國立臺灣大學生命科學學院生化科學研究所 碩士論文
Institute of Biochemical Sciences College of Life Science
National Taiwan University Master Thesis
CRISPR-Cas9 技術應用於粒線體基因編輯之優化設計 Repurposing CRISPR-Cas9 Technology for Mitochondrial
Genome Editing
馮聖富
Michael Sheng-Fu Feng
指導教授:凌嘉鴻 博士 Advisor: Steven Lin, Ph.D.
中華民國一
○八
年六月 June 2019謝辭
感謝楊維元老師與張壯榮老師擔任我的口試委員,並提出客觀建議以及修改方向。
感謝這兩年半以來指導教授凌嘉鴻老師的悉心指導,及實驗室同伴慧瑜學姊、婉君學姊、
George 學長、信安學長、日昇、俞安、欣如、Bella、Max 及幸蓉的陪伴,還有實驗上的
意見交流。雖然起初來到中研院生化所503 實驗室時懷著忐忑不安的心情,經過一個多月
的實驗室組會才終於下定決心加入,但在老師的帶領下,從剛開始實驗上經常出現問題,
一直到後期慢慢學會從問題中自己找出解決辦法,真的在這段日子當中成長很多。尤其在 許多個孤單一人進行實驗的夜晚,抑或面對不如預期的實驗結果而心情低落時,真心感謝 過程當中基督教福音宣教會鄭明析牧師所傳講的寶貴箴言,若這一路上沒有信仰的支持,
這一份碩班論文就不可能完成。最後感謝爸爸媽媽不僅在過程中不斷給予鼓勵,在經濟上 也總是毫不保留地予以支持,讓我可以毫無後顧之憂。妹妹棋棋這段日子也同在台北努力 打拼,雖然我們道路不同,但是看著彼此全力以赴的生活也間接地鼓舞著自己。
原本沒有預期要讀的碩士學位,回過頭卻發現是我神賜下最棒的禮物。這兩年半的 過程中,我學會更謙遜地看待過去的成就,也更感謝每一天生活當下所得到的一切祝福。
也因為這段時間的沉潛,讓我能戳破原本包覆在生活周圍的糖衣泡泡,更能夠看清生命中 最重要的事物與關係,也認清到自己能力不足的事實,而更加鞭策自己不論在實驗上抑或 想法上的層次。如果當初我直接前往美國攻讀博士班,恐怕會因為無法適應而跌入更深的 谷底,為此我真心感謝在碩士班這段時間當中所有願意教導我、讓我了解自己哪裡還做得 不好的人天使們。最後,感謝我神、聖靈、聖子與主透過福音生下了我,僅以此碩士論文
作為一切靈肉祝福的致謝。
中文摘要
粒線體對於維持細胞內的能量及動態恆定扮演極重要的角色,當粒線體基因受損時會 造成粒線體功能部分失活,甚至導致嚴重的遺傳性疾病,例如萊氏症(Leigh syndrome)。
目 前 雖 然 有許 多團 隊嘗 試 利 用 限 制 酶 或 人工 合 成的 DNA 內切 酶, 例如 :ZFN 或 TALEN,來針對突變的粒線體基因進行剪切剔除,但這些方法往往會受到選擇位點不
足、製作不易等缺點所限制。近年來迅速發展的基因編輯技術 CRISPR-Cas9 便可解決上
述兩項問題,目前除了三篇仍具有爭議的研究發表外,尚未有其他人提出相關研究成果或
重複其研究。因此,在此篇研究中,我們分別在Cas9 蛋白及嚮導 RNA (guide RNA)上進
行一系列不同粒線體標的序列(mitochondria-targeting sequence, MTS)的修飾。根據結果,
我們發現帶有MTHFD1L MTS 之 mito-Cas9 蛋白與僅帶有 5’端 10 個核苷酸延長序列之嚮
導 RNA 51 號,這兩者可以最有效率地進入粒線體。我們的結果可望運用 CRISPR-Cas9
技術來建立一套更加完善的粒線體基因編輯工具,未來將可應用於生物學研究與罕見粒線 體疾病之治療。
中文關鍵字
粒線體疾病、CRISPR-Cas9、基因治療、粒線體 RNA、PNPase
Abstract
The mitochondrial genome is responsible for the maintenance of the cellular energy source and homeostasis. Therefore, partial loss of mitochondrial functionality and even devastating diseases happen when the mitochondrial genome is damaged. Nowadays, several strategies like restriction enzymes, ZFNs and TALENs, have been developed to specifically eliminate mutated mitochondrial DNA. However, all methods aforementioned have their own limitations on either limited target site choice or laborious manufactural process. CRISPR-Cas9 is a soaring new DNA editing tool which has not been widely applied in mitochondrial genome engineering, except for three controversial papers published. Consequently, in this study, we aim to establish a more reliable and tractable platform by fusing both Cas9 protein and guide RNA with various mitochondria-targeting sequences (MTSs). Our data show that the mito-Cas9 with the MTS from mitochondrial monofunctional C1-tetrahydrofolate synthase (MTHFD1L) and the mito-guide RNA 51 with only 10-nucleotide extended form the 5’ end both have the best import efficiency into mitochondria. Our study provides a potential technique to edit mitochondrial genome through CRISPR-Cas9 gene tool and may help generate gene therapy for mitochondrial diseases.
Keywords
Mitochondrial genome editing, mitochondria-targeting Cas9, mitocondria RNA import, PNPase
目 錄
謝辭 ... 2
中文摘要 ... 3
Abstract ... 4
Introduction ... 8
1. Significance of mitochondria ... 8
2. Diseases caused by pathogenic mtDNA mutations ... 9
3. Current status in mitochondrial genome manipulation ... 10
4. CRISPR-Cas9 gene editing technology and its application on mtDNA ... 11
5. DNA double-strand break repair mechanism in mammalian mitochondria ... 13
6. Mitochondrial protein import machinery ... 14
7. Mitochondrial RNA import machinery ... 15
8. Specific aim of this study ... 16
Results ... 17
1. Folded protein cannot be transported into mitochondria ... 17
2. A novel MTS is able to import mito-Cas9 into mitochondria ... 18
3. Putative RNA transporter and its counterpart RNAs exist in HeLa mitochondria ... 20
4. Structures from nucleus-encoded mitochondrial RNAs maintain Cas9 cleavage ... 21
5. Mito-sgRNA with only linker on 5’ end performs the best import ... 22
6. Cellular abnormality observed after sorting of mito-Cas9-GFP-positive cells ... 23
Discussion ... 25
1. Previous research on mtDNA editing by CRISPR-Cas9 system ... 25
2. Cas9 can only enter mitochondria with the assistance of a novel MTS ... 26
3. All modifications on guide RNA show no complete impediment on Cas9 cleavage ability ... 27
4. Guide RNA with only linker shows unexpectedly high mitochondrial import rate ... 28
5. Low expression level of mito-Cas9 may hinder downstream experiments ... 29
6. Recommendation for future research ... 29
Materials and Methods ... 31
1. Bacterial strains, primers and plasmids ... 31
2. Competent cell preparation ... 31
3. Plasmid construction ... 32
4. Cell culture ... 35
5. Nucleofection ... 36
6. Immunofluorescence ... 36
7. Synthesis of RNA by T7 in vitro transcription ... 37
8. Calf intestinal alkaline phosphatase (CIP) treatment ... 40
9. In vitro cleavage assay ... 41
10. RNA transfection ... 42
11. Dynabead mitochondrial extraction ... 43
12. MACS mitochondrial extraction ... 44
13. RNA extraction ... 46
14. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) ... 46
Reference ... 98
圖 目 錄 Figure 1. Comparison between current methods for mtDNA manipulation ... 48
Figure 2. Schematic design for mito-EGFP transfection ... 49
Figure 3. The intracellular localization of mito-EGFP ... 50
Figure 4. Co-translational mechanism for mitochondrial protein import ... 51
Figure 5. Plasmid map for pSL127 series... 52
Figure 6. Schematic flow to determine localization of mito-Cas9s ... 53
Figure 7. Common MTSs are not enough for mito-Cas9 to enter mitochondria... 54
Figure 8. Candidate MTSs from large nuclear-encoded mitochondrial proteins on UniProt ... 55
Figure 9. Most candidate MTSs from UniProt are not suitable for mito-Cas9 ... 56
Figure 10. MTHFD1L MTS is suitable for mito-Cas9... 57
Figure 11. PNPase as a putative RNA transporter ... 58
Figure 12. PNPase exists in HeLa mitochondria ... 59
Figure 13. Schematic flow for mitochondrial RNA extraction ... 60
Figure 14. Calculation for mitochondrial translocation rates of PNPase-related RNAs ... 61
Figure 15. PNPase-related nuclear-encoded RNAs exist in HeLa mitochondria ... 62
Figure 16. Reported hairpin-shaped motifs for mitochondrial RNA transportation ... 63
Figure 17. Hypothesis for PNPase-mediated mitochondrial import of mito-sgRNA... 64
Figure 18. Cartoon illustration of predicted mito-sgRNA structures ... 65
Figure 19. Modifications on all mito-sgRNAs maintain Cas9 cleavage ... 66
Figure 20. Schematic flow to determine timing for mito-sgRNA transfection ... 67
Figure 21. Schematic flow for in vivo mito-sgRNA screening and calculation ... 68
Figure 22. Mito-sgRNA with only linker on 5’ end has the highest import efficiency ... 69
Figure 23. Schematic flow for expression of mito-Cas9 in vivo ... 70
Figure 25. Physiological anomaly occurs after transfection of mito-Cas9-GFP ... 72
Figure 26. Plasmid map for pSL296 ... 73
Figure 27. Plasmid map for pSL307 which has not been cloned yet ... 74
Figure 28. Hypothesized influence of PNPase expression on mitochondrial RNA import ... 75
表 目 錄 Table 1. Strains, plasmids and primers used in this study ... 76
Table 2. Humanized SpCas9 sequence and mitochondria-targeting sequences ... 81
Table 3. T7 in vitro transcribed RNAs (IVT RNAs) used in this study ... 86
Table 4. PCR condition and program setting for DNA template of IVT RNA ... 88
Table 5. Amount of Lipofectamine 2000 for transfection of different mito-sgRNAs ... 93
