedls (enhancers of decapping enzymes), Dcp1 and
Dcp2 (decapping enzymes) [15,17]. After the decapping of mRNA, the exposed 5’ end
of mRNA is digested by Xrn-1 (5’-exoribonuclease) which is recruited by Ttp [15]. The
N-terminal domain of Ttp is necessary for associating with Dcp1, Dcp2, and Xrn-1 [15].
For 3’ɦ 5’!exonucleolytic decay, Ttp is also required for recruiting exosome
components, including Rrp4, Rrp40, Rrp41, Rrp42, Rrp43, Rrp46, hCsl4, Mtr3, and
PM-Scl75 [15,18]. Both dacay pathways are shown in Appendix 2 [19].
In addition, Ttp interacts with Ago2 and Ago4, and it helps microRNA 16
(containing an UAAAUAUU sequence which is complementary to the ARE sequence)
targeted to ARE-containing mRNA [20].
1.1.4 Ttp in the Transcriptional Regulation
Ttp serves as a repressor in NF-κB-dependent transcription by associating with
different proteins. It was observed that the amount of nuclear p65, one subunit of NF-κB,
was increased in the mouse embryonic fibroblasts from the Ttp knockout mice [21].
Loss of mRNA binding ability, Ttp still impairs the nuclear import of p65, and the
activity of NF-κB is repressed [21]. Another study indicates that Ttp also associates with
5
histone deacetylases (HDACs) which are transcriptional co-repressors, such as HDAC-1,
HDAC-3, and HDAC-7. The abolished effect of Ttp is recovered in the knockdown of
HDAC-1, HDAC-3, and DHAC-7 or the treatment of histone deacetylase inhibitors [22].
1.1.5 The Phosphorylation Modification of TTP Family Proteins
The modification of phosphorylation alters the functions, subcellular localizations,
protein stability of TTP family proteins. Ser52, Ser80, Ser82, Ser178, Ser249, Ser250,
and Ser264 of Ttp are phosphorylated by mitogen-activated protein kinase
(MAPK)-activated protein kinase 2 (MK2) [23]. Ttp is stabilized and locates in the
cytoplasm when Ser52 and Ser178 are phosphorylated; moreover, phosphorylation of
Ser52 and Ser178 is necessary for Ttp associating with adapter protein 14-3-3. The
complex of 14-3-3 and Ttp abolishes the recruitment of deadenylase complex [24];
therefore, TTP cannot destabilize the mRNA targets [25].
Similarly, Ser54, Ser92, and Ser203 of Zfp36l1 are the phosphorylation sites of
MK2, and the mRNA destabilizing ability of Zfp36l1 is repressed. Phosphorylated
Zfp36l1 can form the complex with 14-3-3, but the association with AREs, Ccr4, and
Dcp2 is still remained [26]. These results indicate that the modification of
phosphorylation of Zfp36l1 regulates its mRNA decay activity by the steps after
6
recruiting mRNA decay enzymes. Other studies showed that Ser92 and Ser203 are
phosphorylated by Protein kinase B (also known as AKT) [23,27]. Phosphorylated
Ser92 and Ser203 of Zfp36l1 binds to 14-3-3 and abolishes its mRNA decay activity;
moreover, the protein stability of Zfp36l1 is up-regulated by the phosphorylation of
those two sites [23]. The phosphorylation of Zfp36l2 is unclear until now.
1.1.6 TTP family Proteins in Cancers
The accumulation of ARE-containing mRNA is observed in the early stage of
tumorigenesis. Abnormal expressions of ARE-binding-proteins may result in the loss of
ARE-mediated mRNA decay [28]. The low expression of TTP had been examined in
breast, colon, cervix, prostate, and lung cancer [8], whereas the expression of HuR
(mRNA stabilizing factor which also associating with AREs) was high in colon
carcinogenesis [29]. Human head and neck squamous cell carcinoma (HNSCC) with
low expression level of ZFP36L1 are insensitive to cisplatin (a common
chemotherapeutic drug) because the decrease of ZFP36L1-mediated destabilization of
cellular inhibitor of apoptosis 2 (cIAP2) mRNA [30].
7
1.2 Innate Immune Response
1.2.1 Mitogen-activated Protein Kinase (MAPK) in the Innate Immune Response
There are three major MAPKs signaling pathways in mammals: p38 MAPK,
extracellular signal-regulated kinase (ERK), and the c-Jun amino-terminal kinases (JNK)
(Appendix 3) [31]. MAPKs are the important regulators in differentiation, survival,
apoptosis, and production of inflammatory mediators [31,32].
The activation of MKK3-p38 pathway induced the expression of inflammatory
mediator, Interleukin-12 (IL-12) [33]. Another inflammatory mediators, Tnf-α, is
up-regulated by p38 MAPK and ERK. p38 MAPK induces the transcriptional increase
of Tnf-α mRNA through NF-NB [34], and the activation of p38 MAPK pathway
up-regulates the translation of Tnf-α mRNA [35]. In addition, TPL2-ERK pathway is
required for the translocation of Tnf-α mRNA from nuclear to cytoplasm [36].
1.2.2 Mitogen-activated Protein Kinase Phosphatases (MKPs) in the Innate
Immune Response
Mitogen-activated protein kinase phosphatases (MKPs) are negative regulators of
MAPKs through de-phosphorylation at both threonine and tyrosine residues. Many
studies reported that the substrate specificity of MKPs was different [37] .
8
MKP-1 is one of the most commonly studied MKP in the innate immune response.
Mkp-1 can be activated by ERK signaling pathway [38]. Overexpression of Mkp-1 may
inactivate Jnk and p38, and then inhibits the expressions of Tnf-α and IL-6 [38].
Therefore, inducers of MKP-1 are potential drugs for autoimmune diseases [38].
1.2.3 TTP Family Proteins in the Innate Immune Response
According to the study from Dr. Baltimore [39], there are three types of gene
expression profiles after inflammatory stimuli. The expressions of type I genes are
rapidly increased and decreased, and they are also known as immediate early gene. The
expressions of type II genes are continuously increased until 2 hour after Tnf-α
treatment and then their expressions are sustained. Last, the expressions of type III
genes are continuously increased, and they do not reach the peak until 24 hour after
Tnf-α treatment (Appendix 5). The quality and timing of those genes expressions are
controlled by mRNA stability. For example, the transcripts of type I genes are unstable
in the resting condition, but they were stable after Tnf-α treatment.
In mouse macrophages RAW264.7 cells, immune signals such as
Lipopolysaccharide (LPS) from gram-negative bacteria and Tnf-α activate MAPK
signaling pathways [40]. The expression of Ttp is also induced after LPS-stimulation
9
[41], and it serves as an anti-inflammatory role through rapidly destabilizing mRNAs of
pro-inflammatory mediators which containing AREs. The mRNA decay of one third
unstable transcripts depends on Ttp, and the decay timing was controlled by the activity
of p38 MAPK [42]. In addition, Ttp is able to interact with mRNA 3’UTR of itself, and
the auto-regulation of Ttp can limit its own expression [41,43].
As mentioned before, many diseases related to the innate immune response are
developed in the Ttp knockout mice due to the expression of Tnf-α is increased. Those
results also indicate that Ttp may be a negative regulator in the innate immune response.
Moreover, Tnf-α mRNA is down-regulated by overexpression of Zfp36l1 and Zfp36l2 in
HEK 293 cells [13]. However, the effects of protein overexpression may be different
with the real effects, and the identified mRNA targets may be considered as
“non-physiological targets”. It is better to analysis the functions and mRNA targets of
TTP family proteins in the knockout/knockdown mice or cells than in the
overexpression systems. Hence, we study the detail mechanisms of Zfp36l1 and
Zfp36l2 in the innate immune response in their knockdown cells.
10
2. Materials and Methods 2.1 Plasmid Constructs
To generate Flag-tagged mouse Ttp, Zfp36l1, and Zfp36l2, oligonucleotides for Ttp,
Zfp36l1, and Zfp36l2 coding sequences were amplified from 2 hours LPS-treated
RAW264.7 cDNA. The sequences of primers used in PCR were listed in Table 1. The PCR
fragments were cloned into pCRII-TOPO vector (Invitrogen, Carlsbad, CA, USA). After
sequences were confirmed, recombinant genes were then subcloned into mammalian
cells expression vector pCMV-Tag-2B (Stratagene, La Jolla, CA, USA) via EcoRI sites.