Table 6. Raw data for qPCR of mito-sgRNA screening ... 94
附 錄 Appendix 1. Setup flow of Urea-PAGE for RNA in vitro transcription ... 96
Appendix 2. Setup for arrangement of RT-qPCR loading ... 97
Introduction
1. Significance of mitochondria
Mitochondria serve as the powerhouse of cells. They not only produce energy, but also participate in several essential cellular processes, like calcium regulation, redox potential, cell cycle control, antiviral response and apoptosis1. According to endosymbiosis hypothesis, mitochondrion originated from a bacterium which was engulfed into a protoeukaryotic host cell during evolution2. Consequently, mitochondria possess a bacterium-like size and contain plasmid-like DNA. Through evolution, mitochondria have kept simplifying its own genome and integrating parts of mitochondrial genome (mtDNA) into the host nuclear genome, creating the so-called nuclear mitochondrial pseudogenes3. Human mtDNA is an intronless circular double- stranded DNA that harbors 16,569 base pairs. Each mtDNA encodes 37 essential genes, including 13 protein subunits, 22 tRNAs and 2 rRNAs4. Due to its crucial biological roles and concise genetic organization, mutations to mtDNA have high probability of disrupting the genetic information and affecting mitochondrial functionality. Single-nucleotide variations and deletions are the top two genomic changes within mtDNA that commonly result in mitochondrial diseases. For years, scientists have attempted to manipulate mtDNA by creating double-strand breaks (DSB) and introducing sequence changes via DNA repair process. Methods including mtDNA-targeting restriction enzymes, zinc-finger nucleases (ZFNs), and transcription activator- like effector nucleases (TALENs) have been applied to trigger DSB on mtDNA for depletion of pathogenic mtDNA5-7. Nevertheless, all the methods mentioned above have their own shortcomings. On the contrary, the utilization of CRISPR-Cas9 on mtDNA has yet been fully explored. The goal of this thesis is to engineer mitochondria-targeting CRISPR-Cas9 system
2. Diseases caused by pathogenic mtDNA mutations
Several mitochondrial diseases are caused by single-nucleotide mutations. The disease symptom varies depending on the cell type in which the mtDNA is mutated. In general, high energy-demanding organs, like brain, heart, and muscle, are usually the first ones to show defective mitochondrial phenotypes. Since mitochondrial phenotypic defects determined by mtDNA are highly correlated with each other, a certain disease symptom can be caused by more than one type of mutation. For instance, Leber hereditary optic neuropathy (LHON) is a common cause of inherited blindness in young males. Point mutations like m.3460G>A, m.11778G>A and m.14484T>C are three sites commonly associated with this syndrome8. On the contrary, point mutations such as m.8344A>G and m.8993T>G/C may lead to Leigh syndrome, a progressive neurodegenerative disorder with early-onset occurrence in infancy and childhood9,10.
However, not every individual who possesses mtDNA mutations succumbs to metabolic abnormality. A single human cell contains on average 1,000 copies of mtDNA11. Wild-type and mutated mtDNA usually co-exist within the same cell, a phenomenon known as mtDNA heteroplasmy12. Thus, it requires elevated ratio of pathogenic mtDNA mutation over the wild- type mtDNA to disrupt mitochondrial functions. Different tissues and diseases may have various thresholds, but the threshold for common mitochondrial defects falls around 80%13. Since human mitochondria lack efficient DSB repair, one strategy to shift the wild-type-to-mutant ratio is to target the mutant mtDNA for degradation and allow repopulation of wild-type mtDNA. Upon linearization, mtDNA is reported to be degraded spontaneously due to the lack of an efficient repair mechanism and undergoes a heteroplasmic shift5,14. A reduction in mutant mtDNA was
observed, followed by an increase in wild-type mtDNA15-17. Furthermore, instead of repairing the damaged mtDNA, cells also tend to induce mitophagy to destroy malfunctioning organelles18.
3. Current status in mitochondrial genome manipulation
In order to specifically trigger DSB on mutant mtDNA, methods including restriction enzyme (RE), zinc-finger nuclease (ZFN), and transcription activator-like effector nuclease (TALEN) have been developed5-7,19. Mitochondria-targeting SmaI and XmaI recognize and degrade m.8399T>G mutation, which is the cause of NARP (neuropathy, ataxia, and retinitis pigmentosa) and Leigh syndromes, to allow restoration of intracellular ATP level15,16,20. Mitochondria-targeting ApaLI induces heteroplasmic shift on well-characterized mouse model possessing two polymorphic mtDNA variants, NZB and BALB/c5,14. In general, mitochondria- targeting REs provide dramatic heteroplasmic shift and show no off-target effect on both mouse model and human culture cell lines20,21.5,15. However, among ~200 pathogenic mutation sites on mtDNA, only two can be targeted by the existing restriction enzymes17.
To overcome the target restriction of REs, programmable DNA endonucleases such as ZFN and TALEN technologies, have been engineered to target and cleave a broader range of mutant mtDNA. Both technologies utilize programmable DNA binding domains, either zinc finger DNA binding modules or transcription activator-like effector (TALE) DNA binding domains, to direct the dimerization of a non-specific nuclease FokI and trigger DNA breaks at the targeted mutation sites7,22-24. Some zinc finger DNA binding modules can recognize three nucleotides which makes up a codon and are possible to target all combinations of 64 codons25. For instance, mitochondria-targeting ZFNs (mito-ZFNs) have been reported to target a 12-nucleotide sequence and specifically remove mutant mtDNA which possesses only one single nucleotide variation
from the wild-type mtDNA26. Mito-ZFNs also demonstrate profound reduction of mtDNA and a heteroplasmic shift toward wild-type mtDNA in human cells by targeting m.8993T>G mutation associated with NARP and Leigh syndromes7. In comparison, one TALE DNA binding domain recognizes one single nucleotide, offering a higher flexibility on DNA targeting by rearranging four different TALE domains. In mouse model, mitochondria-targeting TALENs (mito-TALENs) can reduce successful alteration on mtDNA heteroplasmy ratio on NZB and BALB/c17. Mito- TALENs also demonstrate permanent elimination of both mtDNA harboring single nucleotide variation or common deletion in patient-derived cells6,27.
Nevertheless, DNA targeting by mito-ZFNs and mito-TALENs requires combinations and rearrangements of their DNA-binding domains. It is therefore more laborious to design a new pair of ZFN or TALEN. A more flexible and user-friendly programmable gene-editing tool for mtDNA manipulation is still highly desirable.
4. CRISPR-Cas9 gene editing technology and its application on mtDNA
The clustered regularly interspaced short palindromic repeats (CRISPR)-Cas (CRISPR- associated proteins) systems are adaptive immune systems found in most archaea and many bacteria. Directed by a CRISPR RNA molecule (crRNA), CRISPR-Cas nucleases are capable of sequence-specific recognition and cleavage of target DNA. This simple defense immunity is effective against invading foreign nucleic acids, such as DNA viruses and conjugative elements.