For preparing riboprobes for RNA pull-down assay, the 3’UTR of Mkp-1 was PCR
amplified from 1 hour differentiation-triggered 3T3-L1 cDNA. The sequences of primers
used in PCR were listed in Table 1. The PCR fragment was cloned into pCRII-TOPO
vector, and sequence was confirmed. For luciferase reporter constructs, Mkp-1 3cUTR
was subcloned into pCMV-Tag-2C-Luciferase (Stratagene) reporter using the ApaI and
KpnI sites.
To generate Flag-tagged and Myc-tagged mouse Caf1a (Ccr4-Associated Factor;
CCR4-NOT transcription complex, subunit 7 (Cnot7)), the coding sequence of Caf1a
was amplified from 2 hours LPS-treated RAW264.7 cDNA. The sequences of primers
11
used in PCR were listed in Table 1. The PCR fragments were cloned into pCRII-TOPO
vector. After sequence were confirmed, recombinant gene was then subcloned into
mammalian cells expression vector pCMV-Tag-2B and pCMV-Tag-3 via HindIII and
BamH1 sites.
For glutathione-S-transferase (GST) fusion protein mouse 14-3-3 zeta construct,
14-3-3 zeta coding region were PCR-amplified from 2 hour LPS-treated RAW264.7
cDNA. The sequences of primers used in PCR were listed in Table 1. The PCR fragment
was cloned into pCRII-TOPO vector. After sequences were confirmed, 14-3-3zeta was
subcloned into E.coli expression vector pGEX-3 (GE Healthcare BioScience, Chalfont
St. Giles, UK).
2.2 Cell Culture
RAW264.7 cells (mouse macrophage) were grown in Dulbecco’s modified Eagle
medium (Gibco) containing 1.5 g/L sodium bicarbonate, and supplemented with 10%
fetal bovine serum (Characterized; Hyclone or 12003C; SAFC), 2mM L-glutamine
(Gibco). Human embryonic kidney (HEK) 293T cells were grown in Dulbecco’s
modified Eagle medium (Gibco-BRL) containing 3.7 g/L sodium bicarbonate, and
12
supplemented with 10% fetal bovine serum (Qualified, Gibco). Both RAW264.7 and
HEK 293T cells were cultured in the 37ɗ, humidified, 5% CO2incubator.
2.3 Preparation Whole Cell Extracts and Cytoplasmic/Nuclear Extracts
Cells in a 10-cm dish were washed once with PBS and then harvested. For
preparing whole cell extracts, the harvested cells were lysed in 400 μL of whole cell
extract buffer (25 mM HEPES pH7.7, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT,
0.1% NP-40, 0.3 M NaCl, protease inhibitor cocktail (Sigma) and phosphatase inhibitor
containing 0.01M beta-glycerol phosphate, 0.1 mM Na2MoO4, 0.1 mM Na3VO4 pH10,
0.01M NaF). The cell lysates were shaken at 4ɗ for 30 minutes, and next they were
centrifuged for 5 minutes at 13,000 rpm, 4ɗ. The supernatant was collected as a whole
cell extract. For preparing cytoplasmic/nuclear extracts, harvested cells were lysed in
400 μL hypotonic buffer (10 mM HEPES pH7.5, 10 mM KOAc, 2.5 mM DTT, 0.05%
NP-40, protease inhibitor and phosphatase inhibitor). The cell lysates were shaken at 4
ɗ for 30 minutes, and then they were centrifuged for 30 seconds at 9,000 rpm, 4ɗ.
The supernatant was collected in as a cytosolic extract. The pellets were washed once
with hypotonic buffer, and then they were re-suspended in 50 μL of buffer C (20 mM
HEPES pH7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, protease
13
inhibitor and phosphatase inhibitor). The cell suspension was shaken at 4ɗġ for 30
minutes, and then it was centrifuged for 5 minutes at 13,000 rpm, 4ɗ. The supernatant
was collected as a nuclear extract.
2.4 Alkaline Phosphatase, Calf Intestinal (CIP) Treatment
The total proteins from LPS-stimulated RAW264.7 cells were extracted by
hypotonic buffer. Taking 100 µg of total proteins incubated with 1µL of CIP (NEB;
M0290s) in 37ɗ for 30 minutes.
2.5 Western Blot Assay and Antibodies
4X SDS-PAGE Sample buffer (200 mM Tris pH 6.8, 8% SDS, 0.4% bromopheol
blue, 40% glycerol, 400 mM β-mercaptoethanol) was added to the sample till final
concentration was 1X, and then heated at 100ɗġ for 5 minutes. Proteins were separated
on 10% polyacrylamide gels and transferred onto 0.45 µm-pore-size PVDF membrane
(Millipore) for Western blotting. The specific antibodies were used including
anti-BRF1/2 (Cell signaling; 2119S), anti-hnRNPC1/C2 (Santa Cruz; sc-10037),
anti-MKP-1 (Santa Cruz; sc-1102), anti-COX-2 (Santa Cruz; sc-1745), anti-p-p38
14
MAPK T180/Y182 (Cell signaling; 9211S), anti-total-p38 (Sigma), anti-p-p44/42
MAPK (Cell signaling; 9101S), anti-ERK1 (Santa Cruz; sc-093), anti- p-JNK (Sigma),
anti-JNK1 (Santa Cruz; sc-571), anti-Myc (Genscrip; A00172), anti-Flag M2 (Sigma;
F1804), anti-Ttp, anti-Zfp36l2, anti-beta-tubulin for 1 hour at room temperature. After
washing with PBST (PBS containing 0.1% Tween 20) for an appropriate time,
horseradish peroxidase-conjugated secondary antibodies, goat anti-rabbit IgG (KPL;
474-1516), goat anti-mouse IgG (KPL; 474-1806), or rabbit anti-goat (Sigma) was
incubated for 1 hour at room temperature. Western Lightning enhanced
chemiluminescemce substrate (Perkin Elmer) were used for detection.
2.6 RNA Extraction and Reverse-transcription
RNA isolation was performed with TRIzol reagent (Invitrogen) according to the
suggested procedures of manufacturer. Cells in a six-well plastic culture plate were
washed once with PBS and directly lysed in a well with 1ml TRIzol. The samples were
incubated at room temperature for 5 minutes, and next they were added with 200 μL of
chloroform and vortexed for 15 seconds. After incubated at room temperature for 3
minutes, the samples were centrifuged for 15 minutes at 13,000 rpm, 4ɗ. The upper
layer (colorless) was transferred to a new tube, and then it was add with equal volume
15
of isopropanol and mixed well. After incubated at room temperature for 10 minutes, the
mixture was centrifuged for 15 minutes at 13,000 rpm, 4ɗ. The white RNA pellet was
washed by 75% ethanol, and then it was centrifuged for 10 minutes at 13,000 rpm, 4ɗ.
After the pellet was air-dried for 10 minutes, it was dissolved with 20-30 μL DEPC H2O.
In order to dissolve RNA entirely, the sample was incubated twice at 60 ɗġ for 10
minutes. After quantitated by A260/A280 measurements, 5 μg RNA was taken for
reverse-transcription. 5 μg total RNAs and 0.5 μg oligo dT were annealed at 70 ɗ for
5 minutes, and then the samples were cooled down on ice for 2 minutes. cDNAs were
produced following the recommended procedures of M-MLV reverse transcriptase
(Promega).
2.7 Real-time PCR
RNA quantitative assay was preform with the Applied Biosystems 7300 Real-Time
PCR system (Applied Biosystems) in total volume of 20 μL, containing 10 μL FastStart
Universal SYBR Green Master (Rox) (Roche; 04913914001), 4 μL of 10-times diluted
cDNA, 0.4 μL of 5-20 μM forward and reverse primer (see table 2), and 5.6 μL DEPC
H2O. The result was normalized with beta-actin according to 2-ΔΔCtrelative quantitation
method on the manual of manufacturer.
16
2.8 RNA Pull-down Assay
Cytoplasmic extracts from LPS-stimulated RAW264.7 cells were collected as
previously described. Potassium acetate was adjusted to 90 mM, and 0.1 U/μL RNasin
(Promega) and 20 μg/μL yeast tRNA were add to the lysates. In order to remove the
non-specific binding, heparin-agarose (Sigma-Aldrich) was incubated with the lysates
for 15 minutes at 4ɗ and then centrifuged for 1 minute at 8,000 rpm, 4ɗ. The
supernatant was further cleaned with Streptavidin Sepharose (Invetrogen) for 1 hour at 4
ɗ, and then centrifuged for 1 minute at 8000 rpm, 4ɗ. The supernatant was incubated
with biotinylated Mkp-1 3’UTR or negative control 18S RNA which made by
T7-MEGA shortscriptTM, High Yield Transcription Kit (Ambion) for 1 hour at 4 ɗ.
Next, Streptavidin Sepharose was added to pulled down biotinylated RNA complex for
2 hours at 4ɗ. The pulled down complexes were washed four times with binding buffer
(hypotonic buffer containing 90 mM potassium acetate). Last, the RNA complexes were
separated by 10% SDS-PAGE and detected by Western blotting.
17
2.9. Dual Luciferase Reporter Assay
HEK293T cells were seeded in six-well plastic culture plate, and they were
transfected using calcium phosphate precipitation method with different plasmids
(containing 0.5 μg Renilla luciferase expression vector as a control of transfection rate)
at 30% confluency. At 24 hours post-transfection, the cells were harvested and lysed in
50 μL of passive lysis buffer (Promega). The samples were shaken for 30 minutes at 4ɗ
and centrifuged for 5 minutes at 13000 rpm, 4ɗ. The supernatant was transferred to a
well of black 96-well plate. The firefly luciferase activities were measured by
luminometer (Packard, Downer Grove) after adding 50 μL of LAR II. The renilla
luciferase activities were measured sequentially after adding 50 μL of Stop & Glo®
Reagent. Each treatment group contained duplicate cultures, and each experiment was
repeated more than 3 times. The firefly luciferase activities were normalized with the
renilla luciferase activities. The relative luciferase activity represented the luciferase
activity of reporter carrying 3’UTR normalized with that of reporter alone.
2.10 Short-hairpin RNA (shRNA)
Lentivirus vectors encoding shRNA targeted to mouse Zfp36l1 and Zfp36l2 were
purchased from National RNAi core facility (Academia Sinica). TRCN0000123469
18
clone (5’-CCACATACAATATCTGTGTAA-3’) for mouse Zfp36l1 knockdown,
TRCN0000173172 clone (5’-CCAAACACTTAGGTCTCAGAT-3’) for mouse Zfp36l2
knockdown, TRCN0000072243 clone (5c-CTTCGAAATGTCCGTTCGGTT-3c) for
Luciferase knockdown. Both of TRCN0000123469 and TRCN0000173172 clones
targeted to the 3’UTR of mRNA.
2.11. Lentivirus Knockdown
HEK293T cells were seeded in a 10-cm dish for transfection (calcium phosphate
precipitation method) of virus production vectors, 14 μg of CMV ΔR8.9.1, 2 μg of
pMD.G, and 14 μg of specific shRNA sequence bearing-pLKO.1 plasmids. 8 hours after
transfection, the culture medium was replaced with fresh medium for RAW264.7 cells.
24 and 48 hours later, the virus-containing medium was collected for primary-infection
and super-infection for RAW264.7 cells. 24 hours after super-infection, virus-containing
medium was replaced with fresh medium for RAW264.7 cells. For generation of stable
knockdown clones, puromycin (3 μg/mL) was added and GFP signal was served as a
selection marker. After puromycin-selection for one week, cells were harvested and
analyzed by Western blotting in order to check knockdown efficiency.
19
2.12. Co-immunoprecipitaion (Co-IP)
24 hours after transfection, HEK293T cells from 10-cm dishes were harvested and
re-suspended in NET buffer (50 mM Tris pH 7.4, 350 mM NaCl, 1 mM EDTA, 0.3%
Triton X-100, protease inhibitor, phosphatase inhibitor). The cell lysates were shaken at
4ɗ for 30 minutes, and then they were centrifuged for 5 minutes at 13,000 rpm, 4ɗ.
The supernatant was collected for total proteins measurement using Bradford assay
(Bio-Rad). 20 µL of 30% anti-FLAG M2 affinity agarose beads (Sigma) were added for
2-hour incubation, and then washed 3 times with NET buffer. The protein complexes
were separated by 10% SDS-PAGE, and then they were detected by Western blotting.
2.13. GST Fusion Protein Production and GST Pull-down Assay
pGEX-2T or pGEX-3-14-3-3 zeta plasmid was transformed to E.coli (DH5α), and
then E.coli (DH5α) were grown in 5 mL of LB (1% tryptone, 0.5% yeast extract, 1%
NaCl) overnight in the 37ɗ incubator. The broth was seeded to 100 ml of LB and
cultured for 2 hours in the 37ɗ incubator. 0.1 mM IPTG was added to induce
production of GST fusion protein (GST only or GST-14-3-3) for 2 hours, and then cells
20
were harvested and re-suspended in PBST (1X PBS containing 1% triton X-100). The
cell suspension was frozen (liquid nitrogen) and thawed (37ɗ water bath) 4 times. The
cell suspension was lysed completely using Bioruptor® (Diagenode; UCD-200) with
high voltage for 10 minutes. Last, the cell suspension was centrifuged for 20 minutes at
13,000 rpm, 4ɗ . The supernatant was collected and purified with Glutathione
Sepharose 4B (Amersham Biosciences, GE Healthcare) by incubating at 4ɗ for 1 hour
and washing 3 times with PBST. The pulled down complex was analyzed by 10%
SDS-PAGE and Coomassie blue staining to check the expression of GST-tagged
protein.
LPS-stimulated RAW264.7 cells from 10-cm dishes were harvested and lysed in
LSBT buffer (20 mM HEPES pH 7.9, 100mM NaCl, 0.1 mM EDTA, 2.5 mM MgCl2,
1.5% Triton X-100, 0.05% NP-40, protease inhibitor, phosphatase inhibitor). The cell
lysates were shaken at 4ɗ for 30 minutes, and then centrifuged for 5 minutes at 13,000
rpm, 4ɗ. The lysates of GST-fusion protein were incubated with glutathione beads at 4
ɗ for 1 hour. The beads were washed two times with PBST and then once with LBST
buffer. The beads conjugated with GST fusion protein were incubated with the lysates
form LPS-stimulated RAW264.7 cells at 4ɗ for 2 hours, and then the beads were
washed three times with LBST buffer. The protein complexes were separated by 10%
21
SDS-PAGE and analyzed by Western blotting.
2.14 Statistical Analysis
All of the results were presented as the mean ± SD of at least three independent
experiments. The statistically significant values were calculated by one-tailed Student's
t-test. One asterisk indicated p-value < 0.05, and two asterisks indicated p-value < 0.01.
22
3. Specific Aims
1.Analysis of expression kinetics of TTP family proteins in the period of
LPS-stimulation in RAW264.7 cells.
2. Identification of novel Zfp36l1 and Zfp36l2-targeted mRNAs.
3. Molecular mechanism of which Zfp36l1 and Zfp36l2 regulate Mkp-1 mRNA stability
in LPS-stimulation.
4. Functional mechanism of Zfp36l1 and Zfp36l2 in the innate immune response.
23
4. Results
4.1 The Consistent Expression and Protein Phosphorylation of Zfp36l1 and Zfp36l2 in the Period of LPS-stimulation in RAW264.7 Cells.
The TTP family contains four major members, Ttp, Zfp36l1, Zfp36l2, and rodent
specific Zfp36l3. It is well known that Ttp plays a key role in innate immune response.