Currently, there are six types of CRISPR-Cas systems28. Types I, III and IV systems involve multiple protein subunits to perform functional cleavage of target nucleic acid. In contrast, in type II, V and VI systems, a single nuclease protein is responsible for the same purpose. Among all the systems, type II CRISPR-Cas9 is the best-characterized system. In CRISPR-Cas9 system,
Cas9 is the effector protein that is activated upon binding to two-RNA molecules: crRNA and a trans-activating crRNA (tracrRNA). These two RNA molecules can also be fused by a GAAA tetraloop to form a chimeric single guide RNA (sgRNA). Once Cas9 binds sgRNA, it undergoes conformational change and is able to recognize NGG PAM (protospacer adjacent motif) nearby the target DNA region. Using the sequence information provided by the sgRNA, Cas9 can be loaded onto the target DNA by RNA:DNA basepairing interactions and activate its two endonuclease domains, RuvC and HNH domains, to produce a blunt-ended double-strand break.
Since the establishment of this technique in 201229, CRISPR-Cas9 has emerged as a predominant genetic tool for manipulation of nuclear DNA in a wide variety of organisms. Since the DNA targeting activity is solely defined by the sgRNA sequence, Cas9 can be reprogramed to cleave nearly any sequence possible. Yet, despite of its extensive applications on nuclear genome, the usage of CRISPR-Cas9 on organellar DNA, such as mitochondrial or chloroplast, has not been fully explored.
To date, three published research articles have mentioned mtDNA editing by CRISPR-Cas9 system. In 2015, Jo and colleagues first reported CRISPR-Cas9-mediated mtDNA editing30. Unfortunately, their data was recently under debate in a review paper due to its lack of proof of the mechanism how the Cas9 with nucleus-leading sequence (NLS) and the sgRNA without any modification can localize into mitochondria spontaneously31. In addition, even though in the later part of their paper they engineered a modified Cas9 by adding a specific mitochondria-targeting sequence (MTS), we could not reproduce their results with the same Cas9 construct, suggesting that mitochondria-targeting Cas9 (mito-Cas9) might need a different MTS in order to enter mitochondria efficiently. In 2018, Loutre et al. introduced mitochondria-targeting Cas9 and mitochondria-targeting sgRNA to edit mtDNA in human culture cell line HepG2. However, their
results did not provide any evidence regarding the localization of the sgRNA within mitochondria32. Recently, Bian and his team also published their work using CRISPR-Cas9 technology to edit mtDNA both in human cell line and zebrafish embryo. Similarly, they did not provide solid proof for the mitochondrial localization of their sgRNA33. Therefore, my goal is to establish a more robust CRISPR-Cas9 platform for mtDNA manipulation by providing a measurable efficiency for transport of both Cas9 and sgRNA into mitochondrial matrix.
5. DNA double-strand break repair mechanism in mammalian mitochondria
After a successful colocalization of Cas9 and sgRNA within mitochondrial matrix, a targeted DNA double-strand break (DSB) is expected to occur. Following the DSB, whether the damaged mtDNA undergoes a repair process, like homology-directed repair (HDR) or nonhomologous end joining (NHEJ), is a crucial question. For mtDNA gene editing to occur, DSB repair pathway is needed. For instance, through electroporation of an active Cas9:sgRNA ribonucleoprotein complex (Cas9 RNP), scientists can generate a specific knock-out through introducing random insertion/deletion (indels) by NHEJ repair or a specific knock-in with an exogenous single-stranded DNA oligonucleotide template by HDR in nuclear genome34.
For eukaryotes, studies in yeast and Chlamydomonas reinhardtii confirm functional HDR pathways35,36. In contrast, recombination-mediated DNA repair seems to be an infrequent event in mammalian mitochondria37-39. Although in vitro joining of blunt-end DNA fragments was detected in the protein extract made from mammalian mitochondria40, DSBs by mitochondria- targeting restriction endonucleases generally led to large deletions or reduction in mtDNA copy- number39,41. These experiments suggest that mammalian mitochondria lack an efficient DSB repair mechanism and degradation of the damaged mtDNA seems a simpler solution due to high
abundance of mtDNA42,43. While nuclear genome faces a “repair or die” constraint after DSBs occur, mtDNA exists in hundreds to thousands of copies. Under normal cell physiology, a few copies of damaged mtDNA can undergo degradation without detrimental impact on mitochondrial functions. It may be more efficient to maintain mtDNA level by DNA replication than by importing and maintaining a complete set of the DNA repair machineries in mitochondria. With this in mind, it is more realistic to first focus on DSB-mediated elimination of targeted mtDNA through CRISPR-Cas9 DNA cleavage activity.
6. Mitochondrial protein import machinery
For a successful cleavage of target mtDNA to happen, an active Cas9 protein needs to be translocated into the mitochondrial matrix, where mtDNA locates. Although several essential mitochondrial proteins are encoded by mtDNA, the vast majority of the human mitochondrial proteins (about 1,500) are encoded by nuclear genome and translated as precursor proteins in the cytosol. These precursor proteins enter mitochondria through a series of mitochondrial protein import machineries. Generally, proteins are directed to their intramitochondrial destinations through four principle pathways: the β-barrel pathway into the outer membrane, the redox- regulated import pathway into the intermembrane space, the carrier protein pathway to the inner membrane, and the presequence pathway to the matrix and inner membrane44.
For all four pathways, the translocase of the outer membrane (TOM) serves as the common entrance for precursor proteins. To send Cas9 into mitochondrial matrix, we decided to explore the presequence pathway. Most of the precursor proteins possess mitochondria-targeting sequence (MTS) at their N-termini, which is recognized by TOM. MTSs are a group of peptides which usually consist of 15 to 50 amino acids and can form positively charged amphipathic α
helix. The hydrophobic and positively charged surfaces of the amphipathic α helix are recognized by TOM20 and TOM22, respectively45-47. The precursor proteins cross the outer membrane under the assistance of TOM20 and TOM22. In the intermembrane space, the precursor proteins are further recognized by the translocase of inner membrane 23 (TIM23).
Utilizing two different energy sources, the membrane potential generated through the electron- transfer chain and the adenosine triphosphate (ATP), TIM23 allows the transportation of the precursor proteins across the inner membrane and into the matrix47,48. Once the precursor proteins enter the mitochondrial matrix, their MTS is proteolytically cleaved by the mitochondrial processing peptidase (MPP) and folded into functional mature protein with the help of mitochondrial chaperons44. When constructing mitochondrial-targeting Cas9 (mito-Cas9), we need to take the proteolytic processing of MTS into consideration to avoid clipping off the RuvC nuclease domain at the N-terminal end of Cas9.
7. Mitochondrial RNA import machinery
Several nuclear-encoded non-coding RNAs are imported in human mitochondria49. These non-coding RNAs include 5S ribosomal RNA (5S rRNA), RNase P RNA, RNase MRP RNA, the RNA component of human telomerase (hTERC), a lncRNA called SAMMSON, and various microRNAs. It is also reported that several yeast tRNAs can be imported into human mitochondria50,51. The exact functions of these RNAs in mitochondria and the pathways through which these RNAs are imported from cytosol into mitochondria are still unclear49.
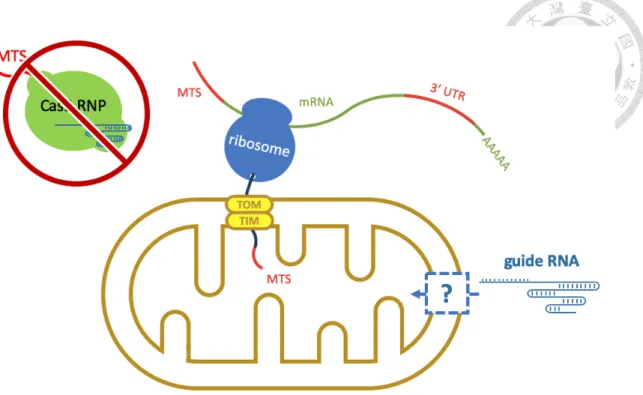
A putative mechanism of RNA translocation involves a multifunctional protein called polynucleotide phosphorylase (PNPase). Encoded by human PNPT1 gene, PNPase was originally recognized as an evolutionarily conserved 3’ to 5’ exonuclease from bacteria to
human52,53. Its function as poly(A) polymerase was also found in E. coli and plant chloroplast54,55. Human PNPase is a homotrimeric complex and locates mainly in the intermembrane space and partially in the matrix56. Besides its exonuclease activity, which may play an important role in the maintenance of mitochondrial RNA decay, PNPase was recently postulated to be a mitochondrial RNA transporter. Researchers found that the stem-loop structures on 5S rRNA, RNase P RNA and RNase MRP RNA might be recognized by PNPase and facilitate their import into mitochondrial matrix from the cytosol52. However, there are still many questions on how these nucleus-encoded RNAs cross the outer membrane and enter intermembrane space and how PNPase helps these RNAs cross the inner membrane and finally reach matrix52. I aim to explore the PNPase pathway and engineer mitochondria-targeting sgRNA (mito-sgRNA) by carrying the mitochondrial importing stem-loop structures.