The mRNA and protein of Ttp were highly induced by LPS (Figure 1A), and it was
suggested to destabilize mRNA targets rapidly. To investigate the roles of other two TTP
family proteins, Zfp36l1 and Zfp36l2, in the innate immune response, we first examined
their RNA and protein expression profiles in LPS-stimulated RAW264.7 cells. The
proteins of Zfp36l1 and Zfp36l2 were maintained in the consistent level in cytoplasm
(Figure 1A), whereas their mRNA expression levels were down-regulated in the period
of LPS-stimulation (Figure 1B). Multiple forms of Zfp36l1 and Zfp36l2 were detected
in Western blotting and LPS treatment resulted in their band shifts (Figure 1A). When
the cytosolic extracts (100μg) from 120 minutes of LPS-stimulation were treated with
alkaline phosphatase (CIP) for 30 minutes, the higher protein bands of Zfp36l1 and
Zfp36l2 were back to the lower positions (Figure 1C), which might indicate a status of
protein hypo-phosphorylation. These observations suggested that the protein levels of
Zfp36l1 and Zfp36l2 were maintained constantly and were phosphorylated under
24
LPS-stimulation.
4.2 Zfp36l1 and Zfp36l2 Destabilize MAPK Phosphotase-1 ( Mkp-1) and Cyclooxygenase-2 ( Cox-2) mRNAs in Resting RAW264.7 Cells.
Since Zfp36l1 and Zfp36l2 were constitutively expressed in macrophages, we
inferred that they play roles to control mRNA stability in the resting condition. The
strategy to identify the mRNA targets of Zfp36l1 and Zfp36l2 was knockdown of
Zfp36l1 and Zfp36l2 using Lentivirus-carrying shRNAs in RAW264.7 cells. As shown
in Figure 2A, the knockdown efficiency of shRNA specific to Zfp36l1 and Zfp36l2 was
confirmed by Western blotting. ARE-containing immediate early genes as well as
inflammatory mediator genes were target candidates of Zfp36l1 and Zfp36l2, such as
Ttp, Mkp-1, Tnf-α, Cox-2, Ccl-2, and Icam-1. The RNA and protein expression of these
candidates were examined by real-time PCR and Western blotting in different
knockdown cells, including Zfp36l1 knockdown, Zfp36l2 knockdown, and dual
knockdown cells. We predicted that the mRNA targets of Zfp36l1 and Zfp36l2 would be
increased in knockdown cells because Zfp36l1 and Zfp36l2 function in mRNA
destabilizing. We found that the RNA (Figure 2B) and protein expressions (Figure 2A)
of Mkp-1 were significantly increased in all knockdown cells, whereas the RNA (Figure
25
2C) and protein expressions (Figure 2A) of Cox-2 were significantly increased in
Zfp3l2 knockdown cells and dual knockdown cells but not in Zfp36l1 knockdown cells.
On the contrary, mRNA levels of Ttp, Tnf-α, Ccl-2, and Icam-1 were decreased in all
knockdown cells (Figure 2F).
Mkp-1 and Cox-2 mRNAs were possible targets of Zfp36l1 and Zfp36l2 in the
resting condition of RAW264.7 cells. To further elucidate whether Mkp-1 and Cox-2
mRNAs were post-transcriptionally regulated by Zfp36l1 and Zfp36l2, we determined
the mRNA half-lives of Mkp-1 and Cox-2 in different knockdown cells. As shown in
Figure 2D, half-lives of Mkp-1 mRNA increased from 15.2 minutes in control cells
(Luciferase knockdown cells) to at least 41.6 minutes in all knockdown cells.
Furthermore, Figure 2E showed that half-lives of Cox-2 mRNA increased from 15.8
minutes in control cells (Luciferase knockdown cells) to at least 55 minutes in the cells
which containing Zfp36l2 knockdown. Collectively, Mkp-1 mRNA stability was
down-regulated by both Zfp36l1 and Zfp36l2, and Cox-2 mRNA stability was only
down-regulated by Zfp36l2 in the resting macrophages.
4.3 Zfp36l1 and Zfp36l2 Down-regulate the Mkp-1 and Cox-2
3’UTR-mediated Luciferase Reporter Activity and Interact with
26
Deadenylase Caf1a.
To delineate the molecular mechanism underlying Zfp36l- and Zfp36l2-regulated
Mkp-1 mRNA stability, the full-length 3’UTR of Mkp-1 mRNA was cloned into
downstream of the luciferase reporter gene for co-transfection analysis. The Mkp-1
3’UTR derived luciferase activity was reduced when co-transfected with Zfp36l1 or
Zfp36l2 expression plasmid in HEK293T cells which did not constitutively express
human ZFP36L1 or ZFP36L2 (Figure 3A). Similarity, the 3’UTR (1-1477 base pairs) of
Cox-2 mRNA was cloned into downstream of the luciferase reporter gene, and
luciferase activity was also decreased when co-transfected with Zpf36l1 and Zfp36l2
expression plasmid in HEK293T cells (Figure 3B). The associated proteins of Cox-2
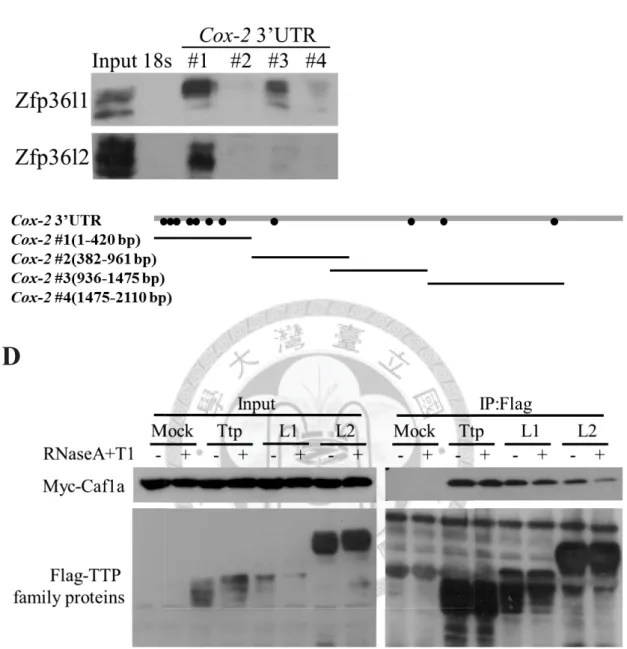
3’UTR were analyzed by the RNA pull-down. The Cox-2 3’UTR was separated into
four fragments (Figure 3C, lower panel). Four biotinylated Cox-2 3’UTR fragments
were incubated with cytosolic lysates from RAW264.7 cells. The streptavidin
sepharoses were used to precipitate the ribonucleoprotein complexes. The complexes
were analyzed by Western blotting with anti-Zfp36l1 and anti-Zfp36l2 (Figure 3C,
upper). Both Zfp36l1 and Zfp36l2 could be pulled down by ARE-rich fragment 1 of
Cox-2 3’UTR.
Deadenylase Caf1a was recruited by Ttp to result in destabilization of target
27
mRNAs [16]. To understand whether Caf1a associated with Zfp36l1 and Zfp36l2 for
their mRNA destabilizing activity, Flag-tagged Zfp36l1or Zfp36l2 was co-expressed
with myc-tagged Caf1a in HEK293T cells. In order to rule out the RNA mediated
interactions, RNase A and RNase T1 were added to the lysates and they were incubated
in 30ɗ for 10 minutes. The lysates were precipitated by anti-Flag M2 affinity agarose
beads. Ttp severed as a positive control, and mock (Flag only) severed as a negative
control. The protein samples were detected by anti-Myc and anti-Flag. According to the
results of Figure 3D, the interaction between TTP family proteins and Caf1a was
RNA-independent. These results suggested that through ARE-containing 3’UTR,
Zfp36l1 and Zfp36l2 could down-regulate Mkp-1 and Cox-2 mRNA expressions, which
might be due to the recruitment of deadenylase Caf1a.