8. Specific aim of this study
The overarching goal of this thesis is to repurpose CRISPR-Cas9 genome editing technology for mtDNA targeting. To achieve this, I seek to utilize the current knowledge of mitochondrial protein and RNA import to develop strategies to transport Cas9 protein and sgRNA into mitochondrial matrix where mtDNA is located. My specific aims are to: (1) identify mitochondria-targeting sequence capable of efficient localization of Cas9 into mitochondria; (2) engineer and test sgRNA carrying mitochondria-importing RNA stem-loops to enable import into mitochondrial matrix; (3) determine target mtDNA cleavage efficiency upon co-introduction of mito-Cas9 and mito-sgRNA. The methods and findings of this thesis will provide a foundation for CRISPR-Cas9-based mitochondrial genome editing in the future.
Results
1. Folded protein cannot be transported into mitochondria
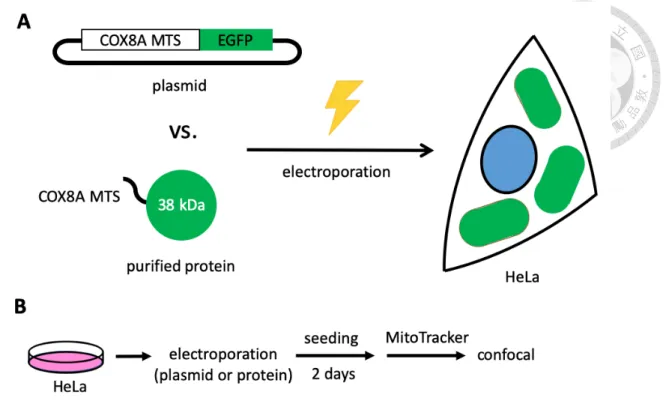
Electroporation of preassembled Cas9:sgRNA ribonucleoprotein (Cas9 RNP) into cells is a well-established protocol in our lab34. It has advantages, like shorter half-life time, lower toxicity, higher expression, and lower possibility of genome-integrating, when compared to vector-based methods. However, since a pure Cas9 protein is as large as about 150 kDa, we need to verify whether a mitochondria-targeting sequence (MTS) is able to import such a huge protein into mitochondria. To make the condition much simpler, we firstly used enhanced green fluorescence protein (EGFP) with double repeats of COX8A MTS (termed mito-EGFP; plasmid was kindly provided by Dr. Wei-Yuan Yang) to test if a folded small protein with MTS can get into mitochondria. If even a folded mito-EGFP cannot enter mitochondria, not to mention a folded Cas9 or Cas9 RNP. (Figure 2)
We expressed the plasmid in E. coli and purified mito-EGFP protein. We transfected either the plasmid or the purified protein forms of mito-EGFP into HeLa cells through nucleofection.
We observed a clear concentration effect of mito-EGFP within mitochondria in plasmid group (Figure 3A-C). The green fluorescent signal of mito-EGFP neatly overlaps the red signal of mitochondria, showing yellow signal in merged figure. By contrast, protein form of mito-EGFP showed no such phenomenon but spread all over the cell, even in the nucleus (Figure 3D-F). This data suggests that electroporation may produce temporary diffusion of desired particles within the whole cells, so both plasmids and proteins can localize into the nucleus. The plasmid can undergo transcription and produce mRNA which is further exported to the cytosol. During translation, the polypeptide generated from the mRNA can enter mitochondria with the help of
MTS. On the contrary, once electroporation finishes, purified proteins stuck in situ and cannot cross the organellar membranes.
Our results were consistent with other scientists’ findings that the translocation of proteins with MTS is coupled with translation (Figure 4)57. Namely, these nuclear-encoded mitochondrial proteins are co-translationally recognized by protein receptors on the mitochondrial outer membrane, like TOM20 and TOM22, and enter mitochondria as polypeptide chains58. The imported polypeptide may undergo different pathways and localize into the correct intramitochondrial site where it is folded into its mature form and perform normal function.
Consequently, a folded protein, such as purified mito-EGFP in this case, can no longer enter mitochondria. This result suggests that a preassembled Cas9:sgRNA RNP cannot be used for mitochondrial genome editing, but a plasmid-based method is needed. Furthermore, according to the mitochondrial protein import mechanisms, Cas9 needs to enter mitochondria as polypeptide through the assistance of mitochondria-targeting sequence. In that condition, unfolded Cas9 polypeptide can no longer hold its single guide RNA (sgRNA). As a result, we need to import Cas9 and sgRNA separately.
2. A novel MTS is able to import mito-Cas9 into mitochondria
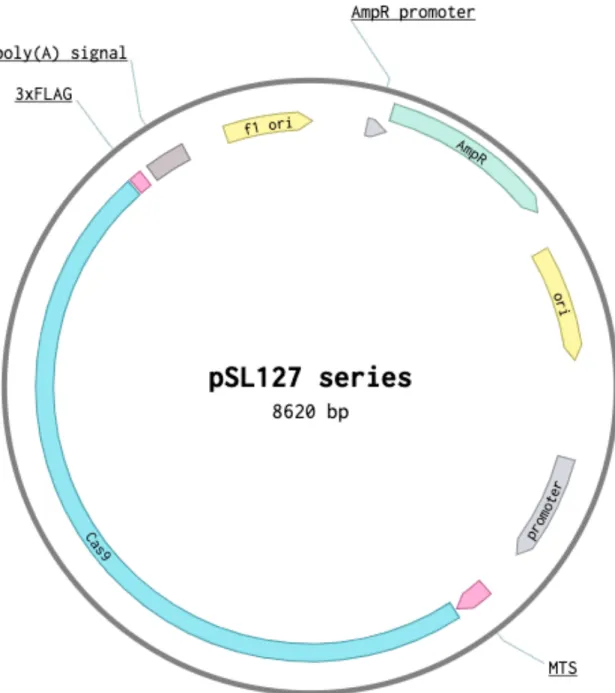
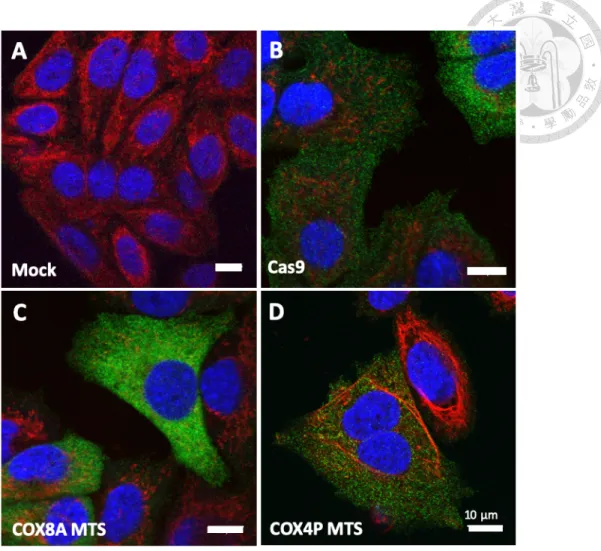
Firstly, we established a series of plasmids (pSL127 series, see Figure 5 and Table 1) to express Cas9 (without or with different MTSs) through electroporation (Figure 6). Since a pure Cas9 cannot localize into mitochondria (Figure 7B), in order to send Cas9 into mitochondrial matrix through transfection of plasmid, we need to seek a suitable mitochondria-targeting sequence (MTS). Although COX8A MTS is a well-known MTS to deliver small proteins, like EGFP (38 kDa), into mitochondria, it may be less efficient to carry a large protein, like Cas9
(~150 kDa)59. Controversially, a study from Jo and colleagues demonstrated that mito-Cas9 (mitochondria-targeting Cas9) bearing a COX8A MTS (1-21 amino acids of cytochrome c oxidase subunit 8A) can localize into mitochondria30. In contrast, according to our result, Cas9 with double repeats of COX8A MTS does not perform such localization within mitochondria (Figure 7C). Another well-studied MTS is from S. cerevisiae COX4P60 (1-24 amino acids of cytochrome c oxidase subunit 4 in Saccharomyces cerevisiae. see Table 2). Nevertheless, fusion of COX4P MTS does not help Cas9 enter mitochondria either (Figure 7D).
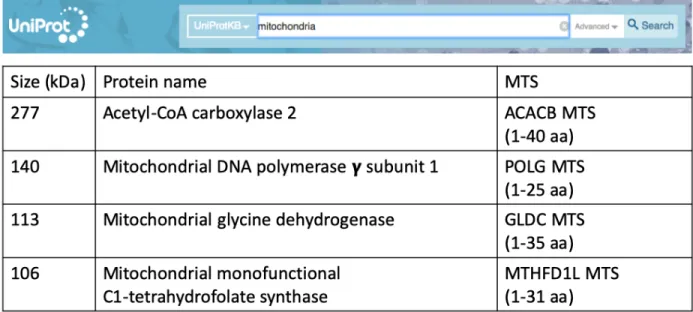
In order to find a more suitable MTS for directing mito-Cas9 into mitochondria, we searched on UniProt website for large mitochondrial proteins which are encoded by nucleus and later transported into mitochondria during translation (Figure 8). I chose the four largest mitochondrial proteins which are over 100 kDa on molecular mass and have been reported to be translocated into mitochondria after their mRNA transcribed from nuclear genome. Four candidate MTSs were chosen either by reported studies, by prediction of UniProt online software, or randomly from their N termini. Their lengths range from the first 25 to 40 nucleotides of their sequences (Table 2).
The largest protein I chose is acetyl-CoA carboxylase 2 (UniProt entry: O00763), which is almost 280 kDa and has been reported to localized into mitochondria after translation61. Its MTS which encodes the first 40 amino acids of acetyl-CoA carboxylase 2 was cloned into pSL127D and marked as ACACB MTS. The second protein is mitochondrial glycine dehydrogenase (UniProt entry: P23378), which is 113 kDa and has been reported to localized into mitochondria62. The first 35 amino acids of mitochondrial glycine dehydrogenase sequence were analyzed by manual assertion as an MTS, so I cloned their presequence into pSL127E and marked it as GLDC MTS. Next protein I chose is mitochondrial DNA polymerase subunit
gamma-1 (UniProt entry: P54098), which is 140 kDa and has been reported to be localized into mitochondria63. The presequence which encodes the first 38 amino acids of mitochondrial DNA polymerase subunit gamma-1 was cloned into pSL127F and marked as POLG MTS. Finally, the last protein is mitochondrial monofunctional C1-tetrahydrofolate synthase (UniProt entry:
Q6UB35), which is 106 kDa and has been reported to be localized into mitochondria64. The first 31 amino acids of mitochondrial monofunctional C1-tetrahydrofolate synthase have been confirmed to be its MTS by experiments65, and thus its presequence was cloned into pSL127G and marked as MTHFD1L MTS. After human codon optimization, Streptococcus pyogenes Cas9 sequence following aforementioned MTSs were cloned into plasmids (Table 1 and 2) and transfected into HeLa cells through nucleofection (Figure 5 and 6).
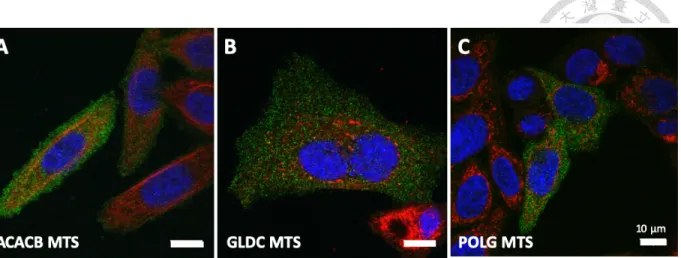
However, three out of the four MTSs do not have the ability to transport Cas9 across the mitochondrial double membrane (Figure 9). Fortunately, we found that the MTS from mitochondrial monofunctional C1-tetrahydrofolate synthase (MTHFD1L MTS) provided the best transport efficiency among all the six MTSs (Figure 10).
3. Putative RNA transporter and its counterpart RNAs exist in HeLa mitochondria However, ApoCas9 (protein alone) does not have DNA cleavage activity. It must be activated by binding to single guide RNA (sgRNA) in order to edit targeted DNA sequence.
After confirmation of mitochondrial localization of mito-Cas9 with MTHFD1L MTS, I further investigated a possible mitochondrial RNA import pathway for sgRNA. In contrast to the well- established study of mitochondrial protein import mechanisms, the mechanisms by which cells import nuclear-encoded RNAs into mitochondria are still under debate, leading to skepticism of applying CRIPSR technology onto mitochondrial genome31.
Despite the lack of research articles on mitochondrial RNA import mechanisms, there are still several groups who mentioned possible pathways nuclear-encoded RNAs may utilize to enter mitochondria50,66-68. In brief, a multifunctional protein called polynucleotide phosphorylase (PNPase) which mainly localizes in the mitochondrial intermembrane space and partially in the matrix was reported to be involved in the mitochondrial import of several nuclear-encoded non- coding RNAs, like 5S rRNA, RNase P RNA and RNase MRP RNA (Figure 11)66,67. When the gene encoding PNPase (PNPT1) was knocked out, these RNA transcripts lost the ability to enter mitochondria52. Although how this protein which mainly localizes in the intermembrane space can recognize RNAs in cytosol and which protein is also involved in this pathway are still unknown, PNPase seems to be the most possible candidate as a putative mediator for mitochondrial RNA pathway52,68.
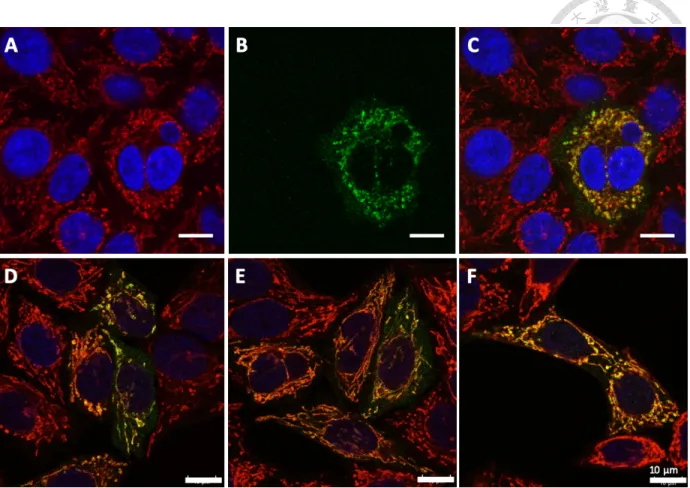
From our data, we confirmed that PNPase indeed localizes at mitochondria, in spite of the fact that we cannot determine its actual intramitochondrial localization due to the limited resolution of confocal microscope (Figure 12). Furthermore, we conducted mitochondria purification with RNase treatment to collect only the RNAs within mitochondria (Figure 13).
Through RT-qPCR, we confirmed that 5S rRNA, RNase P RNA and RNase MRP RNA all exist in the mitochondria of our cultured HeLa cell line (Figure 15).
4. Structures from nucleus-encoded mitochondrial RNAs maintain Cas9 cleavage With the perspective of utilizing PNPase as an RNA transporter for our sgRNAs, we cloned the secondary structures of 5S rRNA (5S), RNase P RNA (RP) and RNase MRP RNA (MRP) in front of our sgRNAs (see Figure 16 and Table 3 and 4). We also collected another two RNA hairpin structures, F1D1 and HD, which originate from tRNALys of yeast and had been reported
to have the ability to import RNA into human mitochondria(Table 3 and 4) 69,70. Finally, since the 3’ untranslated region (3’ UTR) of the mRNA of human mitochondria ribosomal protein S12 (MRPS12) had been studied to confer localization to mitochondrial outer membrane71, I added MRPS12 3’ UTR sequence at the 3’ end of several sgRNAs (Table 3 and 4). In total, 15 sgRNAs were synthesized through T7 in vitro transcription (Figure 18). Before we tested the mitochondrial localization efficiency of these modified sgRNAs, we wondered if the attached motifs may influence the DNA cleavage performance of Cas9. Thus, we conducted in vitro cleavage assay to determine Cas9 activity along with each sgRNAs (Figure 19). According to our result, compared to the original sgRNA36 and sgRNA51 which only possesses 10-nucleotide long linker, sgRNAs with modifications on either 5’ end only or both ends had impeded Cas9 performance (Figure 19C). On the other hand, modifications on the internal scaffold of sgRNA did not hinder Cas9 editing efficiency.
5. Mito-sgRNA with only linker on 5’ end performs the best import
We utilized commercial Lipofectamine 2000 reagent (Thermo Fisher Scientific) to transfect mito-sgRNAs. Each mito-sgRNA was firstly surrounded by Lipofectamine 2000 to form lipid micelles and crosses plasma membrane through either fusion of endocytosis (Figure 20). Within the cytosol, each mito-sgRNA enters mitochondria according to its modification. Before we screened the in vivo mitochondrial translocation efficiency of all the synthesized mito-sgRNAs, the first step is to determine the timing when the most amount of mito-sgRNA can accumulate within the cytosol through Lipofectamine 2000-mediated transfection. In this part, we used mito- sgRNA51 to serve as a representative for all the mito-sgRNAs generated in this study. After transfection with the indicated time period, total RNA was lysed by TRIzol reagent, purified
through commercial Direct-zol RNA recovery kit and finally analyzed by RT-qPCR targeting mito-sgRNA with primers listed in Table 1. As we expanded the transfection period from 3 hours to 9 hours, the amount of mito-sgRNA51 increased and peaked at 9 hours (Figure 20B).