4.4 Regulation of Mkp-1 mRNA Stability by Phosphorylation of Zfp36l1.
To further investigate the regulation of Mkp-1 mRNA during the early
LPS-stimulation in RAW264.7 cells, we examined its RNA expression profiles in
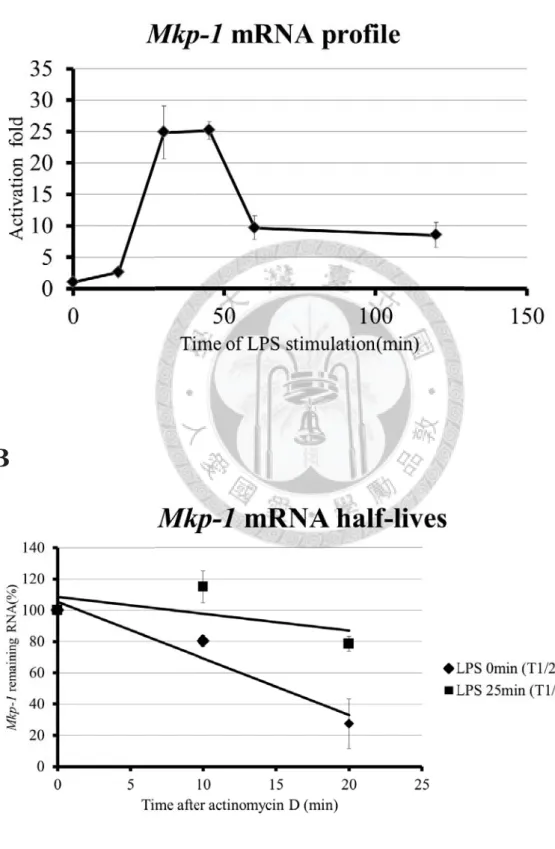
LPS-stimulated RAW264.7 cells (Figure 4A). Mkp-1 mRNA level was increased
significantly after LPS-stimulation from 15 to 30 minutes. However, Mkp-1 mRNA
28
level was decreased rapidly after LPS-stimulation from 45 to 60 minutes. In order to
demonstrate that the rise of Mkp-1 mRNA level at the early LPS-stimulation was
regulated by mRNA stability, we examined the mRNA half-lives of Mkp-1 in the resting
condition and LPS-stimulation for 25 minutes. In Figure 4B, half-lives of Mkp-1 mRNA
increased from 15.3 minutes in the resting condition to 54.7 minutes LPS-stimulation
for 25 minutes. This result suggested that the increase of Mkp-1 mRNA in the early
LPS-stimulation was regulated in part post-transcriptionally.
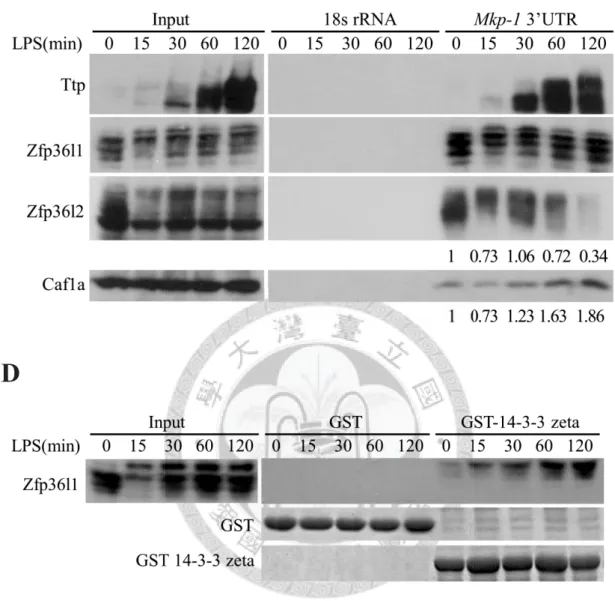
To understand how Zfp36l1 and Zfp36l2 regulate Mkp-1 mRNA stability during
LPS-stimulation, firstly, the RNA pull-down assay was performed to examine the
RNA-protein interaction. The biotinylated Mkp-1 3’UTR was incubated with cytosolic
lysates from LPS-stimulated RAW264.7 cells. The ribonucleoprotein complexes were
precipitated by the streptavidin sepharoses and then subjected to SDS-PAGE for
Western blotting with anti-Zfp36l1 and anti-Zfp36l2 (Figure 4C). The interaction of
Zfp36l1 and Mkp-1 3’UTR was constant during LPS-stimulation, whatever Zfp36l1 was
phosphorylated or not. However, the interaction of Zfp36l2 and Mkp-1 3’UTR was
changeable, and it was depended on the protein amount of Zfp36l2 (Figure 4C). In
addition, the amount of deadenylase Caf1a in the ribonucleoprotein complexes was
decrease in the early stage of LPS-stimulation, and this change might stabilize Mkp-1
29
mRNA.
It had been reported that the interaction between phosphorylated Ser92 of Zfp36l1
and 14-3-3 protein could inhibit the mRNA decay activity of Zfp36l1 after
insulin-stimulation [27,44]. Since LPS-induced Zfp36l1 phosphorylation did not affect
its RNA binding activity (Figure 4C), GST pull-down assay was performed to study
whether hyper-phosphorylated Zfp36l1 in the period of LPS-stimulation interacted with
14-3-3. As shown in Figure 4D, only hyper-phosphorylated Zfp36l1 formed the
complex with 14-3-3. This complex might repress the mRNA destabilization function of
Zfp36l1.
4.5 The Induction of Mkp-1 mRNA in Early LPS-stimulation is Post-transcriptionally Modulated by Zfp36l1 and Zfp36l2.
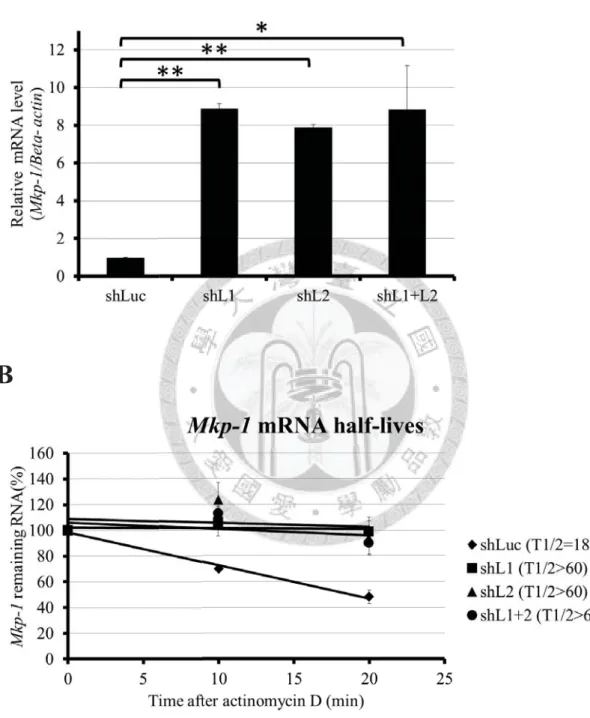
To verify the functions of Zfp36l1 and Zfp36l2 in the initiation stage of Mkp-1
expression, we examined the activation levels of Mkp-1 mRNA in different knockdown
cells after LPS-stimulation for 15 minutes. A marked rise of the activation levels in all
knockdown cells compared with control knockdown cells implied that the decline of
Zfp36l1 and Zfp36l2 protein expressions facilitated the mRNA expression of Mkp-1
30
(Figure 5A); what is more, half-lives of Mkp-1 mRNA in control, Zfp36l1, Zfp36l2, or
dual knockdown cells were 15.2, 41.6, > 60, and > 60, respectively (Figure 5B). This
result indicated that knockdown of Zfp36l1, Zfp36l2, or dual proteins could stabilize
Mkp-1 mRNA in early LPS-stimulation.
4.6 p38 MAPK Activity is Regulated by Zfp36l1 and Zfp36l2 Through Mkp-1.