Afterwards, the amount decreased as the transfection was expanded to 12 hours, suggesting cellular degradation of exogenous RNAs.
Afterwards, I conducted 9-hour transfections of all mito-sgRNAs generated throughout this study by Lipofectamine 2000 reagent to test whether modifications on sgRNA can enhance mitochondrial import. After transfection, a small portion of cells were collected and underwent TRIzol lysis to extract total RNA. The rest of transfected cells firstly went through mitochondrial purification using anti-TOM22-antibody coated nanoparticles and then RNase A/T1 treatment for 20 minutes before mitochondrial RNA recovery by TRIzol. The relative amount of each mito-sgRNA was compared to the endogenous mitochondrial 5S rRNA by the indicated formula (see Figure 21B). According to our data, compared to 5S rRNA, the abundance of mito- sgRNA51 is the highest among all the sgRNAs synthesized in this study (Figure 22 and Table 6).
6. Cellular abnormality observed after sorting of mito-Cas9-GFP-positive cells After identifying that mito-Cas9 possessing MTHFD1L MTS can efficiently localize to mitochondria and that mito-sgRNA51 has the best mitochondrial import rate, we aimed to co- transfect the two cargos through Lipofectamine reagents to determine whether our mito-Cas9 and mito-sgRNA can work together within mitochondria and trigger in vivo ND4 cleavage. However, since the expression level of mito-Cas9 was low by electroporation of pSL127G (Figure 10), we decided to enhance it by utilizing Lipofectamine-3000-mediated transfection of either pSL294, which expresses Cas9 fused with turboGFP, or pSL295, which expresses mito-Cas9 following
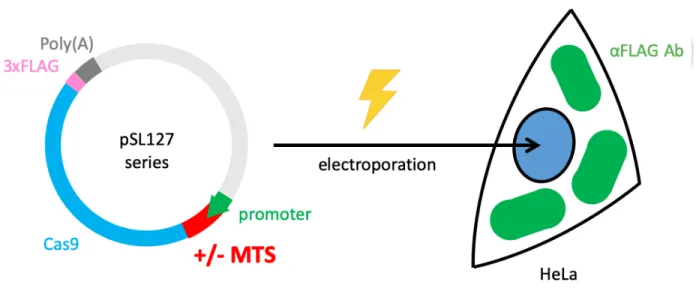
MTHFD1L MTS as well as turboGFP. 2 days after transfection, the turboGFP-positive cells were sorted out and seeded onto cultured dishes for another day. By immunofluorescence assay, we determined the localization of Cas9 or mito-Cas9 by anti-FLAG antibody (Figure 24).
According to our results, several Cas9 proteins without MTS were located at the cytosol. On the other hand, mito-Cas9 with MTHFD1L MTS showed localization within mitochondria as expected. However, when we observed with a broader field, we found that the cellular morphology in both groups appeared unhealthy and may undergo apoptosis at the timing (Figure 25).
Discussion
1. Previous research on mtDNA editing by CRISPR-Cas9 system
CRISPR-Cas9 genome editing technology is a robust system which has been widely used to manipulate nuclear DNA. However, not much attention has been drawn to mitochondrial or chloroplast DNA. So far, only three papers claimed a success by using CRISPR-Cas9 gene editing technology to cleave targeted mtDNA31,33,34.
In Jo’s report, the authors constructed lentiviral vectors which expressed either Cas9 with nuclear localization sequence (NLS) or mitochondria-targeting Cas9 (mito-Cas9) with COX8A MTS, and sgRNA. According to their immunofluorescence and Western blot results, Cas9 with NLS and mito-Cas9 can be partially or specifically localized into mitochondria, respectively.
What made their results less unconvincing was that their sgRNA was cloned onto the viral vector following U6 promoter. Since U6 promoter is coupled with RNA polymerase III system and is responsible for generation of short RNAs (usually between 20 and 30 nt) like short hairpin RNAs (shRNAs) and small interfering RNAs (siRNAs)72. After transcription by RNA polymerase III, unlike mRNA, these RNAs do not undergo nuclear export pathway and can only accumulate within the nucleus. Therefore, the authors’ claim for a successful mtDNA reduction by U6 promoter-generated sgRNA was unsolid without any proof of mitochondrial localization of these sgRNA.
In the report by Loutre et al., they used human HepG2 cells expressing Cas9 with COX8A MTS on the N terminus. For guide RNA, they generated various sgRNA fused with different hairpin structures to serve as import determinants by T7 in vitro transcription. Their sgRNAs were in vitro transcribed and transfected into HepG2 cells. Even though they provided Northern blot result to confirm the mitochondrial localization of their sgRNAs, they did not further offer
any detail about the purity of their mitochondrial fraction, leaving the quality of their subcellular fraction questionable.
Finally, Bian et al. used vectors to express mito-Cas9 which was flanked with 2 COX8A MTSs, one from human and one from zebrafish. On the same vector, mtDNA-targeting sgRNA can be synthesized in vivo through U6 promoter. Their microscopy results were too unclear to claim a colocalization of mito-Cas9 within mitochondria. Secondly, U6-generated sgRNA, as mentioned before, cannot be exported out of nucleus, thereby rendering its transport into mitochondria improbable. Finally, mitochondria do not possess a well-studied DNA import pathway, how the authors claimed a successful import of HEX-tagged ssDNA into mitochondria is controversial.
To sum up, these reports do not validify the localization of their single guide RNAs (sgRNAs). Without an MTS to transport sgRNAs from cytosol to mitochondria and any mean to prove its localization, the authors failed to prove that their sgRNA can truly localize into mitochondria and trigger the activity of Cas9. Consequently, their declaration of successful gRNA-mediated editing on mtDNA by Cas9 did not hold solidly. We were motivated to establish a novel system to co-localize both Cas9 and sgRNA into mitochondria and cleave the targeted mtDNA sequence.
2. Cas9 can only enter mitochondria with the assistance of a novel MTS
In this thesis, we tried to use electroporation to transfect both plasmid and purified EGFP protein with 2xCOX8A MTS, which is the same as the Korean group30, and saw if we can directly send folded protein into mitochondria through electroporation. Unfortunately, the answer was negative. Although cells transfected with plasmid can express EGFP signals in their
mitochondria, cells transfected with purified proteins express EGFP signals all over the cell, indicating that the MTS on EGFP cannot translocate folded protein into mitochondria. Since EGFP is much smaller than Cas9, we believe that transfection of purified Cas9 protein through electroporation would be harder and impracticable. Consequently, we tested several other MTSs collected from other known nucleus-encoded large mitochondrial proteins and saw if these MTSs have better ability to send our mito-Cas9 into mitochondria.
Among the six candidate sequences, the MTS from MTHFD1L (UniProt: Q6UB35) has shown the best localization ability. By mean of confocal images, we observed a clear concentration effect of this MTHFD1L MTS-Cas9 within mitochondria. However, the expression level of MTHFD1L MTS-Cas9 was too low through electroporation or lipofection of plasmids. Consequently, we decided to use lentivirus system to establish a HeLa cell line which is able to express higher level of MTHFD1L MTS-Cas9 within mitochondria. Again, through confocal imaging, we proved that virus-expressed MTHFD1L MTS-Cas9 as well has a concentration effect within mitochondria and the expression level is higher than its plasmid- expressed counterpart. Nevertheless, subcellular fractionation and Western blotting are needed to further prove that MTHFD1L MTS-Cas9 is able to specifically localize into mitochondria and not exist in nucleus, where lots of mitochondrial pseudogenes exist and may trigger off-target effect.
3. All modifications on guide RNA show no complete impediment on Cas9 cleavage ability On the other hand, concerning the activation of Cas9 after binding with sgRNA, we transfected artificially synthesized sgRNAs into Cas9-expressing HeLa cell lines. All the sgRNAs generated in this study have been designed to target a specific sequence site within ND4
gene on mtDNA. ND4 gene encodes the subunit 4 of NADH dehydrogenase which plays a crucial role on oxidative phosphorylation (OXPHOS). We designed a reverse transcription- (RT-) qPCR system to test both the mitochondrial internal control, mitochondrial 16S (M16S) rRNA, and our in vitro transcribed sgRNAs. The efficiency of all primer pairs for both internal control and for guide RNAs surpass 90% with R square around 0.95. In order to transport gRNAs into mitochondria, several MTSs for RNA were collected from reported studies (Figure 16). We transfected these modified sgRNAs with various combinations of 5’ or internal MTSs. Total RNA and RNA from mitochondrial subcellular fraction were extracted to monitor the increase/decrease of sgRNA expression through RT-qPCR.
4. Guide RNA with only linker shows unexpectedly high mitochondrial import rate
From our results, we found the best sg RNA is the one with modifications on both ends.
However, in order to further validate this result, other techniques for RNA localization are considered. For example, we hope to utilize three different state-of-the-art fluorescence in situ hybridization technique and confocal imaging to make sure gRNA localized into mitochondria.
The first one is DFBHI-induced fluorescence, which express fluorescent signal when chemical DFHBI recognizes and binds specific RNA secondary structure. We are designing four pairs of guide RNAs. Each one has different secondary structure for DFHBI recognition. Our preliminary data showed that guide RNA with BoBs sequence within the 1st hairpin area is able to express the greatest fluorescent signal. The second technique is branched DNA (from ACD), which can enlarge the gRNA signal through a series of annealing of complementary sequence. The last technique is rolling amplification, which is able to amplify the guide RNA signal through rolling
extension of targeted sequence. All in all, three of them will prove whether or not our sgRNA can specifically localize into mitochondria.
5. Low expression level of mito-Cas9 may hinder downstream experiments
Ultimately, we still wander the activity of mitoCas9 within mitochondria after its binding with sgRNA. As mentioned above, we have designed a sgRNA to target ND4 gene. Once Cas9 is combined with this sgRNA and thus activated, it should be capable of making a double-strand break within that specific region. As a result, the cleavage will induce the linearization of mitochondrial genome and potentially lead to a natural degradation of this linearized DNA.
Hypothetically, once the linearized mitochondrial genome is degraded, it exhibits a decrease in DNA level through qPCR. After we transfect in vitro transcribed sgRNAs through electroporation into cell lines which express mito-Cas9 within mitochondria, we extract DNA from whole cell lysate and conduct qPCR with mitochondria-specific primers. Expectantly, we anticipate to observe a decrease of expression level of targeted mitochondrial sequence.
Furthermore, in the future, we hope to use Seahorse machine to detect mitochondrial activity after transfection of sgRNA into Cas9-expressing cell lines. Since NADH dehydrogenase play a crucial role in electron transfer chain, once the ND4 gene is cleaved by activated Cas9, NADH dehydrogenase activity will drop and an impeded mitochondrial activity should be observed.
6. Recommendation for future research
Nevertheless, detailed mechanisms of translocation of mito-Cas9 and mito-sgRNA from cytosol into mitochondria are still largely unknown. A better understanding of these mechanisms may help to optimize the mitochondria-targeting sequences and enhance the translocation
efficiency. On the other hand, many single nucleotide mutations on mitochondrial genome can lead to different diseases. It is an urgent need to have a reprogrammable genome editing technique which is user-friendly and changeable to detect even a single nucleotide variable and cleave the targeted sequence. Also, precision and lower rate of off-target influence come from concentrating both Cas9 and guide RNA into mitochondria rather than nucleus. Through our data we have shown the best combination of mitochondria-targeting sequence for both Cas9 and guide RNA delivery into mitochondria.
Materials and Methods
1. Bacterial strains, primers and plasmids
All the E. coli strains used in this study are listed in Table 1. The E. coli strains 93 and 189 was a gift from Dr. Jennifer Doudna (UC Berkeley, USA). Homemade E. coli TOP10 and Stable3 chemically competent cells were prepared from these two strains as described in the following text. All E. coli strains were stored in 40% (v/v) glycerol as stocks at -80°C.
All the primers (Table 1) used in this study were synthesized by Integrated DNA Technologies (IDT, Singapore). After arrival, dried primer molecules were dissolved in 20 mM Tris-HCl (pH8.0) and stored in -20°C.
All the plasmids used in this study are listed in Table 1. The plasmid mito-EGFP was a gift from Dr. Wei-Yuan Yang (Academia Sinica, Taiwan). The plasmids pET28a and pAW006 were gifts from Dr. Jennifer Doudna (UC Berkeley, USA). All the plasmids generated in this work were extracted from the corresponding E. coli strains through ZR Plasmid MiniprepTM Classic kit (Zymo Research) according to the manufacturer’s instructions.
2. Competent cell preparation
Homemade TOP10 and Stable3 chemically competent E. coli strains (Table 1) were used as hosts for plasmid construction. To store stock competent cells, TSS buffer was prepared as follows: For 50 mL TSS buffer, 5 g PEG3350, 0.3 g MgCl2・6H2O, 2.5 mL DMSO and 1.25 g LB powder were mixed up with ddH2O until the final volume reaches 50 mL. Small amount of HCl was added in TSS buffer to adjust pH value to about 6.5. The TSS buffer was filtered through Millipore SteriflipⓇ (50 mL) and stored at 4°C before usage.
Strain 93 or 189 (for TOP10 and Stable3, respectively) was inoculated in 50 mL Terrific Broth (TB) and shaken at 30°C overnight. 500 µL cells were subcultured into 100 mL TB at 37°C until OD600nm reaches 0.5. Cells were transferred into a 50 mL tube and remained on ice for 10 min. Centrifuge the cells at 3,000 x g, 4°C for 10 minutes. Discard the supernatant and resuspend the cell pellet with 10 mL TSS buffer. The cells were distributed as 50 µL aliquots in each 1.5 mL Eppendorf tubes, immediately frozen by liquid nitrogen and finally stored at -80°C.
3. Plasmid construction
Plasmids used in this study are listed in Table 1. Restriction enzymes, like BamHI and BbsI, 10X T4 ligation buffer, T4 polynucleotide kinase (PNK), T4 ligase and HiFi DNA Assembly Master Mix for Gibson assembly were all bought from New England Biolabs and were used according to the manufacturer’s protocol. The constructed plasmid products were transformed into homemade TOP10 or Stable3 competent cells according to the protocol written below.
Plasmid was extracted from subcultured E. coli through ZR Plasmid Miniprep Classic (Zymo Research) according to the manufacturer’s protocol. The extracted plasmid underwent Sanger sequencing conducted by Academia Sinica Core Facility.
The plasmid pSL122 was constructed through Gibson assembly of two PCR amplicons. One was amplified from pET28a with primers SL410 and SL412. The other amplicon was amplified from plasmid mito-EGFP with primers SL411 and SL413. The Gibson assembly was conducted by mixing 1 µL HiFi DNA Assembly Master Mix and 0.5 µL of each amplicon and heated at 50°C for 1 hour. The product was transformed into homemade Stable3 competent cell.
The plasmid pSL127 was constructed through Gibson assembly of three PCR amplicons. All
SL435 and SL436, or SL437 and SL438. The Gibson assembly was conducted by mixing 1 µL HiFi DNA Assembly Master Mix and 0.33 µL of each amplicon and heated at 50°C for 1 hour.
The product was transformed into homemade Stable3 competent cell. The ligation was conducted by
The plasmids pSL127B to pSL127G were all constructed through Gibson assembly of two PCR amplicons. One of the two amplicons is the same for all the six plasmids and was amplified from pSL127 with primers SL448 and SL449. The other amplicons are different but were all amplified from a commercially synthesized gBlock gene fragment SL533 from IDT with various primer pairs. For pSL127B, primers SL456 and SL457 were used to amplify DNA segment of double repeats of COX8A MTS. For pSL127C, primers SL460 and SL461 were used to amplify DNA segment of S. cerevisiae COX4P MTS. For pSL127D, primers SL450 and SL451 were used to amplify DNA segment of ACACB optimized MTS. For pSL127E, primers SL454 and SL455 were used to amplify DNA segment of GLDC optimized MTS. For pSL127F, primers SL452 and SL453 were used to amplify DNA segment of POLG optimized MTS. For pSL127g, primers SL458 and SL459 were used to amplify DNA segment of MTHFD1L optimized MTS.
The Gibson assembly was conducted by mixing 1 µL HiFi DNA Assembly Master Mix and 0.5 µL of each amplicon and heated at 50°C for 1 hour. The product was transformed into homemade Stable3 competent cell.
The plasmids pSL127Ga, pSL127Gb and pSL127Ge were generated through “round-the- horn” technique. Firstly, pSL127G was amplified with primer pairs SL510 and SL509, SL512 and SL511, or SL601 and SL600, respectively. Next, the PCR amplicons were ligated to form the final product. The ligation mixture included 2.5 µL linear PCR product, 0.5 µL molecular ddH2O, 0.5 µL 1 mM ATP, 10x T4 ligation buffer, T4 PNK and T4 ligase. The mixture was
incubated at 37°C for 1 hour. The product was transformed into homemade Stable3 competent cell.
The plasmids pSL127Gc, pSL127Gd and pSL127Gf were generated through Gibson assembly from pSL127Ga, pSL127Gb and pSL127Ge, respectively. In general, pSL127Ga, pSL127Gb and pSL127Ge were all amplified with primer pairs SL563 and SL562 as backbones.