Inflammatory stimuli such as LPS activate the three major MAPKs pathways in
mammals: p38 pathways, ERK, and JNK. MAPK pathways can activate innate immune
response through the transcriptional induction and the post-transcriptional modulation
[37,45]. Inactivation of MAPKs mediated by the de-phosphorylation activity of Mkp-1
[46]. MAPKs was inactivated by Mkp-1 through de-phosphorylation [46]. We examined
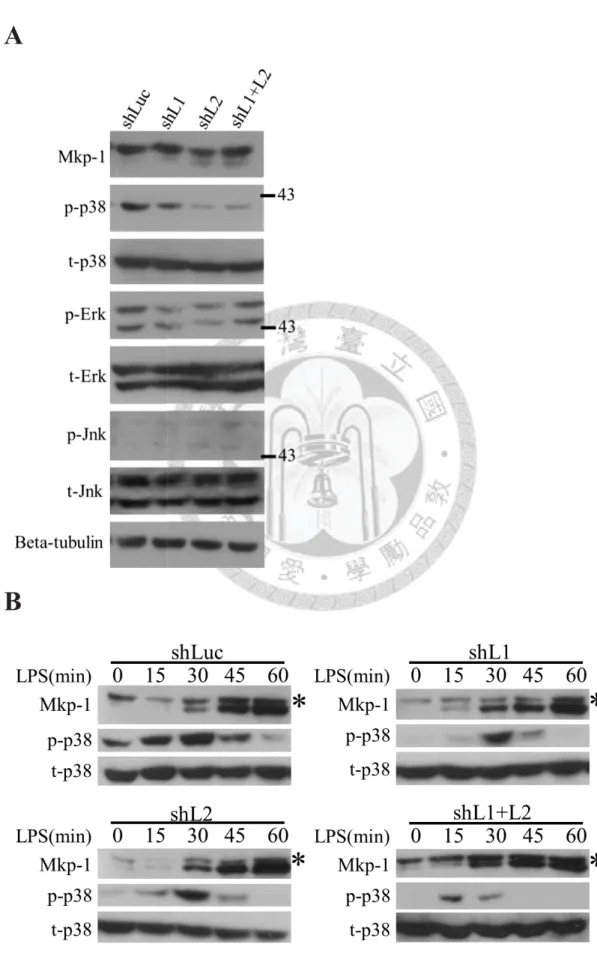
the three MAPK activities in different knockdown cells. In the resting condition, p38
activity was down-regulated by the increase of Mkp-1 in all knockdown cells, but the
activities of the other two MAPKs were consistent (Figure 6A). The time courses of
Mkp-1 expression showed that Mkp-1 expression was detected earlier in all knockdown
cells than in control cells in LPS-stimulation, and it was correlated with p38 activity
(Figure 6B).
31
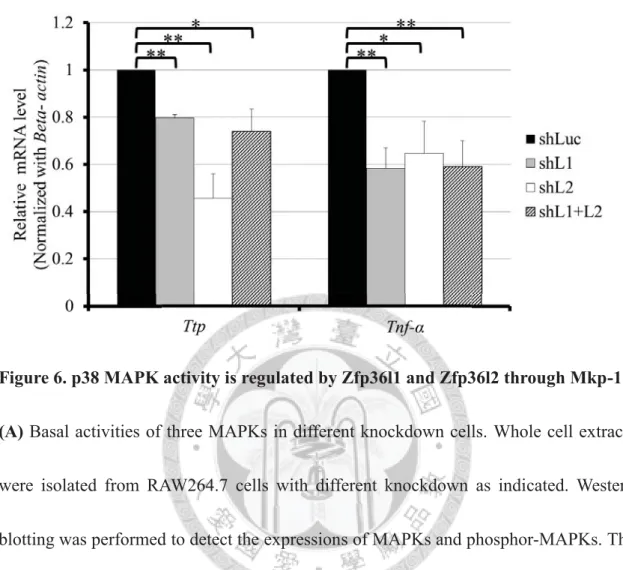
Furthermore, the activation of p38 pathway is required for the mRNA inductions of
Ttp and Tnf-α in LPS stimulated RAW264.7 cells [47,34]. The mRNA expression levels
of Ttp and Tnf-α were reduced in all knockdown cells in LPS-stimulation due to
decrease of p38 activity by Mkp-1 (Figure 6C). In the other word, Zfp36l1 and Zfp36l2
are necessary for macrophages in the initiation of the innate immune response.
32
5. Discussion
The activities of MAPKs are necessary for the initiation of the innate immune
response. Both the activation of transcription factors by phosphorylation and the
stabilization of ARE-containing mRNA can help cells to produce pro-inflammatory
mediators or cytokines. The activations of MAPKs are turned off within several minutes,
and then the mRNAs of those pro-inflammatory mediators or cytokines will be
degraded rapidly. Some negative regulators such as Mkp-1 (phosphatase) and Ttp
(mRNA decay mediator) can help cells to return to the resting condition. Hence, the
gene expressions should be tightly controlled in both transcription and
post-transcription levels in the innate immune response.
We are the first to observe the protein expression profiles of endogenous TTP
family proteins during the period of LPS-stimulation in mouse macrophage RAW264.7
cells. There are two interesting observations. One is the protein expression profiles of
these three TTP family members were different, and the other is the uncorrelated mRNA
and protein expression profiles of Zfp36l1 and Zfp36l2.
Ttp was induced after LPS-stimulation, and Zfp36l1 and Zfp36l2 were constitutive
expressions (Figure 1A). This observation indicates that Zfp36l1 and Zfp36l2 may play
important roles in the resting condition, and the low expressions of pro-inflammatory
33
mediators or cytokines are maintained by the mRNA destabilized abilities of Zfp36l1
and Zfp36l2.
As shown in Figure 1B, the mRNA expressions of Zfp36l1 and Zfp36l2 were
down-regulated after LPS-stimulation, but the protein expressions of Zfp36l1 and
Zfp36l2 were consistent. The mRNA expression profiles of Zfp36l1 and Zfp36l2 were
uncorrelated with their protein profiles after LPS-stimulation (Figure 1B). Some similar
observations of mRNA expression profiles of Zfp36l1 and Zfp36l2 were also reported
by Liang et al. [48] and Cao et al. [49]. This uncorrelated relationship implys the
expressions of Zfp36l1 and Zfp36l2 are regulated in the translational and
post-translational levels. The polysome analysis of their mRNA during LPS-stimulation
can be performed to verify the translational regulation. Furthermore, the
post-translational modifications such as phosphorylation (Figure 1C) may alter protein
stability [25]. Zfp36l1 and Zfp36l2 may be more stable in the hyper-phosphorylated
forms than the hypo-phosphorylated forms. Thus, their protein expression levels are
near consistent, even their mRNAs are decreased after LPS-stimulation.
To identify the possible mRNA targets of Zfp36l1 and Zfp36l2 in resting
macrophages, we knocked down Zfp36l1 and Zfp36l2 by using Lentivirus-carrying
shRNA. Based on previous reports, the candidates of mRNA targets are chosen by the
34
ARE number in their 3’UTR, the associations with Ttp, and their mRNA expression
profiles which were categorized to the immediate-early genes [39,50].
In our study, the RNA and protein levels of Mkp-1 were increased in Zfp36l1,
Zfp36l2, and dual knockdown cells (Figure 2A, 2B). In addition, the RNA and protein
levels of Cox-2 were increased in the cells which Zfp36l2 was knocked down (Figure
2A, 2D). The mRNA expression levels of Mkp-1 and Cox-2 were increased through the
mRNA stabilities when Zfp36l1 and Zfp36l2 were knocked down, which were
correlated with the typical function of Zfp36l1 and Zfp36l2 in mRNA destabilization
(Figure 2C, 2E).
Tnf-α is one of the well-known targets of TTP family proteins. Much to our
surprise, the mRNA expressions of Tnf-α were decreased in all knockdown cells (Figure
2F). This result may be due to the importance of the transcription regulation of Tnf-α
mRNA controlled by activation of p38 MAPK [35]. Therefore, the increase of Mkp-1
expression in Zfp36l1 and Zfp36l2 knockdown cells repressed the activity of p38
MAPK (Figure 6A), which down-regulated the Tnf-D mRNA expression. Similarly, the
mRNA expression of Ccl-2 is also activated by p38 MAPK [51]. However, the mRNA
expression of Ttp and Icam-1 were no significant differences whether Zfp36l1 and
Zfp36l2 were knockdown or not (Figure 2F). The possible explanation is that the
35
expressions of their mRNAs are controlled equally in both transcriptional and
post-transcriptional levels.
The mRNA targets of TTP family proteins are not all the same [52], although their
RNA binding domains are highly conserved. In Figure 2A and 2D, Cox-2 was the
Zfp36l2-specific mRNA target. However, the activity of luciferase containing Cox-2
3’UTR was down-regulated by both over-expressed Zfp36l1 and Zfp36l2 (Figure 3B).
This result indicates that both of Zfp36l1 and Zfp36l2 can recognize and destabilize the
Cox-2 mRNA in HEK 293T cells. However, overexpression of Zfp36l1 and Zfp36l2 in
HEK 293T cells may not reflect their “real” functions and the “real” conditions in
RAW264.7 cells. The RNA pull-down assay was shown that both of Zfp36l1 and
Zfp36l2 could associate with Cox-2 3’UTR in RAW264.7 cells (Figure 3C). Both of
Zfp36l1 and Zfp36l2 do not have enzyme activity of mRNA decay, so the difference of
their associated proteins may alter their functions. Mass spectrometry may be used to
identify the associated proteins of Zfp36l1 and Zfp36l2, and the detailed regulation can
be further studied. In conclusions, the mRNA expression of Cox-2 is Zfp36l2-specific
regulation in the post-transcriptional level.
The rapid mRNA activation of Mkp-1 after the LPS-stimulation was regulated
through mRNA stability (Figure 4A, 4B). In Figure 4C, the interaction between Zfp36l1
36
and Mkp-1 3’UTR was consistent during LPS-stimulation, but the interaction between
Zfp36l2 and Mkp-1 3’UTR was variable with the protein expression of Zfp36l2. This
result suggests that the decline of Zfp36l2 on Mkp-1 3’UTR stabilizes Mkp-1 mRNA.
Knockdown of Zfp36l1 and Zfp36l2 increased the mRNA expression of Mkp-1 through
RNA stability in the early stage of LPS-stimulation (Figure 5A, 5B). This result
confirms that the mRNA activation of Mkp-1 is regulated by Zfp36l1 and Zfp36l2 after
LPS-stimulation.
In addition, the activation of Mkp-1 is also regulated by MAPKs in both
transcriptional and post-translational levels. The activation of ERK signaling pathway
increases the transcription of Mkp-1 [53], and the activation of ERK and p38 MAPK
signal pathway can increase the protein stability of Mkp-1 by phosphorylation [54,55].
Associated proteins may change the functions of Zfp36l1 and Zfp36l2. We were
the first to observe that TTP family proteins, Zfp36l1 and Zfp36l2, could associate with
deadenylase Caf1a and degrade mRNA targets by removing poly-A tails (Figure 4C).
However, Zfp36l1 could be phosphorylated after LPS-stimulation and form the complex
with 14-3-3 (Figure 4D). This complex may repress the function of Zfp36l1 [26], and
Mkp-1 mRNA can be stabilized.
37
Knockdown of Zfp36l1 and Zfp36l2 increased the protein expression of Mkp-1
(Figure 6B), and the activity of p38 MAPK was down-regulated in the resting condition
(Figure 6A). Therefore, p-38 mediated mRNA expressions of Ttp and Tnf-α were
repressed (Figure 6C). According to the results, we proposed the following model for
the mechanism (Figure 7). In the resting condition, Zfp36l1 and Zfp36l2 destabilize
mRNA of Mkp-1, and the cells are sensitive to the stimuli such as LPS under the low
expression of Mkp-1. In LPS-stimulation, the induction of Mkp-1 mRNA is done by
hyper-phosphorylated Zfp36l1 which losing its ability of mRNA decay and decreasing
the expression of Zfp36l2.
A number of important ARE-containing genes are commonly involved in
inflammation and cancer [45]. Both MKP-1 and COX-2 have been reported playing
important roles in human cancers. MKP-1 is over-expressed in many human cancer cell
lines, including breast, lung, prostate, ovarian, pancreatic, liver, and gastric cancer [56],
and due to MKP-1 expression, the lung and ovarian cell lines are resistant to
chemotherapy such as cisplatin [57]. TTP mRNA and protein levels are significantly
decreased in tumors of the thyroid, lung, ovary, uterus, and breast compared to
non-transformed tissues [58,59]. Another study showed that the mRNA expression
levels of TTP family proteins were repressed in lung and ovarian cancers [60]. However,
38
rare studies clearly elucidate the expression and function of three TTP family members
in a cancer cell. Combining previous researches with our observations, the possible
explanation of MKP-1 overexpression is losing of ZFP36L1 and ZPL36L2 in cancer
cells, and the detailed mechanism can be further investigated.
The abnormal expression of COX-2 in breast cancer cells is reported in many
studies[61,62], and it may involve in tumorigenesis and angiogenesis in the breast
cancers [63,64]. The relationship between COX-2 and TTP is confirmed [65], but
whether the overexpression of COX-2 is regulated by ZFP36L2 in breast cancer cells is
unclear.
39
6. Figures
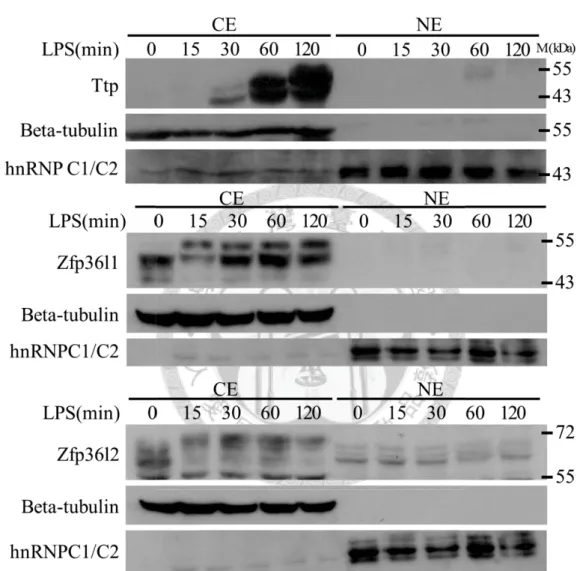
Figure 1.
The consistent expression and protein phosphorylation of Zfp36l1 and Zfp36l2 during the period of LPS-stimulation in RAW264.7 cells.A
40
B
C
41
Figure 1. The consistent expression and protein phosphorylation of Zfp36l1 and
Zfp36l2 in the period of LPS-stimulation in RAW264.7 cells.
(A) Protein expression profiles of TTP Family members in LPS-stimulated RAW264.7
cells for 0, 15, 30, 60, 120 minutes. Beta-tubulin was a positive control for the cytosolic
extract (CE), and hnRNPC1/C2 was a positive control for the nuclear extract (NE). (B)
mRNA expression profiles of TTP Family members in LPS-stimulated RAW264.7 cells
for 0, 15, 30, 60, 120 minutes. (C) The cytosolic extracts from LPS-stimulated
RAW264.7 cells for 120 minutes were treated with alkaline phosphatase (CIP) for 30
minutes. The data indicated both Zfp36l1 and Zfp36l2 were phosphorylated after
LPS-stimulation.
42
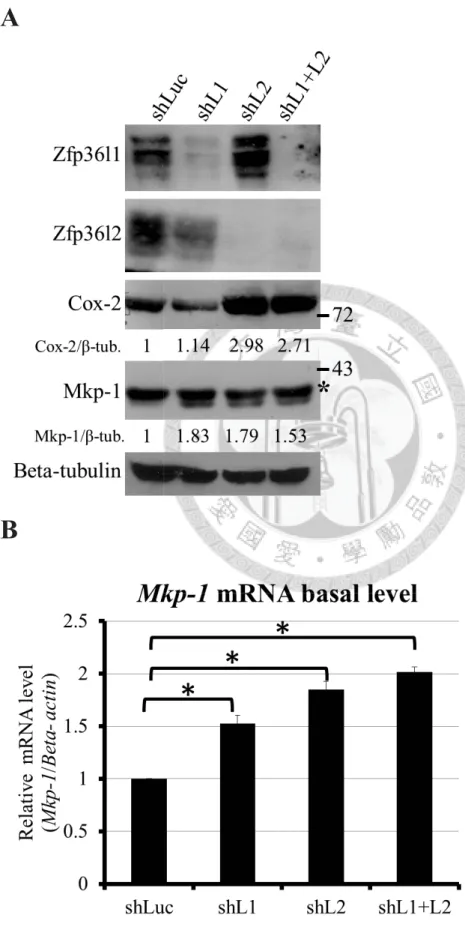
Figure 2.