On the other hand, the insert sequence for the three constructs was the same one which was amplified from SL533 gBlock (IDT) with primer pairs SL551 and SL552. Finally, the Gibson assembly was conducted by mixing 1 µL HiFi DNA Assembly Master Mix, 0.5 µL backbone PCR product and 0.5 µL insert PCR product and heated at 50°C for 1 hour. The product was transformed into homemade Stable3 competent cell.
The plasmids pSL146, pSL147, pSL148, pSL149, pSL187 and pSL188 were generated through ligation of BbsI-digesed pSL127Ga, pSL127Gb, pSL127Gc, pSL127Gd, pSL127Ge and pSL127Gf, respectively, with annealed oligos. For pSL146 and pSL148, the annealed oligos was SL584 and SL585. For pSL147 and pSL149, the annealed oligos was SL586 and SL587. For pSL187 and pSL188, the annealed oligos was MF3 and MF4.
The plasmids pSL245 was generated through “round-the-horn” technique by amplifying pSL146 with primers MF44 and MF45. After mixing 2.5 µL linear PCR product, 0.5 µL molecular ddH2O, 0.5 µL 1 mM ATP, 0.5 µL 10x T4 ligation buffer, 0.5 µL T4 PNK and 0.5 µL T4 ligase, the mixture was incubated at 37°C for 1 hour. The product was transformed into homemade TOP10 competent cell. Similarly, the plasmid pSL297 was also generated through
“round-the-horn” technique by amplifying pSL245 with primers MF94 and MF95. The ligated product was transformed into homemade TOP10 competent cell as well.
The plasmids pSL246 to pSL249 and pSL298 were generated through “round-the-horn”
technique by amplifying pSL179 with various primer pairs. For pSL246, primers MF47 and MF48 were used. For pSL247, primers MF49 and MF50 were used. For pSL248, primers MF51 and MF52 were used. For pSL249, primers MF53 and MF54 were used. For pSL298, primers MF96 and MF97 were used. After mixing 2.5 µL linear PCR product, 0.5 µL molecular ddH2O, 0.5 µL 1 mM ATP, 0.5 µL 10x T4 ligation buffer, 0.5 µL T4 PNK and 0.5 µL T4 ligase, the mixture was incubated at 37°C for 1 hour. The product was transformed into homemade TOP10 competent cell.
4. Cell culture
HeLa cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% heated fetal bovine serum (FBS, 56°C for 30 minutes), 1X Antibiotic-Antimycotic (Thermo Fisher Scientific) and 1% HEPES (v/v) and incubated at 37°C in a humidified atmosphere of 5%
CO2/95% air. For each time cells were passaged, take a 10-cm dish for instance, cells reached about 80% confluency, were added 1 mL 0.25% trypsin and incubated at 37°C for 3 minutes.
After incubation, 9 mL DMEM was added to quench trypsin activity. Cells were then centrifuged at 300 x g for 3 minutes under room temperature and washed with Dulbecco's Phosphate Buffer Saline (DPBS) once. Cell amount was assessed by 0.2% trypan blue staining and CountessTM II automated cell counter (Thermo Fisher Scientific).
5. Nucleofection
To efficiently transfect plasmids or proteins, cells were nucleofected by 4D NucleofectorTM X Unit (Lonza) according the manufacturer’s instructions with modification written in the following.
For small scale nucleofection, 2x105 HeLa cells were mixed with 20 µL DPBS and indicated cargos (1 µg mito-EGFP plasmid or 500 µM purified mito-EGFP protein). The cells were transferred into 16-well 20 µL Nucleocuvette™ Strip and nucleofected with program CN-114.
After nucleofection, 130 µL DMEM was added into each well to resuspend the cells before the cells were transferred into the 35-mm glass bottom dish (#81158, ibidi) with 700 µL prewarmed DMEM at 37°C. The nucleofection well was washed again with 150 µL DMEM, which would be combined into the glass bottom dish.
After nucleofection, in order to reuse the Nucleocuvette Strip, it was firstly washed with a substantial amount of tap water to remove cells, and then was immersed in 20% ethanol. A day before the usage, the strip would be naturally dried out in a biosafety cabinet.
6. Immunofluorescence
2x105 HeLa cells were seeded on 35-mm glass bottom dishes (#81158, ibidi) in 1 mL DMEM. After 24 hours, the cells were washed once with 1 mL DPBS before incubated with 200 nM MitoTracker Deep Red (Invitrogen) in DPBS for 30 minutes at 37°C. After MitoTracker labeling, the cells were washed again with 1 mL DPBS and immersed in 1 mL DMEM. 8 hours after mitochondrial labeling, cells were taken out of the cell culture room. On bench, medium was removed and the cells were fixed with 1 mL 4% (v/v) paraformaldehyde (PFA)/PBS (pH 7.4) for 15 minutes before being washed for three times with 1 mL PBS. Next, cells were penetrated
for 15 minutes by 1 mL 0.1% (v/v) Triton X-100/PBS under room temperature, after which the cells were washed three times with 1 mL PBS.
After penetration, the cells were incubated with either rabbit anti-FLAG antibody (Protintech;
1:2,000 dilution in PBS) to locate Cas9 or rabbit anti-PNPT1 antibody (#ab96176, Abcam; 1:500 dilution in PBS) to locate PNPase at 4°C overnight with aluminum foil covering. After incubation, the cells were firstly washed with 1 mL PBST (PBS containing 0.1% Tween 20) for three times and finally once with 1 mL PBS before being incubated with goat-anti-rabbit IgG antibody which is conjugated with fluorochrome DyLightⓇ 488 (#ab96899, Abcam; 1:250 dilution in PBS) or goat-anti-rabbit IgG antibody with Alexa Fluor 546 (#A-11035, Thermo Fisher Scientific; 1:500 dilution in PBS) for 30 minutes under room temperature with aluminum foil covering. After removing the secondary antibody, the cells underwent nuclear staining by 300 nM DAPI (#D1306, Invitrogen) in PBS for 5 minutes under room temperature with aluminum foil covering. The samples were washed with three times of 1 mL PBST and finally immersed in 1 mL PBS. Images were obtained using confocal microscopy (Olympus FV1000 or Leica SP5 X inverted)
7. Synthesis of RNA by T7 in vitro transcription
The T7 in vitro transcribed RNAs (IVT RNAs) used in this study are listed in Table 2 and their secondary structures are schematically illustrated in Table 3. The DNA templates, including a T7 promoter, a 20-nucleotide target sequence and an optimized sgRNA scaffold73, were either assembled from synthetic oligonucleotides (IDT, Singapore) by overlapping PCR, or directly amplified through PCR of respective plasmids, using KAPA HiFi PCR kit (Kapa Biosystems,
USA) according to the manufacturer’s protocol. PCR conditions and program settings for each DNA template of sgRNA are listed in Table 4.
A day before IVT, 10% polyacrylamide gel containing 6 M urea (Urea-PAGE) should be prepared as following. The front and rear glasses are firstly cleaned by tap water supplemented with Aquet detergent (Scienceware) and Nuclease/EtBr terminator (Protech). After being wiped dry, the two glasses were separated by two plastic strips (0.3 cm thickness) on both sides and fixed with low adhesive yellow tape on the three edges (see Appendix 1A and B). To strengthen the fixation, a shorter piece of tape was used to adhere on the bottom edge (Appendix 1C).
A 500 mL Urea-PAGE premix solution was prepared ahead by mixing 210 g urea, 125 mL acrylamide/bis 29:1 (40%), 50 mL 5X TBE buffer and 165 mL ddH2O, and was stored at 4°C.
To prepare a slide of Urea-PAGE with 0.3-cm thickness, 80 mL Urea-PAGE premix solution is prewarmed in 37°C water bath before being mixed with 160 µL 20% ammonium sulfate (APS) and 80 µL TEMED. The mixture was immediately added into the mold by 25 mL autopipette.
Notice that when adding the mixture, the mold should be tilted to avoid bubbles (Appendix 1D).
Finally, the comb was carefully set onto the mold without forming any bubble. After about 2 hours under room temperature, Urea-PAGE was solidified and stored at 4°C. Before usage, the tapes were torn off from the glasses and the gel mold was set onto the OwlTM Dual-Gel Vertical Electrophoresis System (Thermo Fisher Scientific) (Appendix 1E). The wells were washed for several times by pipette P1000 to ensure the absence of remained gel debris within.
A 300 µL total volume of T7 in vitro transcription (IVT) reaction consists of 60 µL of 25 mM ribonucleotide triphosphate (rNTP) mixture, 30 µL of 10X IVT buffer (30 mM Tris–HCl [pH 8], 20 mM MgCl2, 0.01% Triton X-100, 2 mM spermidine), 30 µL of 50% (w/v) polyethylene glycol 3000 (PEG3000), 6 µL of 1 M dithiothreitol (DTT), 30 µL of 100 µg/mL