Zfp36l1 and Zfp36l2 destabilize MAPK phosphotase-1 (Mkp-1) and cyclooxygenase-2 (Cox-2) mRNAs in resting RAW264.7 cells.A
B
43
C
D
44
E
F
Figure 2. Zfp36l1 and Zfp36l2 destabilize MAPK phosphotase-1 (Mkp-1) and
cyclooxygenase-2 (Cox-2) mRNAs in resting RAW264.7 cells.
(A) shLuc, shL1, shL2, shL1+L2 represented Luciferase knockdown cells, Zfp36l1
knockdown cells, Zfp36l2 knockdown cells, and dual Zfp36l1 and Zfp36l2 knockdown
45
cells, respectively. The upper two panels showed the efficiency of knockdown. The third
and the forth panels showed the protein levels of Mkp-1 and Cox-2 in different
knockdown cells. Beta-tubulin was a loading control. shLuc cells were used as reference
sample to calculate the relative expression which indicated in the panels. The asterisk
indicated the Mkp-2. (B) The basal mRNA levels of Mkp-1 (detected by quantitative
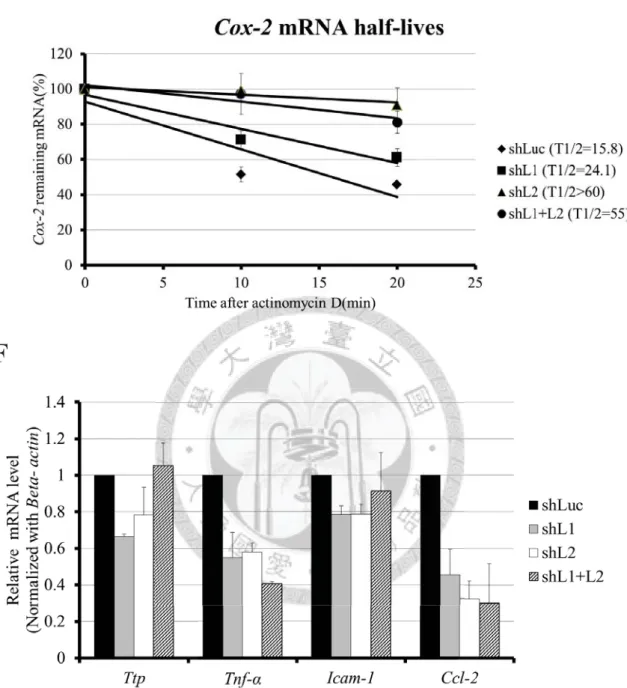
PCR) in different knockdown cells. The asterisk indicated p-value < 0.05. (C) The basal
mRNA levels of Cox-2 (detected by quantitative PCR) in different knockdown cells.
The asterisk indicated p-value < 0.05. (D) Analysis of Mkp-1 mRNA half-lives in
different knockdown cells. The 10 μg/mL of actinomycin D was added to stop
transcription for 0, 10, and 20 minutes. Remaining mRNA was detected by quantitative
PCR. Mkp-1 mRNA half-lives was calculated by linear regression, and the half-life of
shLuc, shL1, shL2, shL1+L2 were 15.2 minutes, 41.6 minutes, > 60 minutes, and > 60
minutes, respectively. (E) Analysis of Cox-2 mRNA half-lives in different knockdown
cells. Cox-2 mRNA half-lives were calculated by linear regression, and the half-lives of
shLuc, shL1, shL2, shL1+L2 were 15.8 minutes, 24.1 minutes, > 60 minutes, and 55
minutes, respectively. (F) The basal mRNA levels of Ttp, Tnf-α, Icam-1, and Ccl-2
(detected by quantitative PCR) in different knockdown cells.
46
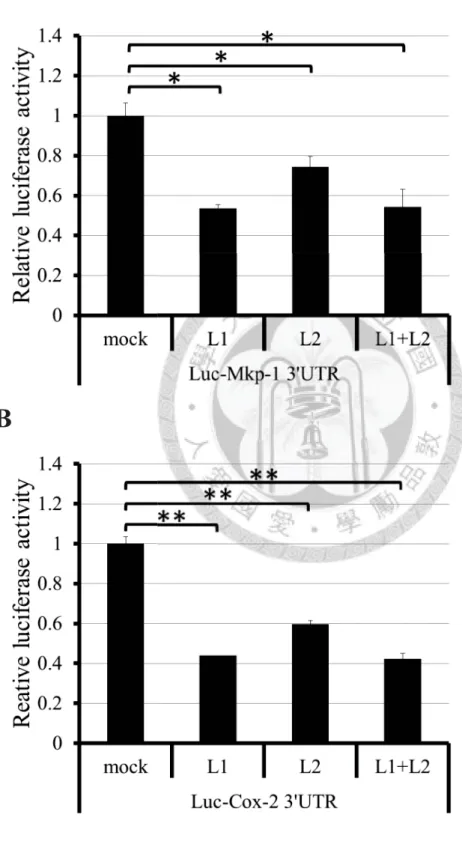
Figure 3.
Zfp36l1 and Zfp36l2 down-regulate the Mkp-1 and Cox-2 3’UTR -mediated luciferase reporter activity and interact with deadenylase Caf1a.A
B
47
C
D
Figure 3. Zfp36l1 and Zfp36l2 down-regulate the Mkp-1 and Cox-2 3’UTR
-mediated luciferase reporter activity and interact with deadenylase Caf1a.
(A)(B) Luciferase reporter assay. HEK293T cells were co-transfected with pCMV-
Flag-Zfp36l1 (L1) or pCMV-Flag-Zfp36l2 (L2) or both together (L1+L2) and pCMV-
Luciferase reporter construct containing Mkp-1 3’UTR or Cox-2 3’UTR. The firefly
48
luciferase reporter activities were normalized with relina luciferase activity to correct
for the transfection efficiency. The relative luciferase activity represents the luciferase
activity of reporter carrying 3’UTR normalized with that of reporter alone. One asterisk
indicated p-value < 0.05, and two asterisks indicated p-value < 0.01. (C) RNA
pull-down assay. Cox-2 3’UTR was separated into four fragments as indicated. Four
Biotinylated-Cox2 3’UTR fragments were incubated with cytosolic extracts from
RAW264.7 cells. The biotinylated RNAs and associated proteins were precipitated by
strepavidin beads. Biotinylated 18s rRNA was a negative control. The lower panel show
the schematics RNA probes of the Cox-2 3’ UTR and the dots indicate AREs. (D)
Co-immunoprecipitation. Flag-tagged TTP family proteins and Myc-tagged Caf1a were
overexpressed in HEK293T cells, and Flag-tagged TTP family proteins were
precipitated by anti-Flag M2 affinity agarose beads. RNase A and RNase T1 were added
to digest RNA. The Co-immunoprecipitated Myc-tagged Caf1a was detected by
anti-Myc. The interaction between TTP family proteins and Caf1a was
RNA-independent.
49
Figure 4.
Regulation of Mkp-1 mRNA stability by phosphorylation of Zfp36l1.A
B
50
C
D
Figure 4. Regulation of Mkp-1 mRNA stability by phosphorylation of Zfp36l1.
(A) mRNA expression profile of Mkp-1 in LPS-stimulated RAW264.7 cells for 0, 15, 30,
45, 60, 120minutes. (B) Analysis of Mkp-1 mRNA half-lives in LPS-stimulated
RAW264.7 cells for 0 minute and 25 minutes. The 10 μg/mL of actinomycin D was
added to stop transcription for 0, 10, and 20 minutes. Remaining mRNA was detected
by quantitative PCR. Mkp-1 mRNA half-lives was calculated by linear regression, and